

Abstract
Parkinson's disease and dementia with Lewy bodies are associated with abnormal neuronal aggregation of α-synuclein. However, the mechanisms of aggregation and their relationship to disease are poorly understood. We developed an in vivo multiphoton imaging paradigm to study α-synuclein aggregation in mouse cortex with subcellular resolution. We used a green fluorescent protein-tagged human α-synuclein mouse line that has moderate overexpression levels mimicking human disease. Fluorescence recovery after photobleaching (FRAP) of labeled protein demonstrated that somatic α-synuclein existed primarily in an unbound, soluble pool. In contrast, α-synuclein in presynaptic terminals was in at least three different pools: (1) as unbound, soluble protein; (2) bound to presynaptic vesicles; and (3) as microaggregates. Serial imaging of microaggregates over 1 week demonstrated a heterogeneous population with differing α-synuclein exchange rates. The microaggregate species were resistant to proteinase K, phosphorylated at serine-129, oxidized, and associated with a decrease in the presynaptic vesicle protein synapsin and glutamate immunogold labeling. Multiphoton FRAP provided the specific binding constants for α-synuclein's binding to synaptic vesicles and its effective diffusion coefficient in the soma and axon, setting the stage for future studies targeting synuclein modifications and their effects. Our in vivo results suggest that, under moderate overexpression conditions, α-synuclein aggregates are selectively found in presynaptic terminals.
Introduction
Alpha-synuclein is a 140 aa protein localized to vertebrate presynaptic terminals. Although its exact function is unclear, α-synuclein binds synaptic vesicle membranes and likely assists vesicle trafficking and SNARE complex formation (Goedert, 2001; Norris et al., 2004; Rizo and Sudhof, 2012). Increases in α-synuclein levels lead to its abnormal aggregation and neuronal degeneration both in vitro (Zach et al., 2007; Outeiro et al., 2008; Jiang et al., 2010) and in vivo (Masliah et al., 2000; Giasson et al., 2002; Emmer et al., 2011). For example, multiplication mutations that increase expression levels by 50–100% cause Parkinson's disease (PD) or dementia with Lewy bodies (DLB; Singleton et al., 2003; Chartier-Harlin et al., 2004). Interestingly, these mutations are also associated with Lewy bodies and neurites, identical to the more common sporadic forms of these diseases (Farrer et al., 2004). Because aggregated α-synuclein is the principal component of Lewy pathology, these observations strongly suggest that α-synuclein aggregation is a critical factor in the etiology of genetic and sporadic PD and DLB. Although much is known about factors that promote α-synuclein aggregation in vitro, including posttranslational modifications such as phosphorylation (Fujiwara et al., 2002), oxidization (Hashimoto et al., 1999), and particular point mutations (Conway et al., 1998), how these factors relate to aggregation in vivo is less well understood.
In cell culture, overexpression of human α-synuclein in hippocampal neurons leads to its presynaptic aggregation (Scott et al., 2010) and deficits in neurotransmitter release (Nemani et al., 2010; Scott et al., 2010). However, examination of α-synuclein aggregation in living brain tissue has been limited. To determine binding and aggregation properties of α-synuclein in vivo, we adapted multiphoton fluorescence recovery after photobleaching (FRAP) paradigms developed for studying fluorescent species in vitro (Brown et al., 1999; Mazza et al., 2008; Schnell et al., 2008; Sullivan and Brown, 2010) for in vivo studies of green fluorescent protein (GFP)-tagged human α-synuclein (Syn-GFP) in mouse cortex using cranial windows (Unni et al., 2010). Using the Syn-GFP transgenic mouse line and in vivo multiphoton imaging, we characterized in vivo Syn-GFP FRAP in mouse cortex and demonstrated that it is an accurate way to measure protein mobility. Under conditions of moderate α-synuclein overexpression (2- to 3-fold), levels similar to those seen in patients with multiplication mutations, the somatic Syn-GFP pool was entirely soluble and freely diffusible. In contrast, presynaptic Syn-GFP existed in at least three different pools, one of which was soluble and freely diffusible, whereas the other two had decreasing levels of mobility. Binding constant measurements for α-synuclein's association and dissociation with presynaptic vesicles and its effective diffusion coefficient in different subcellular compartments in vivo suggest potentially important tools for future studies of the human-disease-associated point mutations (Polymeropoulos et al., 1997; Krüger et al., 1998; Zarranz et al., 2004). Our results demonstrate that in vivo multiphoton FRAP is a powerful tool for studies of protein aggregation and binding in living mouse brain and that, under moderate overexpression conditions mimicking human disease, α-synuclein aggregation is selectively found at presynaptic terminals in vivo and is potentially related to their dysfunction.
Materials and Methods
Animals.
Syn-GFP (PDNG78; Rockenstein et al., 2005) heterozygous male mice were mated to BDF1 females from Charles River Laboratories and housed by Oregon Health and Science University's (OHSU's) Department of Comparative Medicine (DCM). Thy1-GFP M-line animals were used for GFP-only control experiments. Animals were held in a light-dark cycle, temperature- and humidity-controlled animal vivarium and maintained under ad libitum food and water diet supplied by the DCM. All experiments were approved by the OHSU Institutional Animal Care and Use Committee and every effort was made to minimize the number of animals used and their suffering. Animals between 1 and 18 months old were used, as specified in the text. Male and female mice were analyzed separately, but no significant differences were noted so combined results for both sexes are reported.
Cranial window surgery and in vivo imaging.
Cranial window surgery, in vivo imaging, and photobleaching were done similarly to previously published protocols (Unni et al., 2010). Briefly, isoflurane (1–2%)-anesthetized animals were placed in a custom-built stereotaxic frame and surgery was performed to create an ∼5-mm-diameter circular craniotomy overlying primary sensory and motor cortex, which was closed with an 8-mm-diameter coverglass and a custom-built aluminum fixation bar cemented into place. On imaging days, isoflurane-anesthetized animals were mounted again into the stereotaxic frame and imaged using a Zeiss LSM 7MP multiphoton microscope outfitted with dual channel BiG (binary GaAsP) detectors and the Coherent Technologies Chameleon titanium-sapphire femtosecond pulsed laser source (tuned to 860 nm). Zeiss Zen 2011 image acquisition software was used for all in vivo imaging. Baseline images of Syn-GFP-labeled pyramidal-shaped neurons and presynaptic terminals in cortical layers 2/3 were taken using 1–3 mW power (measured at objective exit). Square, circular, or line regions of interest (ROI) were used for photobleaching, with bleach times <5 ms and ∼100–150 mW power (measured at objective exit). Immediately after photobleaching, images were acquired to follow the recovery time course of fluorescence using the baseline settings. For chronic synapse imaging experiments, animals were remounted into the stereotaxic frame and cortical surface blood vessel patterns and Syn-GFP-positive cell body profiles were used to reidentify previously imaged terminals.
Imaging analysis.
Images were analyzed with ImageJ or Fiji software (Schindelin et al., 2012) to obtain mean fluorescence values in relevant ROIs over time. FRAP data were then analyzed in Prism 5 (GraphPad) to obtain single exponential fits to the recovery time course and the immobile and mobile fractions. Calculations of the effective diffusion coefficient (Deff) of Syn-GFP and bleach depth parameter β were made by fitting FRAP recovery curves using the least-squares curve fitting routine “lsqcurvefit” in MATLAB R2013a (The MathWorks) to previously established models for two-photon FRAP recovery kinetics in the soma (equation 8, Schnell et al., 2008) or in the axon (equation 1, Schmidt et al., 2007). Measurements of the bleaching radial and axial radii (ωr and ωz) needed for this fitting were made by photobleaching fixed tissue cortical sections from Syn-GFP mice using the same settings as used for the in vivo FRAP experiments. To check for possible excitation saturation during bleaching in our system, the bleach depth parameter β was measured as a function of bleaching laser power. This relationship was linear throughout the bleach power ranges used (data not shown), indicating the absence of excitation saturation.
Given that the immobile fraction of Syn-GFP present at terminals did not exchange with other Syn-GFP pools on the time scale of <24 h (see below), we calculated the fraction of free Syn-GFP available for binding to vesicles as the ratio of the amplitude of the fast-recovering component (tau ∼100 ms) over the sum of the amplitudes of the fast component and the next slower component (tau ∼2.6 min, representing the vesicle-bound fraction, see below). This free fraction is equal to koff/(kon* + koff), where koff is the first-order “off” rate constant of Syn-GFP unbinding from vesicles and kon* is the pseudo-first-order “on” rate constant for Syn-GFP binding to vesicles, assuming a constant concentration of vesicle-binding sites on this time scale. This equivalency can be used to derive the ratio kon*/koff. Given that diffusion is much faster than the binding event, we fitted the slower component terminal FRAP recovery curves to an established model for extracting koff values (Sprague et al., 2004) and could then calculate kon*. To determine the relationship between the immobile fraction and total Syn-GFP present at a synapse, we developed a metric for assessing total Syn-GFP by comparing the prebleach intensity of a particular synapse to the whole distribution of terminal intensities, which is positively skewed and invariant in shape between animals and at different ages (Unni et al., 2010). This “synapse position” or “Sp” number was calculated for each bleached synapse by comparing the mean prebleach intensity of that synapse to the mode in the synapse intensity distribution as measured by the number of Sp units by which it was brighter than the mode. One Sp unit is defined as the distance between the mode intensity and the point where the mode intensity had decreased by 90%. This metric has the advantage of comparing terminal intensities with the internal distribution from which they are drawn, which was stable between animals (Unni et al., 2010) and therefore useful for comparisons between animals for which absolute measured intensity values vary. For chronic terminal imaging, individual bleached synapse intensities were normalized to the average of the same two to four unbleached terminals in the field on multiple imaging days to control for possible differences in imaging conditions present on different days.
Mouse brain removal, fixation, and sectioning.
Whole brains were dissected from mice and the cerebellum and olfactory bulbs removed. Brains were divided into two along the midsagittal plane. Both hemispheres were placed in plastic scintillation vials with 4% paraformaldehyde and fixed for 1 h, 150 W, at 30°C using a BioWave (Pelco) microwave fixation system. Hemispheres were postfixed in 4% paraformaldehyde overnight at 4°C and then stored in PBS containing sodium azide (0.05%). Fixed hemispheres were mounted on a Leica VT1000 S Vibratome and sagittally sectioned into 50 μm slices. Slices were stored in PBS containing sodium azide at 4°C.
