

Abstract
Context:
The cAMP signaling pathway is implicated in bilateral adrenocortical hyperplasias. Bilateral adrenocortical hyperplasia is often associated with ACTH-independent Cushing syndrome (CS) and may be caused by mutations in genes such as PRKAR1A, which is responsible for primary pigmented nodular adrenocortical disease (PPNAD). PRKAR1A regulates cAMP-dependent protein kinase (PKA), an essential enzyme in the regulation of adiposity. Although CS is invariably associated with obesity, its different forms, including those associated with PKA defects, have not been compared.
Objective:
The purpose of this study was to characterize the phenotypic and molecular differences in periadrenal adipose tissue (PAT) between patients with CS with and without PRKAR1A mutations.
Design and Setting:
Samples from adrenalectomies of 51 patients were studied: patients with CS with (n = 13) and without (n = 32) PRKAR1A mutations and a comparison group with aldosterone-producing adenomas (APAs) (n = 6). In addition, clinical data from a larger group of patients with Cushing disease (n = 89) and hyperaldosteronism (n = 26) were used for comparison.
Methods:
Body mass index (BMI), abdominal computed tomography scans, and cortisol data were collected preoperatively. PAT was assayed for PKA activity, cAMP levels, and PKA subunit expression.
Results:
BMI was lower in adult patients with CS with PRKAR1A mutations. cAMP and active PKA levels in PAT were elevated in patients with CS with PRKAR1A mutations.
Conclusions:
Increased PKA signaling in PAT was associated with lower BMI in CS. Differences in fat distribution may contribute to phenotypic differences between patients with CS with and without PRKAR1A mutations. The observed differences are in agreement with the known roles of cAMP signaling in regulating adiposity, but this is the first time that germline defects of PKA are linked to variable obesity phenotypes in humans.
Cushing syndrome (CS) is induced by long-term hypercortisolemia; obesity with redistribution of fat tissue is among its most distinctive clinical features (1, 2). ACTH-independent CS is most frequently caused by cortisol-producing adenomas (CPAs) (3). Bilateral adrenocortical hyperplasias (BAHs) have been recently recognized as an important cause of ACTH-independent CS, albeit substantially more rarely than CPAs (4).
BAHs are a heterogeneous group of disorders roughly divided in half in terms of frequency and age distribution: massive macronodular adrenocortical disease also known as ACTH-independent macronodular hyperplasia with lesions >1 cm is more frequent in older adults (5) and micronodular forms of BAH (lesions <1 cm) are more frequent in children and young adults (6). Micronodular BAH is further subdivided into a relatively more frequent pigmented variant, primary pigmented nodular adrenocortical disease (PPNAD) (7), and the more recently described (8) isolated micronodular adrenocortical disease that with the exception of pigmentation shares key features with PPNAD, including the expression of neuroendocrine markers (9, 10).
The cAMP-signaling pathway plays a role either directly due to genetic defects of a molecule central to the pathway or indirectly via the up-regulation of ectopically expressed G-protein–coupled receptors in the adrenal cortex in most forms of BAHs and CPAs (5, 11, 12). Even when neither a mutation in a known gene nor ectopic receptor expression could be documented, cAMP-dependent protein kinase A (PKA) activity and/or cAMP levels were increased in various benign adrenocortical lesions (13). Mutations of the PRKAR1A gene that codes for the regulatory subunit 1α of PKA (RIα) cause PPNAD (7, 14, 15).
PKA is known to play an important role in energy balance and fat metabolism. Knockout (KO) of PKA regulatory subunit 2β (RIIβ), which is highly expressed in adipose and brain led to decreased adiposity, improved glucose and insulin sensitivity, and resistance to diet-induced obesity in mice (16–19). These phenomena in Prkar2b KO were in part mediated by increased PKA type I activity due to a substantial compensatory increase in RIα protein, the product of the PRKAR1A gene (16, 17). Obese humans had decreased RIIβ mRNA expression in visceral adipose tissue compared with that in lean individuals and lacked compensatory changes in RIα (20). KO or haploinsufficiency of the PKA catalytic subunit β, the product of the Prkacb gene, caused resistance to diet-induced obesity and attenuated declining metabolic function associated with aging in mice (21).
Changes in PKA function in adipose tissue (AT) have not been previously investigated in different forms of CS. Here we characterized differences in anthropometric and biochemical measures in patients with CS with and without PRKAR1A mutations. Body mass index (BMI) or BMI z-score and fat distribution in patients with CS with PRKAR1A mutations were compared with those in patients with CS lacking PRKAR1A mutations and with non-CS comparison groups. PKA activity and mRNA and protein expression of PKA subunits were quantified. We found that PRKAR1A mutations affected cAMP signaling in AT and were associated with significantly reduced CS-related obesity. Although cAMP signaling in AT may not be the only reason for the observed clinical differences, such cAMP signaling effects in the presence of PRKAR1A defects constitute at least part of the explanation, as demonstrated by our studies of PKA function in AT of patients with CS.
