

Abstract
Purpose
Whereas T2 hyperintensities known as NF-associated bright spots are well described in patients with neurofibromatosis type I (NF-1), there is a paucity of data on incidental findings in patients with neurofibromatosis type II (NF-2). We aim to characterize unexplained imaging findings in the brains of patients with NF-2.
Materials and methods
This study is retrospective, HIPAA-compliant and approved by the institutional review board. 34 patients with NF-2 underwent brain magnetic resonance imaging (MRI) between January 2000 and December 2012. T2 and T1-weighted imaging characteristics, diffusion weighted imaging (DWI) characteristics, and enhancement patterns were analyzed by visual inspection. Clinical information at time of imaging was available for all patients. Neuropathologic data was available for one patient.
Results
We found unexplained T2 hyperintensities present on initial imaging in 23/34 patients (67%). Of the 23 patients with unexplained MRI findings, 15 (65%) had wedge-shaped T2 hyperintensities in the subcortical white matter extending to the cortex suggestive of a cortical dysplasia. 3 additional cases (17%) had a lesion within the cerebellum suggestive of a neuronal migration anomaly. In one patient where the MRI was suggestive of focal cortical dysplasia, histopathologic analysis revealed dysplastic glial foci without other alterations of cortical architecture or other cytologic abnormalities.
Conclusion
Unexplained T2 hyperintensities occur frequently in patients with NF-2. While they may not be the NF-2 equivalent of NF-associated bright spots seen in NF-1, some of these T2 hyperintensities in patients with NF-2 may represent underlying disorders of neuronal migration. Further studies are needed to validate our findings.
Keywords: Neurofibromatosis type 2, MRI, Cortical dysplasia, Migration anomalies, NF-associated bright spots, Micro-hamartomas
Highlights
-
•
34 patients with NF2 underwent consecutive brain magnetic resonance imaging (MRI).
-
•
67% had brain MRI findings suggestive of a focal cortical dysplasia.
-
•
17% had a lesion within the cerebellum suggestive of a neuronal migration anomaly.
-
•
One patient was found to have micro-hamartomas involving the cortex.
-
•
Patients with NF-2 may have radiographic findings suggestive of cortical dysplasia.
1. Introduction
Neurofibromatosis type II (NF-2) is a neurocutaneous disorder that is caused by genetic mutations of the NF-2 gene on chromosome 22 (Rouleau et al., 1993). The NF-2 gene encodes an intracellular membrane-associated protein, a tumor suppressor known as merlin (Trofatter et al., 1993). Aberrant merlin predisposes individuals with NF-2 to the development of multiple tumors of the central nervous system, including meningiomas, schwannomas and ependymomas (Asthagiri et al., 2009, Hagel et al., 2012). The most common of these tumors are bilateral vestibular schwannomas. Spinal cord tumors are also a prominent component of NF-2.
Hyperintensities on T2-weighted images known as NF-associated bright spots are seen in the brain MRIs of most patients with the other type of neurofibromatosis, neurofibromatosis type I (NF-1) (van Engelen et al., 2008). NF-associated bright spots are focal areas of increased signal intensity that occur most often in the basal ganglia, cerebellum, brainstem, and subcortical white matter. Pathologic studies performed in NF-1 so far have revealed intramyelinic vacuolar changes or spongiotic myelinopathy that correlate with the hyperintensities found on T2-weighted images (DiPaolo et al., 1995). However, there is a lack of data on the existence and significance of unexplained imaging findings in the brains of patients with NF-2. One case report suggests that hyperintensities seen on T2-weighted imaging in the brain of a 21-year-old man with NF-2 were likely due to myelin vacuolization (Sener et al., 2003). This assumption was based primarily on MRI appearance as there was no corresponding histopathology. Furthermore, there are no large-scale pathologic studies on these hyperintensities seen on T2-weighted images in the brains of patients with NF-2.
The purpose of this study was to characterize unexplained T2-hyperintense lesions and other incidental brain MRI findings in patients with NF-2 in an effort to elucidate specific patterns.
2. Materials and methods
Our retrospective study was approved by our institutional review board, and informed consent was waived. Our study was compliant with the Health Insurance Portability and Accountability Act.
2.1. Study design and patients
We performed a retrospective analysis of brain imaging, histopathology and clinical data on patients with NF-2 to characterize unexplained brain lesions in this population. Patients were included in this study if they had a definitive clinical and/or genetic diagnosis of NF-2 and had brain MRI imaging performed consecutively anytime between January 2000 and December 2012. The lesion inclusion criteria were lesions present on available initial brain MRI and lesions not otherwise explained by tumor, edema, radiation changes or postoperative changes. Brain MRI scans obtained without gadolinium were excluded from the study. Patients who had only one brain MRI available for review were also excluded from the study. Of 37 patients with NF-2 and available imaging during this time period, 34 had two or more brain MRI scans performed. The remaining 3 patients only had spinal imaging available and were excluded from the study. Neuropathologic data was available for one patient who expired during this time period and had complete autopsy performed.
