

Abstract
Context:
Hypoglycemia-associated autonomic failure (HAAF) limits the ability of patients with diabetes to achieve target glycemia. Animal models have provided insights into the pathogenesis of HAAF, but a robust human model of HAAF in which recurrent hypoglycemia impacts the counterregulatory responses to hypoglycemia days later is lacking.
Objective:
The aim of this study was to determine the impact of two or three episodes of moderate hypoglycemia on counterregulatory responses to subsequent hypoglycemia induced 5 days later.
Design and Subjects:
Six healthy subjects participated in each of the two study protocols. In both protocol 1 and 2, subjects underwent two 2-hour hypoglycemic clamp studies during the morning and afternoon of day 1. In protocol 2, subjects underwent an additional third hypoglycemic clamp during the morning of day 2. All subjects in both protocols underwent a final hypoglycemic clamp on the morning of day 5.
Results:
In protocol 1, there were no significant differences in the hypoglycemia-induced hormone response or in symptoms scores between the mornings of days 1 and 5. In protocol 2, hypoglycemia-induced epinephrine (P = .02) and cortisol (P = .04) secretions were significantly lower on day 5 compared with day 1, whereas glucagon (P = .08) and norepinephrine (P = .59) were not different. Also in protocol 2, neurogenic (P = .02) and neuroglycopenic (P = .04) symptoms during hypoglycemia were decreased on day 5 compared with day 1.
Conclusion:
These results demonstrate that exposure of healthy humans to three 2-hour hypoglycemic episodes over 30 hours leads to significant blunting in counterregulatory and symptom response to subsequent hypoglycemia on day 5.
Iatrogenic hypoglycemia is a major challenge in the treatment of patients with diabetes. It causes significant morbidity and often limits the ability of patients to achieve the level of glycemic control known to prevent the complications of the disease. Patients with diabetes who are exposed to recurrent episodes of iatrogenic hypoglycemia frequently develop hypoglycemia-associated autonomic failure (HAAF) in which the glucose threshold required to elicit the counterregulatory (CR) response is shifted to a lower and lower glucose value, whereas the glucose threshold at which neuroglycopenia occurs remains unchanged. For such patients, the first sign of hypoglycemia may be unconsciousness. The mechanisms responsible for the development of HAAF have been the focus of much research, but the work has been limited by the absence of a robust human model of HAAF in which the failure to mount a CR response lasts for days after exposure to recurrent episodes of experimentally induced hypoglycemia. Such a model would be useful in identifying the specific pathways through which recurrent hypoglycemia leads to HAAF and avoidance of hypoglycemia resolves HAAF over time.
Previous investigation in this area has focused on the impact of one or two episodes of hypoglycemia on a single day on the CR response to hypoglycemia induced 18–24 hours later. Heller and Cryer (1) demonstrated that one 30-minute episode of moderate hypoglycemia (50 mg/dL) was not sufficient to blunt CR response to hypoglycemia induced 24 hours later but that two episodes of hypoglycemia on the same day significantly blunted CR response and symptoms to hypoglycemia induced the following morning (18 h later). However, exposure to short episodes (<30 min) of daily hypoglycemia for 3 consecutive days did not result in the blunting of the CR response (2) on day 4. In addition, two episodes of hypoglycemia on day 1 and one episode on day 3 did not result in significant reductions in hypoglycemia-induced catecholamine secretion or symptoms on day 8 (3).
To date we do not have a validated model of experimentally induced HAAF in humans that can be used to watch the development and resolution of the syndrome over more than a 24-hour period. Therefore, in this study we sought to determine the impact of two different HAAF induction protocols on the response to hypoglycemia 5 days after the study initiation. We hypothesized that three episodes of moderate hypoglycemia (50 mg/dL) in the first 30 hours of the study would result in greater blunting of the CR on day 5 than would two episodes of moderate hypoglycemia in the first 8 hours.