Subjects and Methods
Patients and surgical specimens
This retrospective study included patients with CS and APA enrolled in National Institutes of Health (NIH) protocols 95CH059, 97CH0076, and 00CH160 who were seen and/or operated on at the National Institutes of Health during the last decade. Fifty-one patients (40 female and 11 male; mean age, 35.1 ± 2.6 years; range, 4–74 years) for whom periadrenal adipose tissue (PAT) samples were available were included in the study. Six patients (2 female and 4 male) with aldosterone-producing adenomas (APAs) who underwent adrenalectomy but did not have CS served as a comparison group for the in vitro data. All studies were approved by the Eunice Kennedy Shriver National Institute for Child Health and Human Development institutional review board; adults signed their own consent forms, and younger patients signed assent forms (after the age of 8 years), whereas their legal custodians signed their consent forms.
Clinical data: biochemical and anthropometric measurements
Weight, height, and BMI at the time of diagnosis were recorded. For BMI and biochemical comparisons, additional patients enrolled in the same NIH protocols for whom preoperative anthropometric and biochemical measurements were available were also included (APA, n = 19; CS with PRKAR1A mutation [mtCS], n = 12; CS, n = 24; and APA, n = 9; mtCS, n = 6; CS, n = 26, respectively). For the pediatric patients, there was no non-CS comparison group; instead, we compared patients with PRKAR1A mutations with pediatric patients with (pituitary) Cushing disease (CD) (n = 89). Because the study was retrospective, no other measurements of fat or adiposity were available. Thus, we quantified subcutaneous adiposity by a radiologic method that was available and could be used for this purpose (see below).
CS was confirmed on the basis of elevated midnight serum cortisol values and/or elevated 24-hour urine cortisol or 17-hydroxysteroid excretion, as described previously (1, 4, 5, 22). Serum and/or urine free cortisol (UFC) concentrations during high-dose dexamethasone suppression testing were measured, along with stimulation of serum cortisol or ACTH during CRH stimulation testing, as described previously (4, 5, 22). Serum cortisol was measured by a fluorescence polarization immunoassay (Abbott Laboratories) with intra- and interassay coefficients of variation of 2.1% and 4.1%, respectively; the functional detection limit was 1 to 2 μg/dL (23). UFC was measured by an immunoradiometric assay (Mayo Laboratories and Clinical Center Department of Laboratory Medicine). The functional detection limit was 10 μg/dL. The respective form of CS was diagnosed in all patients after surgery and on the basis of their histopathological analysis, as described previously (1, 4, 5). All samples with micronodular forms of BAH were sequenced for PRKAR1A as described previously (14, 15).
Radiologic assessment of abdominal subcutaneous fat and PAT
Abdominal subcutaneous adipose tissue (ScAT) and PAT were quantified by computed tomography (CT) scans that were read by radiologists who were blinded to the diagnosis and the outcome of the study. Axial images of the abdomen were selected from postcontrast abdominal CT scans. Image analyses were performed using Vue PACS (version 11.3; Carestream Health). Slice thickness ranged from 2.5 to 3 mm. The amount of PAT was assessed by measuring the transverse distance from the medial limb of each adrenal gland to the crux of the adjacent diaphragm. Three consecutive slices were chosen for these measurements. ScAT was assessed by measuring the vertical distance from the posterior aspect of the paraspinal muscles to the skin on both the right and left sides. Slice selection was the same as that for adrenal gland, and slices were identical to those used for periadrenal fat assessment. Individual measurements of ScAT and PAT were analyzed by diagnosis; the ratio of PAT to ScAT was analyzed to examine the relationship between the amount of PAT and overall fatness within each diagnostic group.
Protein extraction and Western blotting
PAT was collected from patients at the time of adrenalectomy, snap-frozen, and stored in liquid nitrogen for later analyses. Total cellular protein was extracted from frozen tissue by homogenization in Tissue Protein Extraction Reagent (Thermo Scientific) with added protease and phosphatase inhibitor cocktails, each at a 1:100 volume ratio (Calbiochem). Total protein concentration was determined by the BCA protein assay (Pierce). For Western blotting, 20 μg of total protein lysate was loaded onto 10% to 20% gradient Tris-HCl gels (Invitrogen), and SDS/PAGE was performed for 2 hours at 100 V using 1× Tris-glycine-SDS running buffer (National Diagnostics). Proteins run on gels were then transferred to nitrocellulose membranes that were blocked in 1× PBS-Tween 20 with 5% nonfat milk and probed with commercially available polyclonal antibodies for the following PKA subunits: RIα, RIIα, RIIβ, Cα, and Cβ. β-Actin served as a loading control. Membranes were incubated with appropriate secondary horseradish peroxidase-linked antibodies and visualized with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Developed films were analyzed qualitatively with SeeBlue Plus2 Pre-Stained Standard (Invitrogen) and quantitatively by densitometry using ImageJ (version 1.44; NIH). Relative quantities are presented as the ratio of the densities of the protein of interest to β-actin.