Proteinase K digestion.
Tissue sections were washed 3 times in PBS to remove azide and then incubated in 0.05% SDS in PBS ± 10 μg/ml proteinase K (Thermo Scientific) for 2 h at 37°C in the dark with shaking. Alternatively, free floating tissue sections were wet mounted on a slide using a Pap pen and incubated ± 10 μg/ml proteinase K in 0.05% SDS/PBS for 5 min at 60°C in a dark, humidified chamber on a hot plate. Sections were washed 3 times, mounted in Vectashield (Vector Laboratories), and imaged on a Zeiss LSM710 confocal microscope. For each condition, four cortical z-stack sections per animal for three animals were imaged and fluorescence was quantified for individual cell bodies and synapses using Fiji. Whole mouse brain was prepared for Western blot analysis via use of Syn-PER Synaptic Protein Extraction Reagent (Thermo Fisher), resulting in both a functional synaptosome fraction and a cytosolic fraction. Equal volumes of each fraction were then incubated with a 1:1 ratio of proteinase K stock solution (1 mg/ml proteinase K, 10 mm Tris-HCl, 20 mm CaCl, 50% glycerol in PBS) for 30 min at 37°C. Digestion was stopped by the addition of Novex Tris-glycine SDS sample buffer (Invitrogen) and subsequently heated to 95°C for 10 min. Samples were run on a 4–12% Tris-glycine gels (Invitrogen) for 2 h and 45 min at 80 V. Gels were transferred onto Immobilon-FL membranes (0.45 μm pore; Millipore) at 25 V for 18 h at 4°C. Membranes were blocked with Odyssey blocking buffer for 1 h at room temperature. Blots were probed with anti-GFP antibody (Abcam) overnight at 4°C at a 1:1000 dilution in blocking buffer. IR800 anti-rabbit IgG secondary antibody (Li-Cor) was used at a 1:15,000 dilution. Blot fluorescence was measured with a LI-COR Odyssey CLx and images analyzed using Image Studio software. The proteinase K-resistant fraction was measured as the ratio of treated versus untreated integrated band intensity.
Immunohistochemistry and confocal imaging.
Two anti-α-synuclein antibodies were used, mouse monoclonal Syn303 (Covance) raised against oxidized α-synuclein species and rabbit monoclonal pSyn (Abcam) raised against serine-129 phosphorylated α-synuclein. Rabbit anti-synapsin polyclonal antibody (Novex) was used to detect the synaptic vesicle protein synapsin 1 and mouse anti-α-synuclein monoclonal antibody Syn1 (BD Biosciences) was used to detect mouse and human α-synuclein. Then 50 μm fixed brain slices were washed 3 times in PBS, and incubated in blocking buffer (0.1% Triton-X, 10% goat serum) for 2 h while shaking in the dark. Primary antibodies were used at 3 μg/ml for Syn303 and 1.5 μg/ml for pSyn in a 1:5 dilution of blocking buffer and incubated with shaking in the dark overnight at 4°C. Tissue was washed in PBS 5 times for 30 min each. Secondary antibodies goat anti-mouse or goat anti-rabbit Alexa Fluor 647 (Invitrogen) were diluted to 1:2000 and subjected to incubated shaking in the dark overnight at 4°C. Tissue was washed in PBS 5 times for 30 min each at room temperature. Brain slices were mounted on slides in CFM-2 (Ted Pella), sealed with CoverGrip (Biotium), and allowed to dry overnight in the dark. Synapsin (0.4 μg/ml) and Syn1 (0.5 μg/ml) staining was performed using microwave-assisted immunohistochemistry as described previously (Ferris et al., 2009) with a modified primary antibody incubation step of 12 min (4 on, 4 off, 4 on), followed by a 5 min rest step to increase antibody binding. For confocal imaging, sections were imaged on a Zeiss LSM710 confocal microscope with a Plan-Apochromat 63×/1.40 oil objective. Laser powers of 1–5% were used to acquire z-stacks through 2–4 regions of cortex per tissue section. Fluorescence images were analyzed in Fiji, ImageJ, and Adobe Photoshop, with only linear scaling of the pixel values. To quantify colocalization at synapses, we used the CoLoc parameter in Imaris data analysis software with a GFP fluorescence threshold of 0.1% to limit the analysis to only synapses and an antibody fluorescence threshold of 5% fluorescence.
Electron microscopy and glutamate immunogold analysis.
Electron microscopy (EM) and glutamate immunogold analysis were done similarly to previously published protocols (Walker et al., 2012). Animals were perfused using a transcardiac approach with 0.5% paraformaldehyde/1% glutaraldehyde/0.1% picric acid in 0.1 m phosphate buffer, pH 7.3, at room temperature. The primary sensory and motor cortex was processed for EM pre-embed diaminobenzidine (DAB) immunolabeling for localization of GFP (GFP rabbit polyclonal antibody, 1:1000; Fitzgerald) using a microwave procedure. Tissue was incubated in the microwave (Pelco BioWave; Ted Pella) for 5 min, 550 W, at 35°C with the vacuum off (all the remaining steps occurred at this temperature) in 10 mm sodium citrate, pH 6.0 (antigen retrieval), rinsed in 0.1 m phosphate buffer (PB) for 2 × 1 min at 150 W with the vacuum off, exposed to 3% hydrogen peroxide at 150 W for 1 min with the vacuum on, rinsed in PB at 150 W for 2 × 1 min with the vacuum off, exposed to 0.5% Triton X-100 for 5 min, 550 W with the vacuum on, washed in PB for 2 × 1 min at 200 W with the vacuum off, then exposed to the primary antibody for 12 min at 200 W 4 times using the following cycle: 2 min on, 2 min off, 2 min on, 5 min off, all on a continuous vacuum. The tissue is then rinsed in PB twice at 1 min each at 150 W with the vacuum off, and then exposed to the secondary antibody (biotinylated goat anti-rabbit, 1:100; Vector Laboratories) for 15 min at 200 W for 2 cycles of the following: 4 min on, 3 min off, 4 min on, 5 min off, all on a continuous vacuum. The tissue is then rinsed in PB, 2 × 1 min, at 150 W with the vacuum off and then exposed to ABC (Vector Elite Kit, 1 μl/ml solution A and B in PB) for 11 min at 150 W under vacuum using the following cycle: 4 min on, 3 min off, 4 min on. The tissue was then rinsed in PB twice at 1 min each, at 150 W with the vacuum off and then exposed to DAB (0.5 μg/ml + 1.5% hydrogen peroxide) for up to 10 min at room temperature.
Postembedding immunogold electron microscopy was performed using a glutamate antibody (non-affinity-purified, rabbit polyclonal; Sigma). Thin sections (60 nm) were cut on an ultramicrotome (EM UC7; Leica) along the leading edge of the cortical tissue block, where layers I-VI were exposed, using a diamond knife (Diatome). The primary glutamate antibody, as described previously (Phend et al., 1992), was diluted 1:250 in TBST 7.6 and aspartate (1 mm) was added to the glutamate antibody mixture 24 h before incubation with the thin-sectioned tissue to prevent any cross-reactivity with aspartate within the tissue. The secondary antibody was goat anti-rabbit IgG (Jackson ImmunoResearch; diluted 1:25 in TBST, pH 8.2) tagged with 12 nm gold particles. We previously reported that incubation of the antibody with 3 mm glutamate resulted in no immunogold labeling, showing the specificity of the glutamate labeling (Meshul et al., 1994). Photographs (10/animal) were taken on a JEOL 1400 transmission electron microscope of Syn-GFP-labeled and Syn-GFP-unlabeled terminals from a single 50 mesh grid (1 thin section/grid, 1 photograph/grid square) throughout the neuropil (an area containing the highest numbers of synapses) of layers II and III at a final magnification of 40,000× by an individual blinded to the experimental groups, using a digital camera (Advanced Microscopy Techniques). For all Syn-GFP-immunolabeled terminals and non-Syn-GFP-labeled terminals, the density of immunogold labeling was determined in each terminal at the ages mentioned.
For quantification of glutamate labeling, the number of immunogold particles located within or at least touching the synaptic vesicle membrane (i.e., vesicular pool), the number located outside the synaptic vesicles (i.e., the cytoplasmic pool), and those associated with mitochondria were counted. The vesicular and cytoplasmic pools were combined because the cytoplasmic pool is very small (<10%) compared with the vesicular pool (Meshul et al., 1999). We have reported that nerve terminals making a symmetrical contact contain GABA (Meshul et al., 1999), the precursor for which is glutamate. Therefore, nerve terminals making a symmetrical contact will naturally contain some glutamate immunolabeling and cannot be considered immunonegative as a way of determining a ratio between glutamatergic and GABAergic terminals (Meshul et al., 1994, 1999). The metabolic pool is also relatively small and thus unlikely to be a major source of variation in labeling intensity. The density of gold particles per square micrometer of nerve terminal area was determined for each animal and the mean density for each group calculated. Background labeling was determined within glial cell processes and was found to be 10 immunogold-labeled particles per square micrometer (Meshul et al., 1994). This was subtracted from the density of presynaptic immunogold-labeled glutamate within the nerve terminals.
Statistics.
All data are reported as the mean ± SD unless otherwise noted. Numbers and statistical tests used are reported in each relevant section.