2.2. Image acquisition
All patients were examined with a 1.5-T whole-body MR imaging unit (Echospeed; GE Medical Systems, Milwaukee, Wisconsin) equipped with high-performance gradients and a manufacturer-supplied quadrature head coil. The following conventional sequences were performed: sagittal T1-weighted (300/14 [repetition time in ms/echo time in ms], one signal acquired), transverse T2-weighted fast spin-echo (3000/91, one signal acquired), transverse fast fluid-attenuated inversion-recovery (10,002/172, inversion time of 2.2 s, one signal acquired), transverse T1-weighted (500/14, one signal acquired), and transverse diffusion-weighted echo-planar (6000/99–100, one signal acquired, b values of 0 and 1000 s/mm2) MR imaging. The transverse sequences usually involved the use of a 5-mm section thickness with an intersection gap of 2.5 mm, a 256 × 192 matrix, the same imaging angle along the orbitomeatal line, and a 22- or 24-cm field of view. Gadopentetate dimeglumine (Multi-Hance; Bracco Diagnostics, Princeton, NJ) was administered with each MRI scan to allow for the evaluation of contrast enhancement. Contrast agent administration was performed intravenously in an identical manner with a power injector at a rate of 2 mL/s with a 20-gauge needle unless patient-related factors (e.g., small veins) necessitated the use of a needle with a different size.
2.3. Data interpretation
All images were independently analyzed by visual inspection by an experienced pediatric neuroradiologist (L.A.H.) with 30 years of experience interpreting brain MRI to exclude tumors, vasogenic edema or post-operative changes within the central nervous system (CNS) and to characterize the findings. In addition to T2-weighted imaging characteristics, T1-weighted imaging characteristics, diffusion weighted imaging (DWI) characteristics, and enhancement patterns were analyzed to characterize associated abnormalities. Clinical data at time of all imaging was available for all patients. Spearman's correlation between patient age and number of unexplained lesions was calculated. The histopathologic data was independently analyzed by an experienced neuropathologist (F.R.) with 11 years of experience in interpreting both neoplastic and nonneoplastic neuropathology.
3. Results
A total of 34 patients were included in this study. 23/34 (68%) were males and 11/34 (32%) were females. The median age at time of unexplained brain MRI finding was 20 years old (range 8–69 years) (Table 1). 23/34 patients (68%) had at least one unexplained finding present on the initial MRI of the brain (Table 2). A total of 73 incidental lesions were identified among the 23 initial patient MRIs. 22/23 patients (96%) had at least one incidental hyperintense finding on T2-weighted imaging. The remaining patient with unexplained abnormal imaging had a purely cortical T1 hypointense and T2 hypointense lesion. The average number of unexplained T2-hyperintensities for all study patients at time of initial brain imaging was 2.2 ± 2.3. The most common location for these T2-hyperintense lesions was the frontal lobe, with a higher frequency appearing in the right frontal lobe. During consecutive brain imaging over a period of at least six months (range up to 12 years), no patient (0%) developed any new unexplained lesions and only two patients were noted to have an increase in incidental lesion size. One patient developed interval increase in the size of his unexplained T2-hyperintense lesion; this patient was among three patients in our study who had received brain radiation. One patient developed an increase in size of a cortical T2 hypointensity (Fig. 1). The remainder of the cases (94%) had no interval change in the size of any of their unexplained lesions. Of the three patients with a history of cranial irradiation, one did not have any unexplained abnormalities on initial or subsequent brain MRI. The other two had incidental findings present on brain MRI prior to the initiation of radiation therapy. The patient with a change in his lesions received conformal radiation therapy with a total of 5400 cGy to the right parietal area for ependymoma completed in December 2007. His unexplained lesions were first noted prior to radiation treatment in May 2007. He developed diffuse white matter T2-hyperintensities in December 2009. A second patient received conformal radiation (dose unknown as performed at an outside institution) to the lower brain stem/upper cervical cord for neurofibroma completed in 1991. His unexplained lesion was first noted in May 2007 and was not observed to change over time. A third patient received conformal radiation (dose unknown as performed at an outside institution) to the right orbit and right skull base for spheno-orbital meningioma completed in April 2004. His unexplained lesions were first noted prior to radiation therapy in May 2002 and were not noted to change over time. Among all 73 incidental lesions, only one was hyperintense on T1-weighted sequences. A total of three unexplained gadolinium-enhancing lesions were identified (4%). Among the three enhancing lesions was leptomeningeal enhancement over a wedge-shaped cortical right anterior temporal lesion (Fig. 2). Seven of the 23 patients (30%) had T2 hyperintense lesions associated with an enlarged Virchow–Robin space (Fig. 3). Nine of 23 patients (39%) had hyperintense cone-shaped lesions with subcortical extension thinning towards the lateral ventricles (the classic so called “transmantle sign”) on T2 and fluid-attenuated inverse recovery (FLAIR) sequences (Fig. 4; Table 3). An additional six of 23 (26%) patients had wedge-shaped cortical and subcortical T2 hyperintense lesions without the classic transmantle sign but suggestive of cortical dysplasia nonetheless (Fig. 5; Table 3). Six of the 23 (26%) patients had well circumscribed lesions that were hypointense on T1 and T2-weighted sequences suggestive of mineralization (Fig. 6). In addition, three patients (13%) had a lesion within the cerebellum suspicious for a disorder of neuronal migration (Fig. 7). Two of these 3 patients (67%) displayed the classic “transmantle sign” in the right cerebellum. The remaining patient had a cleft within the left cerebellum lined with cortical gray matter. None of the patients (0%) had epilepsy or a documented cognitive deficit. Overall we found that younger age at diagnosis of NF-2 correlated inversely with number of incidental lesions found on the initial brain MRI (Fig. 8).