mRNA expression
RNA was extracted from PAT that was snap-frozen after adrenalectomy and had been stored in liquid nitrogen. An RNeasy Lipid Mini Kit (QIAGEN) was used, following the manufacturer's protocol. cDNA was created from 100 ng of RNA that was first screened for quantity and quality (260/280 nm ratio) using a NanoDrop spectrophotometer. cDNA was created with SuperScript-RT III First-Strand Synthesis SuperMix for quantitative (q) RT-PCR (Invitrogen). Relative mRNA expression was measured by qRT-PCR (ViiA 7 real-time system; Applied Biosystems) using Power SYBR Green Master Mix (Applied Biosystems). qRT-PCR primers were designed with Primer3Plus (Whitehead Institute for Biomedical Research) and were tested and optimized before use with the PAT cDNA. mRNA expression was compared between forms of CS with values from patients with APAs serving as the non-CS comparison group. β-Actin served as the housekeeping gene and fold change was determined using the 2−ΔΔCT method (24).
cAMP levels and PKA activity
cAMP was measured in PAT extracts using the cAMP 3H Biotrak Assay System (Amersham Biosciences). Samples were homogenized in ethanol and centrifuged at 1000 × g for 10 minutes. The supernatant was dried and resuspended in 50 μL of assay buffer according to the manufacturer's protocol. Two aliquots were prepared from each sample and assayed. Human PAT was homogenized in ice-cold buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA, 1 mM dithiothreitol, and protease inhibitor cocktail I [EMD Biosciences[). Lysates were centrifuged at 10 000 × g for 10 minutes at 4°C. The total protein concentration of the supernatant was determined using the BCA protein assay. PKA enzymatic activity was measured using a method described previously (25). Assays were performed in a total volume of 50 μL for 15 minutes at 37°C in a reaction mixture containing 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 1 mM dithiothreitol, 25 μM kemptide, and 25 μL of [γ-32P]ATP (0.1 μCi/nmol) with or without 5 μM cAMP and 10 μL of cell extract. After incubation, reaction mixtures were spotted onto 0.23-mm phosphocellulose (Whatman P81) discs and washed 3 times in 0.5% phosphoric acid. Filters were air dried and measured by liquid scintillation counting.
DEAE-cellulose chromatography
All procedures were performed at 0 to 4°C as described by Nesterova et al (25). In brief, after 2 washes with ice-cold isotonic NaCl/P, PAT samples were suspended in 10 mL of 10 mM Tris-HCl, 1 mM EDTA, 1 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, leupeptin (30 μg/mL), aprotinin (5.0 μg/mL), and pepstatin (5 μg/mL), pH 7.1, and kept on ice for 20 minutes, homogenized (70 strokes), and centrifuged at 10 000 × g for 10 minutes. The resulting supernatants were subjected to chromatography. The DEAE column (1 × 10) was equilibrated with 10 mM Tris-HCl, pH 7.1, containing 1 mM EDTA and 1 mM phenylmethylsulfonyl fluoride. Cell extracts (10 mg of protein/sample) were loaded onto the column, and the column was washed with 30 mL of buffer. The column was developed with a 0 to 350 mM NaCl gradient in 10 mM Tris-HCl, pH 7.1, 1 mM EDTA, and 1 mM phenylmethylsulfonyl fluoride at a flow rate of 15 mL/h, and 2-mL fractions were collected on ice and assayed for protein kinase activity.
Statistical analysis
SAS (version 9.3; SAS Institute) was used for data analysis. All patient anthropometric, biochemical, and molecular data are presented as means ± SEM. The normality of distribution of quantitative variables was assessed using a Kolmogorov-Smirnov test with a cutoff value set at P < .05. For normally distributed data, one-way ANOVA was performed to identify differences between diagnostic groups. Matrix grids were used to identify which groups were significantly different from each another. qRT-PCR and Western blot data that were not normally distributed were log-transformed to achieve normal distribution of the data. Data that were not normally distributed after log- transformation were subjected to the Mann-Whitney U nonparametric test. Differences were considered significant at a value of P ≤ .05.
Results
Diagnostic and anthropometric data
Table 1 provides baseline anthropometric and biochemical characteristics for patients that underwent adrenalectomy and were included in the studies.
Table 1.