Subjects and Methods
Subjects
Healthy volunteers on no medications were recruited from the University of Minnesota community. Six subjects participated in each of the two study protocols [protocol 1: two females, four males, aged 41 ± 12 y, body mass index (BMI) 26.5 ± 6.3 kg/m2; protocol 2: two females, four males, aged 26.5 ± 8.1 y, BMI 23.9 ± 5.1 kg/m2, mean ± SD]. Before participation, subjects provided informed consent as governed by the Institutional Review Board at the University of Minnesota. Subjects were asked to avoid strenuous exercise from the day before the start of the protocol through to the completion on day 5. Female subjects were studied in the follicular phase of the menstrual cycle.
Protocol
Subjects participated in one of two protocols (Figure 1). Both protocols 1 and 2 started with a 2-hour hypoglycemic clamp study in the morning (8:00–10:00 am) and afternoon (12:00–2:00 pm) of day 1. In protocol 2, subjects underwent an additional third hypoglycemic clamp on the morning (8:00–10:00 am) of day 2. Both protocols ended with a final hypoglycemic clamp in the morning (8:00–10:00 am) of day 5.
Figure 1.
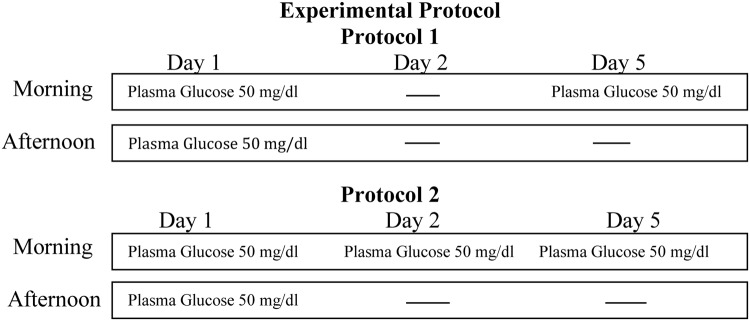
Schematic representation of the experimental protocols. Both morning and afternoon hypoglycemic clamps were for 2 hours.
All hypoglycemic clamp studies were performed in the same way. On the day of study, subjects arrived at the Clinical and Translational Science Institute in the morning after an overnight fast. Upon arrival, an iv catheter was placed antegrade in each forearm for subsequent infusions and for blood sampling. The forearm used for blood sampling was wrapped in heated towels and hot packs to arterialize the venous blood (4). At the start of the clamp study, a continuous infusion of insulin was started at a rate of 2.0 mU/kg·min along with an infusion of potassium phosphate (4 mEq/h). Samples were collected every 5 minutes for measurement of plasma glucose. Plasma glucose was allowed to fall to 50 mg/dL and then maintained at this level by a variable infusion of 20% dextrose. One hundred twenty minutes after starting insulin, the insulin and potassium were discontinued and the blood glucose was normalized with 20% dextrose infusion. Subjects were allowed to eat a snack with less than 10 g of carbohydrate after the first clamp study on day 1 in each protocol. Twenty percent dextrose infusion was continued as needed to keep blood glucose in the range of 80–100 mg/dL until the start of afternoon hypoglycemia. The hypoglycemic clamp in the afternoon on day 1 was started 2 hours after the end of the morning clamp study. At the conclusion of day 1, participants were fed and discharged. Those subjects participating in protocol 2 returned the following morning in a fasting state for a third hypoglycemic clamp. All subjects returned on the morning of day 5 in a fasting state for a final hypoglycemic clamp study.
