

Abstract
Francisella tularensis is a highly infectious bacterial pathogen that invades and replicates within numerous host cell types. After uptake, F. tularensis bacteria escape the phagosome, replicate within the cytosol, and suppress cytokine responses. However, the mechanisms employed by F. tularensis to thrive within host cells are mostly unknown. Potential F. tularensis mutants involved in host-pathogen interactions are typically discovered by negative selection screens for intracellular replication or virulence. Mutants that fulfill these criteria fall into two categories: mutants with intrinsic intracellular growth defects and mutants that fail to modify detrimental host cell processes. It is often difficult and time consuming to discriminate between these two possibilities. We devised a method to functionally trans-complement and thus identify mutants that fail to modify the host response. In this assay, host cells are consistently and reproducibly infected with two different F. tularensis strains by physically tethering the bacteria to antibody-coated beads. To examine the efficacy of this protocol, we tested phagosomal escape, cytokine suppression, and intracellular replication for F. tularensis ΔripA and ΔpdpC. ΔripA has an intracellular growth defect that is likely due to an intrinsic defect and fails to suppress IL-1β secretion. In the co-infection model, ΔripA was unable to replicate in the host cell when wild-type bacteria infected the same cell, but cytokine suppression was rescued. Therefore, ΔripA intracellular growth is due to an intrinsic bacterial defect while cytokine secretion results from a failed host-pathogen interaction. Likewise, ΔpdpC is deficient for phagosomal escape, intracellular survival and suppression of IL-1β secretion. Wild-type bacteria that entered through the same phagosome as ΔpdpC rescued all of these phenotypes, indicating that ΔpdpC failed to properly manipulate the host. In summary, functional trans-complementation using bead-bound bacteria co-infections is a method to rapidly identify mutants that fail to modify a host response. Francisella tularensis is a facultative intracellular bacterial pathogen and is the causative agent of the disease tularemia. F. tularensis enters host cells through phagocytosis, escapes the phagosome, and replicates in the host cell cytosol while suppressing cytokine secretion [1]–[4]. Although substantial progress has been made in understanding the intracellular life cycle of F. tularensis, the F. tularensis proteins responsible for manipulating many host cell pathways are unknown. Identifying novel host-pathogen effector proteins is difficult because there is no rapid method to reliably distinguish between bacterial proteins that modify host processes and proteins that are involved in bacterial processes that are required for the bacteria to survive or replicate in the intracellular environment. The ability to identify mutants that are deficient for host-pathogen interactions is important because it can aid in prioritizing the investigation of genes of interest and in downstream experimental design. Moreover, certain mutant phenotypes, such as decreased phagosomal escape, hinder investigation of other potential phenotypes. A method to specifically complement these phenotypes would allow for further characterizations of certain F. tularensis mutants. Thus we sought to develop a method to easily identify and functionally complement mutants that are deficient for interactions with the host.
Introduction
In order to distinguish whether a phenotype results from a host-pathogen interaction or an intrinsic bacterial defect, we devised a method to functionally complement and thus identify host-pathogen interactions in trans. Wild-type and a mutant strain were tethered to the same magnetic bead to ensure that both bacteria enter the same eukaryotic cell. Since cells are consistently infected with both strains of bacteria via the same phagosome, the wild-type bacteria functionally complement the host-pathogen interactions that the neighboring mutant strain fails to initiate. For example, a mutant deficient for phagosomal escape that co-infects a host cell with wild-type bacteria will escape the phagosome because the wild-type bacteria secrete the effectors required for phagosomal escape. Bacterial mutants that exhibit a phenotype caused by intrinsic deficiencies such as defective metabolite production or acquisition will not be functionally complemented by this method since intrinsic defects cannot be trans-complemented by neighboring bacteria.
To demonstrate the efficacy of this protocol, we functionally complemented cytokine suppression, phagosomal escape, and intracellular survival in F. tularensis subsp. holartica live vaccine strain (LVS) ΔpdpC and ΔripA. The pdpC gene is located in the Francisella pathogenicity island (FPI), which is proposed to encode a secretion and effector system that facilitates phagosomal escape [5]–[13]. PdpC contributes to phagosomal escape, intracellular survival and cytokine suppression [5], [13]. We therefore used ΔpdpC as a model of a mutant that contributes to a host-pathogen interaction.
ΔripA can escape the phagosome but is defective for intracellular growth and cytokine suppression [2], [14]. ΔripA replication is reduced in defined media at a pH of 7.5 compared to a pH of 6.5, which implies that cytosolic pH, rather than a host-pathogen interaction, is responsible for decreased intracellular proliferation [15]. Furthermore, RipA regulates the activity of LpxA, a protein required for F. tularensis lipid A synthesis (our unpublished data). The ΔripA strain does not proliferate within host cells, but ΔripA strains encoding lpxA suppressors are able to replicate within host cells (our unpublished data). These data imply that ΔripA fails to replicate inside of host cells due to irregular regulation of lipid A synthesis and therefore intracellular proliferation of this mutant should not be restored by co-infection with wild-type organisms.
Results
Two bacterial strains consistently bind to the same bead
Reliable functional complementation of mutants within infected cells requires that both the mutant and complementing strain consistently enter host cells together. To achieve this result we tethered two different F. tularensis strains to individual magnetic beads. To test if the two different strains consistently bound to the same bead, we combined anti- F. tularensis lipopolysaccharide (LPS) antibody coated beads with a 1∶1 mixture of F. tularensis expressing either GFP or DsRed. By microscopy, virtually every observed bead had GFP and DsRed bacteria bound to it (Figure 1 A, B, C). We quantified the amount of beads that bound to both GFP and CellTrace Far Red labeled F. tularensis by flow cytometry and found that 97.3+/−0.7% (mean +/− SD) of the beads bound to both GFP and CellTrace Far Red labeled bacteria (Figure 1 D, E, F). This indicates that in infection experiments where wild-type and mutant bacteria are bound to beads, the majority of cells will be infected with both bacterial strains.