Results
Somatic α-synuclein exists in a single soluble pool
To measure potential fast dynamics of Syn-GFP within the cell bodies of layer 2/3 primary sensory and motor cortex neurons in our mice, we performed a series of FRAP experiments in line scan mode that allowed us to acquire recovery data with a time resolution of up to 1 ms. Syn-GFP demonstrated a fast recovery tau that was well fit by a single exponential (23.4 ± 7.4 ms, n = 7 cells, 4 animals; Fig. 1A,B), similar to recovery in a different mouse line expressing soluble, monomeric GFP within cortical neurons (26.7 ± 8.2 ms, n = 12 cells, 2 animals, p > 0.40; Fig. 1A,B). This suggests that somatic Syn-GFP, like free GFP, exists predominantly in a soluble, freely diffusible state without appreciable binding to somatic membranous compartments. We also measured FRAP recovery curves for Syn-GFP using square ROIs to estimate Deff in vivo. Square ROIs were used because this simplifies the assessment of model parameters needed for estimating Deff. We fit recovery curves using established methods for extracting Deff from multiphoton FRAP data (Brown et al., 1999; Schnell et al., 2008; Sullivan and Brown, 2010; Fig. 1C) to calculate the in vivo Deff of Syn-GFP (see Materials and Methods). The in vivo Deff for somatic Syn-GFP was 8.6 ± 3.8 μm2/s (n = 14 cells, 4 animals), which was somewhat less than predicted for a soluble, monomeric Syn-GFP fusion protein (∼13–34 μm2/s) based on the molecular weight of the fusion protein and Deff of cytoplasmic GFP in cultured cells (Yokoe and Meyer, 1996; Arrio-DuPont et al., 2000; Sprague et al., 2004; ∼15–40 μm2/s).
Figure 1.

Neuronal cell body in vivo multiphoton FRAP. A, Left, Top, Individual cortical neuron soma expressing Syn-GFP (Left) or GFP-only (Right) is shown with the soma outlined in red and a yellow line representing location of line scan used for imaging and bleaching. Scale bar, 2.5 μm. Bottom, X–T plot of line scan data showing baseline, bleach (arrow), and postbleach recovery. Middle and Right, FRAP recovery curves with single exponential fits and calculated tau values for cells shown on left. Each black circle represents the average line scan intensity value from a single time point from a single cell. B, Group data of calculated tau values for Syn-GFP and GFP-only cell bodies. C, Square ROI bleach data from the soma of an individual Syn-GFP neuron fit to extract the value for Deff (see Materials and Methods).
In vivo FRAP accurately measures α-synuclein mobility in mouse cortex
Our previous work suggested that FRAP could be a useful tool for studying mechanisms of α-synuclein aggregation in the living mouse brain on the time scale of minutes (Unni et al., 2010). However, to assess FRAP recovery of Syn-GFP on a millisecond time scale, one must consider the possibility of reversible dark state recovery of GFP. In addition to irreversible GFP photobleaching, under certain conditions, photobleaching GFP temporarily converts it into a reversible “dark state” (Swaminathan et al., 1997; Mueller et al., 2012) that recovers on the order of hundreds of microseconds to milliseconds.
We performed several experiments to test for possible dark state recovery. In FRAP experiments in which signal recovery is dominated by protein movement from unbleached regions into the bleached region, recovery will be slower with larger bleach ROIs. In contrast, reversible dark state recovery of GFP involves an intramolecular reaction that is independent of the size of the bleached area (Swaminathan et al., 1997). Therefore, we bleached different-sized ROIs within the same neuron sequentially, measuring the rate of recovery for each bleach size (Fig. 2A). Syn-GFP FRAP showed a slower recovery tau when a 4-fold larger bleach area was compared with the smaller bleach area (mean tau ratio large ROI/small ROI = 1.79, 95% confidence interval = 1.41–2.16, range = 1.03–3.02, n = 13 cells, 4 animals; Fig. 2B). A plot of the absolute recovery tau versus bleach area also showed a significant positive correlation, as expected for movement of unbleached Syn-GFP into the bleached area (r2 = 0.38, p < 0.001, n = 26, 4 animals; Fig. 2C). Finally, we saw no recovery of Syn-GFP FRAP in fixed tissue sections of cortex (data not shown). Dark state recovery of GFP can occur in fixed tissue (Mueller et al., 2012) in which protein movement on the micron scale is precluded, strongly suggesting that the rapid recovery of somatic Syn-GFP FRAP signal that we measured represents true protein movement within neurons.
Figure 2.

FRAP recovery as a function of bleached area. A, Left, Individual cortical neuron soma expressing Syn-GFP is shown before and after a small (top) and four times larger (bottom) square ROI bleach pulse (red square). Scale bar, 3 μm. Right: FRAP recovery curves from cell shown at left fit with single exponentials. B, Ratio of the large over small bleach area tau values calculated for each cell all demonstrate values >1. C, Recovery tau values as a function of bleach area show a significant linear relationship.
Presynaptic terminal α-synuclein exists in at least three different pools, each with different mobility
To measure Syn-GFP mobility in vivo within presynaptic terminals, as defined as fluorescent neuropil puncta (Unni et al., 2010), we used FRAP in line scan mode. Unlike the soma, individual terminals showed slower recovery that was not well fit by a single exponential. The fastest component in terminals was well fit by a single exponential with a tau of 102 ± 51 ms (n = 11 synapses, 4 animals; Fig. 3A). This component comprised only 23 ± 9% of total terminal Syn-GFP. In contrast, bleaching of free GFP in presynaptic terminals recovered completely with a tau of 122 ± 62 ms (n = 9 synapses, 2 animals, p > 0.43, t test; Fig. 3B). This suggests that only a small fraction of Syn-GFP in presynaptic terminals is soluble and freely diffusible. Although the rate of recovery for the fast component in presynaptic terminals was slower than the soma, this difference is likely due to geometrical barriers to diffusion present in thin axonal processes rather than binding interactions. Further support for this idea came from measuring Deff in the axonal compartment using multiphoton FRAP recovery (Schmidt et al., 2007) compared with the measured value of Deff in the soma. Measuring axonal Deff in this way requires assuming one-dimensional diffusion along thin axonal processes, which was validated for parvalbumin in axons of cerebellar Purkinje neurons in acute slices (Schmidt et al., 2007). We measured an axonal Deff for Syn-GFP of 9.0 ± 6.6 μm2/s (n = 11 synapses, 3 animals; Fig. 3C,D), which was very similar to the measured Deff in the soma (8.6 ± 3.8 μm2/s, p > 0.85, t test; Fig. 3D). This similarity suggests that a portion of terminal Syn-GFP is freely diffusible and that the one-dimensional diffusion approximation for soluble protein mobility in the axon is valid in vivo, as it is in slices (Schmidt et al., 2007). Although measurements from small axonal processes potentially suffer from lower signal-to-noise levels, our data show that multiphoton imaging provides high-quality data from even these small volume structures. A rough estimate of the magnitude of the constraint that axonal geometry places on the diffusion of a soluble protein of this size in vivo can be made by taking the ratio of the recovery tau measured using line scan FRAP in the presynaptic terminal and the soma, which demonstrates a ∼5-fold slower recovery in the axonal compartment.
Figure 3.

Fast synaptic terminal in vivo multiphoton FRAP imaging. A, Left, Top, Set of cortical terminals expressing Syn-GFP is shown with line representing location of line scan used for imaging (yellow) and bleaching (red). Scale bar, 1.5 μm. Bottom, X–T plot of line scan data showing baseline, bleach (arrow), and postbleach recovery. Right, Individual terminal fluorescence before and after bleaching pulse with single exponential fit to FRAP recovery curve (inset). B, Group data of calculated tau values for Syn-GFP and GFP-only expressing terminals. C, Individual terminal Syn-GFP FRAP recovery data fit to extract the value for Deff (see Materials and Methods). D, Group data of calculated Deff values for Syn-GFP in the soma and terminals.
Using FRAP with circular ROIs encompassing individual terminals (Unni et al., 2010), we measured two additional components of Syn-GFP recovery, one well fit by a single exponential recovery tau of 2.6 ± 2.5 min (n = 22 synapses, 7 animals) comprising 45 ± 21% of total terminal Syn-GFP (n = 19 synapses, 5 animals; Fig. 4A,B). The remaining 32 ± 21% of total terminal Syn-GFP (n = 19 synapses, 5 animals) did not recover over the course of 10 min, comprising essentially an immobile fraction (IF) on this time scale. In contrast, circular ROI bleaching studies of individual terminals in GFP transgenic animals demonstrated a much faster recovery tau of 5.1 ± 3.7 s (n = 6 synapses, 2 animals), consistent with a homogenous population of soluble GFP. The recovery of soluble GFP was slower than in our line scan experiments, likely because of differences in bleach area and time when using the two different bleach ROI conditions. The IF of Syn-GFP in each terminal (ranged from 0 to 83%) and was correlated with the total amount of Syn-GFP present before bleaching (r2 = 0.26, p < 0.03, n = 19 synapses, 5 animals; Fig. 4C), suggesting that synapses with greater Syn-GFP have more immobilized protein. In contrast, there was no correlation between the recovery tau and IF size at each synapse (r2 = 0.001, p > 0.89, n = 19 synapses, 5 animals). Next we used a slightly modified technique that bleached a larger square area ROI (53×53 μm2) encompassing 50–100 terminals simultaneously. This allowed us to measure FRAP recovery data on a pooled group of synapses more efficiently to get better estimates of the population as opposed to bleaching each terminal one-by-one. The terminal recovery parameters measured in this way were indistinguishable from our individual terminal bleaching experiments (individual terminal FRAP recovery tau = 2.6 ± 2.5 min, n = 22 synapses, 7 animals; area FRAP recovery tau = 1.9 ± 1.3 min, n = 78,652 μm2, 9 animals; t test p > 0.47; individual terminal FRAP IF = 42 ± 21%, n = 19 synapses, 5 animals; area FRAP IF = 49 ± 11%, n = 78,652 μm2, 9 animals; t test p > 0.42). Using this approach, we measured FRAP parameters from animals of different ages, including 1, 3–6, and 12–18 months of age (1 month IF: 45 ± 10%, 3–6 month IF: 48 ± 1%, 12–18 month IF: 55 ± 17%, one-way ANOVA p > 0.62; Fig. 4D; 1 month tau: 1.2 ± 0.9 min, 3–6 month tau: 1.8 ± 1.0 min, 12–18 month tau: 2.8 ± 1.4 min, one-way ANOVA p > 0.18; Fig. 4E; n = 3 animals per age group). Interestingly, the IF and recovery tau values measured did not vary significantly over this age range.
Figure 4.