Analytical methods
Plasma glucose concentration was measured in replicate using an Analox machine (Analox Instruments). Baseline samples for glucagon, epinephrine, norepinephrine, and cortisol were drawn 30 minutes after the placement of the last iv catheter in the first and last hypoglycemic clamp in both protocols. Additional samples for measurement of these hormones were collected at minutes 30, 45, 60, 75, 90, 105, and 120 after starting insulin during the first and final hypoglycemic clamps. Preservatives aprotinin and glutathione were added to sample collection tubes for glucagon and catecholamine, respectively. Samples were placed on dry ice immediately after collection. Hormone assays were performed at the Vanderbilt Diabetes Research and Training Center Core Laboratory. Plasma epinephrine and norepinephrine were measured by HPLC (Dionex, formerly ESA, Inc). Plasma cortisol was measured by RIA (Diagnostic Products Corporation, Inc). Plasma glucagon was measured by RIA (modified; Millipore, Merck).
Symptoms of hypoglycemia were quantified by using a previously validated questionnaire (5) at baseline and at minutes 30, 45, 60, 75, 90, 105, and 120. Subjects were asked to score from 0 (none) to 6 (severe) each of 12 symptoms, including six autonomic (neurogenic) symptoms (heart pounding, shaky/tremulous, nervous/anxious, sweaty, hungry, tingling) and six neuroglycopenic symptoms (difficulty thinking, tired/drowsy, weak, warm, faint, dizzy).
Statistical analysis
Data are expressed as mean ± SEM unless otherwise stated. Postbaseline glucose, hormone, and symptom score values were summarized using within-person averages over the last 30 minutes of hypoglycemia (min 90–120) and using within-person area under the curve (AUC) over minutes 45–120. One between-protocol analysis was carried out: two-sample t tests on the average over the last 30 minutes for morning clamp day 1 to compare protocol 1 to protocol 2. Three within-protocol analyses were carried out: 1) paired t tests comparing the average over the last 30 minutes for the morning clamp day 1 with the morning clamp day 5; 2) paired t tests comparing the AUC for the morning clamp day 1 with the morning clamp day 5; and 3) repeated-measures ANOVA to test the day 1 vs day 5 clamp difference at each postbaseline time point. A value of P < .05 indicated significant difference.
Results
Glucose
Target plasma glucose concentrations were achieved in all hypoglycemic clamp experiments (Figure 2). In protocol 1, mean plasma glucose concentrations during the morning hypoglycemic clamps on day 1 and day 5 were 53 ± 0.5 mg/dL and 53 ± 0.5 mg/dL, respectively. In protocol 2, mean plasma glucose concentrations during the morning hypoglycemic clamps on day 1 and day 5 were 56 ± 0.5 mg/dL and 54 ± 0.5 mg/dL. In both study protocols, target glycemia was reached by minute 45 during the morning hypoglycemia on day 1 and day 5. The mean glucose concentrations achieved during the hypoglycemic plateaus were similar during all hypoglycemic clamps.
Figure 2.

Plasma glucose levels (mean ± SEM) during hypoglycemic clamps on days 1 and 5 in protocol 1 and on days 1, 2, and 5 in protocol 2.
CR hormones
There was no statistically significant difference in the CR hormone response to hypoglycemia during the morning hypoglycemic clamps on day 1 between protocols 1 and 2. The CR hormone response to hypoglycemia on day 5, as compared with the response measured on day 1, is shown in Figure 3 for both protocols. In protocol 1, there was no statistically significant difference in the hypoglycemia induced hormone response between the mornings of days 1 and 5 using an AUC analysis. The mean concentration of CR hormones achieved during the last 30 minutes of the hypoglycemia also showed no statistically significant difference between the mornings of days 1 and 5 (Table 1).
Figure 3.

Comparison of CR hormones (mean ± SEM) including epinephrine, norepinephrine, glucagon, and cortisol during morning hypoglycemia on days 1 and 5 of the study. In protocol 1, there was no significant blunting of CR hormones on day 5 as compared with day 1. In protocol 2, epinephrine and cortisol were significantly blunted, whereas norepinephrine and glucagon were not significantly reduced on day 5 compared with day 1. *, P < .05; **, P < .01.
Table 1.