Baseline Characteristics for Patients With CS and APAs for Whom PAT Samples Were Available
| Sex | Age | Diagnosis | BMI | Diurnal Cortisol |
Chol | TG | Glucose | Mutations | |
|---|---|---|---|---|---|---|---|---|---|
| MN | 8 am | ||||||||
| F | 46 | APA | 27.9 | NA | NA | NA | NA | NA | |
| F | 59 | APA | 21.4 | NA | NA | 189 | 111 | 82 | |
| M | 54 | APA | 26.4 | 5.3 | 15.1 | 196 | NA | 103 | |
| M | 58 | APA | 36.4 | 8.9 | NA | 189 | 216 | 94 | |
| M | 63 | APA | 27.6 | NA | NA | 162 | 74 | 126 | MEN1 |
| M | 64 | APA | NA | 16.5 | 3.9 | 220 | NA | 93 | |
| F | 14 | CPA | 42.6 | 32.4 | 30.4 | 174 | 126 | 114 | |
| F | 14 | CPA | 24.5 | 22.3 | 20 | 224 | 81 | 84 | |
| F | 15 | CPA | NA | 19.6 | 17.9 | 159 | 158.5 | 0.57 | |
| F | 16 | CPA | 20.5 | 21.3 | 18.8 | 172 | 124.5 | 85 | |
| F | 27 | CPA | 36.4 | 6 | 8.5 | 289 | 61 | 102 | |
| F | 36 | CPA | 29.7 | NA | NA | 207 | 114 | NA | |
| F | 44 | CPA | 24.4 | NA | NA | 169 | 197 | 105 | |
| F | 51 | CPA | 32.7 | 13 | 18.8 | 204 | NA | 89 | |
| F | 53 | CPA | 36.8 | 8.9 | 10.4 | 163 | 81 | 85 | |
| F | 66 | CPA | 26.1 | NA | NA | 203 | 80 | 104 | |
| F | 8 | iMAD | 28 | 32.9 | 31.1 | 161 | 61 | 79 | |
| F | 10 | iMAD | 37.1 | 20.6 | 19.8 | NA | 91 | NA | PDE11A |
| F | 37 | iMAD | NA | NA | NA | NA | NA | NA | |
| F | 30 | MMAD | 39.9 | 21.5 | 23.3 | 351 | NA | 107 | |
| F | 42 | MMAD | NA | 30.5 | 21.8 | 273 | 125.5 | 120 | |
| F | 46 | MMAD | 33.5 | 24.4 | 22.8 | NA | 87 | 129 | |
| F | 49 | MMAD | NA | 29.3 | 32.5 | NA | NA | NA | |
| F | 49 | MMAD | 29.9 | 16.7 | 14.3 | 152 | NA | 90 | |
| F | 50 | MMAD | 40.9 | 9.5 | 8.3 | 143 | 101.5 | 90 | |
| F | 53 | MMAD | 35.7 | 22.5 | 20.6 | 234 | 161.5 | 134 | |
| F | 57 | MMAD | 19.4 | 22.6 | 31.4 | 217 | 122 | 94 | |
| F | 66 | MMAD | 37.2 | 12 | 30.5 | 224 | 147.5 | 80 | |
| M | 45 | MMAD | 38.2 | 10 | 6.7 | 269 | 169.5 | 142 | |
| M | 47 | MMAD | 39.5 | 10.4 | 8.6 | 192 | 89 | NA | |
| M | 54 | MMAD | 34.6 | 15.4 | 5.5 | NA | 190 | 87 | |
| F | 10 | mtPPNAD | 22.2 | 23.9 | 21.1 | 167 | 71 | 92 | PRKAR1A |
| F | 10 | mtPPNAD | 22.8 | 2.7 | 17.2 | 162 | 69.5 | 91 | PRKAR1A |
| F | 17 | mtPPNAD | 26.8 | 4.3 | 12.3 | 162 | 88.5 | 80 | PRKAR1A |
| F | 19 | mtPPNAD | 30.6 | 13 | 10.6 | 201 | 104 | 124 | PRKAR1A |
| F | 20 | mtPPNAD | 26.6 | 30.1 | 25.4 | 200 | 81 | 67 | PRKAR1A |
| F | 22 | mtPPNAD | 30.4 | 30.8 | 34.5 | 171 | 33 | NA | PRKAR1A |
| F | 23 | mtPPNAD | 37.2 | 31 | 31 | NA | NA | 110 | PRKAR1A |
| F | 25 | mtPPNAD | 38.4 | 28.1 | 29.1 | 223 | NA | 79 | PRKAR1A |
| F | 28 | mtPPNAD | 25.7 | 13.6 | 12.8 | 123 | 61 | 88 | PRKAR1A |
| F | 31 | mtPPNAD | 23.7 | 8.5 | 3.1 | 161 | 84.5 | 78 | PRKAR1A |
| F | 51 | mtPPNAD | 23 | 10.4 | 11.7 | NA | 91 | 103 | PRKAR1A |
| M | 13 | mtPPNAD | 31.4 | 3.8 | 7.5 | 165 | 96.5 | 81 | PRKAR1A |
| M | 58 | mtPPNAD | 40.7 | 5.8 | 16.2 | 159 | 193 | 104 | PRKAR1A |
| F | 3 | PPNAD | 19.1 | 20 | 21 | 184 | 82 | 83 | |
| F | 4 | PPNAD | 16 | 6.5 | 14.1 | 143 | 40 | 79 | |
| F | 6 | PPNAD | 22.6 | 52.5 | 40.7 | 157 | 44 | 86 | |
| F | 17 | PPNAD | 25.3 | 1.1 | 6.2 | 212 | 78 | 88 | |
| M | 16 | PPNAD | 28.1 | 15.1 | 17.1 | 275 | 97.5 | 94 | |
| M | 19 | PPNAD | 24.3 | 22.3 | 21.9 | 233 | 62 | 77 | |
Abbreviations: Chol, cholesterol; F, female; Gluc, glucose; iMAD, isolated micronodular adrenocortical hyperplasia; M, male; MMAD, massive macronodular adrenocortical disease; MN, midnight; mtPPNAD, primary pigmented nodular adrenocortical disease (with PRKAR1A mutations); NA, not applicable; TG, serum triglycerides. All measurements were performed preoperatively within the week before adrenalectomy.