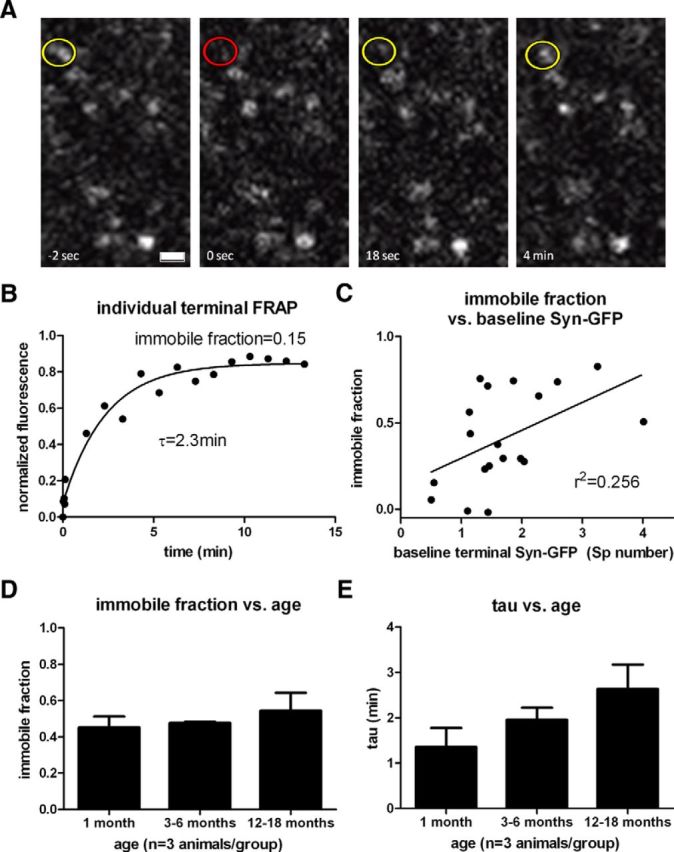
Slow synaptic terminal in vivo multiphoton FRAP imaging. A, Set of cortical terminals expressing Syn-GFP is shown with circle demonstrating individual terminal before (yellow) and immediately after (red) bleach pulse and during recovery phase (yellow). Scale bar, 1.5 μm. B, Individual terminal FRAP recovery curve with single exponential fit and immobile fraction. C, Immobile fraction plotted as a function of the prebleach Syn-GFP terminal intensity metric (see Materials and Methods) demonstrates a significant linear relationship. D, Group data of terminal IF measured at different ages. E, Group data of terminal recovery tau measured at different ages.
Using the percent free and vesicle-bound Syn-GFP in the terminal, the ratio of free/(free + vesicle bound) Syn-GFP was 34 ± 17%. Assuming a simple second-order binding reaction for association of Syn-GFP with vesicles (see Materials and Methods), we calculated the ratio of kon*/koff to be 1.96 ± 1.10 in vivo. This calculation assumes a constant concentration of vesicle binding sites on this time scale. Given that diffusion is fast compared with the binding rate, we used established FRAP analysis methods (Sprague et al., 2004) to obtain koff = 6.47 ± 3.74 × 10−3 sec−1 and kon* = 12.7 ± 10.2 × 10−3 sec−1.
Long-term imaging of individual bleached Syn-GFP-positive terminals over the time scale of several hours to 1 week revealed that there was no further recovery of FRAP signal after the first several minutes or over the course of 1–2 h after the bleach nor was further fluorescence recovery seen 24 h later. However, 7–9 d after bleaching, some terminals demonstrated full recovery (Fig. 5A,B), whereas other terminals showed only partial or no recovery of the IF. On average, presynaptic Syn-GFP signal had recovered to 67 ± 25% of total Syn-GFP by 1 d after bleaching and was significantly higher (79 ± 34% of total Syn-GFP) at 7–9 d after bleaching (n = 20 synapses, 3 animals, paired t test p < 0.02; Fig. 5C). Therefore, ∼36% of the IF from the first 10 min after the bleach eventually recovered after 7–9 d. This indicates that the IF component is heterogeneous, with more than one Syn-GFP exchange rate.
Figure 5.

Long-term synaptic terminal in vivo multiphoton FRAP imaging. A, Set of cortical terminals expressing Syn-GFP is shown with circle demonstrating individual terminal before (yellow), immediately after (red) bleach pulse, and during recovery phase (yellow). Scale bar, 2 μm. B, Individual terminal FRAP recovery curve measured over seconds, hours, and days time scales. C, Group terminal FRAP data over multiple time scales demonstrates significant recovery of a portion of the immobile fraction between 1 and 7–9 d postbleach. Error bars indicate SEM.
Pool of presynaptic α-synuclein is proteinase K resistant
Although in vivo multiphoton FRAP is a useful tool for measuring the mobility of different Syn-GFP species at presynaptic terminals, the molecular nature of these different pools is unclear. To better characterize Syn-GFP in cortical tissue, we treated fixed tissue sections and synaptosome-fractionated fresh brain tissue with proteinase K. Then, tissue sections were imaged with confocal microscopy and synaptosome fractions assayed by Western blotting to determine whether a proteinase K-resistant fraction was present. Proteinase K-resistance is a well established hallmark of certain aggregated species of α-synuclein (Takeda et al., 1998; Miake et al., 2002) and other neurodegeneration-associated proteins (Neumann et al., 2002). Although both somatic staining and the cytosolic fraction of Syn-GFP were almost completely abolished by proteinase K treatment (normalized somatic Syn-GFP signal in fixed tissue after proteinase K = 0.042 ± 0.041, n = 3 animals; normalized cytosolic fraction Syn-GFP signal in freshly prepared tissue after proteinase K = 0.015 ± 0.013, n = 3 animals), a significant fraction of presynaptic terminal Syn-GFP was not degraded by proteinase K (normalized terminal Syn-GFP signal after proteinase K = 0.44 ± 0.21, n = 3 animals, paired t test p < 0.02; normalized synaptosome fraction Syn-GFP signal after proteinase K = 0.35 ± 0.13, n = 3 animals, t test p < 0.02), suggesting that aggregates were present only in presynaptic terminals (Fig. 6A–C). Mean Syn-GFP terminal intensity comparing slices from the same animal showed that proteinase K treatment resulted in a decrease in the frequency of “dim” terminals, with a relative sparing of the “bright” terminals (Fig. 6D), suggesting that terminals with high levels of Syn-GFP contain more of the aggregate form. We also measured the average terminal volume of Syn-GFP labeling under control conditions and after proteinase K treatment. Interestingly, proteinase K treatment reduced terminal Syn-GFP volume by ∼25% (average control volume = 0.41 ± 0.54 μm3, n = 46,117 terminals, 3 animals; average proteinase K-treated volume = 0.31 ± 0.17 μm3, n = 24,009 terminals, 3 animals). Next, we measured the fraction of proteinase K-resistant Syn-GFP in terminals in different age groups. When comparing mice aged 1, 3–6, and 12–18 months, we detected no significant differences in the proteinase K-resistant fraction (data not shown) in these age groups, in agreement with our in vivo imaging data showing a similar IF across the ages tested.
Figure 6.
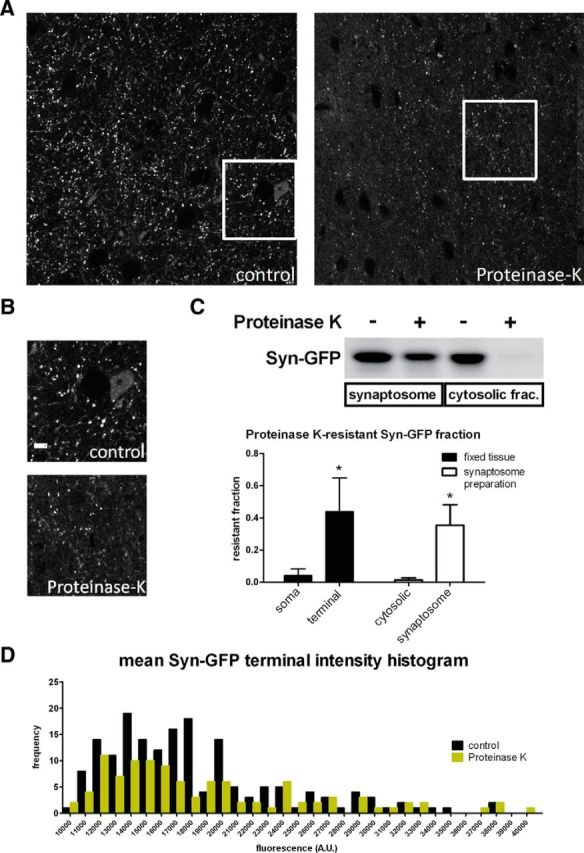
Proteinase K digestion of fixed and fresh tissue. A, Region of cortex from fixed tissue demonstrates Syn-GFP signal in terminals, axons, and neuronal cell bodies under control conditions (Left) and after exposure to proteinase K (Right). Scale bar, 10 μm. B, Boxed regions in A are shown at higher magnification. Scale bar, 6 μm. C, Top, Western blot showing Syn-GFP levels in both synaptosome and cytosolic fractions with and without proteinase K treatment. Bottom, Group data of proteinase K-resistant Syn-GFP fractions in neuronal somata and terminals from fixed tissue confocal image analysis and in cytosolic and synaptosome fractions from Western blot analysis. D, Histogram of mean Syn-GFP intensity in individual terminals is shown for control conditions and with proteinase K treatment. High-Syn-GFP expressing terminals (toward right side of x-axis) are relatively preserved in frequency compared with lower expressing terminals (left side of axis).
Pool of presynaptic α-synuclein is phosphorylated at serine-129, oxidized, and associated with decreases in the presynaptic vesicle protein synapsin
To characterize the Syn-GFP aggregates in terminals, we stained fixed tissue for several molecular markers of specific aggregates. Serine-129 phosphorylation is a marker of α-synuclein aggregation and serine-129 phospho-antibodies stain Lewy pathology from human tissue (Fujiwara et al., 2002) and aggregated α-synuclein in some mouse models (Emmer et al., 2011; Luk et al., 2012a,b). However, recent work has suggested that serine-129 phosphorylation is required for autophagy-mediated α-synuclein degradation and marks a pool that is not necessarily aggregated (Oueslati et al., 2013). In addition, in some mouse models (Tanji et al., 2010), there is evidence that presynaptic terminal proteinase K-resistant α-synuclein aggregates can exist without being phosphorylated at serine-129. Oxidization of α-synuclein is another modification associated with human disease and this form is also present in Lewy pathology (Giasson et al., 2000). Therefore, we next measured the serine-129 phosphorylation and oxidation status of terminal Syn-GFP aggregates in our system.