Comparison of CR Hormones and Symptom Response During Final 30 Minutes of Hypoglycemia on Days 1 and 5
| Protocol 1 |
Protocol 2 |
|||||
|---|---|---|---|---|---|---|
| Morning Day 1 | Morning Day 5 | P Value | Morning Day 1 | Morning Day 5 | P Value | |
| Plasma glucose | 51 ± 5 | 52 ± 3 | .60 | 54 ± 3 | 55 ± 2 | .77 |
| Epinephrine, pg/mL | 720 ± 362 | 619 ± 229 | .33 | 496 ± 201 | 246 ± 224 | .02a |
| Norepinephrine, pg/mL | 393 ± 193 | 397 ± 140 | .91 | 372 ± 148 | 397 ± 230 | .59 |
| Glucagon, pg/mL | 154 ± 63 | 160 ± 67 | .39 | 144 ± 52 | 109 ± 37 | .08 |
| Cortisol, mg/dL | 42 ± 9 | 38 ± 6 | .10 | 45 ± 22 | 31 ± 11 | .04a |
| Neurogenic symptom score | 13 ± 6 | 14 ± 6 | .71 | 14 ± 3 | 11 ± 3 | .02a |
| Neuroglycopenic symptoms core | 14 ± 6 | 11 ± 7 | .14 | 13 ± 4 | 9 ± 5 | .04a |
Data are expressed as mean ± SD. Final 30 minutes of CR hormone symptom score data represent an average of three measurements taken during this time period.
P < .05.
In protocol 2, hypoglycemia induced epinephrine secretion was significantly lower during hypoglycemia during the morning of day 5 than hypoglycemia during the morning of day 1, whether assessed by AUC analysis or mean hormone values during the final 30 minutes of hypoglycemia (Figure 3 and Table 1). The mean cortisol level measured during the final 30 minutes of hypoglycemia on day 5 was significantly lower than the mean values measured during the same time period on day 1 (Table 1), but statistically significant differences were not found using an AUC analysis. Mean glucagon levels measured during the final 30 minutes of hypoglycemia on day 5 trended to be lower than the mean values measured during the same time period on day 1, but this did not reach statistical significance. Hypoglycemia induced norepinephrine secretion was similar on both days.
Symptom scores
Neurogenic and neuroglycopenic symptoms during hypoglycemia were not attenuated on day 5 compared with day 1 in protocol 1 (Table 1). In protocol 2 both neurogenic and neuroglycopenic symptoms during hypoglycemia were significantly decreased on day 5 compared with day 1 (Table 1).
Discussion
In this investigation, we found that hypoglycemia-induced epinephrine secretion and symptoms during hypoglycemia were blunted on the morning of day 5 after an induction protocol consisting of exposure to three 2-hour periods of moderate hypoglycemia within a 30-hour period but were not blunted after an induction protocol consisting of exposure to two 2-hour periods of moderate hypoglycemia within an 8-hour period. Thus, protocol 2 with three episodes of hypoglycemia in the first 30 hours offers investigators a new model in which to study the development and resolution of HAAF in humans.
Most previous studies have looked at the effects of antecedent hypoglycemia on subsequent near-term CR responses. Results from previous studies have suggested that greater depth, duration, and frequency of antecedent episodes of hypoglycemia results in a greater degree of the blunting of CR response to subsequent hypoglycemia (6, 7). Therefore, we hypothesized that three 2-hour episodes of moderate hypoglycemia within 30 hours would result in a greater blunting of the CR response on day 5, as compared with two 2-hour episodes of moderate hypoglycemia within 8 hours. The objective of this study was to develop a robust model of HAAF in healthy humans that can be used to watch the development and resolution of the syndrome over several days. Mechanisms underlying the development of HAAF are not known, but in animals studies alterations in brain metabolism and neurotransmission including changes in brain glycogen content have been suggested. Magnetic resonance spectroscopy in which the incorporation of administered 13C-glucose into brain glycogen is followed over time can be used to measure brain glycogen turnover and content but currently require the infusion of isotope over 80+ hours to detect the impact of metabolic events on brain glycogen metabolism. Therefore, to determine how changes in brain glycogen metabolism might contribute to HAAF, it was critical to develop a robust model in which HAAF persists for 80+ hours after the initiation of the study. Because HAAF is a significant problem in patients with type 1 and advanced type 2 diabetes, the use of this model in future studies will provide mechanistic insights into how and why recurrent hypoglycemia independent of diabetes itself leads to HAAF in these patient groups.