Table 1.
Patient demographics.
| Number of patients in the study (N) | 34 |
| Median age at time of first incidental MRI finding (years) | 20 |
| Female, no. (%) | 11 (32) |
| Male, no. (%) | 23 (68) |
| Received cranial irradiation, no. (%) | 3 (9) |
Table 2.
Results.
| Incidental brain MRI finding | Number of patients with finding, no. (%) |
|---|---|
| Any incidental finding | 23 (68) |
| Nonspecific T2 hyperintensities | 17 (74) |
| Wedge shaped cortical/subcortical T2 hyperintense lesions | 15 (65) |
| Transmantle sign | 9 (39) |
| Cortical T2 hypointensities | 8 (34) |
| Well circumscribed cortical T2 hypointensity with associated T1 hypointensity | 6 (26) |
| Migrational cerebellar anomalies | 3 (13) |
| T2 hyperintense lesions associated with an enlarged Virchow–Robin space | 7 (30) |
| More than one of the above abnormalities | 15 (65) |
Fig. 1.
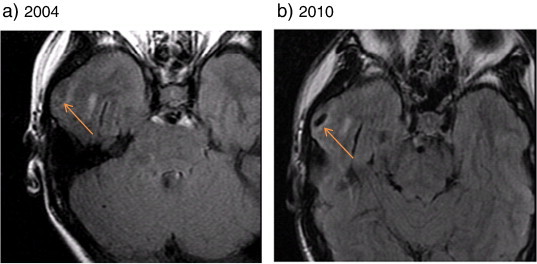
Cortical T2-hypointense lesion seen on MRI imaging of a 13-year-old boy with NF-2. Transverse fast fluid-attenuated inversion-recovery (FLAIR) images in a boy with NF-2 demonstrate a cortical hypointense lesion in the right anterior temporal lobe (orange arrows) present on initial imaging in 2004 (a) that grew in size when imaged 6 years later (b).
Fig. 2.
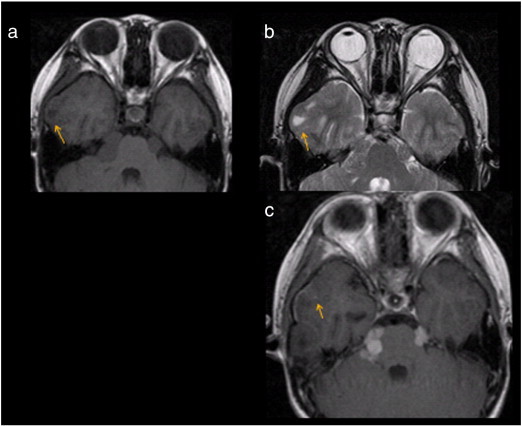
Leptomeningeal enhancement associated with a cortical T2-hyperintense wedge shaped lesion in a 13-year-old boy with NF-2. (a) Transverse T1-weighted sequence shows a wedge shaped hypointense lesion situated in the right anterior temporal cortex (arrow). (b) Transverse T2-weighted fast spin-echo sequence shows that the lesion is T2-hyperintense (arrow) and demonstrates mild mass effect with erosion of the inner skull table. (c) Transverse T1-weighted post-gadolinium sequence reveals mild leptomeningeal enhancement (arrow) over the wedge-shaped right temporal cortical lesion.
Fig. 3.
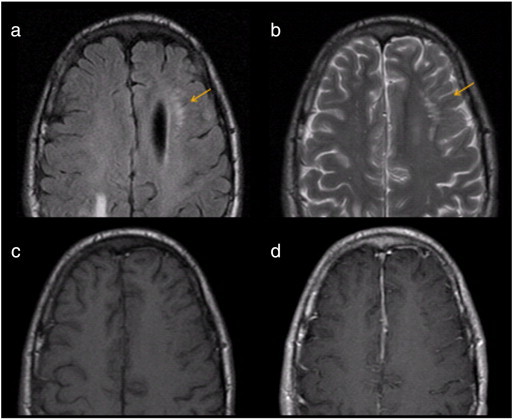
Periventricular T2-hyperintense lesion associated with enlarged Virchow–Robin space in a 20-year-old man with NF-2. (a) Transverse fast fluid-attenuated inversion-recovery (FLAIR) sequence shows left periventricular hyperintensity surrounding a dilated Virchow–Robin space (arrow). (b) Transverse T2-weighted fast spin-echo sequence shows that the lesion is T2-hyperintense (arrow). Transverse T1-weighted sequence pre-gadolinium (c) and post-gadolinium (d) administration reveal that the lesion does not contrast-enhance.