Figure 1. Multiple F. tularensis bacteria reliably bind to the same bead.

Representative flourecence micrographs of beads bound to (A) GFP LVS, (B) DsRed LVS, or (C) a mixture of GFP LVS and DsRed LVS. All scale bars represent 2 µm. Representative flow cytometry histograms of three independent experiments depicting beads bound to (D) GFP LVS, (E) CellTrace Far Red labeled LVS, or (F) a one to one mixture of GFP LVS and CellTrace Far Red labelled LVS. Histograms were pregated on size to exclude aggregates and non-specific events. Quantification was compiled from all 3 experiments and represents the mean +/− the standard deviation.
Although this method requires multiple bacteria to be present on each bead, too many bacteria infecting the same cell may skew results or phenotypes compared to a normal infection. To quantify the number of bacteria per bead, F. tularensis LVS containing a luciferase plasmid was bound to beads and the amount of luminescence per bead was compared to a standard curve. The median bacteria per bead was 3.43 (SEM = 6.69, 4 independent experiments). Additionally, 6.66+/−2.52 intracellular wild-type bacteria per cell were present at 4 hours post inoculation (average +/− SEM, n = 23 from 9 independent experiments, assumptions described in materials and methods). Taken together, we estimate that the average cell is infected with 3 to 8 bacteria using bead co-infections.
F. tularensis is transiently linked to the bead
Tethering the bacteria to the same bead ensures that cells are co-infected with different strains, but irreversible binding of F. tularensis to the bead could affect the intracellular life cycle of F. tularensis. Thus, we took advantage of a binding mechanism that should allow the bacteria to detach from the bead over time by linking the bacteria to beads coated with antibodies to F. tularensis LPS. Each bacterium should initially link to the bead by binding to several anti-LPS antibodies, which should create a high avidity between the bead and the bacterium. The advantage of binding F. tularensis to the bead by an anti- LPS antibody is that Gram-negative bacteria, including F. tularensis, shed LPS. Thus, viable F. tularensis should detach from the bead over time. Indeed, the majority of bacteria were bound to beads immediately prior to infection as determine by flow cytometry (Figure 2A). Intracellular bacteria dissociated from beads within 2 hours during an infection of J774A.1 macrophage-like (J774) cells, presumably due to the bacteria shedding LPS (Figure 2A). Furthermore, microscopy of cells infected with GFP LVS bound to beads shows that some bacteria are spatially separated from the bead by 4 hours post inoculation (Figure 2B). These data demonstrate that the described methodology results in bacteria bound to beads upon initial infection, but that the bacteria detach from the bead following host cell entry. The bacteria also released from beads at similar rates when left in PBS for 2 hours, suggesting that bacterial release from the bead is not mediated by infection (data not shown).
Figure 2. F. tularensis separates from the beads inside host cells.
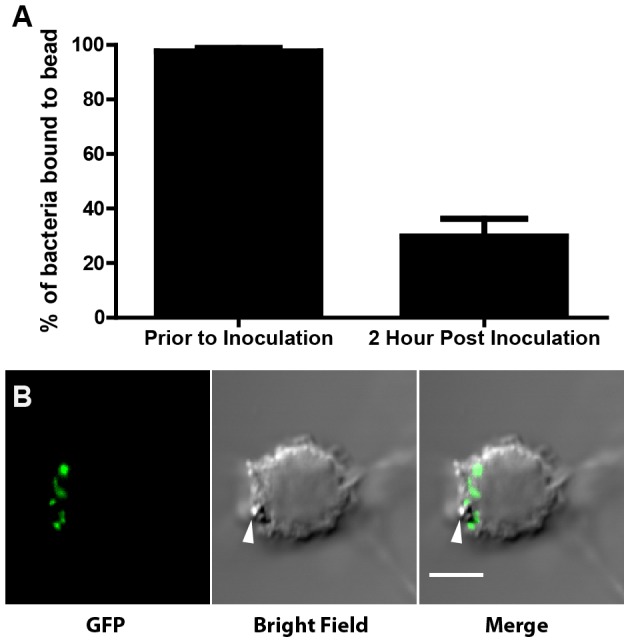
(A) Percentage of events representing bacteria that were bound to a bead prior to infection (n = 3) and the percentage of events representing intracellular bacteria bound to a bead 2 hours post inoculation (n = 9), as quantified by flow cytometry. Bar graph represents the mean +/− the standard deviation. (B) Representative fluorscence micrograph of a cell infected with beads bound to GFP (green) expressing LVS. The white arrow indicates the location of the bead. The image was taken 4 hours post inoculation. The scale bar represents 5 uM.
Bead co-infections functionally complement cytokine suppression
F. tularensis suppresses host cell production or secretion of several different cytokines, including interleukin 1β (IL-1β) by both active (such as MAPK inhibition) and passive mechanisms (such as via LPS modifications) [2], [4], [5], [16]–[18]. Since wild-type F. tularensis actively suppresses inflammatory responses, co-infection of cells with wild-type bacteria should complement a mutant that fails to suppress the immune response. To test the efficacy of bead co-infections on rescuing immune suppression, we co-infected murine bone marrow derived macrophages (mBMDM) with wild-type F. tularensis bound to beads with either ΔpdpC or ΔripA and measured IL-1β secretion.
Infections of mBMDMs with ΔripA results in increased IL-1β secretion compared to wild-type [2]. Similarly, cells infected with ΔripA bound to beads also induced increased IL-1β secretion compared to cells infected with bead-bound wild-type F. tularensis (Figure 3). However, co-infecting cells with wild-type bacteria and ΔripA resulted in reduced IL-1β secretion compared to ΔripA alone (Figure 3). ΔripA bound to beads and typical ΔripA infections elicit similar levels of IL-1β, even though the cells infected with beads should contain more bacteria initially (data not shown). Thus the decrease of IL-1β secretion during co-infections was not due to fewer ΔripA infecting each cell. Together, these data demonstrate that active immune suppression mechanisms expressed by wild-type F. tularensis was sufficient to inhibit the inflammatory response induced by ΔripA.