In our experiments, a subset of Syn-GFP-positive presynaptic terminals in the cortex colabeled with serine-129 phosphorylated α-synuclein (Fig. 7A). However, the phosphorylated form was not detected in neuronal cell bodies (data not shown). Likewise, staining for oxidized α-synuclein colabeled with some Syn-GFP-positive presynaptic terminals (Fig. 7A), but not in cell bodies. There was also a significant positive correlation between the amount of serine-129-phosphorylated α-synuclein and the total Syn-GFP at each terminal, indicating that terminals with greater amounts of Syn-GFP contained more phosphorylated protein (r2 = 0.34, p < 0.0003, n = 37 synapses; Fig. 7B). These results are consistent with the correlation between total Syn-GFP levels and the IF measured by in vivo imaging (Fig. 4C), indicating the presence of more aggregated protein in brightly labeled Syn-GFP terminals. To quantify the amount of colocalization between Syn-GFP and phospho-synuclein, we calculated the Pearson's colocalization coefficient using Imaris software and found a significant amount of colocalization in Syn-GFP-positive presynaptic terminals (Pearson's coefficient = 0.194 ± 0.042, n = 3 animals, 4 cortical areas per animal; Fig. 7C). Next, we measured serine-129-phosphorylated α-synuclein levels in cortical terminals treated with proteinase K to determine whether the proteinase K-resistant fraction is phosphorylated. Using our protocol, phospho-synuclein staining was abolished by proteinase K treatment (data not shown). This suggests that these two aggregate pools are separate and that the proteinase K-resistant fraction is not phosphorylated at this residue, as is seen for presynaptic terminal aggregates in other human α-synuclein overexpression transgenic models (Tanji et al., 2010). Furthermore, markers of amyloid-like aggregate species, thioflavine S (ThS), and mature Lewy pathology (anti-ubiquitin antibody staining) showed no clear colocalization with Syn-GFP-positive terminals (data not shown). Combined, these data suggest that the proteinase K-resistant, serine-129-phosphorylated, oxidized Syn-GFP presynaptic aggregate pools are not ubiquitinated or in an amyloid (ThS-binding) conformation.
Figure 7.
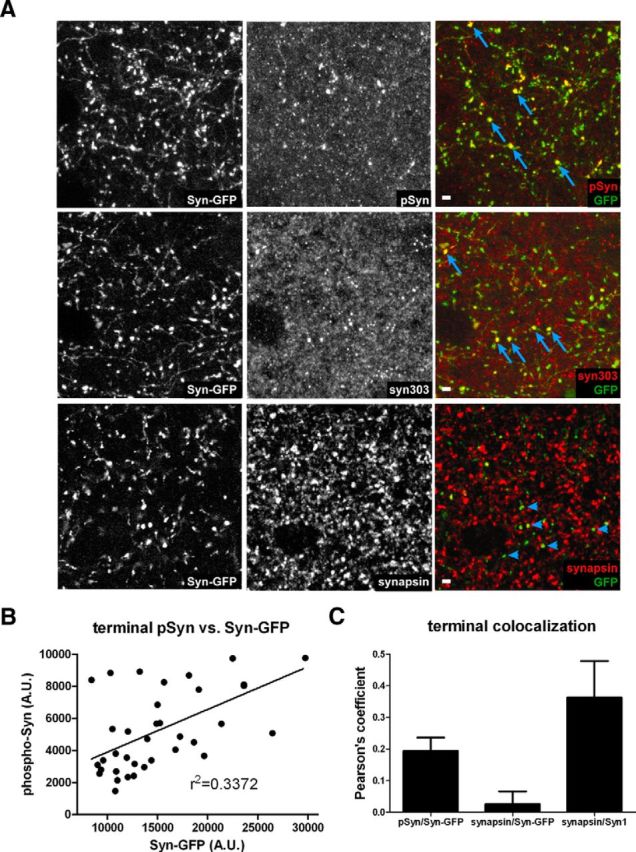
Immunohistochemistry of terminals. A, Top, Three panels showing Syn-GFP expression in axons and terminals (Left), staining for serine-129 phosphorylated α-synuclein (pSyn, Middle), and the overlay (Right). Middle, Three panels showing Syn-GFP expression in axons and terminals (Left), staining for oxidized α-synuclein (syn303, Middle), and the overlay (Right). Bottom, Three panels showing Syn-GFP expression in axons and terminals (Left), staining for the synaptic vesicle protein synapsin (synapsin, Middle), and the overlay (Right). Individual terminals demonstrating colocalization of Syn-GFP and antibody are marked with arrows and lack of colocalization is marked with arrowheads. Scale bar, 5 μm. B, Serine-129-phosphorylated α-synuclein (pSyn) levels at each terminal plotted as a function of the Syn-GFP terminal intensity at that synapse demonstrates a significant positive linear relationship. C, Pearson's colocalization coefficients for pSyn/Syn-GFP, synapsin/Syn-GFP, and synapsin/Syn1.
Our data presented above from in vivo imaging and proteinase K digestion suggests that an aggregated pool of Syn-GFP in presynaptic terminals forms as early as 1 month after birth in this line and is present at relatively constant levels until the animals are >1 year-old. We next investigated whether these aggregates might be associated with evidence of terminal dysfunction, as has been suggested to occur when α-synuclein is overexpressed (Scott et al., 2010; Larson et al., 2012; Scott and Roy, 2012). For example, Roy et al. demonstrated what they termed “vacant synapses” in a subset of terminals expressing Syn-GFP aggregates, which contained greatly reduced levels of several presynaptic vesicle proteins involved in exocytosis (Scott et al., 2010). We determined the levels of the vesicle protein synapsin in Syn-GFP-expressing versus Syn-GFP-nonexpressing cortical terminals (Fig. 7A) and found that Syn-GFP-expressing terminals had a large decrease in synapsin staining in all age groups tested (1, 3–6, or 12–18 months old; data not shown). The Pearson's coefficient for colocalization between Syn-GFP-positive terminals and synapsin was close to zero, indicating that the proteins do not colocalize (Pearson's = 0.026 ± 0.040, n = 3, 1 year-old animals, 2 cortical areas per animal; Fig. 7C). In contrast, synapsin did colocalize with an antibody that recognizes mouse α-synuclein (Syn1), indicating that adjacent neurons that do not express the transgene likely have healthy synaptic function (Pearson's = 0.36 ± 0.12, n = 3, 1-year-old animals, 2 cortical areas per animal; Fig. 7C). These data suggest that presynaptic Syn-GFP aggregates are toxic to the normal exocytotic apparatus.
Ultrastructural analysis demonstrates vesicle-bound and granular aggregate forms of terminal α-synuclein
To characterize the ultrastructural properties of Syn-GFP in cortical presynaptic terminals, we used EM pre-embedding DAB immunolabeling (Meshul and McGinty, 2000) with an anti-GFP antibody and a newly developed microwave fixation/processing procedure (Walker et al., 2012). Syn-GFP-containing terminals showed two different patterns of DAB staining (Fig. 8A). The more common pattern was perivesicular labeling of Syn-GFP, which likely corresponds to the presumed synaptic vesicle binding pool of Syn-GFP detected with in vivo FRAP. Association of α-synuclein with synaptic vesicles has been described previously (Maroteaux and Scheller, 1991; Shibayama-Imazu et al., 1993). We also observed a more granular pattern of Syn-GFP staining not associated with vesicles in a subset of terminals making both an inhibitory (symmetrical; Fig. 8A) and excitatory (asymmetrical; Fig. 8B) synaptic contacts. This appearance resembled the granular aggregates of α-synuclein seen in human (Sone et al., 2005) and mouse (Rockenstein et al., 2005; Rieker et al., 2011) tissue. This granular pattern may represent the aggregated, immobile Syn-GFP pool observed in our in vivo FRAP and ex vivo proteinase K experiments.
Figure 8.
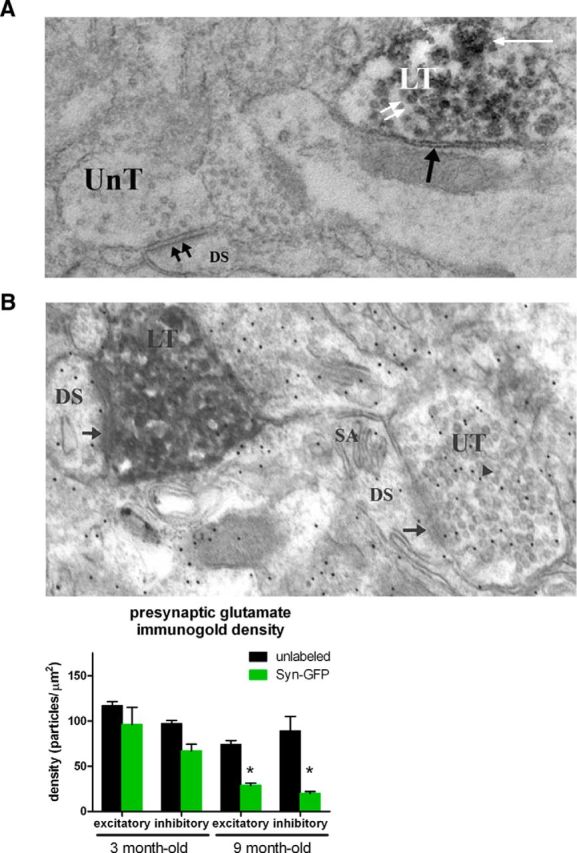
Electron microscopy of terminals. A, Electron photomicrograph showing unlabeled terminal (UnT) making an asymmetrical (double black arrows) synaptic contact onto a dendritic spine (DS), indicating that the synapse is excitatory. Also in the field is a DAB-labeled terminal (LT) making a symmetrical (inhibitory) synaptic contact (single black arrow) and containing a perivesicular (double white arrows) and granular (single white arrow) pattern of labeling. B, Top, Electron photomicrograph showing an unlabeled nerve terminal (UT) making an asymmetrical synaptic contact (arrow) onto a DS containing a spine apparatus (SA). Within the unlabeled nerve terminal are numerous 12 nm gold particles (arrowhead) indicating the location of an antibody against glutamate. There is also a DAB/α synuclein-GFP-labeled nerve terminal (LT) making an asymmetrical synaptic contact (arrow) onto a DS. Bottom, Group data from animals of two different age groups, 3 and 9 months old, showing a significant decrease in presynaptic glutamate levels in 9-month-old Syn-GFP-positive excitatory (asymmetrical) and inhibitory (symmetrical) nerve terminals.