Fig. 4.
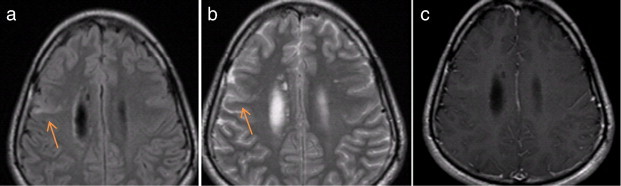
Possible transmantle sign in a 20-year-old man with NF-2. (a) Transverse fast fluid-attenuated inversion-recovery (FLAIR) image reveals a wedge-shaped cortical hyperintense lesion (arrow) extending towards the ventricle in a 20-year-old man with NF-2, suggestive of the “transmantle sign”. (b) Transverse T2-weighted fast spin-echo sequence shows that the lesion is T2-hyperintense (arrow). (c) Post-gadolinium transverse T1-weighted sequence fails to show contrast-enhancement.
Table 3.
Characteristics of those patients found to have MRI suspicious for cortical/cerebellar dysplasias.
| Number of cases | 16 |
| Female | 5 (31%) |
| Male | 11 (69%) |
| Median age at MRI finding of cortical dysplasia (years) | 19.5 |
| Finding present on initial available brain MRI | 16 (100%) |
| Change over time (# pts) | 2 (1%) |
Fig. 5.
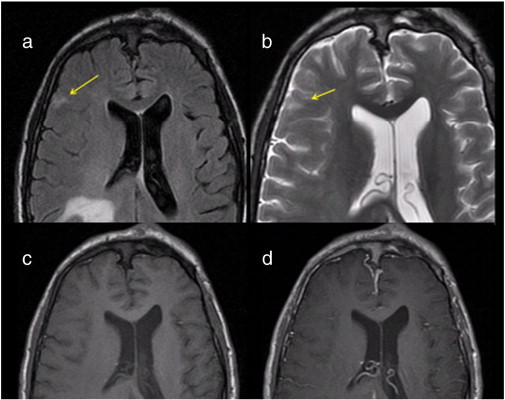
Cortical T2-hyperintense lesion suggestive of cortical dysplasia in a 19-year-old woman with NF-2. Transverse fast fluid-attenuated inversion-recovery (FLAIR) sequence (a) and transverse T2-weighted fast spin-echo sequence (b) demonstrate a hyperintense lesion in the right anterior frontal cortex which is wedge-shaped. Transverse T1-weighted sequence pre-gadolinium (c) and post-gadolinium (d) administration reveal that the lesion does not contrast-enhance.
Fig. 6.
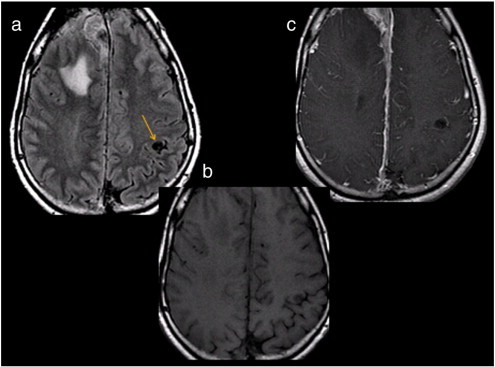
Purely cortical circular lesion in a 46-year-old man with NF-2. (a) Transverse fast fluid-attenuated inversion-recovery (FLAIR) sequence shows a well-circumscribed hypointense lesion in the left post-central gyrus (arrow). Transverse T1-weighted sequence pre-gadolinium (b) and post-gadolinium (c) administration reveal that the lesion is T1-hypointense (b) and does not enhance with contrast (c).
Fig. 7.
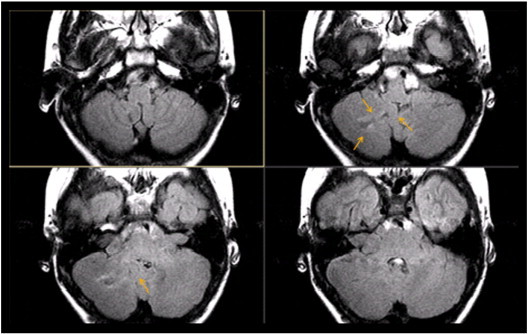
Transverse fast fluid-attenuated inversion-recovery (FLAIR) sequences in a 13-year-old boy with NF-2 show a hyperintense lesion within the right cerebellum suspicious for a disorder of neuronal migration (arrows).
Fig. 8.
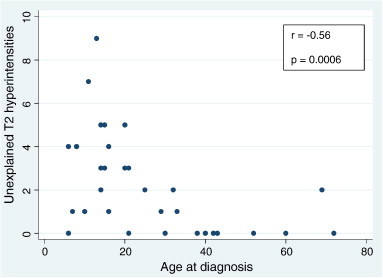
Scatterplot analysis of unexplained T2 hyperintensities as a function of age in 34 patients with NF-2. Younger age at diagnosis strongly correlated with higher number of incidental lesions (Spearman's r = − 0.56; p = 0.0006).