Figure 3. Functional trans-complementation via bead-bound bacteria complements suppression of cytokine secretion.
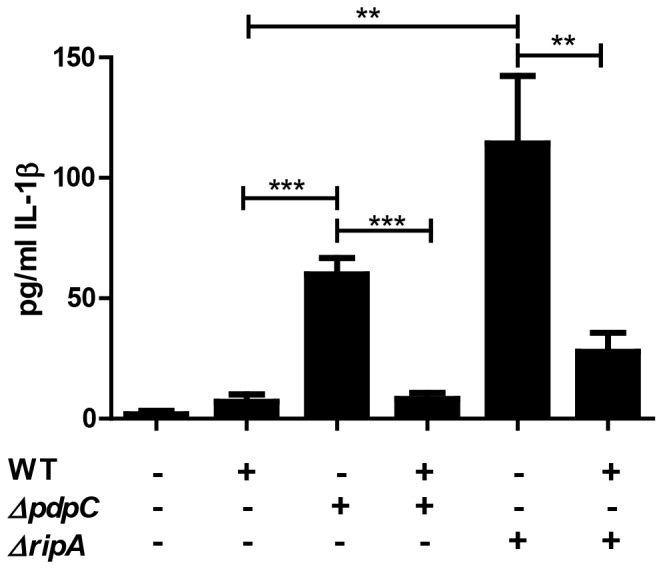
IL-1β ELISA of murine bone marrow derived macrophages that were inoculated with single or mixed inoculations of wild-type, ΔpdpC or ΔripA LVS (triplicates, n = 3). All samples were bound to beads prior to infection. Bar graph represents the mean +/− SEM. **p<.01, ***p<.001
We also measured IL-1β secretion of ΔpdpC infected mBMDMs and found that cells infected with ΔpdpC bound to beads also had slightly increased IL-1β secretion when compared to wild-type infected cells (Figure 3). Co-infecting cells with wild-type and ΔpdpC bacteria resulted in decreased IL-1β secretion compared to ΔpdpC bacteria alone (Figure 3). Thus, ΔpdpC failed to entirely suppress the host immune response but suppression could be rescued by the presence of wild-type bacteria. Wild-type bacteria fully complemented suppression of IL-1β secretion during mixed infections of wild-type and ΔpdpC (p = 0.77) but there was a slight increase in the wild-type and ΔripA mixed infection when compared to wild-type infections alone (p = 0.01). We hypothesize that the observed increase in IL-1β secretion during co-infections with ΔripA is due to a small subset of cells that are infected by beads bound only to ΔripA bacteria combined with the increased magnitude of IL-1β secretion observed during ΔripA infections compared to ΔpdpC infections. However, we cannot rule out the possibility that ΔripA may stimulate the immune response via a pathway that cannot be entirely suppressed by wild-type bacteria. In summary, co-infections of mutant and wild-type bacteria can rescue a phenotype caused by the inability of the mutant to properly modify the host cell.
Phagosomal escape is functionally complemented during bead co-infections
Several FPI mutants, including ΔpdpC, as well as killed F. tularensis do not effectively escape the phagosome [5]–[13]. Therefore, F. tularensis phagosomal escape is a F. tularensis mediated process. As a result, wild-type bacteria should facilitate release of escape-defective mutants so long as the mutant and wild-type bacteria are within the same phagosome. To test this hypothesis, we bound ΔpdpC containing a GFP-expressing plasmid (GFPΔpdpC) to beads and quantified the amount of GFPΔpdpC present in the cytosol of J774 cells. We found that GFPΔpdpC had reduced phagosomal escape at 2 hours post inoculation compared to GFP expressing wild-type bacteria and GFPΔpdpC phagosomal escape was rescued by the presence of wild-type bacteria within the same phagosome (Figure 4). This result is consistent with other published analyses of ΔpdpC mutant phenotypes and indicates that PdpC contributes to, but is not absolutely required for, F. tularensis LVS phagosomal escape [5], [13]. More importantly, wild-type bacteria export the necessary effector protein(s) to allow both wild-type and ΔpdpC to escape the phagosome.
Figure 4. Functional trans-complementation via bead-bound bacteria complements phagosomal escape.

J774 cells were inoculated with either GFP wild-type LVS or mixtures of GFP ΔpdpC, wild-type LVS, or ΔripA (n = 4 independent experiments). The amount of cytosolic GFP positive bacteria was quantified by flow cytometry and normalized based on the amount of permeabilized cells, as determine by calnexin staining controls. All samples were bound to beads prior to infection. Data includes bacteria attached and detached from beads in the same sample. Bar graph represents the mean +/− SEM. * p<.05, **p<.01
Bead co-infections achieve functional complementation of phagosomal escape across an entire population. This allows for the further characterization of phagosomal escape deficient mutants in the cytoplasmic environment. One caveat to using wild-type bacteria for complementation is that wild-type bacteria may out-compete certain mutants in the cytosol or obscure additional host-pathogen interactions. We hypothesized that any mutant strain that escapes the phagosome can be used to complement phagosomal escape of escape deficient mutants. To test this hypothesis, we co-infected cells with GFPΔpdpC and ΔripA. The ΔripA strain escapes the phagosome with similar kinetics as wild-type bacteria, but does not replicate inside the host cell [14]. Indeed, ΔripA functionally complemented GFPΔpdpC phagosomal escape when GFPΔpdpC entered through the same phagosome as ΔripA (Figure 4). Our data demonstrate that infecting cells with ΔpdpC and a phagosome escape competent F. tularensis bacterium results in phagosomal escape of both bacteria. Thus, pairing a phagosomal escape deficient mutant with an escape competent mutant will allow for further characterization of cytoplasmic phenotypes associated with the phagosomal escape deficient mutant.
