

Abstract
Selective strengthening of specific glutamatergic synapses in the mammalian hippocampus is critical for encoding new memories. This is most commonly achieved by input-specific Hebbian-type plasticity involving glutamate-dependent coincident presynaptic and postsynaptic depolarization. Our results demonstrate a novel mechanism by which nicotinic signaling, independently of coincident fast glutamatergic transmission, increases synaptic strength in the hippocampus. Electrophysiological recordings from rat hippocampal neurons in culture revealed that 1–3 h of exposure to 1 μm nicotine, even with action potentials being blocked, produced increases in both the frequency and amplitude of miniature EPSCs. Possible mechanisms were analyzed both in mouse organotypic slice culture and in rat cell culture by inducing the cells to express super-ecliptic pHluorin-tagged GluA1-containing AMPA receptors, which fluoresce only on the cell surface. Pharmacological and genetic manipulation of the cells, in combination with fluorescence-recovery-after-photobleaching experiments, revealed that nicotine, acting through α7-containing nicotinic acetylcholine receptors on the postsynaptic neuron, induces the stabilization and accumulation of GluA1-containing AMPA receptors on dendritic spines. The process relies on intracellular calcium signaling, PDZ [postsynaptic density-95 (PSD-95)/Discs large (Dlg)/zona occludens-1 (ZO-1)] interactions with members of the PSD-95 family, and lateral diffusion of the GluA1 receptors on the cell surface. These findings define a new avenue by which nicotinic signaling modulates synaptic mechanisms thought to subserve learning and memory.
Keywords: FRAP, GluA1, hippocampus, nAChR, nicotine, plasticity
Introduction
Long-lasting synaptic changes induced by experience are widely viewed as the cellular basis for learning and memory (Mayford et al., 2012). Such changes have been found previously to depend on coincident presynaptic and postsynaptic activity at the synapse, generating input-specific Hebbian-type plasticity (Feldman, 2012). Best characterized in this respect is long-term potentiation (LTP) at glutamatergic synapses in the hippocampus (Bliss and Collingridge, 1993; Whitlock et al., 2006). Induction of LTP requires depolarization-triggered release of transmitter from the presynaptic terminal, coupled with postsynaptic depolarization to relieve the Mg2+ block on NMDA-type glutamate receptors (NMDARs). AMPA-type glutamate receptors (AMPARs) laterally diffusing on the cell surface then become restrained at the postsynaptic site and constitute the early phase of LTP (Liao et al., 1995; Nicoll and Malenka, 1999; Makino and Malinow, 2009; Opazo and Choquet, 2011).
Nicotinic cholinergic signaling is also extensive in the hippocampus and has profound behavioral effects on cognitive function, including attention, learning, and memory (Peeke and Peeke, 1984; Levin et al., 1998; Kenney and Gould, 2008; Thiel and Fink, 2008). One of the most abundant nicotinic acetylcholine receptors (nAChRs) is the homopentameric α7-containing subtype (α7-nAChR), which has a high relative permeability to calcium and is found both presynaptically and postsynaptically at most glutamatergic synapses in the hippocampus (Bertrand et al., 1993; Séguéla et al., 1993; Zarei et al., 1999; Fabian-Fine et al., 2001). Activation of presynaptic α7-nAChRs facilitates glutamate release and can promote LTP, but does so in a synapse-specific manner requiring coincident glutamatergic transmission at the synapse (McGehee et al., 1995; Gray et al., 1996; Maggi et al., 2003; Placzek et al., 2009; Yakel, 2012).
Here, we report a novel mechanism for nicotinic enhancement of glutamatergic transmission. Surprisingly, the enhancement can be induced in the absence of coincident action potentials (APs) at the synapse, and depends instead on α7-nAChRs acting in a cell-autonomous manner on the postsynaptic cell. The enhancement results from a stabilization and accumulation of GluA1-containing AMPARs (GluA1s) at synaptic sites on dendritic spines. The nicotine-induced entrapment of GluA1s depends on intracellular free calcium, the availability of PDZ [postsynaptic density-95 (PSD-95)/Discs large (Dlg)/zona occludens-1 (ZO-1)]-binding scaffold proteins, and the lateral mobility of surface GluA1s. The findings demonstrate an unexpected form of synaptic plasticity in which nicotinic signaling can directly and independently enhance glutamatergic synaptic efficacy. Such a mechanism could explain some of the effects that nicotinic signaling is known to have on higher-level cognitive function.
Materials and Methods
Experiments.
Experiments were conducted according to the National Institutes of Health guidelines for animal research and were approved by the Institutional Animal Care and Use Committee at the University of California–San Diego.
DNA constructs.
The super-ecliptic pHluorin (SEP)-tagged GluA1 construct (FUGW-GluA1-SEP) was made by removing green fluorescent protein (GFP) from FUGW (plasmid 14883; Addgene ; Lois et al., 2002) with the restriction enzymes AgeI and BsrG1 (New England BioLabs) and inserting the PCR-amplified GluA1-SEP from pCI-GluA1-SEP with SgrAI- and BsiWI-cut (New England BioLabs) sticky ends. Sequence integrity for the GluA1-SEP was confirmed using primer-walk sequencing (Integrated DNA Technologies). In FUGW, GluA1-SEP is expressed under a ubiquitin promoter, which allows for higher expression levels in neurons than pCI-GluA1-SEP. The pCI-GluA1-SEP (pCI-GluR1-SEP) and pCI-GluA2-SEP (pCI-GluR1-SEP) constructs were generous gifts from Roberto Malinow (University of California–San Diego) and were cloned as described previously (Kopec et al., 2006). The RNAi-C-red-fluorescent-protein (RNAi-c-RFP) and pHcRED-CRIPT vectors were generated as described for other constructs (Conroy et al., 2003; Neff et al., 2009).
The short-hairpin RNA (shRNA) α7-shRNA and scrambled Scr-shRNA used the sequences 5′-AGGCAGATATCAGCAGCTATA-3′ and 5′-GAGAGTACGCTAAGATCCTAA-3′, respectively (Campbell et al., 2010; Lozada et al., 2012a), contained within the plasmid FG12 (plasmid 14884; Addgene; Qin et al., 2003). The H1-shRNA cassette from these constructs was excised using the restriction enzymes PacI and SnaBI (New England BioLabs) and ligated into a similarly cut FUGW-GluA-SEP to generate constructs driving shRNA expression under the promoter H1 and driving GluA1-SEP expression under a ubiquitin promoter.
Rat hippocampal dissociated cell cultures.
Dissociated hippocampal cultures were prepared from embryonic day 18–19 Sprague Dawley rat embryos (Harlan Laboratories) of both sexes as described previously (Kawai et al., 2002; Gómez-Varela et al., 2012). Briefly, cells were plated on a 12 mm glass coverslip in 500 μl of medium at 106 cells per well in a 24-well plate (Falcon), maintained in a humidified tissue culture incubator at 37°C with 5% CO2, and fed twice weekly by replacing half the volume with fresh medium. All cultures were used between 14 and 17 days in vitro (DIV) and, when necessary, transfected on DIV 6–7 using a calcium-phosphate precipitation kit (CalPhos 631312; Clontech; Goetze et al., 2004). SEP and RFP constructs were cotransfected at a ratio of between 3:1 and 12:1. In some cases, SEP constructs were transfected alone. The pHcRED-CRIPT construct was cotransfected with FUGW-GluA1-SEP and RNAiC-RFP constructs at a ratio of 5:10:3, respectively.
Mouse hippocampal slice cultures.
Postnatal day 1–2 mouse pups of both sexes (C57BL/6; Charles River Laboratories) were quickly decapitated and their brains were rapidly excised into ice-cold sucrose-buffered artificial CSF (sucrose-ACSF) saturated with 95% O2/5% CO2 containing the following (in mm): 110 sucrose, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2, 2.5 CaCl2,25 NaHCO3, and 10 glucose. Transverse hippocampal slices including a portion of the entorhinal cortex were made at 300–400 μm using a Vibratome (Series 1000 Plus; Technical Products International). Slices were kept in culture as described previously (Stoppini et al., 1991; Lozada et al., 2012b). Briefly, 1–4 slices were plated per Millicell insert (PICMORG50; Millipore) and fed once weekly by replacing half the volume with fresh medium. Cultures were biolistically transfected (Gene-gun; Bio-Rad) on DIV 8 as described previously (Woods and Zito, 2008), and used for experiments on DIV14–16. Transfection parameters were as follows: 4–5 mg of Au, 35 μg of pCI-GluA1-SEP or FUGW-GluA1-SEP, and 15 μg of RNAiC-RFP per round of bullets made. Standard barrels were covered with a fine mesh (CMN-0090-D; 90 μm opening, 30 μm thread diameter, 48.5% open area; Sefar Nitex) and slices were shot using 80–100 PSI of helium.
Imaging.
Fluorescence-recovery-after-photobleaching (FRAP) experiments were performed on a Leica SP5 confocal microscope using the FRAP-Wizard Leica software plugin (LAS-AF version 2.6.0.7266). Time-lapse images were taken as simultaneously scanned, multichannel single planes in the z dimension. Excitation and absorption parameters were designed to maintain signal ranges below saturation and were tested to minimize crosstalk between channels. The following settings were used for OT slice image acquisition: 40% argon laser power with 13% of 488 nm excitation; 15% 543 nm HeNe excitation; PMT1 absorption 500–525 nm, gain 1100, offset −1%; PMT2 absorption 588–683; gain 700–100, offset −1%; 63× (numerical aperture, 0.9) water-immersion objective; pinhole at 1 AU; 512 × 512 pixels; 700 Hz; 15× zoom; 2× frame average; 32.1 nm × 32.1 nm pixel size; 16.4 μm × 16.4 μm image size. The OT slice FRAP protocol was as follows: 2 baseline images per 10 s; two zoom-in bleach images for a total of 3 s with 543 at 0% and 458, 476, and 488 at 100%; 5 recovery images/30 s; manually refocused recovery images taken 2, 4, 9, 14, 19, and 29 min after the automatic image acquisition. Slices were continuously perfused for a maximum of 2 h at 33°C in 95% O2/5% CO2-saturated ACSF containing the following (in mm): 119 NaCl, 2.5 KCl, 1 NaH2PO4, 1.3 MgSO4, 2.5 CaCl2,26 NaHCO3, and 11 glucose.