Microscopic examination of the deep gray matter of one patient in whom there was a “transmantle sign” on brain MRI suggestive of cortical dysplasia revealed micro-hamartomas without the classic histologic features of cortex dysplasia (Fig. 9).
Fig. 9.
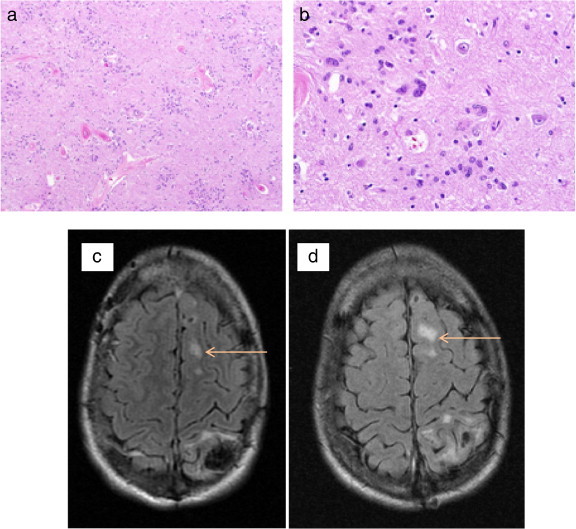
Micro-hamartomas in the brain of a 20-year-old man with NF-2. Low (a) and high (b) quality magnification of an area of the left thalamic deep gray matter in this patient with radiographic evidence of cortex dysplasia reveal dysplastic foci of glial cells without alterations of cytologic abnormalities or cortical architecture. Transverse fast fluid-attenuated inversion-recovery (FLAIR) sequences (c and d) demonstrate a cortical hyperintense lesion (arrows) suggestive of a cortical dysplasia in the left superior frontal gyrus in the same patient.
4. Discussion
In our study, we characterized unexplained T2 hyperintensities and other incidental findings seen in the brains of patients with NF-2. Our data suggest that in some patients with NF-2 and unexplained T2 hyperintensities, these lesions may represent an underlying disorder of neuronal migration. This finding has never been elucidated in humans with NF-2, though it has been previously reported (Ruggieri et al., 2005, Ruggieri et al., 2013, Wiestler et al., 1989). Interestingly though, animal studies have suggested that merlin may play a role in neuronal migration (Huynh et al., 1996). Huynh et al. (1996) sequenced the NF-2 gene in mice embryos and used in situ hybridization and anti-merlin antibodies to determine the developmental expression of the NF-2 gene. They found that in cells migrating from the ventricular zone to the cortical plate on embryonic days 15 and 16, merlin was present only in the cells of the intermediate zone and absent at both the ventricular zone and the cortical plate. The authors suggest that the tightly regulated expression of merlin during migration of cells to the cortical plate might predict cortical abnormalities in patients with NF-2 gene mutations (Huynh et al., 1996). A history of seizures has been reported in 4 to 24% of patients with NF-2 (Evans et al., 1999, Ruggieri et al., 2005), and it is tempting to attribute this epileptic tendency to cortical dysplasia. In our cohort, we did not find an associated tendency towards seizures or learning disabilities in individuals with imaging characteristics of cortex dysplasia, but this lack of correlation may reflect our small sample size.
Merlin is a cytoskeleton membrane scaffolding protein, and many protein factors are known to interact with merlin. Protection offered by the regulatory mechanisms associated with these protein factors may offer a possible mechanism for the lack of seizures in our patients with NF-2 and radiographic evidence of a FCD. Histopathologic differences between true focal cortical dysplasias and the cortical lesions we have observed in the brains of patients with NF-2 provide another possible explanation. Furthermore, cases of cortical dysplasia without seizures and with normal neurodevelopmental outcomes have been reported (Packard et al., 1997).