Bead co-infections complement intracellular survival
F. tularensis mutants can be deficient for intracellular survival or replication due to the inability of the mutant to perform a required interaction with the host or due to an intrinsic intracellular survival or replication defect. Co-infections with both mutant and wild-type bacteria can be used to determine whether the mutant fails to properly control a host-pathogen interaction
The ripA gene encodes a hypothetical protein of unknown function that is required for F. tularensis intracellular proliferation, likely through regulation of lipid A synthesis [14] (our unpublished data). We found that co-infecting J774 cells infected with wild-type F. tularensis did not complement ΔripA intracellular growth (Figure 5A). Likewise, individual cells had similar numbers of ΔripA expressing GFP regardless of whether or not wild-type F. tularensis was present in the same cell (Figure 5D, 5E). These data further indicate that ripA is involved in an intrinsic bacterial process essential for intracellular bacterial proliferation.
Figure 5. Functional trans-complementation via bead-bound bacteria complements intracellular proliferation of ΔpdpC but not ΔripA.

(A) Kanamycin resistant wild-type LVS or hygromycin resistant ΔripA were individually or co-inoculated into J774 cells and assayed for intracellular proliferation at 4 and 22 hours post inoculation. (B) Kanamycin resistant wild-type LVS or hygromycin resistant ΔpdpC were individually or co-inoculated into J774 cells and assayed for intracellular proliferation at 4 and 22 hours post inoculation. (C) Kanamycin resistant wild-type LVS, kanamycin resistant ΔripA or hygromycin resistant ΔpdpC were individually or co-inoculated into J774 cells and assayed for intracellular proliferation at 4 and 22 hours post inoculation. Results are from 3 independent experiments performed in duplicate or triplicate. Bar graphs represent the mean +/− the standard deviation. (D) Representative fluorescence micrographs of J774 cells inoculated for 22 hours with GFP wild-type, GFPΔpdpC or GFPΔripA bacteria attached to beads. (E) Representative fluorescence micrographs of J774 cells inoculated with beads bound to DsRed LVS and either GFP WT, GFPΔpdpC or GFPΔripA. Blue represents the nucleus (DAPI), green represents the indicated GFP LVS mutants, red represents DsRed wild-type LVS, and white represents the plasma membrane stain wheat germ agglutinin (WGA). All scale bars represent 10 µm. All samples were bound to beads prior to infection. Not significant (ns), p>.05, * p<0.05, ***p<0.005.
The PdpC protein is encoded on the FPI and ΔpdpC bacteria are deficient for intracellular proliferation [5], [13]. Intracellular proliferation of ΔpdpC is not consistent across the entire population, as this strain replicates to high numbers in a small subset of cells [13]. Since the FPI is proposed to encode a secretion system, the failure of FPI gene deletion strains to replicate in the host cell is likely due to the inability to initiate host-pathogen interactions [19]. Thus ΔpdpC replication may be rescued by the presence of wild-type bacteria. Consistent with previous reports, the amount of viable intracellular LVS ΔpdpC bacteria decreased over time (Figure 5B) [5], [13]. On average, the number of ΔpdpC organisms decreased over 2 orders of magnitude between 4 and 22 hours post inoculation (9 independent experiments). However, when J774 cells were co-infected with a mixture of wild-type and ΔpdpC bound to beads, ΔpdpC survival was functionally complemented by the wild-type bacteria (Figure 5B). Specifically, we observed a slight increase in ΔpdpC replication during co-infections with wild type between 4 and 22 hours (2.0+/−0.3 fold, n = 7 independent experiments, p = 0.0014) (Figure 5B, 5D, 5E). We conclude that wild-type bacteria complemented the defect of ΔpdpC by secreting the effector(s) necessary to manipulate the host cell into being permissive for F. tularensis survival. Thus, F. tularensis LVS ΔpdpC does not survive inside of J774 cells due to its inability to properly manipulate an interaction(s) with the host that F. tularensis requires for intracellular survival.
Although ΔpdpC replicated slightly when wild-type bacteria were present in the same cell, ΔpdpC replication did not achieve wild-type levels. This is interesting because ΔpdpC escaped the phagosome, albeit at lower levels than wild-type, but was still inhibited by the host cell during individual infections (Figure 4, 5B) [5], [13]. Thus, wild-type bacteria may promote ΔpdpC survival in the host cytosol (Figure 5B) but wild-type F. tularensis either out-competes ΔpdpC or pdpC is also required for a cell intrinsic process to fully restore intracellular replication. To distinguish between these possibilities, we co-infected cells with ΔpdpC and ΔripA. The ΔripA strain has an intact FPI and rescues ΔpdpC phagosmal escape, but since ΔripA does not replicate, it should not out-compete ΔpdpC. We found that co-infections with ΔripA and ΔpdpC resulted in ΔpdpC survival but proliferation did not increase to wild-type levels (Figure 5C). Thus, we conclude that PdpC is required for intracellular survival through a host-mediated process but has a secondary function that enhances bacterial proliferation.
Together, the ΔpdpC and ΔripA intracellular replication data demonstrate that co-infecting cells with wild-type and mutant bacteria can differentiate between mutants that fail to replicate due to defective host-pathogen interactions and mutants that are defective for growth due to intrinsic replication defects.
Discussion
We developed and optimized a method to reliably deliver two distinct F. tularensis bacteria into the same host cell. Co-infection of cells with bacteria bound to beads allows for functional complementation of host-pathogen interactions, which can be used to identify mutants that fail to induce a host-pathogen interaction or to complement a specific host-pathogen interaction.