Unless otherwise noted, images of dissociated hippocampal cell cultures were acquired using similar parameters with the following differences: 30–40% argon laser power; pinhole opened to 4AU; PMT1 absorption 500–530, gain 1000, offset −10%; PMT2 absorption 571–683 offset −5%. The FRAP protocol included 4 baseline images per 10 s; 2 zoom-in bleach images for a total of 3 s with 543 at 0% and 458, 476, and 488 at 100%; 6 recovery images/30 s; 7 recovery images/120 s. Dissociated cell cultures were imaged at 33°C in continuously perfused HEPES-buffered saline designed to mimic the Neurobasal (21103–049; Life Technologies Invitrogen) growth medium (mock NB) containing the following (in mm): 51.72 NaCl, 26.19 Na-gluconate, 0.906 NaH2PO4, 5.33 KCl, 0.814 MgCl2, 1.8 CaCl2, 10.92 HEPES, and 25 glucose, pH 7.4 with NaOH. Once on perfusion, cultures were imaged for no longer than 1.5 h. All images were obtained with age-matched controls.
Drug and antibody treatments.
All stock solutions were dissolved in double-distilled water and diluted in recording/growth medium to the final concentration unless otherwise indicated. The stock and final concentrations for drugs were as follows: BAPTA-AM (B6769; Invitrogen) 10 mm stock (in 50/50 DMSO/Pluronic F-127, P3000MP; Invitrogen), 10 μm final; brefeldin-A (BFA, 1231; Tocris Bioscience) 5 mg/ml in DMSO, 10 μg/ml (35.67 μm); dihydro-β-erythroidine (DHβE, D149; Sigma) 10 mm, 1 μm; DL-AP5 (APV, 0105; Tocris Bioscience) 10 mm, 50 μm; methyllycaconitine (MLA, M168; Sigma) 10 mm, 100 nm; NBQX (0373; Tocris Bioscience) 20 mm in DMSO, 20 μm; nicotine (N3876; Sigma) 100 mm, 1 μm; tetrodotoxin (TTX, 1069; Tocris Bioscience) 1 mm, 1 μm. Antibodies used for x-linking were as follows: an Alexa Fluor 647 conjugate of rabbit anti-GFP (A-31852; Invitrogen), 2 mg/ml stock in provided solution, 0.4 μg/ml final; rabbit anti-GluA1 (PC246; Calbiochem) designed against the n terminus of the receptor subunit, 100 μg/ml stock in provided solution; 2 μg/ml final.
For FRAP and electrophysiological experiments in slices, nicotine was added to the medium below the filter coincident with a feeding, and slices were submerged in 10–50 μl of the nicotine-containing medium. Cultures were then kept in the incubator for 1–2 h before being transferred to the perfusion chamber also containing nicotine, where they were imaged for no longer than 2 h, or, in the case of electrophysiological experiments, recorded from for no longer than 1 h.
For FRAP experiments in dissociated cell cultures, unless otherwise stated, nicotine was added directly to the culture medium. Cells were then incubated with nicotine under growing conditions for 1–1.5 h. Coverslips were transferred to a perfusion chamber containing nicotine and imaged for no longer than 1.5 h. The antagonists MLA, DhβE, and APV + NBQX were added just before nicotine treatment. BAPTA-AM, BFA, and the anti-GFP antibody were added 30 min before nicotine treatment. All were present throughout incubation and imaging except the antibody, which was washed out before imaging. In this case, imaging was limited to 30 min. All electrophysiological experiments in dissociated cell cultures were limited to 30 min in the perfusion chamber and were performed in the presence of drugs but not antibodies. Controls were always performed on day-matched sister cultures that were handled and treated with vehicle in the same manner as the corresponding experimental groups.
Electrophysiology.
For miniature EPSC (mEPSC) recordings, dissociated cell cultures (DIV 14–15) were immersed in HEPES-buffered saline containing the following (in mm): 0.001 TTX, 125 NaCl, 2 KCl, 4 MgCl2, 1 CaCl2, 10 HEPES, and 20 glucose, pH 7.4 with NaOH. Recordings were done at 20–23°C in a perfusion chamber mounted on a Zeiss Axiovert. Microelectrodes (2–5 MΩ) were pulled from thin-walled glass capillaries (G86150T-4; Warner Instruments) with a P-97 pipette puller (Sutter Instrument) and contained the following (in mm): 130 CsMeSO3, 3 CsCl, 10 HEPES, 1 EGTA, 10 NA2-phosphocreatine, 2 MgATP, and 0.3 NaGTP, pH 7.25 with CsOH. Whole-cell recordings were made with an Axopatch 200A and Clampex 8.2 software (Molecular Devices) using the whole-cell patch-clamp configuration in voltage-clamp mode held at −70 mV near the reversal potential of Cl− to eliminate miniature IPSCs. Adding the glutamate receptor blockers APV (50 μm) plus NBQX (20 μm) to the solution completely blocked the events, as expected for mEPSCs (data not shown). Data were acquired at 5 kHz and filtered at 2 kHz at a gain of 5×. Whole-cell capacitance was canceled in all recordings and only recordings with a series resistance of ≤25MΩ were used. Average resistance for each experimental group was 16–17 MΩ with no significant difference between groups (data not shown). Recordings were taken between 1 and 10 min after going whole cell. All recordings were obtained with age-matched controls.
Recordings of mEPSCs in organotypic slice cultures were performed in much the same way except for the following differences: slices (DIV14–16) were immersed in ACSF (305 mosmol l−1) containing the following (in mm): 0.001 TTX, 126 NaCl, 3 KCl, 2 MgSO4, 2 CaCl2, 1.25 NaH2PO4, 24 NaHCO3, and 10 glucose, pH 7.4, and perfused at a rate of 2.4 ml/min. Recordings were done at 24°C in a perfusion chamber mounted on a Olympus scope (BX51W1). Cells were held at −70 mV near the reversal potential of Cl− to eliminate inhibitory currents. Again, 50 μm APV plus 20 μm NBQX eradicated all events, confirming that they were mEPSCs. Microelectrodes (4–6 MΩ) were pulled from glass capillaries (BF150–86-10; Sutter Instrument) and contained the following (in mm): 140 K gluconate, 5 KCl, 2 MgCl2, 10 HEPES, 0.1 EGTA, 0.025 CaCl2, 2 ATP-Na, and 0.4 GTP-Na, pH 7.37 with KOH (295 mosmol l−1). Whole-cell recordings were made with a Multiclamp 700B and Clampex 10.1 software (Molecular Devices). Series resistance was monitored throughout recordings and neurons exhibiting >20% change were rejected.
Data analysis.
Images were analyzed using ImageJ software with the MBF plugin bundle (http://rsbweb.nih.gov/ij/plugins/mbf/index.html). For FRAP experiments, x–y drift was corrected by using a rigid body transformation. Fluorescence signal intensity within a region of interest (ROI) was measured as the optical density, the sum intensity of all the pixels divided by the number of pixels. Fluorescence recovery was calculated as a fraction of prebleach baseline fluorescence and was normalized to both the RFP spine signal to account for changes in spine size and to a separate section of the neuron to correct for incidental bleaching. Background fluorescence was measured from three separate areas with no cellular process and subtracted from the SEP signal as follows:
 |
where signal at an ROI was calculated as the SEP-channel optical density minus background optical density (BG) over the corresponding RFP-channel optical density; Rt is the relative fluorescence recovery at spines for any given time point t; Et is the signal at the experimental spine; Ct is the signal at a control spine or region of dendrite; and E0 and C0 are the corresponding baseline signals calculated as the average of all the prebleach frames. All FRAP experiments were analyzed in this manner except those shown in Figures 2D and 6, A and E, because the low transfection efficiency of pCI-GluA1-SEP did not allow for RFP cotransfection in dissociated cultures. In these cases, spine data were discarded if the spine substantially changed in size during the recovery. In those FRAP experiments in which the images went out of focus during the automatic acquisition, a manually focused image was obtained at the end to measure the last recovery point; all out-of-focus images were discarded.
Figure 2.

Nicotine stabilizes GluA1s on dendritic spines in hippocampal cell culture. A, Depiction of an AMPAR containing a transgenic GluA subunit tagged with SEP, a pH-sensitive GFP that only fluoresces while on the cell surface. B, GluA1-SEP fluorescence on spines of neurons treated with 1 μm nicotine for 1–3 h in cell culture recovers less in 16.5 min after photobleaching than do corresponding controls, indicating decreased mobility (Ctrl vs Nic: 96 ± 3 vs 77 ± 2%, n = 24,24; p = 0.00002, WC). C, Control condition images of GluA1-SEP in dissociated culture with accompanying cytosolic-RFP images. Top and bottom arrows indicate bleached spines. Scale bar, 2 μm. D, The nicotine effect on GluA1-SEP FRAP occurs gradually over 2 h. Cultures were transferred into a perfusion chamber with 1 μm nicotine and imaged for 2 h. Data points represent the final recovery point from different FRAP experiments. Values were fit with a linear regression demonstrating a trend in nicotine-treated cultures, but in not control cultures (control: slope −0.03 ± 0.12%, y-intercept 87.18 ± 8.42%, n = 25, r2 = 0.0026, p = 0.8087; nicotine: slope −0.22 ± 0.10%, y-intercept 85.85 ± 6.86%, n = 27, r2 = 0.1429, p = 0.0519). E, Control condition images of GluA2-SEP FRAP in dissociated culture with accompanying cytosolic-RFP images. F, Nicotine has no effect on GluA2-SEP mobility, yielding a recovery equivalent to control (Ctrl vs Nic: 81 ± 3 vs 84 ± 3%; n = 15,17; p = 0.49).
Figure 6.