Glutamate immunogold labeling demonstrates progressive dysfunction of α-synuclein-overexpressing terminals
Several groups of investigators have suggested that α-synuclein overexpression can lead to synaptic dysfunction that can be measured at the ultrastructural level in different systems (Scott et al., 2010, Scott and Roy, 2012; Larson et al., 2012). We performed quantitative immunogold analysis in the cortex of our transgenic mice to detect presynaptic glutamate in Syn-GFP-labeled versus Syn-GFP-unlabeled terminals at different ages. Glutamate immunogold labeling can be used to detect this neurotransmitter in excitatory and inhibitory nerve terminals, because glutamate is the direct precursor to GABA, the main inhibitory neurotransmitter in mammalian cortex (Meshul et al., 1994; Bamford et al., 2004). Our immunogold labeling in 3-month-old animals showed no significant differences in the density of particles in both asymmetric (unlabeled: 117 ± 4.6 particles/μm2, Syn-GFP-labeled: 96 ± 19, t test p > 0.78, n = 3 animals; Fig. 8C, error = SEM) and symmetric (unlabeled: 97 ± 3.5 particles/μm2, Syn-GFP-labeled: 69 ± 7.4, t test p > 0.21, n = 3 animals) Syn-GFP-positive terminals. In 9-month-old animals, however, a significant decrease in presynaptic glutamate immunogold labeling in Syn-GFP-positive asymmetric terminals (unlabeled: 74 ± 4.2 particles/μm2, Syn-GFP-labeled: 29 ± 2.3, t test p < 0.001, n = 3 animals; Fig. 8B, error = SEM) was seen, which was also the case for terminals making a symmetrical synaptic contact (unlabeled: 89 ± 16 particles/μm2, Syn-GFP-labeled: 20 ± 2, t test p < 0.01, n = 3 animals). These data suggest that Syn-GFP overexpression, which we have shown is associated with presynaptic terminal microaggregate formation at an early age, is also associated with an age-dependent decrease in glutamate immunogold labeling in excitatory (asymmetrical) and inhibitory (symmetrical) nerve terminals.
Discussion
In previous work, we showed the feasibility of using in vivo multiphoton FRAP to measure recovery of Syn-GFP in mouse cortex (Unni et al., 2010). In our current work, we expanded these methods to characterize Syn-GFP FRAP rigorously in vivo and demonstrate that it is an accurate way to measure protein mobility free from confounding artifacts seen in other systems. We demonstrate that, under conditions of moderate α-synuclein overexpression, similar to multiplication mutations that lead to human disease, in vivo aggregation is selectively found at presynaptic terminals starting at an early age (at least by 1 month of age), is not detectable in cell bodies, and is associated with significant decreases in the important presynaptic vesicle protein synapsin. In addition, we measured the effective diffusion coefficient of Syn-GFP within different subcellular compartments, the relative fraction within free and bound terminal pools, the vesicle binding constants kon* and koff, and the exchange rate within terminal microaggregates. All of these numbers have been impossible to measure in vivo in the brain until now. Our EM analysis demonstrates that Syn-GFP overexpression leads to progressive dysfunction, as measured by a decrease in presynaptic glutamate immunogold labeling in both excitatory and inhibitory synapses.
Our results suggest that somatic Syn-GFP is soluble and freely diffusible, whereas in presynaptic terminals, at least three different pools exist, one of which is soluble and freely diffusible. The other two lower mobility pools have very different recovery kinetics from each other, one on the order of ∼2.6 min and the other over several days or longer. Given substantial evidence that α-synuclein normally binds presynaptic vesicles (Maroteaux and Scheller, 1991; Shibayama-Imazu et al., 1993; Davidson et al., 1998), including our own EM data showing perivesicular staining and data from cell-free systems demonstrating synaptosomal binding on the minutes time scale (Wislet-Gendebien et al., 2006, 2008), the pool with a ∼2.6 min recovery tau in our experiments likely represents soluble Syn-GFP binding to synaptic vesicles. The final presynaptic Syn-GFP species measured is more heterogeneous, with individual terminals showing no, partial, or complete recovery within 7–9 d. This pool likely represents an α-synuclein microaggregate form. Its different recovery characteristics may be due to different maturation states, with more compact microaggregates exhibiting decreased Syn-GFP exchange. This kind of sequential compaction has been hypothesized to occur in human disease as part of a pathway toward Lewy pathology formation (Dale et al., 1992; Arima et al., 1998; Gómez-Tortosa et al., 2000). It is important to note that our in vivo FRAP imaging is limited for technical reasons to cells and synapses within superficial cortical layers (all data presented are from layers 2/3). Therefore, the potential somatic aggregation profile in deeper brain regions may not be the same as we measure here. It will be interesting in future studies to use alternative techniques, potentially including fiber-optic approaches (Flusberg et al., 2005; Murray and Levene, 2012), to probe the somatic aggregation profile of deeper structures. Pairing this with multicolor labeling approaches could help determine the IF in cortical terminals originating from different neuronal cell types throughout the brain.
Our biochemical characterization also supports the presence of terminal Syn-GFP microaggregate species because proteinase K-resistant, serine-129-phosphorylated, and oxidized forms of Syn-GFP are present only in presynaptic terminals. These three specific α-synuclein modifications are common in PD and DLB human tissue (Takeda et al., 1998; Giasson et al., 2000; Fujiwara et al., 2002). We find that, depending on technique, 44 ± 21% (confocal imaging in fixed tissue) or 35 ± 13% (Western blotting of fresh tissue) of Syn-GFP in the synapse is proteinase K-resistant, which closely matches the 32 ± 21% IF we detected with in vivo FRAP experiments, suggesting that this may be the same aggregate pool. Our measurements of the age dependence of aggregate formation suggest that a proteinase K-resistant, relatively immobile, Syn-GFP aggregate pool, which is not clearly phosphorylated at serine-129, develops as early as 1 month of age. This fraction is associated with toxicity because there is coincident reduction in the presynaptic vesicle protein synapsin. In addition, our quantitative immunogold measurements show an age-dependent decrease in terminal glutamate labeling in Syn-GFP-containing synapses. We speculate, based on this evidence, that terminal microaggregates represent an early stage in the synucleinopathy pathologic cascade, forming first at presynaptic terminals because of the high local concentration of this protein here. These proteinase K-resistant aggregates could be causing immediate presynaptic abnormalities, as evidenced by the lack of synapsin in these synapses, but may also trigger a second slower process of degeneration requiring months to develop, which leads to decreased presynaptic neurotransmitter levels. Ultimately, this chronic synaptic dysfunction may lead to a “dying back” of affected axonal processes, as has been postulated to occur in several synucleinopathies (Adalbert and Coleman, 2012; Yasuda et al., 2013). Alternatively, it is possible that the presynaptic microaggregates we measure may be beneficial because evidence suggests that certain α-synuclein aggregate species may be cytoprotective, potentially sequestering other more toxic species such as small oligomers (Nakamura et al., 2011) and therefore reducing neurodegeneration (Tofaris and Spillantini, 2005; Bodner et al., 2006). Finally, it is also possible that the presynaptic microaggregates are an epiphenomenon that is not linked directly to presynaptic dysfunction in an either helpful or harmful way. In this case, the microaggregates may be marking terminals where another toxic species is mediating detrimental effects on synapsin and glutamate levels. Therefore, exploring these mechanisms further, including the distinct roles of posttranslationally modified and proteinase K-resistant α-synuclein terminal species in this system, will be an important avenue for future research. Interestingly, recent work suggests that serine-129 phosphorylation, specifically by Polo-like kinase 2, marks a pool which is in the process of being disaggregated and degraded by autophagy (Oueslati et al., 2013).
Several groups of investigators have reported α-synuclein FRAP data in different systems (Fortin et al., 2005; van Ham et al., 2008). Most recently, Roberti et al. (2011) reported results from SHY-SY5Y neuroblastoma cell cultures using the biarsenical labeling compound ReAsH. Somatic aggregates and unaggregated protein were imaged by confocal microscopy. Within aggregates, one immobile and one mobile species were present. The IF within aggregates was ∼85% of total tagged synuclein, whereas the mobile fraction had a Deff of ∼0.035 μm2/s. These results differ substantially from ours because we detected a single, freely diffusible Syn-GFP population within cortical neuron somata in vivo (Deff = 8.6 ± 3.8 μm2/s) and 3 pools of Syn-GFP with differing levels of mobility ranging from milliseconds to days in synaptic terminals. It will be interesting to determine the nature of the α-synuclein species in these different systems and whether they share significant overlap or not.
Our measurements of free Syn-GFP mobility, both in the soma and terminals, are experimentally indistinguishable from those we made for free GFP in these compartments, suggesting that free Syn-GFP may be monomeric. However, previous work has shown limitations of FRAP to resolve small differences in molecular weight. For example, it has been suggested that molecular weight changes of ∼10-fold are required before being clearly detected by FRAP (Lippincott-Schwartz et al., 2001). If this is the case, the freely soluble pool of Syn-GFP we detect could exist as small (<10-mer) multimers. We can confidently say, however, that they are not larger multimers (>10-mers). Determining whether small multimers such as tetramers exist in vivo will be an interesting avenue for future studies because their presence is controversial (Bartels et al., 2011; Fauvet et al., 2012; Dettmer et al., 2013). Other fluorescence-based techniques adapted for in vivo use, such as fluorescent protein complementation, fluorescence correlation spectroscopy, or fluorescence resonance energy transfer, may be useful to answer this question.