The focal cortical dysplasias (FCDs) can be classified into three major categories based on histopathologic findings (Blumcke et al., 2011). When a focal cortical dysplasia is either clinically or radiologically suspected but the lesion is not available for microscopic inspection, this is termed FCD Type III not otherwise specified (FCD Type III NOS). Therefore, almost all of our subcategory of patients with radiographic findings suspicious for FCD could be classified as FCD Type III NOS under the most recent classification system for focal cortical dysplasias (Bailey and Hermann, 1938). One patient for whom neuropathology was available was noted to have foci of dysplastic, immature neuroectodermal cells within the cerebral cortical deep gray matter, termed micro-hamartomas. Glial micro-hamartomas are common in NF-2, being found in at least all cases in one series and correspond to dysplastic foci of immature neuroectodermal cells in the cerebral cortex of these patients (Wiestler et al., 1989). These cells have been found at autopsy in the cerebral cortex and basal ganglia of patients with NF-2 (Wiestler et al., 1989, Bailey and Hermann, 1938, Orzechowski and Nowicki, 1912, Rubenstein, 1963, Rubenstein, 1986). These cells, although often polymorphic and multinuclear, have not shown mitotic activity or a tendency for neoplastic transformation (Wiestler et al., 1989). While our patient's MRI showed the classic “transmantle sign”, his histopathologic findings were not in keeping with FCDs that are typically associated with the “transmantle sign”. The “transmantle sign” refers to white matter signal alterations that taper from the crown of a gyrus or bottom of a sulcus towards the ventricle, reflecting the involvement of radial glialneuronal units. This “transmantle sign”, first described by Barkovich in 1997, is almost exclusively found in FCD Type IIb (Barkovich et al., 1997). Under the new FCD classification, FCD Type IIb is an isolated lesion characterized by cortical dyslamination and dysmorphic neurons with balloon cells (Blumcke et al., 2011). Balloon cells present with a large cell body and opalescent glassy eosinophilic cytoplasm (using H&E stain), which lacks Nissl substance. Our patient did not have any evidence of cytologic abnormalities or cortical architecture despite having the classic MRI finding, confirming the well accepted notion that the neuroimaging characteristics of the FCDs are neither consistent nor reliable (Blumcke et al., 2011). Among the reported MRI findings of FCDs are increased cortical thickness, blurring of the cortical-white matter junction, increased signal on T2 weighted images, a radially-oriented linear or conical transmantle stripe of T2 hyperintensity, cortical thinning, and localized brain atrophy (Blumcke et al., 2011). In keeping with previous descriptions of patients with NF-2 and radiographic evidence of FCD (Wiestler et al., 1989, Ruggieri et al., 2005, Ruggieri et al., 2013), our patients with radiographic evidence of cortex dysplasia did not have seizures or cognitive dysfunction. This lack of clinical correlation to what seemingly appears to be a focal cortical dysplasia on MRI may be explained by the lack of cytologic abnormalities or cortical architecture observed in one patient in this study with available neuropathology.
It is interesting to note that six patients with lesions suspicious for focal cortical dysplasias displayed a pattern of well circumscribed lesions with T1 and T2 hypointensity suggestive of mineralization. This radiologic pattern has been described in patients with focal cortical dysplasia type IIb (FCDIIb) with distinct allelic variants of the tuberous sclerosis complex 2 (TSC2) gene as identified by histopathologic genetic sequencing (Schonberger et al., 2009). It should also be noted that merlin has been shown to interact with hamartin, one of the protein products of the gene mutation seen in tuberous sclerosis (Rosner et al., 2008). The other diagnostic possibility for these 6 cases of well circumscribed lesions with T1 and T2 hypointensity suggestive of mineralization is meningioangiomatosis. Some pathology-proven cases of meningioangiomatosis look identical to our six cases (Izycka-Swieszewska et al., 2000a), and the association of meningioangiomatosis with NF-2 is well documented (Omeis et al., 2006).
The MRI pattern of mineralization within focal cortical dysplasias is atypical for FCDIIb as it differs from the typical MRI characteristic for FCDIIb of a funnel-shaped lesion tapering towards the lateral ventricle on T2 and FLAIR sequences, the so-called “transmantle sign” (Barkovich et al., 1997). Eight of our 15 patients (53%) with imaging characteristics of disorders of neuronal migration did display this classic “transmantle sign” suggestive of FCDIIb. It is interesting to note that two out of three (67%) patients with cerebellar migrational anomalies also displayed cortical migrational abnormalities.
We found a number of lesions which appeared to be T2 hyperintensities surrounding enlarged perivascular (Virchow–Robin) spaces. Dilated perivascular spaces are a normal variant on MR imaging, and they appear as well-defined round or linear lesions, isointense to CSF on all pulse sequences, and usually with no mass effect. Although the exact mechanism for the development of enlarged perivascular spaces remains unknown, mechanisms such as increased CSF pulsation, vascular ectasia or abnormality of arterial wall permeability may relate to their formation. Unlike NF-1, vascular malformations such as arteriovenous fistulas are not common in NF-2. Our findings of dilated Virchow–Robin spaces in NF-2 however have been described in the NF-2 literature (Fedi et al., 2009). Some authors have proposed that an obstructive mechanism for CSF accumulation is the basis of the enlarged perivascular spaces in NF-2 patients. Alternatively, it is possible that the enlarged perivascular spaces and meningioangiomatosis are both the result of an aberrant form of angiogenesis during development (Izycka-Swieszewska et al., 2000b).
In our study, we noted that younger age at time of diagnosis correlated with greater number of incidental brain lesions. This finding may be reflective of several processes. First, these children present at an earlier age presumably because of more severe phenotype which may mirror an aggressive overall radiological pattern. Alternatively, there may be a pubertal influence on the development of these brain MRI anomalies. Dermal neurofibromas have been found to increase during puberty (Duong et al., 2011) in patients with NF-1, and NF-related bright spots show a progressive increase during adolescence (Kraut et al., 2004) in NF-1. To our knowledge, the influence of puberty and other hormonal factors in NF-2 has not been studied.