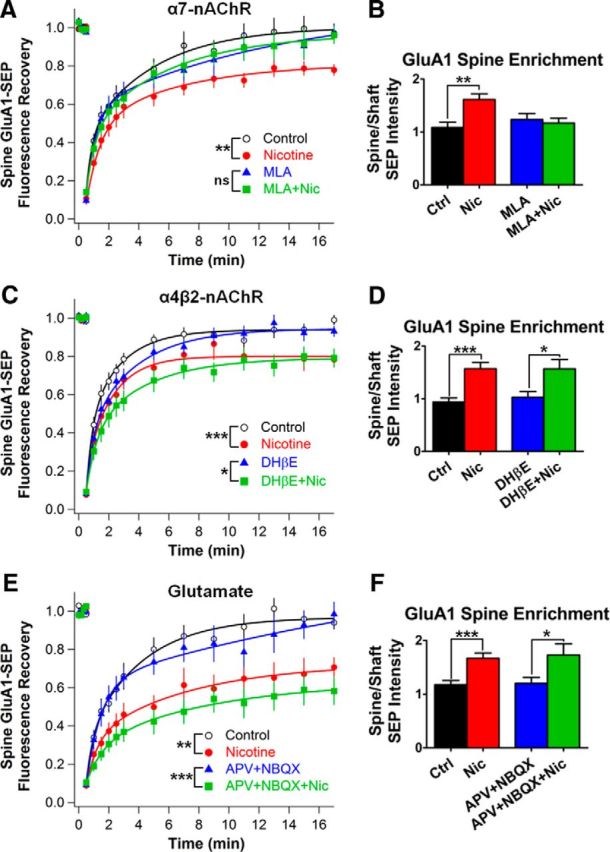
Receptor blockade shows that nicotine acts via α7-nAChRs, not α4β2-nAChRs, to stabilize GluA1s on spines independent of AMPA and NMDA receptor activation. A, Time course of GluA1-SEP FRAP after 1–3 h in nicotine with and without 100 nm MLA to block α7-nAChRs (MLA vs MLA + Nic: 97 ± 5 vs 96 ± 5%, n = 12,15; p = 0.86; Ctrl vs Nic: 97 ± 5% vs 78 ± 3%, n = 14,14; p = 0.0038, WC). B, Relative GluA1-SEP enrichment on spines under conditions from A (MLA vs MLA + Nic: 1.23 ± 0.11 vs 1.17 ± 0.09; n = 16,16; p = 0.66; Ctrl vs Nic: 1.09 ± 0.10 vs 1.61 ± 0.11; n = 16,16; p = 0.0013). C, Blockade of α4β2-nAChRs with 1 μm DHβE during the nicotine treatment had no effect on GluA1-SEP mobility (DHβE vs DHβE + Nic: 93 ± 3 vs 79 ± 5%, n = 11,12; p = 0.024, WC; Ctrl vs Nic: 99 ± 3 vs 78 ± 4%, n = 12,11; p = 0.0004). D, DHβE did not prevent nicotine-induced GluA1-SEP enrichment on spines (DHβE vs DHβE + Nic: 1.03 ± 0.11 vs 1.57 ± 0.18; n = 1 2,12; p = 0.0168; Ctrl vs Nic: 0.94 ± 0.08 vs 1.57 ± 0.12; n = 12,12; p = 0.0002). E, Blockade of AMPARs and NMDARs with 50 μm APV and 20 μm NBQX, respectively, during the nicotine treatment had no effect on GluA1-SEP mobility on spines (APV + NBQX vs APV + NBQX + Nic: 98 ± 6 vs 58 ± 7%, n = 10,11; p = 0.0006; Ctrl vs Nic: 94 ± 4 vs 71 ± 5%, n = 10,11; p = 0.0017). F, APV and NBQX failed to block GluA1-SEP enrichment on spines (APV + NBQX vs APV + NBQX + Nic: 1.2 ± 0.11 vs 1.73 ± 0.21; n = 11,11; p = 0.0405, WC; Ctrl vs Nic: 1.18 ± 0.08 vs 1.67 ± 0.1; n = 11,11; p = 0.001).
The first baseline image from a corresponding FRAP series was used to measure spine enrichment. An ROI was drawn tightly around the SEP signal of a spine and another around the entire length of the dendrite. The SEP optical density of the spine was divided by the SEP optical density of the dendrite to yield the enrichment value.
For mEPSC analysis, 100 events were counted from each cell. Events were analyzed from the first available section of recordings having a sufficiently stable baseline to permit collection of 100 events. Events were detected using Mini Analysis by Synaptosoft. The detection parameters were as follows: amplitude threshold was four times the root mean square of the noise, area threshold was 10–20, and complex peak detection was turned on. Automatic detection was manually checked for accuracy.
Statistics.
All data are presented as means ± SEM, except for cumulative frequency plots. The mean values for each FRAP plot were fit with a double-exponential curve (IGOR Pro; WaveMetrics). Statistical significance (p-value) was assessed for final FRAP points and enrichment values using a two-tailed, unpaired t test (GraphPad Prism 4) between matching control and experimental groups. When necessary, Welch's correction was used for unequal variances and is noted as WC. Unless otherwise stated, experiments were repeated ≥3 times, with each time representing cultures from a separate plating; n represents the number of spines, with 1–3 spines being taken from a single cell. The two-sample Kolmogorov–Smirnov test (KS; Mini Analysis; Synaptosoft) was used to compare cumulative frequency distributions. Grubbs' outlier test (GraphPad QuickCalcs) was used on all mean mEPSC amplitude and frequency data to exclude outliers from cumulative frequency distributions. For figures, *p ≤ 0.05; **p ≤ 0.01; and ***p ≤ 0.001.
Results
Nicotinic signaling enhances glutamatergic synapses independently of APs
To determine whether nicotinic activity can promote plasticity at glutamatergic synapses without requiring coincident APs at the synapse, we first examined mEPSCs. Hippocampal neurons maintained in cell culture for at least 2 weeks were incubated with 1 μm nicotine for 1–3 h before recording mEPSCs. TTX (1 μm) was included to block APs and the cells were clamped at −70 mV (near the Cl− reversal potential calculated from solution ion concentrations) to eliminate miniature IPSCs. The mEPSCs were fully blocked by 50 μm APV plus 20 μm NBQX (data not shown; Atallah and Scanziani, 2009), confirming that they represented glutamatergic events. The nicotine treatment increased both the frequency and size of mEPSCs, as seen in cumulative histograms of interevent intervals and event amplitudes (Fig. 1A–C). This concentration of nicotine represents the upper limit of concentrations found in smokers (Rose et al., 2010). The increase in mEPSC amplitude likely reflects the nicotine-driven addition of AMPARs to postsynaptic sites, whereas the increase in mEPSC frequency could reflect both presynaptic and postsynaptic effects at the synapse.
Figure 1.
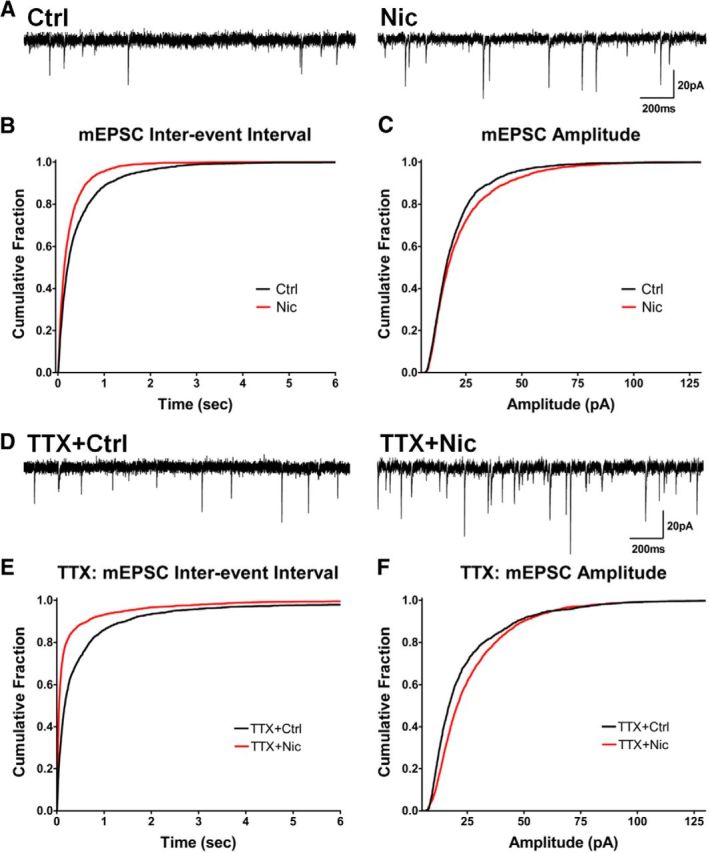
Nicotine treatment increases mEPSC frequency and amplitude generated by native AMPARs independently of AP-driven activity. A, Representative traces of mEPSCs in cells treated with nicotine for 1–3 h (Nic) versus controls (Ctrl). B, Cumulative histograms of mEPSC interevent intervals showing that the nicotine treatment increased the frequency of events (decreased the interevent intervals; Ctrl vs Nic, n = 2800 events for both, p < 0.0001, KS). C, Amplitude histograms showing that the nicotine treatment increases mEPSC amplitude (Ctrl vs Nic, n = 2800 events for both, p < 0.0001, KS). D, Representative traces of mEPSCs in cells treated with TTX to block APs while incubated with nicotine or control solution. E, Reduced interevent intervals, meaning increased mEPSC frequency, in cells treated with nicotine + TTX for 1–3 h (Ctrl + TTX vs Nic + TTX, n = 2500, 2600 events, p < 0.0001, KS). F, TTX did not prevent nicotine from increasing mEPSC amplitude (Ctrl + TTX vs Nic + TTX, n = 2400, 2600 events, p < 0.0001, KS).
The fact that the nicotine-induced changes in mEPSCs occurred without exogenous electrical stimulation raised the possibility that the nicotinic effect did not depend on AP-driven transmitter release at the synapse. To test this, we treated cultures with 1 μm TTX to block APs during the entire 1–3 h incubation with 1 μm nicotine. The TTX did not block the nicotine-induced increase in mEPSC frequency, nor did it prevent the increase in mEPSC amplitude (Fig. 1D–F). These results suggest an unexpected new mechanism by which nicotinic activity can directly increase the efficacy of a glutamatergic synapse without requiring coincident AP-driven transmission at the synapse.