The ΔripA strain fails to replicate inside of cells and induces IL-1β secretion [2], [14], [15]. With the described co-infection method, neighboring wild-type bacteria functionally complement IL-1β suppression in ΔripA infected cells but not ΔripA intracellular proliferation. The Francisella novicida ΔripA strain has also been shown to have increased intracellular lysis compared to wild-type F. novicida bacteria, which leads to increased cytokine secretion via the AIM2 inflammasome [20]. Herein we show that wild-type F. tularensis LVS bacteria can suppress cytokine secretion in cells infected with ΔripA bacteria. Assuming that RipA has an identical function between the different Francisella species, these data indicate that wild-type LVS bacteria, but not ΔripA, suppress the AIM2 inflammasome.
Interestingly, wild-type F. tularensis bacteria replicated in cells that contained ΔripA bacteria, which indicates that the cell cytosol remains permissive for F. tularensis replication and that the proliferation defect is specific to ΔripA bacteria. These data also indicate that increased cytokine secretion and intracellular proliferation phenotypes seen in ΔripA infections occur through different mechanisms because one phenotype is functionally complemented while the other is not. Additionally, we were able to genetically distinguish between ΔripA cytokine suppression and intracellular replication. A chromosomal ripA N21A point mutant of LVS proliferated in the cytosol at a rate comparable to wild-type bacteria, but failed to suppress IL-1β secretion (data not shown). Taken together, the described co-infection method indicates that RipA is involved in cytokine suppression (a host-pathogen interaction) and a separate, intrinsic bacterial process for intracellular replication.
The ΔpdpC strain was functionally complemented for phagosomal escape, intracellular survival, and IL-1β suppression. Thus, PdpC contributes to those host-pathogen interactions. Interestingly, ΔpdpC replication never reached the same level as wild-type bacteria during co-infections even though ΔpdpC escaped the initial phagosome with the wild-type bacteria. These data hint that PdpC is involved in an intrinsic bacterial replication process that is required for optimal growth. It is also possible that a local host-pathogen interaction is required that is not reliably complemented by neighboring bacteria. Further investigation is needed to determine if the intracellular growth defect is specific to ΔpdpC or if other FPI genes are required for replication in the host cell cytosol.
The antibody used throughout these experiments has been used to identify F. tularensis Schu S4 by microscopy and for purifying Schu S4 from infected cell lysates [21], [22]. As described, this method should be compatible with a range of F. tularensis species with O-antigen structures similar to F. tularensis LVS. More importantly, the described co-infection method is compatible with any biotinylated antibody, so antibodies specific to surface molecules of F. tularensis novicida or other bacterial species can be used to link bacterial cells to beads.
Binding bacteria to beads does alter some aspects of infection that must be taken into consideration when designing experiments. The beads are dense and sink to the bottom of the well, allowing for a substantially lower multiplicity of infection (MOI) while maintaining a high infection frequency. More bacteria are phagocytosed per cell when bacteria are bound to beads compared to a typical infection. This may require time points to be taken earlier than a typical infection and the magnitude of certain phenotypes, particularly intracellular proliferation, are slightly different. Lastly, we expect that phagocytosis of beads coated in bacteria will primarily occur through typical phagocytic routes of a given bacteria because bacteria on the bead surface still interact with the cell. But phagocytosis may occasionally occur through a different mechanism than in a typical infection. For example, some Fc receptor mediated phagocytosis likely occurs in some cells due to antibodies bound to the beads. Despite these potential pitfalls, our individual infection controls for IL-1β secretion, phagosomal escape, and intracellular proliferation resulting from infection with bead-bound bacteria are consistent with previously published data based on infection by free bacterial cells [2], [5], [13], [14].
Bead co-infections provide a consistent method of functionally complementing most intracellular bacterial manipulations of the host cell. However, there are a few specialized cases in which further characterization beyond bead co-infections is necessary. We expect that some gain of function mutants and host-pathogen interactions that require local manipulation will not be reliably complemented. For example, immune signaling phenotypes that are not complemented by wild-type bacteria are likely gain of function mutations because the mutants stimulate the immune system in a manner that wild-type bacteria cannot suppress. Likewise, xenophagy evasion and re-entry into Francisella containing vacuoles likely requires local host-pathogen interactions. As a result, a lack of complementation for mutants that are strongly suspected of manipulating the host cell can also narrow down potential functions for a given protein. Altogether, bead co-infections are a reliable method to gain insight into the function of a gene of interest, identify mutants that fail to initiate a host-pathogen interaction, and to aid in downstream experimental design.
Materials and Methods
Ethics Statement
All animal studies were approved by and conducted according to the Institutional Animal Care and Use Committee (NIH/PHS Animal Welfare Assurance Number: A3410-01, USDA Animal Research Facility Registration Number: 55-R-004, AAALAC Institutional Number: #329) guidelines of the University of North Carolina- Chapel Hill (IACUC ID 13-213).
Bacteria and plasmids
Francisella tularensis subsp holartica LVS was obtained from the CDC Atlanta, GA. A pdpC deletion construct was made by splice overlap extension (SOE) PCR as described previously [14]. Primers were used to delete all but the first five amino acid (MNDKY) and the last five codons (KISS stop) while keeping the deletion in frame. After blunt end cloning into pCR BLUNT II (Invitrogen), the SOED pdpC fragment was removed by BamHI-Not I digest and cloned into suicide vector pMP812 [23]. Integrants were selected on kanamycin (10 ug/ml). Resolved integrants were selected by growth overnight in brain heart infusion (BHI) broth (BD Biosciences) supplemented with isovitalex followed by plating on chocolate containing 10% sucrose. Resolved integrants were sequenced to confirm in-frame deletion of pdpC and integrity of the flanking sequence. Deletion of both pdpC genes was verified by PCR. Genetic complementation of pdpC by constitutively expressing the pdpC gene on pMP822 restored intracellular proliferation (Figure S1) [24]. The ΔripA in-frame deletion and GFPΔripA strains were previously generated [25].