Diurnal cortisol (midnight and 8:00 am) and 24-hour UFC were measured preoperatively. Mean midnight cortisol levels were above the normal range (2.9–13 μg/dL) in the CS groups and within the normal range in the non-CS (APA) group as expected (Supplemental Figure 1A published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org.). The difference in midnight cortisol between the mtCS and CS groups was not significant (P = .15). No differences in 8:00 am cortisol levels were observed (Supplemental Figure 1A). The 24-hour UFC was lower in the non-CS group than in either the mtCS or CS group (P = .003 and .05, respectively); mean 24-hour UFC for the mtCS and CS groups did not differ (Supplemental Figure 1B).
Among adult patients with CS, the mean BMI of those with PRKAR1A mutations was lower than that of patients with CS without PRKAR1A mutations (P < .05) (Figure 1A) and did not differ from that for the comparison group with hyperaldosteronism. Among pediatric patients with adrenal CS, PRKAR1A mutation status did not affect the mean BMI z-score, but compared with pediatric patients with pituitary CD, pediatric patients with CS with PRKAR1A mutations had significantly lower BMI z-scores (P < .05) (Figure 1B).
Figure 1.
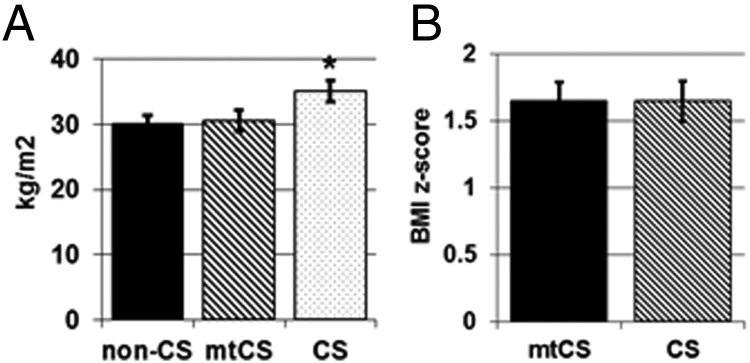
Mean BMIs of adult patients with CS and the non-CS comparison group (A) and the BMI z-score for pediatric patients with CS (B). BMI and BMI z-scores were calculated from anthropometric measurements taken at the time of diagnosis and before adrenalectomy. All values are means ± SEM. *, P < .05. The data set for adult patients includes the following: non-CS, n = 24; mtCS, n = 14; and CS, n = 29. The data set for pediatric patients includes the following: mtCS, n = 11; and CS, n = 23.
Measurement of ScAT and PAT by CT
The CS group without PRKAR1A mutations had significantly more PAT and ScAT than the non-CS group (P < .05) (Figures 2, A–D). There was a trend toward an increased ratio of PAT to ScAT, a measure of abdominal fat distribution, in the mtCS and CS groups, but the group sizes were small and differences did not achieve statistical significance (Figure 2E).
Figure 2.

ScAT and PAT were quantified from preoperative abdominal CT scans. A–C, Representative preoperative abdominal CT scans of patients are shown and include a patient with APA (non-CS) (A), a patient with CS with a PRKAR1A mutation (mtCS) (B), and a patient with CS without a PRKAR1A mutations (CS) (C). Red arrows indicate the right and left adrenal glands, yellow bars show the distance between the medial limb of each adrenal gland and the crux of the diaphragm that was measured as PAT, and green bars indicate the thickness of ScAT measured in each patient. D and E, Mean values for PAT and ScAT (D) and the ratio of PAT to ScAT (E) were calculated in non-CS, mtCS, and CS groups. The data set includes patients for whom digital CT scans were available: APA, n = 4; mtCS, n = 4; and CS, n = 9. *, P ≤ .05. Comparisons were made in reference to the non-CS group and within classification only.
mRNA expression of PKA subunits in PAT
mRNA levels for PRKAR2A and PRKACA were higher in PAT from the mtCS group than in that from the non-CS group (P < .05) (Figure 3). PRKAR1A, PRKAR1B, PRKAR2B, and PRKACB mRNA did not differ between the mtCS or CS group and the non-CS group (Figure 3); however, a trend toward increased levels of all regulatory subunits was seen in the mtCS group.
Figure 3.

mRNA expression of PKA subunits RIα, RIIα, RIβ, RIIβ, Cα, and Cβ in PAT of groups: non-CS (APA) and CS with (mtCS) and without PRKAR1A mutations (CS). qRT-PCR data were log-transformed to achieve normality, and values are presented as means (log) ± SEM. *, P < .05. The data set includes samples from the following: non-CS (n = 8), mtCS (n = 12), and CS (n = 25).