Nicotine stabilizes GluA1s on dendritic spines
To test whether the nicotine-induced increase in synaptic efficacy was the result of a postsynaptic change in AMPAR stability, we measured the mobility of AMPARs using FRAP of fluorescently tagged AMPARs on the cell surface of neurons. For this purpose, hippocampal cell cultures were transfected with constructs expressing SEP-tagged GluA1 and RFP. The GluA1-SEP subunit of AMPARs fluoresces only when on the cell surface because of a pH dependence (Miesenböck et al., 1998; Kopec et al., 2006; Fig. 2A). Prebleach baseline fluorescence was measured at spines, followed by photobleaching with high-intensity light. FRAP was assessed by repeated imaging over the next 16 min. The signal was normalized both to the RFP spine signal to account for changes in spine size and to a separate section of the neuron to correct for incidental bleaching (see Materials and Methods). Under control conditions, the GluA1-SEP signal completely recovered to prebleach levels within 15–20 min, but in cultures treated with nicotine, the recovery levels were significantly reduced (Fig. 2B,C). These results demonstrate that recombinant GluA1 is relatively mobile on spines, as described previously (Makino and Malinow, 2009, 2011), but becomes stabilized by nicotinic stimulation. The reduction in mobility occurred slowly over the 2 h incubation with nicotine (Fig. 2D). In contrast, the mobility of GluA2-SEP-containing receptors was unchanged by nicotine (Fig. 2E,F). GluA2-SEP receptors appear more stable on spines than does GluA1-SEP, again as described previously (Makino and Malinow, 2009, 2011), which is consistent with stability being a correlate of synaptic incorporation.
Nicotine enhances glutamatergic synapses and stabilizes GluA1 on the spines of CA1 pyramidal neurons in organotypic slices
To determine whether nicotinic activity had the same effects on glutamatergic synapses within more ordered pathways, we repeated the mEPSC and FRAP experiments in organotypic (OT) slice cultures. OT hippocampal slices preserve much of the local connectivity, cytoarchitecture, and developmental progression observed in vivo (De Simoni et al., 2003). Similar to cell culture, nicotine treatment (1 μm, 1.5–3 h) of CA1 pyramidal neurons in hippocampal OT slices (DIV 14–16) increased both the frequency and size of mEPSCs, as seen in cumulative histograms of interevent intervals and event amplitudes (Fig. 3A–C). OT slices, however, required a somewhat longer protocol to reach 100% FRAP on spines in control cells (Fig. 3D,E), yet nicotine treatment (1 μm, 2–3 h) still significantly reduced GluA1-SEP mobility on spines of the basal dendrites of CA1 pyramidal neurons (Fig. 3E). These results affirm the robust nature of the nicotine-driven stabilization of GluA1s on spines and enhancement of glutamatergic signaling.
Figure 3.

In organotypic slice culture, nicotine increases mEPSC frequency and amplitude generated by native AMPARs on CA1 pyramidal neurons and stabilizes GluA1 on dendritic spines. A, Representative traces of mEPSCs from CA1 pyramidal neurons in OT slices treated with nicotine for 1.5–3 h (Nic) versus controls (Ctrl). B, Cumulative histograms of mEPSC interevent intervals showing that the nicotine treatment increased the frequency of events (decreased the interevent intervals; Ctrl vs Nic, n = 2400,2200 events, p = 0.0017, KS). C, Amplitude histograms showing that the nicotine treatment increased mEPSC amplitude (Ctrl vs Nic, n = 2500,2400 events, p < 0.0001, KS). D, Images of CA1 pyramidal neuron basal dendrites depicting GluA1-SEP FRAP under control conditions with accompanying cytosolic-RFP images. Arrows indicate bleached spines. Scale bar, 2 μm. E, GluA1-SEP fluorescence on spines treated with 1 μm nicotine for 2–3 h recovers to a lesser extent in 31 min after photobleaching than do corresponding controls, indicating decreased mobility (Ctrl vs Nic: 98 ± 4 vs 77 ± 5%; n = 12,14; p = 0.0104).
Nicotinic control of GluA1 lateral mobility does not occur on the dendrite, but correlates with receptor enrichment on spines
To test whether nicotine causes a pervasive stabilization of GluA1-SEP on the surface of hippocampal neurons, we bleached spine-sized areas on dendrites and measured FRAP in that area (Fig. 4A). As expected, under control conditions, fluorescence recovery was much faster on the dendrite compared with the spine (Ashby et al., 2006). No difference, however, was observed between control and nicotine-treated cultures, suggesting that the effect of nicotine on stabilizing GluA1 is restricted to spines (Fig. 4B).
Figure 4.

The nicotine effect on GluA1-SEP FRAP is restricted to spines and represents a change in the rate of exchange between spine and dendrite surface receptors. A, Representative images depicting the bleaching of a spine-sized area of GluA1-SEP on the dendrite. The bleached region and region measured for FRAP were the same, represented by the outlined area. Scale bar, 2 μm. B, FRAP is quite rapid on the dendrite, and there is no difference in mobility of GluA1-SEP on the dendrite between control and nicotine-treated cells (Ctrl vs Nic: 102 ± 3 vs 101 ± 2%; n = 14,15; p = 0.87). C, Representative images depicting bleach of GluA1-SEP signal along 10 μm of the dendrite surrounding a bleached spine. Solid outline indicates the region used to measure FRAP. Dotted outline represents the entire area that was bleached. Scale bar, 3 μm. D, When the reserve pool of surface fluorescent receptors is depleted, GluA1-SEP recovery at the spine is greatly attenuated both in control and nicotine-treated cells, indicating that previously observed recoveries represented exchange between receptor pools on spines and dendrites (Ctrl vs Nic: 55 ± 3 vs 50 ± 3%; n = 12,12; p = 0.28).
To determine whether the observed FRAP on spines represented lateral movement of surface GluA1-SEP rather than receptor cycling with intracellular pools, we depleted the potential reserve pool of fluorescent surface receptors by bleaching an extended region of the dendrite. The extent of GluA1-SEP dendrite bleach after a 15 s bleach protocol was 60–70% (Ctrl vs Nic having equivalent remaining fluorescence; 30 ± 4 vs 36 ± 3%, n = 12,12; p = 0.18). The amount of recovery on spines subsequently was markedly attenuated under such conditions for both control and nicotine-treated cells, rendering them equivalent (Fig. 4C,D). Accordingly, the differences observed in FRAP on spines must represent changes in mobility of preexisting surface receptors.
The incomplete recovery of GluA1-SEP fluorescence at spines in nicotine-treated cultures likely represents a population of additional receptors that are less mobile. To test whether nicotine increased the number of receptors at spines, we used baseline images taken before photobleaching to measure spine size and relative GluA1-SEP fluorescence at spines compared with the surrounding dendrite. The cytosolic-RFP signal was used to measure spine size (Fig. 5A) and revealed no difference between control and nicotine, indicating that nicotine-induced reductions in GluA1-SEP mobility were not accompanied by changes in spine size (Ctrl vs Nic: 1.57 ± 0.08 vs 1.63 ± 0.11 μm2; n = 33,33; p = 0.66). Measuring the ratio of spine-to-dendrite GluA1-SEP intensity, however, revealed a significant enrichment of GluA1-SEP at spines on nicotine-treated cells (Fig. 5B,C). As with the reduction in GluA1-SEP mobility, spine enrichment of GluA1-SEP occurred gradually over a 2 h exposure to nicotine (Fig. 5D). Analysis of a larger area of the dendritic arbor revealed a shift in the population distribution of GluA1-SEP spines toward greater enrichment in nicotine-treated cells and included the appearance of a subset of spines with high levels of enrichment (Fig. 5E–G). No change was seen in spine size or in SEP enrichment at spines for GluA2-SEP (Ctrl vs Nic spine size: 1.39 ± 0.14 vs 1.6 ± 0.11 μm2, n = 15,18, p = 0.25; Ctrl vs Nic SEP enrichment: 1.89 ± 0.18 vs 2.04 ± 0.14, n = 15,18, p = 0.51). These results indicate that nicotinic activity selectively immobilizes and enriches GluA1s on spines without affecting spine dimensions.
Figure 5.

The nicotine-induced decrease in GluA1-SEP mobility on spines correlates with an increase in receptor density on spines. A, Representative prebleach images from FRAP experiments in which spine area was calculated from the RFP signal. B, Representative prebleach images from FRAP experiments demonstrating nicotine-induced enrichment of GluA1-SEP at spines. C, Quantification of GluA1-SEP enrichment on spines by measuring spine/shaft optical density yielded a significant increase in cells exposed to nicotine, suggesting that nicotine leads to the accumulation of receptors on spines. (Ctrl vs Nic: 0.99 ± 0.05 vs 1.67 ± 0.1; n = 29,28; p = 0.00000014, WC). D, The nicotine effect on GluA1-SEP spine enrichment occurs gradually over 2 h. Cultures were transferred into a perfusion chamber with 1 μm nicotine and imaged for 2 h. Data points represent the spine enrichment of GluA1-SEP measured from the baseline images of corresponding FRAP experiments of different spines. Values were fit with a linear regression demonstrating a trend in nicotine-treated cultures, but not control cultures (control: slope −0.003 ± 0.002, y-intercept 1.52 ± 0.15 n = 27, r2 = 0.0554, p = 0.2373; nicotine: slope 0.007 ± 0.003, y-intercept 1.446 ± 0.21, n = 27, r2 = 0.1572, p = 0.0406). E, Representative images depicting the effect of nicotine on the population distribution of GluA1-SEP spine enrichment on dendritic arbors. Images were taken at 5× zoom to visualize multiple spines from a single dendrite (enrichment images were usually at 15×). Z-stacks were collapsed as a sum of all slices in ImageJ. Arrows indicate enriched spines. Scale bar, 5 μm. F, Frequency histogram of GluA1-SEP enrichment at spines from multiple neuronal dendritic arbors revealing a shift toward greater enrichment in nicotine-treated cells and the appearance of a heavily enriched subpopulation (Ctrl, n = 85 spines from 5 cells; Nic, n = 71 spines from 5 cells, 1 week of plating). G, Cumulative frequency plot of data from B demonstrating the statistically significant difference between the two distributions (Ctrl vs Nic, p = 0.0007, KS).