GFP wild-type LVS (GFP-wt) was generated by Hall et al [26] and GFP-ΔpdpC was generated using the same GFP plasmid. DsRed LVS and luciferase expressing LVS were generated with the plasmids from DsRed Schu S4 and luciferase expressing Schu S4 respectively [22]. Hygromycin B resistant ΔpdpC and ΔripA were generated by transforming the deletion mutant bacteria with the hygromycin B resistance plasmid pMP831 [24]. The kanamycin resistant wild-type strain was generated by transforming wild-type bacteria with the kanamycin resistance plasmid pMP828 and kanamycin resistant ΔripA with the pkkMCS plasmid [14], [24].
Cell Culture
J774A.1 Macrophage like cells were obtained from ATCC and grown in DMEM with 4.5 g/L glucose and supplemented with 10% FBS, L-glutamine and sodium pyruvate (all from Gibco). Mouse bone marrow derived macrophages were generated from a C57Bl/6 as previously described [27].
Binding F. tularensis to the magnetic beads
800 nm streptavidin coated magnetic beads (Solulink) were blocked with sterile filtered Tris buffered saline (TBS) containing 0.1% casein for 20 minutes. The beads were washed 4 times with antibody wash buffer (100 mM Tris, 150 mM NaCl and 0.05% tween 20 in distilled water, pH 8.0). For every 10 ug of beads, we added 2.5 µg of anti-Francisella tularensis LPS antibody (US Biological) that was previously biotinylated using a FluoReporter Mini-Biotin-XX Protein Labeling Kit (Invitrogen) following the manufacturer's instructions. The antibody was suspended in antibody wash. After 30 minutes of rocking at 4°C, the beads were washed twice in antibody wash buffer and twice in PBS. 10 µg of magnetic beads coated with anti- LPS antibody were mixed with 8×108 bacteria for 20 minutes. Each sample was then washed twice in PBS to remove unbound F. tularensis. After the final wash, the beads were suspended in cell culture media and ready to use for the given experiment. All washes were performed by placing a 12×75 mm round bottom tube containing the sample on a BD IMagnet (BD biosciences) and waiting approximately 2 minutes for the beads to move toward the magnet. After all wash steps, approximately 1×107 beads bound to bacteria were present for every 10 ug of beads initially added (based on plating bacteria bound to beads on chocolate agar). The amount varied up to 4×106 beads between experiments. All experiments were inoculated assuming 1×107 bacteria bound to beads per 10 ug of starting beads.
For samples where multiple strains were added together, 4×108 of each strain was added. For all mixed infections, the inoculum of each type of bacteria was plated and compared to ensure both types of bacteria were equally represented. Typically the ratio of one type of bacteria to the other was between 1: 0.8 and 1: 1.2 (data not shown). Infecting cells at a MOI higher than 1 can result in each cell taking up a substantial number of beads, which may impact results (data not shown). Bead aggregates can form over time which may impact the number of bacteria that enter each cell.
Microscopy
For micrographs of bacteria on beads, GFP-LVS and/or DsRed labeled LVS were coated onto the beads as described above. The bacteria bound beads were fixed in 4% paraformaldehyde and were placed into an 8 well chamber slide (Nunc). The beads were allowed to settle and the fixative was carefully removed.
For images of infected cells, beads were prepared as above with the indicated bacteria. J774 cells were inoculated for 2 hours and then the media was removed and replaced with media containing 25 µg/ml of gentamicin. The cells were washed and fixed with 4% paraformaldehyde at 4 or 22 hours post inoculation. Cells were treated with 50 mM ammonium chloride for 10 minutes and then stained with 10 µg/ml of AF647 wheat germ agglutinin (Invitrogen) where indicated.
All samples were mounted using a DAPI containing mounting media (Vector Shield). Images were acquired using a Zeiss 700 confocal laser scanning microscope (Zeiss) using Zen image acquisition software (Zeiss) or an Olympus FV500 confocal scanning laser microscope (Olympus). Images were cropped and scale bars were added using ImageJ [28].
Flow Cytometry
To assess the frequency of both types of bacteria binding to a single bead, beads were prepared as above with GFP-LVS and/or CellTrace Far Red DDAO-SE (Invitrogen) labeled wild-type LVS. Immediately following the final wash, the beads were stained with pacific blue conjugated anti- F. tularensis LPS antibody (US biological) (Pacific blue antibody labeling kit [Invitrogen] following the manufacturers protocol). The beads were washed once more and then immediately fixed using 4% paraformaldehyde. The event trigger on the flow cytometer was set to only record Pacific Blue positive events.
Phagosomal escape assays were performed as previously described [29]. Briefly, J774 cells were inoculated with beads coated with the indicated wild-type or deletion mutants bacteria (prepared as above). 2 hours post inoculation, the cells were suspended and stained with pacific blue conjugated anti- Francisella tularensis LPS antibody to label extracellular bacteria. After the antibody was washed away with KHM buffer, the cells were permeabilized with 100 ul of 10 µg/ml digitonin and the cells were stained with AF647 conjugated anti- Francisella tularensis LPS antibody to stain cytosolic bacteria. The cells were lysed and the samples were then analyzed by flow cytometry. We compared the ratio of intracellular cytosolic bacteria (GFP+ AF647+ Pac Blue-) to bacteria in the phagosome (GFP+ AF647- Pac Blue-). The data from these experiments includes both bacteria bound to beads and unattached bacteria. No significant difference in localization was seen between F. tularensis attached to beads and unbound bacteria, although bacteria bound to beads tended to escape the phagosome at higher rates. Control cells were stained with calnexin to assay the percentage of cells that were permeabilized. The percentage of permeabilized cells was used to determine the total percentage of bacteria that escaped the phagosome.
To assay for bacterial dissociation from the beads prior to inoculation, GFP-LVS was bound to beads as previously described and stained with pacific blue conjugated anti- F. tularensis LPS antibody. The fixed sample was then analyzed for the percentage of bacteria bound to beads. To assess the amount of dissociation from beads inside of cells, GFP-LVS was bound to beads as previously described. The cells were removed from the plate, washed in KHM buffer [29], and permeabilized with digitonin 2 hours post inoculation. The permeabilized cells were stained with an AF647 conjugated anti- F. tularensis LPS antibody (US biological). The antibody was washed away and then the infected cells were lysed by vortexing the cells in distilled water. AF647+, GFP+ events were analyzed for attachment to a bead based on size.