It is important to recognize that HAAF is not a permanent alternation in autonomic function. HAAF can be reversed in patients with type 1 diabetes by the meticulous avoidance of hypoglycemia, something that is very challenging for insulin-treated patients who are striving to maintain optimal glucose control. Improvement in hypoglycemia symptom response has been shown as soon as after 3 days of hypoglycemia avoidance, whereas 2–3 weeks of hypoglycemia avoidance can restore hypoglycemia awareness and improve the neuroendocrine responses to hypoglycemia (8, 9). Approximately 20% of patients with type 1 diabetes are reported to have hypoglycemia unawareness, which increases the risk of severe hypoglycemia by 6-fold (10). Severe hypoglycemia in patients with diabetes is associated with an increased risk of morbidity and death (11). It has been reported that 6%–10% of patients with type 1 diabetes die from hypoglycemia (12, 13). Consequently, it is critical that additional strategies other than strict avoidance of hypoglycemia be developed to prevent and treat HAAF in patients with type 1 diabetes. Use of the model presented in this report will be important in defining what metabolic changes occur with the development and resolution of HAAF in healthy humans. Such insights can subsequently be used to treat diabetic patients with this problem.
One limitation of this approach is that the induction of HAAF in healthy humans may not replicate the hypoglycemia exposure experienced by patient with diabetes in their daily lives, thereby limiting the applicability to this patient population. However, studies using continuous glucose monitoring have shown that frequent and prolonged episodes of hypoglycemia similar to what is used in the induction protocol are common in subjects with type 1 diabetes. Subjects with diabetes can have multiple episodes of hypoglycemia during the same day and hypoglycemia episodes can be prolonged (>2 h), especially during the night (14–16). Relying only on patients with diabetes and HAAF to define the underlying mechanisms of HAAF may be problematic because the severity of hypoglycemia symptom unawareness and defective counterregulation in patients with diabetes can fluctuate based on exposure to recent antecedent hypoglycemia. This makes controlling for antecedent exposure to hypoglycemia difficult. Using a HAAF induction protocol in healthy humans overcomes this problem because the only hypoglycemia to which the participants are exposed is experimentally induced. Arbelaez et al (17) have used a HAAF induction protocol in healthy volunteers consisting of a 2-hour hypoglycemia on day 1 followed by 12–14 hours of interval interprandial hypoglycemia to identify an association between attenuated hormonal counterregulation and activation of the thalamus during hypoglycemia on day 2, but a longer protocol like the one identified here may provide greater opportunity to clarify the individual steps that link recurrent hypoglycemia to HAAF.
Exposure to hypoglycemia over 2 or more days is a protocol used to induce HAAF in animals. Using such a protocol, Herzog et al (18) recently found that cerebral glucose metabolism was maintained in these animals during hypoglycemia as compared with controls, perhaps because of increased glucose availability, and that increased lactate flux supported this change in glucose metabolism. Other investigators have found that rats exposed to recurrent hypoglycemia have increased γ-aminobutyric acid level in the ventromedial hypothalamus, which leads to the suppression of the CR response to acute hypoglycemia (19); these rats also have increased brain glycogen content (20), which could potentially support energy metabolism during hypoglycemia and lead to the development of HAAF. Future investigation with our protocol will help us understand whether these metabolic pathways contribute to the development of HAAF in healthy humans.