Nicotine-induced enrichment and stabilization of GluA1s on spines rely on α7-nAChRs on the postsynaptic cell, not on AMPARs or NMDARs
In addition to α7-nAChRs, the hippocampus contains substantial numbers of heteropentameric β2-containing nAChRs (β2*-nAChRs), with α4β2-nAChRs being the most common version (Wada et al., 1989; Zarei et al., 1999). To identify the nAChR subtype responsible for the effect on AMPAR mobility, we used antagonists specific for individual nicotinic receptor subtypes. The α7-nAChR-specific antagonist MLA (100 nm) prevented nicotine from altering GluA1-SEP mobility (Fig. 6A) and from enriching GluA1-SEP on spines (Fig. 6B). In contrast, the α4β2-nAChR-specific antagonist DhβE (1 μm) failed to prevent either nicotinic effect (Fig. 6C,D). The results indicate that nicotine acts via α7-nAChRs, not α4β2-nAChRs, to exert the observed changes in GluA1 mobility.
To demonstrate that nicotinic stimulation induces the changes in GluA1 mobility directly and independently of fast excitatory transmission, as inferred from mEPSC recordings above, we used the antagonists APV (50 μm) and NBQX (20 μm) to block activity through NMDARs and AMPARs, respectively. The antagonists were unable to prevent either the nicotine-induced reduction of GluA1-SEP mobility (Fig. 6E) or the GluA1-SEP enrichment on spines (Fig. 6F). This indicates that glutamatergic signaling through AMPARs and NMDARs is not necessary for the effects, raising the possibility that postsynaptic α7-nAChRs act in a cell-autonomous manner.
To determine whether α7-nAChRs on the postsynaptic cell mediate nicotinic control of GluA1 trafficking, we used an shRNA targeted against the rat and mouse α7-nAChR sequences (Campbell et al., 2010; Lozada et al., 2012a; α7-shRNA). Sparse transfection conditions, together with RFP labeling, allowed us to avoid spines contacted by transfected axons, thereby ensuring that only the postsynaptic cell expressed the α7-shRNA (Fig. 7A–C). In neurons expressing the α7-shRNA and therefore lacking α7-nAChRs, nicotine had no effect on the mobility of GluA1-SEP (Fig. 7D), nor could it increase the enrichment of GluA1-SEP on spines (Fig. 7E). A scrambled shRNA had no such effects in similarly treated sister cultures (Fig. 7D,E). The shRNA results indicate a novel mechanism by which nicotine, apparently in a cell-autonomous manner, can drive synaptic plasticity at glutamatergic synapses independently of coincident excitatory glutamate transmission.
Figure 7.

Knock-down of α7-nAChRs by shRNA shows that the receptors are required on the postsynaptic cell for the nicotinic effect. A, Representative image of a neuron in dissociated hippocampal culture expressing cytosolic RFP (red) and a SEP-tagged GluA subunit (green). Individual channels are in gray-scale in the images at the top and a merged image is shown on the bottom. GluA-SEP fluorescence, representing surface receptors, is found on the cell body, along the dendritic shaft, and on dendritic spines. It is lacking from putative axons, which only express RFP, indicated by arrows. Scale bar, 5 μm. B, Representative RFP image of an experimentally acceptable scenario free of transfected axons. Scale bar, 2 μm. C, RFP image exemplifying an unacceptable experimental scenario in which a transfected axon runs juxtaposed to a dendritic spine, possibly forming a synapse. The arrow indicates the putative axon. Scale bar, 2 μm. D, E, Nicotine had no effect on GluA1-SEP trafficking on neurons expressing α7-shRNA, although it remained effective on cells expressing the control Scr-shRNA. Nicotine acts directly through α7-nAChRs on the postsynaptic neuron to stabilize and enrich GluA1-SEP at spines (α7-shRNA vs α7-shRNA + Nic, FRAP: 92 ± 5 vs 94 ± 5%, n = 12,12; p = 0.79; enrichment: 1.08 ± 0.08 vs 1.2 ± 0.1; n = 12,12; p = 0.37; Scr-shRNA vs Scr-shRNA + Nic, FRAP: 97 ± 4, vs 76 ± 4%, n = 11,12, p = 0.0007; enrichment: 1.13 ± 0.09 vs 1.91 ± 0.23, n = 11,12, p = 0.0065, WC).
Nicotine-induced effects on GluA1 trafficking depend on calcium, PDZ-scaffold interactions, and lateral diffusion, but not on exocytosis of GluA1
Local rises in intracellular calcium can significantly reduce AMPAR mobility (Borgdorff and Choquet, 2002; Heine et al., 2008) and ligand-induced activation of α7-nAChRs can elevate intracellular calcium (Séguéla et al., 1993; Dajas-Bailador and Wonnacott, 2004). To determine whether elevation of intracellular calcium is necessary for the nicotine effects on AMPAR trafficking, we treated cultures with BAPTA-AM, a calcium chelator that only buffers calcium intracellularly. BAPTA-AM (10 μm) added 30 min before the 1–3 h incubation with 1 μm nicotine prevented the decrease in GluA1-SEP mobility, rendering it equivalent to corresponding controls (Fig. 8A). BAPTA-AM also prevented the increase in the GluA1-SEP enrichment on spines (Fig. 8B), indicating that α7-nAChR stimulation acts via calcium to decrease GluA1 mobility and position more of the receptors on spines.
Figure 8.
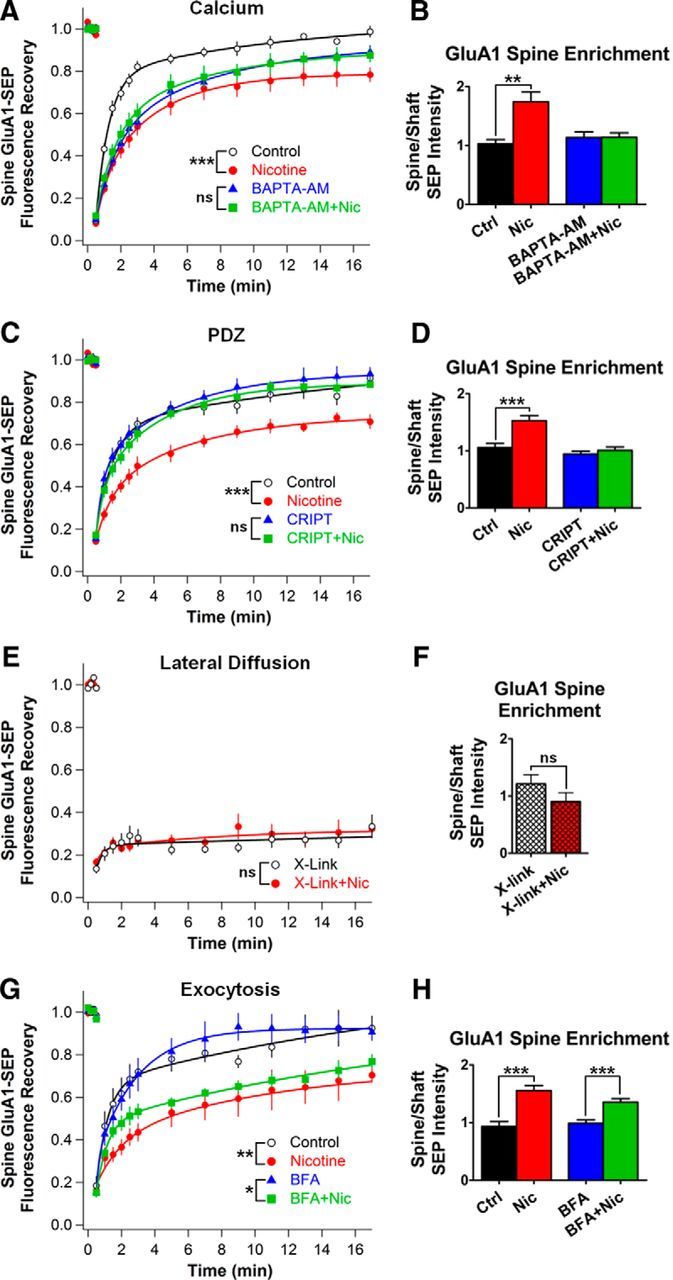
The nicotine-induced effects on GluA1 trafficking can be blocked by chelating intracellular calcium, disrupting PSD-95 family member PDZ-interactions, or restricting lateral diffusion of surface GluA1s with antibody x-linking. A, B, GluA1-SEP FRAP and enrichment on spines in controls versus nicotine with and without the calcium chelator BAPTA-AM applied 30 min before and throughout the nicotine treatment (BAPTA-AM vs BAPTA-AM + Nic, FRAP: 89 ± 3 vs 88 ± 3%, n = 15,15; p = 0.75; enrichment: 1.14 ± 0.1 vs 1.14 ± 0.08; n = 15,15; p = 0.98; Ctrl vs Nic, FRAP: 99 ± 3, vs 78 ± 3%, n = 14,13, p = 0.000087; enrichment: 1.03 ± 0.07 vs 1.74 ± 0.18, n = 14,13, p = 0.0014, WC). C, D, GluA1-SEP FRAP and enrichment on spines in controls veresus nicotine with and without transfection with CRIPT, a dominant-negative protein that disrupts PDZ-interactions with PSD-95 family members (CRIPT vs CRIPT+Nic, FRAP: 93 ± 3 vs 88 ± 2%, n = 14,13; p = 0.18, WC; Enrichment: 0.94 ± 0.05 vs 1.01 ± 0.06; n = 14,14; p = 0.42; Ctrl vs Nic, FRAP: 91 ± 3, vs 71 ± 4%, n = 12,12, p = 0.0004; enrichment: 1.06 ± 0.07 vs 1.53 ± 0.09, n = 12,12, p = 0.00052). E, GluA1-SEP FRAP on spines in controls vs nicotine after x-linking GluA1-SEP with a GFP-specific antibody (x-link vs x-link+Nic; 33 ± 6 vs 32 ± 4%, n = 10,10; p = 0.88). F, GluA1-SEP enrichment on spines in controls vs nicotine after antibody x-linking of GluA1-SEP (x-link vs x-link + Nic; 1.21 ± 0.16 vs 0.9 ± 0.15, n = 10,10; p = 0.19). G, H, GluA1-SEP FRAP and enrichment on spines in controls vs nicotine with and without BFA treatment 30 min before and during the nicotine incubation to block receptor exocytosis (BFA vs BFA+Nic, FRAP: 90 ± 4 vs 77 ± 4%, n = 13,12, p = 0.0135; enrichment: 0.99 ± 0.06 vs 1.36 ± 0.06, n = 13,13, p = 0.0002; Ctrl vs Nic, FRAP: 93 ± 6, vs 70 ± 4%, n = 10,10, p = 0.0048; enrichment: 0.93 ± 0.08 vs 1.56 ± 0.09, n = 10,10, p = 0.00008). Existing surface GluA1-SEP are sufficient for the calcium- and PDZ-dependent effects of nicotine without requiring exocytosis of additional receptors.