All samples were analyzed using a Cyan Flow Cytometer (Dako) with the event trigger set to record only pacific blue positive events or GFP positive events depending on the experiment. All histograms were pre-gated so that only single events were analyzed.
Intracellular bacterial proliferation assay
The indicated LVS mutants were coated on beads as previously described. J774 cells were seeded at approximately 2.5×105 cells per well the night before infection and were infected with an approximate multiplicity of infection (MOI) of 1 bead per cell (assuming 5×105cells prior to infection). The media was replaced with media containing 25 µg/ml of gentamicin 2 hours post inoculation. At 4 and 22 hours post inoculation, the cells were scraped off the plate, lysed, serially diluted and plated on chocolate agar containing isovitalex and either kanamycin or hygromycin to assess bacterial proliferation. As expected, hygromycin resistant colonies did not form under kanamycin selection and vice versa (data not shown).
Quantification of bacteria per bead
F. tularensis LVS containing a luciferase –expressing plasmid was bound to beads following the previously described method. Half of the sample was plated on chocolate agar to assay for the number of beads bound to bacteria and the other half was placed into an Infinite M200 Pro plate reader (Tecan) and the amount of light produced was quantified and compared to a standard curve consisting of know quantities of F. tularensis LVS containing the luciferase plasmid.
The number of bacteria per cell was determined based on the results at 4 hours post inoculation of the wild-type bacteria from the intracellular proliferation assay described previously. We assumed that 60% of the cells were infected based on flow cytometry of similar samples examined 2 hours post inoculation. Since gentamicin is added 2 hours post inoculation, additional cells should not be infected. We also assumed that the J774 cells doubled overnight, resulting in 5×105 cells, that each cell was infected with 1 bead, that 100% of bacteria were bound to beads and no bacterial death or replication occurred between 0 and 4 hours post inoculation.
IL-1β ELISA
Murine bone marrow derived macrophages were seeded at 5×105 cells per well in 24-well plates. Beads coated with the appropriate bacterial strains and were used to inoculate the cells at an MOI of approximately 1 bead per cell. The cell supernatant was collected 22 hours post inoculation and the amount of IL-1β was measured using a BD OptEIA mouse IL-1β ELISA set (BD biosciences) following the manufacturers protocol.
Statistics
All statistics were performed using an unpaired Student t-test using the compiled data from all experiments performed. Intracellular proliferation assays were log10 transformed prior to statistical analysis.
Supporting Information
Genetic complementation of Δ pdpC intracellular proliferation. Intracellular proliferation assay of J774 cells infected with wild-type, ΔpdpC or ΔpdpC with a complementation plasmid containing the pdpC gene. The bar graph represents the mean +/− the standard deviation. Data is a compilation of 3 independent experiments performed in triplicate. ***p<0.005
(TIF)
Acknowledgments
We would like to thank Dr. Jean Celli for assistance with the phagosomal escape assay protocol, Dr. Cheryl Miller for all ripA N21A data, Melanie Kibler for technical assistance, the UNC Flow Cytometry Core Facility for flow cytometry assistance and the UNC Microscopy Service Laboratory for microscopy assistance.
Funding Statement
This work was supported by NIH grant A1082870 to THK. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Clemens DL, Lee BY, Horwitz MA (2004) Virulent and avirulent strains of francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect Immun 72: 3204–3217 10.1128/IAI.72.6.3204-3217.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang MT, Mortensen BL, Taxman DJ, Craven RR, Taft-Benz S, et al. (2010) Deletion of ripA alleviates suppression of the inflammasome and MAPK by francisella tularensis. J Immunol 185: 5476–5485 10.4049/jimmunol.1002154; 10.4049/jimmunol.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bosio CM, Bielefeldt-Ohmann H, Belisle JT (2007) Active suppression of the pulmonary immune response by francisella tularensis Schu4. J Immunol 178: 4538–4547. [DOI] [PubMed] [Google Scholar]
- 4. Hajjar AM, Harvey MD, Shaffer SA, Goodlett DR, Sjostedt A, et al. (2006) Lack of in vitro and in vivo recognition of francisella tularensis subspecies lipopolysaccharide by toll-like receptors. Infect Immun 74: 6730–6738 10.1128/IAI.00934-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindgren M, Eneslatt K, Broms JE, Sjostedt A (2013) Importance of PdpC, IglC, IglI, and IglG for modulation of a host cell death pathway induced by francisella tularensis LVS. Infect Immun. 10.1128/IAI.00275-13. [DOI] [PMC free article] [PubMed]
- 6. Barker JR, Chong A, Wehrly TD, Yu JJ, Rodriguez SA, et al. (2009) The francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol Microbiol 74: 1459–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bonquist L, Lindgren H, Golovliov I, Guina T, Sjostedt A (2008) MglA and igl proteins contribute to the modulation of francisella tularensis live vaccine strain-containing phagosomes in murine macrophages. Infect Immun 76: 3502–3510 10.1128/IAI.00226-08; 10.1128/IAI.00226-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Broms JE, Lavander M, Sjostedt A (2009) A conserved alpha-helix essential for a type VI secretion-like system of francisella tularensis. J Bacteriol 191: 2431–2446 10.1128/JB.01759-08; 10.1128/JB.01759-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Broms JE, Lavander M, Meyer L, Sjostedt A (2011) IglG and IglI of the francisella pathogenicity island are important virulence determinants of francisella tularensis LVS. Infect Immun 79: 3683–3696 10.1128/IAI.01344-10; 10.1128/IAI.01344-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lindgren H, Golovliov I, Baranov V, Ernst RK, Telepnev M, et al. (2004) Factors affecting the escape of francisella tularensis from the phagolysosome. J Med Microbiol 53: 953–958. [DOI] [PubMed] [Google Scholar]