In this study, the two protocols examined to induce HAAF were different not only with regard to the number of antecedent episodes of hypoglycemia but also to the time course in relationship to the final hypoglycemia on day 5. In protocol 1, the final episode of hypoglycemia for induction of HAAF was about 90 hours before the study on day 5. In comparison the final episode of hypoglycemia to induce HAAF in protocol 2 was about 72 hours prior the final study on day 5. It is possible that along with number and duration of hypoglycemia episodes, these differences in relative time courses may have affected the outcomes between the two protocols. In our study, we did not find hypoglycemia-induced glucagon or norepinephrine secretion to be altered by prior exposure to recurrent hypoglycemia. Impaired hypoglycemia induced glucagon secretion is seen in patients with type 1 diabetes with and without HAAF, presumably because they lack the endogenous source of insulin that must be suppressed by hypoglycemia to remove the tonic inhibitory effect of this hormone on glucagon release. Other studies in healthy humans have shown reduction in glucagon secretion after exposure to recurrent hypoglycemia (1, 17).
In our study we observed a trend toward a decrease in glucagon, which did not reach statistical significance (P =.08). In this study, the power to detect differences in glucagon may be limited due to small sample size. Serum norepinephrine levels measured during hypoglycemia reflect activation of the sympathetic nervous system and have been shown to be unchanged by exposure to preceding hypoglycemia in other human models of HAAF (6, 17), perhaps because serum concentrations correlate poorly with intrasynaptic concentrations of norepinephrine. We did find hypoglycemia-induced epinephrine secretion to be significantly reduced on day 5 in subjects participating in protocol 2, which by definition shows we induced HAAF.
Limitations of our study include the possibility that exercise done before the start of the study may have had an impact on our measurements. We asked participants to avoid strenuous exercise from the day before the start of the protocol through to the completion on day 5, but exercise done 48 or even 72 hours before the start of the study could potentially have long-standing metabolic effects. Another limitation is that the age of subjects in protocol 2 is lower compared with protocol 1, which could limit their comparability. Subjects were comparable in other characteristics including sex and BMI.
In summary, this study demonstrates that exposure to a protocol with three episodes of moderate hypoglycemia over 30 hours on days 1 and 2 results in continued blunting of the CR and symptom response to the subsequent hypoglycemia on day 5. This robust model of HAAF in healthy humans can be used in future studies to examine the effects of recurrent hypoglycemia on pathways hypothesized to contribute to the development of HAAF in patients with diabetes.
Acknowledgments
The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This work was supported by Grant RO1NS35192 (to E.R.S.), Clinical and Translational Science Award 5KL2TR000113 (to A.M.). Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the National Institutes of Health Award UL1TR000114.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AUC
- area under the curve
- BMI
- body mass index
- CR
- counterregulatory
- HAAF
- hypoglycemia-associated autonomic failure.
References
- 1. Heller S, Cryer P. Reduced neuroendocrine and symptomatic responses to subsequent hypoglycemia after 1 episode of hypoglycemia in nondiabetic humans. Diabetes. 1991;40:223–226 [DOI] [PubMed] [Google Scholar]
- 2. Peters A, Rohloff F, Kerner W. Preserved counterregulatory hormone-release and symptoms after short-term hypoglycemic episodes in normal men. J Clin Endocrinol Metab. 1995;80:2894–2898 [DOI] [PubMed] [Google Scholar]
- 3. George E, Harris N, Bedford C, Macdonald I, Hardisty C, Heller S. Prolonged but partial impairment of the hypoglycemic physiological-response following short-term hypoglycemia in normal subjects. Diabetologia. 1995;38:1183–1190 [DOI] [PubMed] [Google Scholar]