The canonical mechanism by which AMPARs are held at synapses involves interactions between their TARP auxiliary subunits and PDZ-scaffold proteins (Chen et al., 2000; Bats et al., 2007; Opazo et al., 2010, 2012; Opazo and Choquet, 2011). To determine whether PDZ interactions with scaffold proteins are necessary for nicotine-induced immobilization of GluA1 on spines, we used a form of the protein CRIPT that exerts a dominant-negative effect by blocking PDZ3 binding of all PSD-95 family members (Passafaro et al., 1999; Conroy et al., 2003). Neurons expressing CRIPT showed no change in GluA1-SEP mobility when treated with nicotine (Fig. 8C) and did not display increased numbers of GluA1-SEP on spines (Fig. 8D). The results indicate that a PDZ-scaffold is likely to be required for nicotinic stabilization of AMPARs at synapses. This is consistent with current models for synaptic potentiation in which passively diffusing surface receptors are trapped by postsynaptic scaffold anchoring slots (Opazo et al., 2010, 2012; Mondin et al., 2011).
The photobleaching experiments described earlier indicated that lateral diffusion of surface GluA1-SEP was essential to obtain FRAP on spines. To determine whether the same is true for nicotine-induced increases in receptor density on spines, we restricted the lateral diffusion of GluA1-SEP by antibody cross-linking (x-linking). This procedure greatly hinders the lateral surface mobility of AMPARs while leaving exocytosis largely unaffected (Ashby et al., 2006). Indeed, x-linking GluA1-SEP with an anti-GFP antibody before and throughout the nicotine treatment almost completely abolished GluA1-SEP FRAP, indicating a marked reduction in the mobility of surface GluA1s (Fig. 8E). Under these conditions, nicotine was unable to induce the enrichment of GluA1-SEP at spines (Fig. 8F), demonstrating that lateral diffusion of GluA1-SEP is required for this effect.
To test the possibility that exocytosis is needed to replenish extrasynaptic AMPARs as a source of receptors for the nicotinic effect, we blocked exocytosis of internal receptors by treating cells with 10 μg/ml BFA (Galan et al., 2004). In this case, nicotine was still able to reduce the mobility of GluA1-SEP and increase receptor density on spines (Fig. 8G,H). This suggests that, contrary to other forms of synaptic potentiation, exocytosis of AMPARs to replenish surface-receptor reserve pools is not required for the nicotinic effects over this time frame (Kopec et al., 2006; Makino and Malinow, 2009; Petrini et al., 2009; Opazo and Choquet, 2011).
Stabilization of AMPARs at spines results in strengthened synapses
To confirm that the nicotine-induced increases in mEPSC amplitude are due to increased numbers of AMPARs at synapses via lateral diffusion, we tested the effects of antibody x-linking. As reported above, antibody x-linking of surface GluA1-SEP prevented lateral diffusion and, as a consequence, the nicotine-induced enrichment of the receptors at spines. We treated untransfected cultures with an antibody that recognizes an extracellular epitope on native GluA1. In these cultures, nicotine exposure was unable to increase the number of large-amplitude events, although it still increased mEPSC frequencies, albeit to a lesser extent (Fig. 9A–C). Accordingly, the change in amplitude is likely the result of additional AMPARs being incorporated into postsynaptic sites, whereas the increase in frequency is likely to be due, at least in part, to a presynaptic effect such as an increase in probability of release.
Figure 9.

Preventing nicotine-induced GluA1 enrichment with antibody x-linking blocks the corresponding increase in mEPSC amplitude. A, Representative traces from control and nicotine-treated cells after antibody x-linking of surface GluA1s. B, Cumulative histograms of mEPSC interevent intervals in control versus nicotine-treated cells after antibody x-linking of surface GluA1s (Ctrl vs Nic, p < 0.0001, n = 2700, 2600 events; KS). C, Cumulative histograms of mEPSC amplitudes in controls and nicotine-treated cells after antibody x-linking (Ctrl vs Nic, n = 2600, 2800 events; p < 0.0001, KS, due to more small events in nicotine-treated cells; no difference was seen in larger events, unlike results obtained before x-linking as in Fig. 1).
Discussion
The results demonstrate a novel mechanism by which nicotinic activity can drive synaptic potentiation independent of fast excitatory transmission. Stimulation of α7-nAChRs stabilizes GluA1s at postsynaptic sites on spines, drawing them from the surface pool of mobile extrasynaptic receptors. The stabilization is calcium dependent, requires functional PSD-95 family members, and reflects both an increase in the number of such receptors on spines and a decrease in their mobility. The outcome is an increased signaling capacity for the synapse, reflected in larger mEPSCs. Remarkably, the changes rely upon the activation of α7-nAChRs on the postsynaptic cell and do not require coincident fast excitatory transmission through AMPA and NMDARs, suggesting a cell-autonomous mechanism (Fig. 10). As a result, extended nicotinic activity could promote synaptic plasticity at multiple synapses across a region independent of concomitant rapid synaptic transmission at those synapses.
Figure 10.

Model diagramming the nicotine-dependent changes in GluA1 mobility and stabilization on dendritic spines. Most GluA1s are mobile and not synaptically incorporated. Nicotine activates α7-nAChRs on the postsynaptic cell to trap surface-diffusing GluA1s on spines independently of fast, excitatory glutamatergic transmission. The process requires intracellular calcium and the availability of PDZ-binding scaffold slots. The enrichment and stabilization of GluA1s on spines represents synaptic incorporation of the receptors, strengthening synaptic transmission, as indicated by the influx arrows and by the large depolarizing trace within the spine head.
Nicotinic cholinergic modulation of glutamatergic synaptic plasticity in the hippocampus has frequently been ascribed to the presynaptic or postsynaptic enhancement of glutamatergic transmission, enabling it to produce long-term changes in synaptic efficacy (Fujii et al., 1999; Ji et al., 2001; Kenney and Gould, 2008; Placzek et al., 2009; Gu et al., 2012; Yakel, 2012). The present results differ from those precedents in two important respects. First, the enhancement did not require AP-induced glutamatergic transmission, which, in combination with the necessity of α7-nAChRs located on the postsynaptic cell, suggests a cell-autonomous mechanism. Cell-autonomous actions have been proposed for nAChRs in regulating network integration and synapse formation of developing neurons (Campbell et al., 2010; Lozada et al., 2012b). Second, the time course of nicotine-induced changes was intermediate between acute and chronic treatments, requiring 1–2 h of continued nicotine exposure to become significant. This amount and duration of exposure mimic the systemic administration and brain tissue accumulation of nicotine that occur during nicotine consumption, for example, via repetitive cigarette smoking, transdermal patches, or smokeless tobacco products (Hukkanen et al., 2005; Rose et al., 2010). It is also consistent with persistent ambient levels of ACh thought to occur in the CNS as a result of volume transmission, which has been shown to fluctuate during memory and attention-dependent tasks (Umbriaco et al., 1995; Descarries et al., 1997; Pepeu and Giovannini, 2004). As a result, the nicotinic mechanisms found here may be relevant not only for nicotine exposure, but also for endogenous nicotinic cholinergic signaling via ACh.
A widely accepted model of LTP is that GluA1s are inserted into the plasma membrane, freely diffuse to the synaptic site, and become tethered to a PSD-95 scaffold due to an increase in binding affinity elicited by a calcium-dependent, activity-driven process. GluA1-lacking receptors subsequently replace the GluA1 place-holders in a constitutive, non-activity-dependent manner (Liao et al., 1995; Hayashi et al., 2000; Shi et al., 2001; Malinow and Malenka, 2002; Makino and Malinow, 2009, 2011; Opazo et al., 2010, 2012; Opazo and Choquet, 2011; Tanaka and Hirano, 2012). Consistent with this view are the findings here that the nicotine-driven increase in AMPAR localization and stabilization on spines is specific for GluA1-SEP, as opposed to GluA2-SEP, is calcium-dependent, requires PDZ interactions of the PSD-95 family, and relies on the free lateral diffusion of GluA1s. No increase was seen in spine size with nicotine, although increases are often, but not always, associated with synaptic potentiation (Kopec et al., 2006; Makino and Malinow, 2009, 2011). This scaffold-dependent receptor entrapment may therefore account for both the increase in GluA1 number on spines and their simultaneous immobilization there.
An alternative possibility is that nicotine leads to an increase in the total number of PDZ scaffold sites at spines rather than an increase in the affinity of GluA1 for them. Consistent with this are studies showing that overexpression of PSD-95 can lead to synaptic potentiation (Ehrlich and Malinow, 2004; Elias et al., 2006). The excess of PSD-95 molecules at spines compared with AMPARs under normal conditions and the nonspecific nature of such an increase, however, make this an unlikely physiological scenario for synaptic plasticity (Sheng and Hoogenraad, 2007; Opazo and Choquet, 2011). Notably, α7-nAChRs have been shown to interact with members of the PSD-95 scaffold family (Farías et al., 2007; Gómez-Varela et al., 2012). The reliance on PDZ interactions, therefore, may also reflect a requirement for α7-nAChR anchoring and localization at postsynaptic sites.