We did not observe the development of new incidental lesions over time in our patients with baseline brain MRI abnormalities. In addition, with the exception of one patient who had received radiation therapy, the majority of patients in this study did not experience an increase in the size of T2 hyperintensities. While it is likely that radiation treatment worsened the size of T2 hyperintensities in this one patient out of the three total patients treated with radiation, overall we do not feel that radiation was the inciting factor for the development of incidental lesions in our cohort, as the majority of lesions were present prior to radiation therapy. Given these findings of relative lesion stability over time, we feel that brain MRI scanning outside of standard of care guidelines is unnecessary in patients with NF-2. The clinical utility of our work lies in the reassurance that can be given to patients and parents of children with NF-2 much in the same manner that NF-related bright spots are handled in NF-1. Our findings show that these incidental lesions in patients with NF-2 do not grow over time nor is there a change in other imaging characteristics, which would be expected if these abnormalities indeed represented neoplasms. This information is quite valuable given that patients with NF-2 are inherently predisposed to the formation of CNS neoplasms.
Our study had some limitations. First, the study was conducted in a retrospective manner. The retrospective nature precluded exact MRI-histopathologic correlation for the one study patient in whom autopsy was performed. Second, the study may have been subject to selection bias. Our centers are large, comprehensive neurofibromatosis centers and we see many patients with aggressive forms of NF-2. Third, we did not use quantitative MRI measures such as DTI or MTR which can probe the macromolecular and microstructural properties of brain tissue and would have allowed for better imaging specificity. Most importantly, the histopathologic data was available for only one study patient in whom the MRI was suggestive of a focal cortical dysplasia. Thus, broad conclusions about the significance of these MRI findings in patients with NF-2 cannot be drawn based on our study.
In conclusion, MRI findings suspicious for focal cortical dysplasias occur commonly in patients with NF-2, and these imaging findings may have histopathologic evidence of disruption of neuronal migration. This disruption is unlikely to contain the classic cyto-architectural hallmarks of true focal cortical dysplasia. Our study is the first to characterize unexplained T2 hyperintensities and other incidental findings in patients with NF-2 and the first to report MRI patterns typical of focal cortical dysplasia with histopathologic evidence of dysplastic cortical glial cells in a group of patients with neurofibromatosis type II. Further animal and histopathologic studies are needed to further delineate the role of merlin in cortical migration and the frequency of MRI findings suspicious for focal cortical dysplasias in patients with NF-2.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- Asthagiri A.R., Parry D.M., Butman J.A. Neurofibromatosis type 2. Lancet. 2009;373:1974–1986. doi: 10.1016/S0140-6736(09)60259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P., Hermann J.D. The role of the cells of Schwann in the formation of tumors of the peripheral nerves. Am. J. Pathol. 1938;14:1–37. [PMC free article] [PubMed] [Google Scholar]
- Barkovich A.J., Kuzniecky R.I., Bollen A.W., Grant P.E. Focal transmantle dysplasia: a specific malformation of cortical development. Neurology. 1997;49:1148–1152. doi: 10.1212/wnl.49.4.1148. [DOI] [PubMed] [Google Scholar]
- Blumcke I., Thom M., Aronica E. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc task force of the ILAE diagnostic methods commission. Epilepsia. 2011;52:158–174. doi: 10.1111/j.1528-1167.2010.02777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPaolo D.P., Zimmerman R.A., Rorke L.B., Zackai E.H., Bilaniuk L.T., Yachnis A.T. Neurofibromatosis type I: pathologic substrate of high-signal-intensity foci in the brain. Radiology. 1995;195:721–724. doi: 10.1148/radiology.195.3.7754001. [DOI] [PubMed] [Google Scholar]
- Duong T.A., Bastuji-Garin S., Valeyrie-Allanore L., Sbidian E., Ferkal S., Wolkenstein P. Evolving pattern with age of cutaneous signs in neurofibromatosis type 1: a cross-sectional study of 728 patients. Dermatology. 2011;222:269–273. doi: 10.1159/000327379. [DOI] [PubMed] [Google Scholar]
- Evans D.G.R., Birch J.M., Ramsden R.T. Paediatric presentation of type 2 neurofibromatosis. Arch. Dis. Child. 1999;81:496–499. doi: 10.1136/adc.81.6.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedi M., Kalnins R.M., Shuey N., Fitt G.J., Newton M., Mitchell L.A. Cystic meningioangiomatosis in neurofibromatosis type 2: an MRI-pathological study. Br. J. Radiol. 2009;82:e129–e132. doi: 10.1259/bjr/56536580. [DOI] [PubMed] [Google Scholar]