- 4. Seaquist E. Comparison of arterialized venous sampling from the hand and foot in the assessment of in vivo glucose metabolism. Metabolism. 1997;46:1364–1366 [DOI] [PubMed] [Google Scholar]
- 5. Towler D, Havlin C, Craft S, Cryer P. Mechanism of awareness of hypoglycemia—perception of neurogenic (predominantly cholinergic) rather than neuroglycopenic symptoms. Diabetes. 1993;42:1791–1798 [DOI] [PubMed] [Google Scholar]
- 6. Davis S, Mann S, Galassetti P, et al. Effects of differing durations of antecedent hypoglycemia on counterregulatory responses to subsequent hypoglycemia in normal humans. Diabetes. 2000;49:1897–1903 [DOI] [PubMed] [Google Scholar]
- 7. Davis S, Tate D. Effects of morning hypoglycemia on neuroendocrine and metabolic responses to subsequent afternoon hypoglycemia in normal man. J Clin Endocrinol Metab. 2001;86:2043–2050 [DOI] [PubMed] [Google Scholar]
- 8. Fanelli C, Epifano L, Rambotti A, et al. Meticulous prevention of hypoglycemia normalizes the glycemic thresholds and magnitude of most of neuroendocrine responses to, symptoms of, and cognitive function during hypoglycemia in intensively treated patients with short-term IDDM. Diabetes. 1993;42:1683–1689 [DOI] [PubMed] [Google Scholar]
- 9. Dagogojack S, Rattarasarn C, Cryer P. Reversal of hypoglycemia unawareness, but not defective glucose counterregulation, in IDDM. Diabetes. 1994;43:1426–1434 [DOI] [PubMed] [Google Scholar]
- 10. Geddes J, Schopman JE, Zammitt NN, Frier BM. Prevalence of impaired awareness of hypoglycaemia in adults with type 1 diabetes. Diabet Med. 2008;25:501–504 [DOI] [PubMed] [Google Scholar]
- 11. Moheet A, Seaquist E. Hypoglycemia as a driver of cardiovascular risk in diabetes. Curr Atheroscler Rep. 2013;15:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cryer PE. Death during intensive glycemic therapy of diabetes: mechanisms and implications. Am J Med. 2011;124:993–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feltbower RG, Bodansky HJ, Patterson CC, et al. Acute complications and drug misuse are important causes of death for children and young adults with type 1 diabetes—results from the Yorkshire Register of Diabetes in Children and Young Adults. Diabetes Care. 2008;3:922–926 [DOI] [PubMed] [Google Scholar]
- 14. Boland E, Monsod T, Delucia M, Brandt C, Fernando S, Tamborlane W. Limitations of conventional methods of self-monitoring of blood glucose—lessons learned from 3 days of continuous glucose sensing in pediatric patients with type 1 diabetes. Diabetes Care. 2001;24:1858–1862 [DOI] [PubMed] [Google Scholar]
- 15. Buckingham B, Wilson DM, Lecher T, Hanas R, Kaiserman K, Cameron F. Duration of nocturnal hypoglycemia before seizures. Diabetes Care. 2008;31:2110–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wiltshire EJ, Newton K, McTavish L. Unrecognised hypoglycaemia in children and adolescents with type 1 diabetes using the continuous glucose monitoring system: prevalence and contributors. J Paediatr Child Health. 2006;42:758–763 [DOI] [PubMed] [Google Scholar]
- 17. Arbelaez AM, Powers WJ, Videen TO, Price JL, Cryer PE. Attenuation of counterregulatory responses to recurrent hypoglycemia by active thalamic inhibition—a mechanism for hypoglycemia-associated autonomic failure. Diabetes. 2008;57:470–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Herzog RI, Jiang L, Herman P, et al. Lactate preserves neuronal metabolism and function following antecedent recurrent hypoglycemia. J Clin Invest. 2013;123:1988–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chan O, Cheng H, Herzog R, et al. Increased GABAergic tone in the ventromedial hypothalamus contributes to suppression of counterregulatory responses after antecedent hypoglycemia. Diabetes. 2008;57:1363–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Canada SE, Weaver SA, Sharpe SN, Pederson BA. Brain glycogen supercompensation in the mouse after recovery from insulin-induced hypoglycemia. J Neurosci Res. 2011;89:585–591 [DOI] [PMC free article] [PubMed] [Google Scholar]