Protein expression of PKA subunits in PAT
RIα protein in PAT was barely detectable by Western blot and therefore not quantifiable but appeared to be decreased in patients with PRKAR1A mutations, as one would expect from the germline inactivating defects of this gene (Supplemental Figure 2). Differences in PKA subunits RIIα, RIIβ, Cα, and Cβ were small and did not achieve statistical significance because of the small number of samples (data not shown).
PKA activity and cAMP levels in PAT
The ratio of basal to total (cAMP-stimulated) PKA activity was significantly lower in the mtCS group, indicating increased concentrations of PKA in its active form (P < .005) (Figure 4C). Although the increase in total cAMP-stimulated PKA activity in PAT from the mtCS group did not achieve statistical significance (Figure 4A) and basal PKA activity was not different among the groups (Figure 4B), the cAMP concentration in PAT was higher in the patients with CS with PRKAR1A mutations (P < .05) (Figure 4D), consistent with the higher ratio of basal to total PKA activity. Indeed, the cAMP-stimulated DEAE assay revealed higher kinase activity in PAT from a patient with mtCS compared with PAT from a patient without a PRKAR1A mutation (Figure 4E), again consistent with the higher ratio of basal to total PKA activity.
Figure 4.

Effects of CS or CS with PRKAR1A mutations (mtCS) on the PKA signaling pathway were assessed in PAT by measuring PKA activity and cAMP levels. A–E, cAMP-stimulated PKA activity (A), basal PKA activity (B), the ratio of basal to total PKA activity (C), cAMP concentrations (D), and cAMP-stimulated DEAE assay (E) of PAT samples from CS and non-CS groups. Data presented are means ± SEM. *, P < .05.
Discussion
Abdominal obesity has long been established as one of the primary characteristics of CS. However, phenotypic variants other than those associated with race (26, 27), as well as differences in adipose tissue distribution and differences at the molecular level in different forms of CS, have not been explored. In addition, clinical experience has shown that patients with CS and PPNAD typically have a milder obesity phenotype than those with other forms of CS, even when their cortisol levels are comparable and their CS is not atypical or periodic (1, 4, 28).
Based on this clinical observation, we undertook this study to test the hypothesis that patients with CS and PPNAD with PRKAR1A mutations have altered PKA activity in visceral AT and to further investigate the possible molecular etiology of this phenomenon. Certainly the study has limitations: numbers in groups are small (but these are rare diseases), it is retrospective both in its conception and execution and, thus, desirable measures that examine adiposity are lacking, and, finally, some of the findings may be confounded by comorbidities, duration of CS, age, or sex.
Nonetheless, the data point to one interesting finding that appears to confirm the clinical observation and provide some evidence for its molecular etiology: adult patients with CS with PRKAR1A mutations had a BMI that was similar to that of the patients with APA (the non-CS comparison group). Whereas the BMI z-score was not lower in pediatric patients with CS than in pediatric patients with CS without PRKAR1A mutations, it should be noted that the pediatric CS group used for this anthropometric analysis consisted primarily of patients with PPNAD lacking PRKAR1A mutations (15 of 22). An ancillary analysis comparing these pediatric patients with mtCS and CS with a large cohort of pediatric patients with CD, both CS groups had significantly lower mean BMI z-scores than that of the pituitary CD pediatric cohort (Supplemental Figure 3). This evidence in conjunction with the clinical observations suggests that pediatric patients with CS and PPNAD with or without PRKAR1A mutations have a less obese phenotype than other pediatric patients with CS. What is interesting is that PKA activity was not similarly elevated in the PAT of patients with PPNAD lacking PRKAR1A mutations as in those with mutations. This finding suggests that although pediatric patients with PPNAD share very similar clinical manifestations, there may be some other defect in the cAMP signaling pathway that is responsible for the less obese phenotype compared with that in other pediatric patients with CS.
The data presented are based on few patients for whom digital preoperative CT scans were available; thus, trends in mtCS patients could not be confirmed. It appears that patients with CS with PRKAR1A mutations may have less ScAT, but, interestingly, have levels of PAT similar to those of patients with CS without PRKAR1A mutations. Whether by analyzing abdominal CT or magnetic resonance imaging scans, further analyses are warranted in a larger group of patients with CS to identify potential differences in adiposity and fat distribution that can add to our characterization of phenotypic differences between patients with CS with and without PRKAR1A mutations. In addition, in this study, we examined PAT, a depot of visceral fat that has not been extensively investigated. Although a specific role for PAT is unclear, it is possible that increased PAT deposition in CS may play a role by providing substrate for steroidogenesis, specifically for glucocorticoids, that are increased in CS.