- 11. Schmerk CL, Duplantis BN, Wang D, Burke RD, Chou AY, et al. (2009) Characterization of the pathogenicity island protein PdpA and its role in the virulence of francisella novicida. Microbiology 155: 1489–1497 10.1099/mic.0.025379-0; 10.1099/mic.0.025379-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Straskova A, Cerveny L, Spidlova P, Dankova V, Belcic D, et al. (2012) Deletion of IglH in virulent francisella tularensis subsp. holarctica FSC200 strain results in attenuation and provides protection against the challenge with the parental strain. Microbes Infect 14: 177–187 10.1016/j.micinf.2011.08.017; 10.1016/j.micinf.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 13. Long ME, Lindemann SR, Rasmussen JA, Jones BD, Allen LA (2013) Disruption of francisella tularensis schu S4 iglI, iglJ, and pdpC genes results in attenuation for growth in human macrophages and in vivo virulence in mice and reveals a unique phenotype for pdpC. Infect Immun 81: 850–861 10.1128/IAI.00822-12; 10.1128/IAI.00822-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fuller JR, Craven RR, Hall JD, Kijek TM, Taft-Benz S, et al. (2008) RipA, a cytoplasmic membrane protein conserved among francisella species, is required for intracellular survival. Infect Immun 76: 4934–4943 10.1128/IAI.00475-08; 10.1128/IAI.00475-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fuller JR, Kijek TM, Taft-Benz S, Kawula TH (2009) Environmental and intracellular regulation of francisella tularensis ripA. BMC Microbiol 9: 216–2180-9-216 10.1186/1471-2180-9-216; 10.1186/1471-2180-9-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bosio CM, Bielefeldt-Ohmann H, Belisle JT (2007) Active suppression of the pulmonary immune response by francisella tularensis Schu4. J Immunol 178: 4538–4547. [DOI] [PubMed] [Google Scholar]
- 17. GREISMAN SE, HORNICK RB, CAROZZA FA, Jr, WOODWARD TE (1963) The role of endotoxin during typhoid fever and tularemia in man. I. acquisition of tolerance to endotoxin. J Clin Invest 42: 1064–1075 10.1172/JCI104792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kieffer TL, Cowley S, Nano FE, Elkins KL (2003) Francisella novicida LPS has greater immunobiological activity in mice than F. tularensis LPS, and contributes to F. novicida murine pathogenesis. Microbes Infect 5: 397–403. [DOI] [PubMed] [Google Scholar]
- 19. Barker JR, Chong A, Wehrly TD, Yu JJ, Rodriguez SA, et al. (2009) The francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol Microbiol 74: 1459–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peng K, Broz P, Jones J, Joubert LM, Monack D (2011) Elevated AIM2-mediated pyroptosis triggered by hypercytotoxic francisella mutant strains is attributed to increased intracellular bacteriolysis. Cell Microbiol 13: 1586–1600 10.1111/j.1462-5822.2011.01643.x; 10.1111/j.1462-5822.2011.01643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chong A, Wehrly TD, Nair V, Fischer ER, Barker JR, et al. (2008) The early phagosomal stage of francisella tularensis determines optimal phagosomal escape and francisella pathogenicity island protein expression. Infect Immun 76: 5488–5499 10.1128/IAI.00682-08; 10.1128/IAI.00682-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Steele S, Brunton J, Ziehr B, Taft-Benz S, Moorman N, et al. (2013) Francisella tularensis harvests nutrients derived via ATG5-independent autophagy to support intracellular growth. PLoS Pathog 9: e1003562 10.1371/journal.ppat.1003562; 10.1371/journal.ppat.1003562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. LoVullo ED, Molins-Schneekloth CR, Schweizer HP, Pavelka MS Jr (2009) Single-copy chromosomal integration systems for francisella tularensis. Microbiology 155: 1152–1163 10.1099/mic.0.022491-0; 10.1099/mic.0.022491-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. LoVullo ED, Sherrill LA, Pavelka MS Jr (2009) Improved shuttle vectors for francisella tularensis genetics. FEMS Microbiol Lett 291: 95–102 10.1111/j.1574-6968.2008.01440.x; 10.1111/j.1574-6968.2008.01440.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fuller JR, Craven RR, Hall JD, Kijek TM, Taft-Benz S, et al. (2008) RipA, a cytoplasmic membrane protein conserved among francisella species, is required for intracellular survival. Infect Immun 76: 4934–4943 10.1128/IAI.00475-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hall JD, Woolard MD, Gunn BM, Craven RR, Taft-Benz S, et al. (2008) Infected-host-cell repertoire and cellular response in the lung following inhalation of francisella tularensis schu S4, LVS, or U112. Infect Immun 76: 5843–5852 10.1128/IAI.01176-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mortensen BL, Fuller JR, Taft-Benz S, Kijek TM, Miller CN, et al. (2010) Effects of the putative transcriptional regulator IclR on francisella tularensis pathogenesis. Infect Immun 78: 5022–5032 10.1128/IAI.00544-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider CA, Rasband WS, Eliceiri KW (2012) NIH image to ImageJ: 25 years of image analysis. Nat Meth 9: : 671–675. Available: http://dx.doi.org/10.1038/nmeth.2089 via the Internet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Checroun C, Wehrly TD, Fischer ER, Hayes SF, Celli J (2006) Autophagy-mediated reentry of francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci U S A 103: 14578–14583 10.1073/pnas.0601838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Genetic complementation of Δ pdpC intracellular proliferation. Intracellular proliferation assay of J774 cells infected with wild-type, ΔpdpC or ΔpdpC with a complementation plasmid containing the pdpC gene. The bar graph represents the mean +/− the standard deviation. Data is a compilation of 3 independent experiments performed in triplicate. ***p<0.005
(TIF)