Expression levels of Syn-GFP in this mouse line are two to three times that seen in normal human brain (Rockenstein et al., 2005), similar to patient tissue with multiplication mutations (Singleton et al., 2003). Therefore, these Syn-GFP mice may represent a good model for the neuropathology that underlies early stages of synucleinopathy. Using a variety of techniques, including in vivo multiphoton imaging, proteinase K digestion, immunohistochemistry, and EM, we have demonstrated the presence of human α-synuclein aggregates specifically in presynaptic terminals with a positive dose-aggregate relationship. In addition, we have not found evidence for somatic aggregates. This suggests that an early step underlying synucleinopathies is the development of specifically presynaptic aggregates. It will be interesting in future studies to pair this model with other factors implicated in disease pathogenesis, including mitochondrial toxins (Betarbet et al., 2000; Uversky et al., 2001), point mutations (Polymeropoulos et al., 1997; Krüger et al., 1998; Zarranz et al., 2004), or inoculation with potentially prion-like α-synuclein aggregates (Luk et al., 2012a, 2012b; Masuda-Suzukake et al., 2013). This will allow testing of the hypothesis that presynaptic aggregate pathology, given specific triggers, can cause spreading of aggregation to the soma, as predicted by several groups of investigators (Galvin et al., 1999; Marui et al., 2002; Kramer and Schulz-Schaeffer, 2007). In addition, this model, with aggregation limited to presynaptic terminals, may be useful for the study of therapies thought to only be beneficial early in the course of disease.
Footnotes
This work was supported in part by the National Institutes of Health (Grants NS069625, AG024978, AT002688, and NS061800), a Department of Veterans Affairs Merit Review Award, and the Murdock Charitable Trust. We thank Eliezer Masliah and Edward Rockenstein for providing PDNG78 mice; Anthony Paul Barnes for providing Thy1 GFP M-line mice; Lisa Dirling Vecchiarelli for technical assistance with microwave fixation; and Gary Westbrook for helpful comments on this manuscript.
The authors declare no competing financial interests.
References
- Adalbert R, Coleman MP. Axon pathology in age-related neurodegenerative disorders. Neuropathol Appl Neurobiol. 2012 doi: 10.1111/j.1365-2990.2012.01308.x. [DOI] [PubMed] [Google Scholar]
- Arima K, Uéda K, Sunohara N, Hirai S, Izumiyama Y, Tonozuka-Uehara H, Kawai M. Immunoelectron-microscopic demonstration of NACP/alpha-synuclein-epitopes on the filamentous component of Lewy bodies in Parkinson's disease and in dementia with Lewy bodies. Brain Res. 1998;808:93–100. doi: 10.1016/S0006-8993(98)00734-3. [DOI] [PubMed] [Google Scholar]
- Arrio-Dupont M, Foucault G, Vacher M, Devaux PF, Cribier S. Translational diffusion of globular proteins in the cytoplasm of cultured muscle cells. Biophys J. 2000;78:901–907. doi: 10.1016/S0006-3495(00)76647-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford NS, Robinson S, Palmiter RD, Joyce JA, Moore C, Meshul CK. Dopamine modulates release from corticostriatal terminals. J Neurosci. 2004;24:9541–9552. doi: 10.1523/JNEUROSCI.2891-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels T, Choi JG, Selkoe DJ. Alpha-synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011;477:107–110. doi: 10.1038/nature10324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Bodner RA, Housman DE, Kazantsev AG. New directions for neurodegenerative disease therapy: using chemical compounds to boost the formation of mutant protein inclusions. Cell Cycle. 2006;5:1477–1480. doi: 10.4161/cc.5.14.2929. [DOI] [PubMed] [Google Scholar]
- Brown EB, Wu ES, Zipfel W, Webb WW. Measurement of molecular diffusion in solution by multiphoton fluorescence photobleaching recovery. Biophys J. 1999;77:2837–2849. doi: 10.1016/S0006-3495(99)77115-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A. Alpha-synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364:1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Conway KA, Harper JD, Lansbury PT. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- Dale GE, Probst A, Luthert P, Martin J, Anderton BH, Leigh PN. Relationships between Lewy bodies and pale bodies in Parkinson's disease. Acta Neuropathol. 1992;83:525–529. doi: 10.1007/BF00310030. [DOI] [PubMed] [Google Scholar]
- Davidson WS, Jonas A, Clayton DF, George JM. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 1998;273:9443–9449. doi: 10.1074/jbc.273.16.9443. [DOI] [PubMed] [Google Scholar]
- Dettmer U, Newman AJ, Luth ES, Bartels T, Selkoe D. In vivo cross-linking reveals principally oligomeric forms of alpha-synuclein and beta-synuclein in neurons and non-neural cells. J Biol Chem. 2013;288:6371–6385. doi: 10.1074/jbc.M112.403311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmer KL, Waxman EA, Covy JP, Giasson BI. E46K human alpha-synuclein transgenic mice develop Lewy-like and tau pathology associated with age-dependent, detrimental motor impairment. J Biol Chem. 2011;286:35104–35118. doi: 10.1074/jbc.M111.247965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with Parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- Fauvet B, Mbefo MK, Fares MB, Desobry C, Michael S, Ardah MT, Tsika E, Coune P, Prudent M, Lion N, Eliezer D, Moore DJ, Schneider B, Aebischer P, El-Agnaf OM, Masliah E, Lashuel HA. Alpha-synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J Biol Chem. 2012;287:15345–15364. doi: 10.1074/jbc.M111.318949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferris AM, Giberson RT, Sanders MA, Day JR. Advanced laboratory techniques for sample processing and immunolabeling using microwave radiation. J Neurosci Methods. 2009;182:157–164. doi: 10.1016/j.jneumeth.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Flusberg BA, Cocker ED, Piyawattanametha W, Jung JC, Cheung EL, Schnitzer MJ. Fiber-optic fluorescence imaging. Nat Methods. 2005;2:941–950. doi: 10.1038/nmeth820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin DL, Nemani VM, Voglmaier SM, Anthony MD, Ryan TA, Edwards RH. Neural activity controls the synaptic accumulation of alpha-synuclein. J Neurosci. 2005;25:10913–10921. doi: 10.1523/JNEUROSCI.2922-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, Shen J, Takio K, Iwatsubo T. Alpha-synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Galvin JE, Uryu K, Lee VM, Trojanowski JQ. Axon pathology in Parkinson's disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc Natl Acad Sci U S A. 1999;96:13450–13455. doi: 10.1073/pnas.96.23.13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290:985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron. 2002;34:521–533. doi: 10.1016/S0896-6273(02)00682-7. [DOI] [PubMed] [Google Scholar]
- Goedert M. Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci. 2001;2:492–501. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- Gómez-Tortosa E, Newell K, Irizarry MC, Sanders JL, Hyman BT. Alpha-synuclein immunoreactivity in dementia with Lewy bodies: Morphological staging and comparison with ubiquitin immunostaining. Acta Neuropathol. 2000;99:352–357. doi: 10.1007/s004010051135. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Hsu LJ, Xia Y, Takeda A, Sisk A, Sundsmo M, Masliah E. Oxidative stress induces amyloid-like aggregate formation of NACP/alpha-synuclein in vitro. Neuroreport. 1999;10:717–721. doi: 10.1097/00001756-199903170-00011. [DOI] [PubMed] [Google Scholar]
- Jiang M, Porat-Shliom Y, Pei Z, Cheng Y, Xiang L, Sommers K, Li Q, Gillardon F, Hengerer B, Berlinicke C, Smith WW, Zack DJ, Poirier MA, Ross CA, Duan W. Baicalein reduces E46K alpha-synuclein aggregation in vitro and protects cells against E46K alpha-synuclein toxicity in cell models of familiar Parkinsonism. J Neurochem. 2010;114:419–429. doi: 10.1111/j.1471-4159.2010.06752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer ML, Schulz-Schaeffer WJ. Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci. 2007;27:1405–1410. doi: 10.1523/JNEUROSCI.4564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schöls L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- Larson ME, Sherman MA, Greimel S, Kuskowski M, Schneider JA, Bennett DA, Lesné SE. Soluble alpha-synuclein is a novel modulator of Alzheimer's disease pathophysiology. J Neurosci. 2012;32:10253–10266. doi: 10.1523/JNEUROSCI.0581-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lippincott-Schwartz J, Snapp E, Kenworthy A. Studying protein dynamics in living cells. Nat Rev Mol Cell Biol. 2001;2:444–456. doi: 10.1038/35073068. [DOI] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroteaux L, Scheller RH. The rat brain synucleins; family of proteins transiently associated with neuronal membrane. Brain Res Mol Brain Res. 1991;11:335–343. doi: 10.1016/0169-328X(91)90043-W. [DOI] [PubMed] [Google Scholar]
- Marui W, Iseki E, Nakai T, Miura S, Kato M, Uéda K, Kosaka K. Progression and staging of Lewy pathology in brains from patients with dementia with Lewy bodies. J Neurol Sci. 2002;195:153–159. doi: 10.1016/S0022-510X(02)00006-0. [DOI] [PubMed] [Google Scholar]
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: Implications for neurodegenerative disorders. Science. 2000;287:1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DM, Hasegawa M. Prion-like spreading of pathological alpha-synuclein in brain. Brain. 2013;136:1128–1138. doi: 10.1093/brain/awt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazza D, Braeckmans K, Cella F, Testa I, Vercauteren D, Demeester J, De Smedt SS, Diaspro A. A new FRAP/FRAPa method for three-dimensional diffusion measurements based on multiphoton excitation microscopy. Biophys J. 2008;95:3457–3469. doi: 10.1529/biophysj.108.133637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshul CK, McGinty JF. Kappa opioid receptor immunoreactivity in the nucleus accumbens and caudate-putamen is primarily associated with synaptic vesicles in axons. Neuroscience. 2000;96:91–99. doi: 10.1016/S0306-4522(99)90481-5. [DOI] [PubMed] [Google Scholar]