For LTP, the tethering of AMPARs to the postsynaptic scaffold usually results from a calcium-dependent phosphorylation of their TARP auxiliary subunits (Chen et al., 2000; Bats et al., 2007; Opazo et al., 2010, 2012; Opazo and Choquet, 2011). The calcium dependence of the nicotine-induced effects reported here may reflect a similar mechanism. The slow time course of the nicotinic effect, however, is also consistent with a requirement for early transcriptional events (Greenberg et al., 1992). Activation of α7-nAChRs by prolonged exposure to nicotine is known to alter gene transcription (Chang and Berg, 2001; Dajas-Bailador and Wonnacott, 2004), and signaling through α7-nAChRs on hippocampal neurons has been shown to activate the transcription factor CREB in a manner at least partly independent of NMDA and AMPARs (Hu et al., 2002).
The FRAP experiments indicated a reduction in the mobility and a 50% increase in the amount of GluA1-SEP on spines after nicotine treatment, suggesting that the receptors became synaptically incorporated (Makino and Malinow, 2009, 2011). Consistent with this, patch-clamp recordings indicated an increase in the fraction of mEPSCs having large amplitudes. Importantly, the recorded mEPSCs were generated by native AMPARs and, like the nicotine-induced increases in GluA1-SEP on spines, the increases in mEPSC amplitude were prevented by x-linking AMPARs on the neuron surface. The amplitude increase, however, was less dramatic than the increase in GluA1-SEP accumulation on spines. Part of the reason for this may be that no change was seen in GluA2-SEP over this time frame, yet the majority of synaptic AMPARs contain GluA2, presumably contributing to a large portion of synaptic transmission (Sans et al., 2003; Lu et al., 2009). A second reason, however, may be that the nicotine treatment recruited GluA1s, at least in part, to synapses with an insufficient number of AMPARs to generate mEPSCs, so-called “silent synapses” (Isaac et al., 1995; Liao et al., 1995). Consistent with this, x-linking AMPARs prevented some of the increase in mEPSC frequency induced by nicotine, as if part of this increase represented the postsynaptic unsilencing of previously silent synapses. These mEPSCs, if small in size, would detract from the net increase in mean mEPCS amplitude recorded for nicotine-treated cells. The observation that x-linking AMPARs only prevented a portion of the nicotine-induced increase in mEPSC frequency also suggests that some of the increased frequency is due to an increase in the probability of release of presynaptic glutamate-containing vesicles. Supporting this latter possibility are the reports that presynaptic nAChRs can facilitate neurotransmitter release (McGehee et al., 1995; Gray et al., 1996; Maggi et al., 2003).
Nicotine is known to enhance contextual learning, as evidenced by the role cued and context-evoked cravings play in nicotine addiction and relapse behavior (Kenney and Gould, 2008, Placzek et al., 2009; De Biasi and Dani, 2011). It is likely to do so, at least in part, by Hebbian plasticity, as documented for activity-dependent nicotinic enhancement of synaptic plasticity (Dani et al., 2001; Mansvelder and McGehee, 2002; Kenney and Gould, 2008; Placzek et al., 2009; Yakel, 2012). Unexpectedly, the present results demonstrate a new form of nicotine-driven synaptic plasticity at glutamatergic synapses, namely one that occurs more slowly and does not require coincident fast, excitatory transmission at those same synapses. Such a mechanism raises the prospect of nicotinic signaling either from systemic nicotine or from endogenous cholinergic input, affecting large numbers of synapses in a region without being constrained by patterns of ongoing activity. An alternative possibility is that the nicotinic effect may be confined to synapses having specific properties, for example, sufficient GluA1s in reserve pools on the membrane surface that otherwise may be rate limiting (Makino and Malinow, 2009; Petrini et al., 2009). This possibility is suggested by the fact that the nicotinic effect occurs independently of exocytosis and relies instead on preexisting reserve pools of surface receptors. A further constraint may be the specific cellular location and level of α7-nAChRs, which are known to be limited and subject to activity-dependent regulation (Zarei et al., 1999; Fabian-Fine et al., 2001; Kawai et al., 2002; Fernandes et al., 2010; Gómez-Varela et al., 2012). Compatible with both scenarios is the observed change in population distribution of spines based on their levels of GluA1-SEP. The apparent shift in population distribution is consistent with a cell-wide effect, but the appearance of a subset of highly enriched spines suggests that the nicotinic effect may be more prominent on specific subsets of synapses that were primed by prior events.
The findings presented here describe a novel and robust mechanism by which nicotinic signaling can modulate the cellular and molecular mechanisms controlling the strength of glutamatergic synapses. This is fundamental to understanding how the nicotinic cholinergic signaling system exerts its effects on higher cognitive function involved in learning and memory, addiction, and neurological diseases.
Footnotes
This work was supported by the National Institutes of Health (Grants NS035469, NS012601, and NIH NS068016) and the Tobacco-Related Disease Research Program (Grants 17FT-0053, 19XT-0072, and 22XT-0016). We thank Roberto Malinow for the pCI-GluA1-SEP and pCI-GluA2-SEP constructs, Xulong Wang for computer programming assistance, Xiao-Yun Wang for expert technical assistance, and Catarina Fernandes for drawing the figure illustrations. A.W.H is a National Research Service Award predoctoral fellow and D.G.V. is a Tobacco-Related Disease Research Program postdoctoral fellow.
The authors declare no competing financial interests.
References
- Ashby MC, Maier SR, Nishimune A, Henley JM. Lateral diffusion drives constitutive exchange of AMPA receptors at dendritic spines and is regulated by spine morphology. J Neurosci. 2006;26:7046–7055. doi: 10.1523/JNEUROSCI.1235-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atallah BV, Scanziani M. Instantaneous modulation of gamma oscillation frequency by balancing excitation with inhibition. Neuron. 2009;62:566–577. doi: 10.1016/j.neuron.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bats C, Groc L, Choquet D. The interaction between Stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron. 2007;53:719–734. doi: 10.1016/j.neuron.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Galzi JL, Devillers-Thiéry A, Bertrand S, Changeux JP. Mutations at two distinct sites within the channel domain M2 alter calcium permeability of neuronal alpha 7 nicotinic receptor. Proc Natl Acad Sci U S A. 1993;90:6971–6975. doi: 10.1073/pnas.90.15.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Borgdorff AJ, Choquet D. Regulation of AMPA receptor lateral movements. Nature. 2002;417:649–653. doi: 10.1038/nature00780. [DOI] [PubMed] [Google Scholar]
- Campbell NR, Fernandes CC, Halff AW, Berg DK. Endogenous signaling through alpha7-containing nicotinic receptors promotes maturation and integration of adult-born neurons in the hippocampus. J Neurosci. 2010;30:8734–8744. doi: 10.1523/JNEUROSCI.0931-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KT, Berg DK. Voltage-gated channels block nicotinic regulation of CREB phosphorylation and gene expression in neurons. Neuron. 2001;32:855–865. doi: 10.1016/S0896-6273(01)00516-5. [DOI] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Conroy WG, Liu Z, Nai Q, Coggan JS, Berg DK. PDZ-containing proteins provide a functional postsynaptic scaffold for nicotinic receptors in neurons. Neuron. 2003;38:759–771. doi: 10.1016/S0896-6273(03)00324-6. [DOI] [PubMed] [Google Scholar]
- Dajas-Bailador F, Wonnacott S. Nicotinic acetylcholine receptors and the regulation of neuronal signalling. Trends Pharmacol Sci. 2004;25:317–324. doi: 10.1016/j.tips.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Dani JA, Ji D, Zhou FM. Synaptic plasticity and nicotine addiction. Neuron. 2001;31:349–352. doi: 10.1016/S0896-6273(01)00379-8. [DOI] [PubMed] [Google Scholar]
- De Biasi M, Dani JA. Reward, addiction, withdrawal to nicotine. Annu Rev Neurosci. 2011;34:105–130. doi: 10.1146/annurev-neuro-061010-113734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Descarries L, Gisiger V, Steriade M. Diffuse transmission by acetylcholine in the CNS. Prog Neurobiol. 1997;53:603–625. doi: 10.1016/S0301-0082(97)00050-6. [DOI] [PubMed] [Google Scholar]
- De Simoni A, Griesinger CB, Edwards FA. Development of rat CA1 neurones in acute versus organotypic slices: role of experience in synaptic morphology and activity. J Physiol. 2003;550:135–147. doi: 10.1113/jphysiol.2003.039099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, Fine A. Ultrastructural distribution of the 7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci. 2001;21:7993–8003. doi: 10.1523/JNEUROSCI.21-20-07993.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farías GG, Vallés AS, Colombres M, Godoy JA, Toledo EM, Lukas RJ, Barrantes FJ, Inestrosa NC. Wnt-7a induces presynaptic colocalization of alpha 7-nicotinic acetylcholine receptors and adenomatous polyposis coli in hippocampal neurons. J Neurosci. 2007;27:5313–5325. doi: 10.1523/JNEUROSCI.3934-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. The spike-timing dependence of plasticity. Neuron. 2012;75:556–571. doi: 10.1016/j.neuron.2012.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes CC, Berg DK, Gómez-Varela D. Lateral mobility of nicotinic acetylcholine receptors on neurons is determined by receptor composition, local domain, and cell type. J Neurosci. 2010;30:8841–8851. doi: 10.1523/JNEUROSCI.6236-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Ji Z, Morita N, Sumikawa K. Acute and chronic nicotine exposure differentially facilitate the induction of LTP. Brain Res. 1999;846:137–143. doi: 10.1016/S0006-8993(99)01982-4. [DOI] [PubMed] [Google Scholar]