When we studied PAT in vitro in these patients with CS, the possible molecular etiology for these differences was shown to be overall increased PKA signaling. Although the increase in cAMP-stimulated activity did not achieve statistical significance, the ratio of basal to total PKA activity indicated increased active PKA in patients with mtCS (Figure 4, A and C). This finding was supported by elevated cAMP concentrations and data from DEAE chromatography showing clear increases in both type I and type II kinase activity in PAT in patients with mtCS compared with those with CS (Figure 4, D and E). Increased PKA activity in the PAT of patients with mtCS was accompanied by up-regulation of PKA subunits RIIα and, more important, Cα, the main catalytic subunit of PKA and the effector of deficient RIα (29). The increased expression of RIIα in patients with CS with PRKAR1A mutations was in parallel with the findings of Mantovani et al (21), who showed increased RIIα expression in lean compared with obese subjects, as patients with mtCS were leaner than other patients with CS. It is possible that in both cases, higher Cα activity drives the higher RIIα expression.
These findings have obvious implications for the establishment of the diagnosis of CS in patients with PRKAR1A mutations: these patients may be leaner than other patients with CS. Perhaps more significantly, our findings point to the importance of cAMP and/or PKA signaling in the regulation of adiposity. Unpublished data from our laboratory showed that when PKA activity is measured in omental fat from otherwise healthy obese and lean adults, both basal and total PKA activities were suppressed in obesity (London, E., and C. A. Stratakis, unpublished observation). More studies are needed, some of which are being pursued in animal models in our laboratory, but the data point to a critical role of the regulation of Cα (rather than RIα), as also was supported here by the relatively higher mRNA expression of this catalytic subunit in patients with PRKAR1A mutations.
In conclusion, patients with CS and PRKAR1A mutations are less obese than other patients with CS. Similarly, patients with CS with other defects in cAMP/PKA signaling may be less obese than other patients with CS. The reason may lie in the increased cAMP and PKA activity in adipose tissue (as well as in other organs and tissues). The role of PKA as a regulator of adiposity in the general population should be investigated further.
Acknowledgments
This work was supported by The Eunice Kennedy Shriver National Institute of Child Health and Human Development intramural program.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- APA
- aldosterone-producing adenoma
- AT
- adipose tissue
- BAH
- bilateral adrenocortical hyperplasia
- BMI
- body mass index
- CD
- Cushing disease
- CPA
- cortisol-producing adenoma
- CS
- Cushing syndrome
- CT
- computed tomography
- KO
- knockout
- mtCS
- CS with PRKAR1A mutation
- NIH
- National Institutes of Health
- q
- quantitative
- PAT
- periadrenal adipose tissue
- PKA
- protein kinase A
- PPNAD
- primary pigmented nodular adrenocortical disease
- ScAT
- subcutaneous adipose tissue
- UFC
- urine free cortisol.
References
- 1. Stratakis CA. Cushing syndrome in pediatrics. Endocrinol Metab Clin North Am. 2012;41:793–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Almeida MQ, Stratakis CA. Carney complex and other conditions associated with micronodular adrenal hyperplasias. Best Pract Res Clin Endocrinol Metab. 2010;24:907–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Keil MF, Graf J, Gokarn N, Stratakis CA. Anthropometric measures and fasting insulin levels in children before and after cure of Cushing syndrome. Clin Nutr. 2012;31:359–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stratakis CA, Boikos SA. Genetics of adrenal tumors associated with Cushing's syndrome: a new classification for bilateral adrenocortical hyperplasias. Nat Clin Pract Endocrinol Metab. 2007;3:748–757 [DOI] [PubMed] [Google Scholar]
- 5. Hsiao HP, Kirschner LS, Bourdeau I, et al. Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J Clin Endocrinol Metab. 2009;94:2930–2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bertherat J, Horvath A, Groussin L, et al. Mutations in regulatory subunit type 1A of cyclic adenosine 5′-monophosphate-dependent protein kinase (PRKAR1A): phenotype analysis in 353 patients and 80 different genotypes. J Clin Endocrinol Metab. 2009;94:2085–2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Powell AC, Stratakis CA, Patronas NJ, et al. Operative management of Cushing syndrome secondary to micronodular adrenal hyperplasia. Surgery. 2008;143:750–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gunther DF, Bourdeau I, Matyakhina L, et al. Cyclical Cushing syndrome presenting in infancy: an early form of primary pigmented nodular adrenocortical disease, or a new entity? J Clin Endocrinol Metab. 2004;89:3173–3182 [DOI] [PubMed] [Google Scholar]