- Meshul CK, Stallbaumer RK, Taylor B, Janowsky A. Haloperidol-induced morphological changes in striatum are associated with glutamate synapses. Brain Res. 1994;648:181–195. doi: 10.1016/0006-8993(94)91117-7. [DOI] [PubMed] [Google Scholar]
- Meshul CK, Emre N, Nakamura CM, Allen C, Donohue MK, Buckman JF. Time-dependent changes in striatal glutamate synapses following a 6-hydroxydopamine lesion. Neuroscience. 1999;88:1–16. doi: 10.1016/S0306-4522(98)00189-4. [DOI] [PubMed] [Google Scholar]
- Miake H, Mizusawa H, Iwatsubo T, Hasegawa M. Biochemical characterization of the core structure of alpha-synuclein filaments. J Biol Chem. 2002;277:19213–19219. doi: 10.1074/jbc.M110551200. [DOI] [PubMed] [Google Scholar]
- Mueller F, Morisaki T, Mazza D, McNally JG. Minimizing the impact of photoswitching of fluorescent proteins on FRAP analysis. Biophys J. 2012;102:1656–1665. doi: 10.1016/j.bpj.2012.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray TA, Levene MJ. Singlet gradient index lens for deep in vivo multiphoton microscopy. J Biomed Opt. 2012;17 doi: 10.1117/1.JBO.17.2.021106. 021106. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Nemani VM, Azarbal F, Skibinski G, Levy JM, Egami K, Munishkina L, Zhang J, Gardner B, Wakabayashi J, Sesaki H, Cheng Y, Finkbeiner S, Nussbaum RL, Masliah E, Edwards RH. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J Biol Chem. 2011;286:20710–20726. doi: 10.1074/jbc.M110.213538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W, Müller V, Odoy S, Fujiwara H, Hasegawa M, Iwatsubo T, Trojanowski JQ, Kretzschmar HA, Haass C. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J Clin Invest. 2002;110:1429–1439. doi: 10.1172/JCI15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris EH, Giasson BI, Lee VM. Alpha-synuclein: Normal function and role in neurodegenerative diseases. Curr Top Dev Biol. 2004;60:17–54. doi: 10.1016/S0070-2153(04)60002-0. [DOI] [PubMed] [Google Scholar]
- Oueslati A, Schneider BL, Aebischer P, Lashuel HA. Polo-like kinase 2 regulates selective autophagic alpha-synuclein clearance and suppresses its toxicity in vivo. Proc Natl Acad Sci U S A. 2013;110:E3945–E3954. doi: 10.1073/pnas.1309991110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, Hyman BT, McLean PJ. Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS One. 2008;3:e1867. doi: 10.1371/journal.pone.0001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phend KD, Weinberg RJ, Rustioni A. Techniques to optimize post-embedding single and double staining for amino acid neurotransmitters. J Histochem Cytochem. 1992;40:1011–1020. doi: 10.1177/40.7.1376741. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Rieker C, Dev KK, Lehnhoff K, Barbieri S, Ksiazek I, Kauffmann S, Danner S, Schell H, Boden C, Ruegg MA, Kahle PJ, van der Putten H, Shimshek DR. Neuropathology in mice expressing mouse alpha-synuclein. PLoS One. 2011;6:e24834. doi: 10.1371/journal.pone.0024834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, Sudhof TC. The membrane fusion enigma: SNARES, Sec/Munc18 proteins, and their accomplices–guilty as charged? Annu Rev Cell Dev Biol. 2012;28:279–303. doi: 10.1146/annurev-cellbio-101011-155818. [DOI] [PubMed] [Google Scholar]
- Roberti MJ, Jovin TM, Jares-Erijman E. Confocal fluorescence anisotropy and FRAP imaging of alpha-synuclein amyloid aggregates in living cells. PLoS One. 2011;6:e23338. doi: 10.1371/journal.pone.0023338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Schwach G, Ingolic E, Adame A, Crews L, Mante M, Pfragner R, Schreiner E, Windisch M, Masliah E. Lysosomal pathology associated with alpha-synuclein accumulation in transgenic models using an eGFP fusion protein. J Neurosci Res. 2005;80:247–259. doi: 10.1002/jnr.20446. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: An open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt H, Arendt O, Brown EB, Schwaller B, Eilers J. Parvalbumin is freely mobile in axons, somata and nuclei of cerebellar purkinje neurones. J Neurochem. 2007;100:727–735. doi: 10.1111/j.1471-4159.2006.04231.x. [DOI] [PubMed] [Google Scholar]
- Schnell EA, Eikenes L, Tufto I, Erikson A, Juthajan A, Lindgren M, de Lange Davies C. Diffusion measured by fluorescence recovery after photobleaching based on multiphoton excitation laser scanning microscopy. J Biomed Opt. 2008;13 doi: 10.1117/1.3042274. 064037. [DOI] [PubMed] [Google Scholar]
- Scott D, Roy S. Alpha-synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci. 2012;32:10129–10135. doi: 10.1523/JNEUROSCI.0535-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DA, Tabarean I, Tang Y, Cartier A, Masliah E, Roy S. A pathologic cascade leading to synaptic dysfunction in alpha-synuclein-induced neurodegeneration. J Neurosci. 2010;30:8083–8095. doi: 10.1523/JNEUROSCI.1091-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibayama-Imazu T, Okahashi I, Omata K, Nakajo S, Ochiai H, Nakai Y, Hama T, Nakamura Y, Nakaya K. Cell and tissue distribution and developmental change of neuron specific 14 kDa protein (phosphoneuroprotein 14) Brain Res. 1993;622:17–25. doi: 10.1016/0006-8993(93)90796-P. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. Alpha-synuclein locus triplication causes Parkinson's disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Sone M, Yoshida M, Hashizume Y, Hishikawa N, Sobue G. Alpha-synuclein-immunoreactive structure formation is enhanced in sympathetic ganglia of patients with multiple system atrophy. Acta Neuropathol. 2005;110:19–26. doi: 10.1007/s00401-005-1013-9. [DOI] [PubMed] [Google Scholar]
- Sprague BL, Pego RL, Stavreva DA, McNally JG. Analysis of binding reactions by fluorescence recovery after photobleaching. Biophys J. 2004;86:3473–3495. doi: 10.1529/biophysj.103.026765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KD, Brown EB. Measuring diffusion coefficients via two-photon fluorescence recovery after photobleaching. J Vis Exp. 2010;36:1636. doi: 10.3791/1636. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan R, Hoang CP, Verkman AS. Photobleaching recovery and anisotropy decay of green fluorescent protein GFP-S65T in solution and cells: Cytoplasmic viscosity probed by green fluorescent protein translational and rotational diffusion. Biophys J. 1997;72:1900–1907. doi: 10.1016/S0006-3495(97)78835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda A, Hashimoto M, Mallory M, Sundsumo M, Hansen L, Sisk A, Masliah E. Abnormal distribution of the non-abeta component of alzheimer's disease amyloid precursor/alpha-synuclein in Lewy body disease as revealed by proteinase K and formic acid pretreatment. Lab Invest. 1998;78:1169–1177. [PubMed] [Google Scholar]
- Tanji K, Mori F, Mimura J, Itoh K, Kakita A, Takahashi H, Wakabayashi K. proteinase K-resistant alpha-synuclein is deposited in presynapses in human Lewy body disease and A53T alpha-synuclein transgenic mice. Acta Neuropathol. 2010;120:145–154. doi: 10.1007/s00401-010-0676-z. [DOI] [PubMed] [Google Scholar]
- Tofaris GK, Spillantini MG. Alpha-synuclein dysfunction in Lewy body diseases. Mov Disord. 2005;20:S37–S44. doi: 10.1002/mds.20538. [DOI] [PubMed] [Google Scholar]
- Unni VK, Weissman TA, Rockenstein E, Masliah E, McLean PJ, Hyman BT. In vivo imaging of alpha-synuclein in mouse cortex demonstrates stable expression and differential subcellular compartment mobility. PLoS One. 2010;5:e10589. doi: 10.1371/journal.pone.0010589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN, Li J, Fink AL. Pesticides directly accelerate the rate of alpha-synuclein fibril formation: A possible factor in Parkinson's disease. FEBS Lett. 2001;500:105–108. doi: 10.1016/S0014-5793(01)02597-2. [DOI] [PubMed] [Google Scholar]
- van Ham TJ, Thijssen KL, Breitling R, Hofstra RM, Plasterk RH, Nollen EA. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008;4:e1000027. doi: 10.1371/journal.pgen.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RH, Moore C, Davies G, Dirling LB, Koch RJ, Meshul CK. Effects of subthalamic nucleus lesions and stimulation upon corticostriatal afferents in the 6-hydroxydopamine-lesioned rat. PLoS One. 2012;7:e32919. doi: 10.1371/journal.pone.0032919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wislet-Gendebien S, D'Souza C, Kawarai T, St George-Hyslop P, Westaway D, Fraser P, Tandon A. Cytosolic proteins regulate alpha-synuclein dissociation from presynaptic membranes. J Biol Chem. 2006;281:32148–32155. doi: 10.1074/jbc.M605965200. [DOI] [PubMed] [Google Scholar]
- Wislet-Gendebien S, Visanji NP, Whitehead SN, Marsilio D, Hou W, Figeys D, Fraser PE, Bennett SA, Tandon A. Differential regulation of wild-type and mutant alpha-synuclein binding to synaptic membranes by cytosolic factors. BMC Neurosci. 2008;9:92. doi: 10.1186/1471-2202-9-92. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yasuda T, Nakata Y, Choong CJ, Mochizuki H. Neurodegenerative changes initiated by presynaptic dysfunction. Transl Neurodegener. 2013;2:16. doi: 10.1186/2047-9158-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoe H, Meyer T. Spatial dynamics of GFP-tagged proteins investigated by local fluorescence enhancement. Nat Biotechnol. 1996;14:1252–1256. doi: 10.1038/nbt1096-1252. [DOI] [PubMed] [Google Scholar]
- Zach S, Bueler H, Hengerer B, Gillardon F. Predominant neuritic pathology induced by viral overexpression of alpha-synuclein in cell culture. Cell Mol Neurobiol. 2007;27:505–515. doi: 10.1007/s10571-007-9141-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gómez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–173. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]