- Galan A, Laird JM, Cervero F. In vivo recruitment by painful stimuli of AMPA receptor subunits to the plasma membrane of spinal cord neurons. Pain. 2004;112:315–323. doi: 10.1016/j.pain.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Goetze B, Grunewald B, Baldassa S, Kiebler M. Chemically controlled formation of a DNA/calcium phosphate coprecipitate: application for transfection of mature hippocampal neurons. J Neurobiol. 2004;60:517–525. doi: 10.1002/neu.20073. [DOI] [PubMed] [Google Scholar]
- Gómez-Varela D, Schmidt M, Schoellerman J, Peters EC, Berg DK. PMCA2 via PSD-95 controls calcium signaling by α7-containing nicotinic acetylcholine receptors on aspiny interneurons. J Neurosci. 2012;32:6894–6905. doi: 10.1523/JNEUROSCI.5972-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Greenberg ME, Thompson MA, Sheng M. Calcium regulation of immediate early gene transcription. J Physiol Paris. 1992;86:99–108. doi: 10.1016/S0928-4257(05)80013-0. [DOI] [PubMed] [Google Scholar]
- Gu Z, Lamb PW, Yakel JL. Cholinergic coordination of presynaptic and postsynaptic activity induces timing-dependent hippocampal synaptic plasticity. J Neurosci. 2012;32:12337–12348. doi: 10.1523/JNEUROSCI.2129-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Heine M, Groc L, Frischknecht R, Béïque JC, Lounis B, Rumbaugh G, Huganir RL, Cognet L, Choquet D. Surface mobility of postsynaptic AMPARs tunes synaptic transmission. Science. 2008;320:201–205. doi: 10.1126/science.1152089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Lie QS, Chang KT, Berg DK. Nicotinic regulation of CREB activation in hippocampal neurons by glutamatergic and nonglutamatergic pathways. Mol Cell Neurosci. 2002;21:616–625. doi: 10.1006/mcne.2002.1202. [DOI] [PubMed] [Google Scholar]
- Hukkanen J, Jacob P, 3rd, Benowitz NL. Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 2005;57:79–115. doi: 10.1124/pr.57.1.3. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Nicoll RA, Malenka RC. Evidence for silent synapses: implications for the expression of LTP. Neuron. 1995;15:427–434. doi: 10.1016/0896-6273(95)90046-2. [DOI] [PubMed] [Google Scholar]
- Ji D, Lape R, Dani JA. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron. 2001;31:131–141. doi: 10.1016/S0896-6273(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Kawai H, Zago W, Berg DK. Nicotinic alpha 7 receptor clusters on hippocampal GABAergic neurons: regulation by synaptic activity and neurotrophins. J Neurosci. 2002;22:7903–7912. doi: 10.1523/JNEUROSCI.22-18-07903.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenney JW, Gould TJ. Modulation of hippocampus-dependent learning and synaptic plasticity by nicotine. Mol Neurobiol. 2008;38:101–121. doi: 10.1007/s12035-008-8037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R. Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci. 2006;26:2000–2009. doi: 10.1523/JNEUROSCI.3918-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Conners CK, Silva D, Hinton SC, Meck WH, March J. Transdermal nicotine effects on attention. Psychopharmacology (Berl) 1998;140:135–141. doi: 10.1007/s002130050750. [DOI] [PubMed] [Google Scholar]
- Liao D, Hessler NA, Malinow R. Activation of postsynaptically silent synapses during pairing-induced LTP in CA1 region of hippocampal slice. Nature. 1995;375:400–404. doi: 10.1038/375400a0. [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- Lozada AF, Wang X, Gounko NV, Massey KA, Duan J, Liu Z, Berg DK. Glutamatergic synapse formation is promoted by α7-containing nicotinic acetylcholine receptors. J Neurosci. 2012a;32:7651–7661. doi: 10.1523/JNEUROSCI.6246-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozada AF, Wang X, Gounko NV, Massey KA, Duan J, Liu Z, Berg DK. Induction of dendritic spines by β2-containing nicotinic receptors. J Neurosci. 2012b;32:8391–8400. doi: 10.1523/JNEUROSCI.6247-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. 2009;62:254–268. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi L, Le Magueresse C, Changeux JP, Cherubini E. Nicotine activates immature “silent” connections in the developing hippocampus. Proc Natl Acad Sci U S A. 2003;100:2059–2064. doi: 10.1073/pnas.0437947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. Compartmentalized versus global synaptic plasticity on dendrites controlled by experience. Neuron. 2011;72:1001–1011. doi: 10.1016/j.neuron.2011.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol. 2002;53:606–617. doi: 10.1002/neu.10148. [DOI] [PubMed] [Google Scholar]
- Mayford M, Siegelbaum SA, Kandel ER. Synapses and memory storage. Cold Spring Harb Perspect Biol. 2012;4:a005751. doi: 10.1101/cshperspect.a005751. pi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- Miesenböck G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394:192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- Mondin M, Labrousse V, Hosy E, Heine M, Tessier B, Levet F, Poujol C, Blanchet C, Choquet D, Thoumine O. Neurexin-neuroligin adhesions capture surface-diffusing AMPA receptors through PSD-95 scaffolds. J Neurosci. 2011;31:13500–13515. doi: 10.1523/JNEUROSCI.6439-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff RA, 3rd, Conroy WG, Schoellerman JD, Berg DK. Synchronous and asynchronous transmitter release at nicotinic synapses are differentially regulated by postsynaptic PSD-95 proteins. J Neurosci. 2009;29:15770–15779. doi: 10.1523/JNEUROSCI.4951-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC. Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann N Y Acad Sci. 1999;868:515–525. doi: 10.1111/j.1749-6632.1999.tb11320.x. [DOI] [PubMed] [Google Scholar]
- Opazo P, Choquet D. A three-step model for the synaptic recruitment of AMPA receptors. Mol Cell Neurosci. 2011;46:1–8. doi: 10.1016/j.mcn.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron. 2010;67:239–252. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Opazo P, Sainlos M, Choquet D. Regulation of AMPA receptor surface diffusion by PSD-95 slots. Curr Opin Neurobiol. 2012;22:453–460. doi: 10.1016/j.conb.2011.10.010. [DOI] [PubMed] [Google Scholar]
- Passafaro M, Sala C, Niethammer M, Sheng M. Microtubule binding by CRIPT and its potential role in the synaptic clustering of PSD-95. Nat Neurosci. 1999;2:1063–1069. doi: 10.1038/15990. [DOI] [PubMed] [Google Scholar]
- Peeke SC, Peeke HV. Attention, memory and cigarette smoking. Psycopharmacology (Berl) 1984;84:205–215. doi: 10.1007/BF00427447. [DOI] [PubMed] [Google Scholar]
- Pepeu G, Giovannini MG. Changes in acetylcholine extracellular levels during cognitive processes. Learn Mem. 2004;11:21–27. doi: 10.1101/lm.68104. [DOI] [PubMed] [Google Scholar]
- Petrini EM, Lu J, Cognet L, Lounis B, Ehlers MD, Choquet D. Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation. Neuron. 2009;63:92–105. doi: 10.1016/j.neuron.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placzek AN, Zhang TA, Dani JA. Nicotinic mechanisms influencing synaptic plasticity in the hippocampus. Acta Pharmacol Sin. 2009;30:752–760. doi: 10.1038/aps.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin XF, An DS, Chen IS, Baltimore D. Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc Natl Acad Sci U S A. 2003;100:183–188. doi: 10.1073/pnas.232688199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JE, Mukhin AG, Lokitz SJ, Turkington TG, Herskovic J, Behm FM, Garg S, Garg PK. Kinetics of brain nicotine accumulation in dependent and nondependent smokers assessed with PET and cigarettes containing 11C-nicotine. Proc Natl Acad Sci U S A. 2010;107:5190–5195. doi: 10.1073/pnas.0909184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sans N, Vissel B, Petralia RS, Wang YX, Chang K, Royle GA, Wang CY, O'Gorman S, Heinemann SF, Wenthold RJ. Aberrant formation of glutamate receptor complexes in hippocampal neurons of mice lacking the GluR2 AMPA receptor subunit. J Neurosci. 2003;23:9367–9373. doi: 10.1523/JNEUROSCI.23-28-09367.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 2007;76:823–847. doi: 10.1146/annurev.biochem.76.060805.160029. [DOI] [PubMed] [Google Scholar]
- Shi S, Hayashi Y, Esteban JA, Malinow R. Subunit-specific rules governing AMPA receptor trafficking to synapses in hippocampal pyramidal neurons. Cell. 2001;105:331–343. doi: 10.1016/S0092-8674(01)00321-X. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Hirano T. Visualization of subunit-specific delivery of glutamate receptors to postsynaptic membrane during hippocampal long-term potentiation. Cell Rep. 2012;1:291–298. doi: 10.1016/j.celrep.2012.02.004. [DOI] [PubMed] [Google Scholar]
- Thiel CM, Fink GR. Effects of the cholinergic agonist nicotine on reorienting of visual and special attention and top-down attentional control. Neuroscience. 2008;152:381–390. doi: 10.1016/j.neuroscience.2007.10.061. [DOI] [PubMed] [Google Scholar]
- Umbriaco D, Garcia S, Beaulieu C, Descarries L. Relational features of acetylcholine, noradrenaline, serotonin and GABA axon terminals in the stratum radiatum of adult rat hippocampus (CA1) Hippocampus. 1995;5:605–620. doi: 10.1002/hipo.450050611. [DOI] [PubMed] [Google Scholar]
- Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, Swanson LW. Distribution of alpha2, alpha3, alpha4, and beta2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol. 1989;284:314–335. doi: 10.1002/cne.902840212. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Woods G, Zito K. Preparation of gene gun bullets and biolistic transfection of neurons in slice culture. J Vis Exp. 2008;12:675. doi: 10.3791/675. pii. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakel JL. Nicotinic ACh receptors in the hippocampus: role in excitability and plasticity. Nicotine Tob Res. 2012;14:1249–1257. doi: 10.1093/ntr/nts091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarei MM, Radcliffe KA, Chen D, Patrick JW, Dani JA. Distributions of nicotinic acetylcholine receptor alpha7 and beta2 subunits on cultured hippocampal neurons. Neuroscience. 1999;88:755–764. doi: 10.1016/S0306-4522(98)00246-2. [DOI] [PubMed] [Google Scholar]