- 9. Stratakis CA, Carney JA, Kirschner LS, et al. Synaptophysin immunoreactivity in primary pigmented nodular adrenocortical disease: neuroendocrine properties of tumors associated with Carney complex. J Clin Endocrinol Metab. 1999;84:1122–1128 [DOI] [PubMed] [Google Scholar]
- 10. Carney JA, Gaillard RC, Bertherat J, Stratakis CA. Familial micronodular adrenocortical disease, Cushing syndrome, and mutations of the gene encoding phosphodiesterase 11A4 (PDE11A). Am J Surg Pathol. 2010;34:547–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bourdeau I, Matyakhina L, Stergiopoulos SG, Sandrini F, Boikos S, Stratakis CA. 17q22–24 chromosomal losses and alterations of protein kinase a subunit expression and activity in adrenocorticotropin-independent macronodular adrenal hyperplasia. J Clin Endocrinol Metab. 2006;91:3626–3632 [DOI] [PubMed] [Google Scholar]
- 12. Bertherat J, Groussin L, Sandrini F, et al. Molecular and functional analysis of PRKAR1A and its locus (17q22–24) in sporadic adrenocortical tumors: 17q losses, somatic mutations, and protein kinase A expression and activity. Cancer Res. 2003;63:5308–5319 [PubMed] [Google Scholar]
- 13. Bimpaki EI, Nesterova M, Stratakis CA. Abnormalities of cAMP signaling are present in adrenocortical lesions associated with ACTH-independent Cushing syndrome despite the absence of mutations in known genes. Eur J Endocrinol. 2009;161:153–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kirschner LS, Carney JA, Pack SD, et al. Mutations of the gene encoding the protein kinase A type I-α regulatory subunit in patients with the Carney complex. Nat Genet. 2000;26:89–92 [DOI] [PubMed] [Google Scholar]
- 15. Horvath A, Bertherat J, Groussin L, et al. Mutations and polymorphisms in the gene encoding regulatory subunit type 1-α of protein kinase A (PRKAR1A): an update. Hum Mutat. 2010;31:369–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cummings DE, Brandon EP, Planas JV, Motamed K, Idzerda RL, McKnight GS. Genetically lean mice result from targeted disruption of the RIIβ subunit of protein kinase A. Nature. 1996;382:622–626 [DOI] [PubMed] [Google Scholar]
- 17. Planas JV, Cummings DE, Idzerda RL, McKnight GS. Mutation of the RIIβ subunit of protein kinase A differentially affects lipolysis but not gene induction in white adipose tissue. J Biol Chem. 1999;274:36281–36287 [DOI] [PubMed] [Google Scholar]
- 18. Schreyer SA, Cummings DE, McKnight GS, LeBoeuf RC. Mutation of the RIIβ subunit of protein kinase A prevents diet-induced insulin resistance and dyslipidemia in mice. Diabetes. 2001;50:2555–2562 [DOI] [PubMed] [Google Scholar]
- 19. Nolan MA, Sikorski MA, McKnight GS. The role of uncoupling protein 1 in the metabolism and adiposity of RIIβ-protein kinase A-deficient mice. Mol Endocrinol. 2004;18:2302–2311 [DOI] [PubMed] [Google Scholar]
- 20. Mantovani G, Bondioni S, Alberti L, et al. Protein kinase A regulatory subunits in human adipose tissue: decreased R2B expression and activity in adipocytes from obese subjects. Diabetes. 2009;58:620–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Enns LC, Morton JF, Mangalindan RS, et al. Attenuation of age-related metabolic dysfunction in mice with a targeted disruption of the Cβ subunit of protein kinase A. J Gerontol A Biol Sci Med Sci. 2009;64:1221–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Batista DL, Riar J, Keil M, Stratakis CA. Diagnostic tests for children who are referred for the investigation of Cushing syndrome. Pediatrics. 2007;120:e575–586 [DOI] [PubMed] [Google Scholar]
- 23. Carney JA, Ho J, Kitsuda K, Young WF, Jr, Stratakis CA. Massive neonatal adrenal enlargement due to cytomegaly, persistence of the transient cortex, and hyperplasia of the permanent cortex: findings in Cushing syndrome associated with hemihypertrophy. Am J Surg Pathol. 2012;36:1452–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408 [DOI] [PubMed] [Google Scholar]
- 25. Nesterova M, Yokozaki H, McDuffie E, Cho-Chung YS. Overexpression of RIIβ regulatory subunit of protein kinase A in human colon carcinoma cell induces growth arrest and phenotypic changes that are abolished by site-directed mutation of RIIβ. Eur J Biochem. 1996;235:486–494 [DOI] [PubMed] [Google Scholar]
- 26. Hsiao HP, Iglesias ML, Keil MF, Boikos S, Robinson-White A, Stratakis CA. Differences in cortisol levels and body mass index between East Asians and Caucasians with Cushing's syndrome: an 'East Asian' phenotype for Cushing syndrome. Clin Endocrinol (Oxf). 2007;66:753–755 [DOI] [PubMed] [Google Scholar]
- 27. Jeyaraman K, Ammini AC, Nandita G, Dwivedi SN. Late-night salivary cortisol in normal subjects and in patients with Cushing's syndrome. Postgrad Med J. 2010;86:399–404 [DOI] [PubMed] [Google Scholar]
- 28. Sarlis NJ, Chrousos GP, Doppman JL, Carney JA, Stratakis CA. Primary pigmented nodular adrenocortical disease: reevaluation of a patient with carney complex 27 years after unilateral adrenalectomy. J Clin Endocrinol Metab. 1997;82:1274–1278 [DOI] [PubMed] [Google Scholar]
- 29. Meoli E, Bossis I, Cazabat L, et al. Protein kinase A effects of an expressed PRKAR1A mutation associated with aggressive tumors. Cancer Res. 2008;68:3133–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]