- Hagel C., Stemmer-Rachamimov A., Bornemann A. Clinical presentation, immunohistochemistry and electron microscopy indicate neurofibromatosis type 2-associated gliomas to be spinal ependymomas. Neuropathology. 2012;32:611–616. doi: 10.1111/j.1440-1789.2012.01306.x. [DOI] [PubMed] [Google Scholar]
- Huynh D.P., Tran T., Nechiporuk T., Pulst S. Expression of neurofibromatosis 2 transcript and gene product during mouse fetal development. Cell Growth Differ. 1996;7:1551–1561. [PubMed] [Google Scholar]
- Izycka-Swieszewska E., Rzepko R., Kopczynski S., Franc Z., Szurowska E., Borowska-Lehman J. Meningioangiomatosis with a predominant fibrocalcifying component. Neuropathology. 2000;1:44–48. doi: 10.1046/j.1440-1789.2000.00278.x. [DOI] [PubMed] [Google Scholar]
- Izycka-Swieszewska E., Rzepko R., Kopczynski S., Franc Z., Szurowska E., Borowska E. Meningioangiomatosis with a predominant fibrocalcifying component. Neuropathology. 2000;20:44–48. doi: 10.1046/j.1440-1789.2000.00278.x. [DOI] [PubMed] [Google Scholar]
- Kraut M.A., Gerring J.P., Cooper K.L., Thompson R.E., Denckla M.B., Kaufmann W.E. Longitudinal evolution of unidentified bright objects in children with neurofibromatosis-1. Am. J. Med. Genet. 2004;129A:113–119. doi: 10.1002/ajmg.a.20656. [DOI] [PubMed] [Google Scholar]
- Omeis I., Hillard V.H., Braun A., Benzil D.L., Murali R., Harter D.H. Meningioangiomatosis associated with neurofibromatosis: report of 2 cases in a single family and review of the literature. Surg. Neurol. 2006;65:595–603. doi: 10.1016/j.surneu.2005.09.034. [DOI] [PubMed] [Google Scholar]
- Orzechowski K., Nowicki W. Zur Pathogenese und pathologischen Anatomie der multiplen Neurofibromatose und der Sclerosis tuberosa (Neurofibromatosis universalis) Z. Gesamte Neurol. Psychiatr. 1912;11:16–307. [Google Scholar]
- Packard A.M., Miller V.S., Delgado M.R. Schizencephaly: correlations of clinical and radiologic features. Neurology. 1997;48:1427–1434. doi: 10.1212/wnl.48.5.1427. [DOI] [PubMed] [Google Scholar]
- Rosner M., Hanneder M., Siegel N., Valli A., Hengstschlager M. The tuberous sclerosis gene products hamartin and tuberin are multifunctional proteins with a wide spectrum of interacting partners. Mutat. Res. 2008;658:234–246. doi: 10.1016/j.mrrev.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Rouleau G.A., Merel P., Lutchman M. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363:515–521. doi: 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- Rubenstein L.J. In: Les Phakomatoses cerebrales. Miehaux L., Feld M., editors. SPEI Editeurs; Paris: 1963. Tumeurs et hamartomes dans la neurofibromatose central; pp. 427–451. [Google Scholar]
- Rubenstein L.J. The malformative central nervous system lesions in the central and peripheral forms of neurofibromatosis. A neuropathological study of 22 cases. Ann. N. Y. Acad. Sci. 1986;486:14–29. doi: 10.1111/j.1749-6632.1986.tb48058.x. [DOI] [PubMed] [Google Scholar]
- Ruggieri M., Iannetti P., Polizzi A. Earliest clinical manifestations and natural history of neurofibromatosis type 2 (NF2) in childhood: a study of 24 patients. Neuropediatrics. 2005;36:21–34. doi: 10.1055/s-2005-837581. [DOI] [PubMed] [Google Scholar]
- Ruggieri M., Gabriele A.L., Polizzi A. Natural history of neurofibromatosis type 2 with onset before the age of 1 year. Neurogenetics. 2013 May;14(2):89–98. doi: 10.1007/s10048-013-0354-0. [DOI] [PubMed] [Google Scholar]
- Schonberger A., Niehusmann P., Urbach H. Increased frequency of distinct TSC2 allelic variants in focal cortical dysplasias with balloon cells and mineralization. Neuropathology. 2009;29:559–565. doi: 10.1111/j.1440-1789.2009.01018.x. [DOI] [PubMed] [Google Scholar]
- Sener R.N., Dzelzite S., Migals A., Raits U., Veinbergs A.A. Prominent myelin vacuolization in neurofibromatosis type 2. J. Clin. Imaging. 2003;27:11–13. doi: 10.1016/s0899-7071(02)00501-6. [DOI] [PubMed] [Google Scholar]
- Trofatter J.A., MacCollin M.M., Rutter J.L. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72:791–800. doi: 10.1016/0092-8674(93)90406-g. [DOI] [PubMed] [Google Scholar]
- van Engelen S.J., Krab L.C., Moll H.A. Quantitative differentiation between healthy and disordered brain matter in patients with neurofibromatosis type I using diffusion tensor imaging. AJNR Am. J. Neuroradiol. 2008;29:816–822. doi: 10.3174/ajnr.A0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiestler O.D., von Siebenthal K., Schmitt H.P., Feiden W., Kleihues P. Distribution and immunoreactivity of cerebral micro-hamartomas in bilateral acoustic neurofibromatosis (neurofibromatosis 2) Acta Neuropathol. 1989;72:137–143. doi: 10.1007/BF00294370. [DOI] [PubMed] [Google Scholar]