

Abstract
Environmental insults during sensitive periods can affect hippocampal development and function, but little is known about peripheral infection, especially in humans and other animals whose brain is gyrencephalic and experiences major perinatal growth. Using a piglet model, the present study showed that inoculation on postnatal day 7 with the porcine reproductive and respiratory syndrome virus (PRRSV) caused microglial activation within the hippocampus with 82% and 43% of isolated microglia being MHC II+ 13 and 20 d after inoculation, respectively. In control piglets, <5% of microglia isolated from the hippocampus were MHC II+. PRRSV piglets were febrile (p < 0.0001), anorectic (p < 0.0001), and weighed less at the end of the study (p = 0.002) compared with control piglets. Increased inflammatory gene expression (e.g., IL-1β, IL-6, TNF-α, and IFN-γ) was seen across multiple brain regions, including the hippocampus, whereas reductions in CD200, NGF, and MBP were evident. In a test of spatial learning, PRRSV piglets took longer to acquire the task, had a longer latency to choice, and had a higher total distance moved. Overall, these data demonstrate that viral respiratory infection is associated with a marked increase in activated microglia in the hippocampus, neuroinflammation, and impaired performance in a spatial cognitive task. As respiratory infections are common in human neonates and infants, approaches to regulate microglial cell activity are likely to be important.
Keywords: brain, cognition, inflammation, PRRSV, sickness behavior
Introduction
Infectious disease remains the most common cause of illness in children, with acute respiratory infection constituting the most prevalent reason for medical intervention in children under one year of age (Hall et al., 2009). Given the well-characterized immune-to-brain signaling pathways that lead to neuroinflammation (Dantzer et al., 2008), serious peripheral infection in the neonatal period is a concern. Although brain areas are formed prenatally, the brain growth spurt continues for the first 2 years after birth due to dendritic growth, synaptogenesis, and glial cell proliferation. Neurogenesis in the hippocampal dentate gyrus, as well as different cortical regions, persists in the neonatal period in infants (Sanai et al., 2011; Feliciano and Bordey, 2013). Developing and mature neurons, as well as glia, have numerous cytokine receptors, and the hippocampus is especially rich with receptors for proinflammatory cytokines (Parnet et al., 2002). Overall, studies on the effects of neonatal infection on neuroinflammation are lacking.
Rodent studies designed to mimic prenatal infections in humans suggest that untimely neuroinflammation can produce structural and functional changes to the brain that are measurable in adulthood (Fatemi et al., 1998, 1999, 2002, 2008; Shi et al., 2003; Boissé et al., 2004; Ikeda et al., 2005; Bilbo et al., 2006; Meyer et al., 2006; Kohman et al., 2008). However, little is known about peripheral infection in the neonatal period, especially in humans and other animals whose brain is gyrencephalic and experience major perinatal growth. Furthermore, because rodent models are not amenable to behavioral testing as neonates, the impact of infection on hippocampal function during this period is largely unknown. To overcome several of these obstacles, the present study used a neonatal piglet model. Several attributes make the piglet an excellent translational model for psychoneuroimmunological studies. For example, pigs and humans experience a major brain growth spurt from the late prenatal to early postnatal period, which differs from other common animal models (Dobbing and Sands, 1979). Gross brain anatomical features, including gyral pattern and distribution of gray and white matter, are similar in piglets and human infants (Dickerson and Dobbing, 1967; Thibault and Margulies, 1998). Additionally, piglets can undergo behavioral testing at a young age (Dilger and Johnson, 2008; Elmore et al., 2012) and show far more overlap with humans concerning genes involved in immunity compared with rodents (Dawson, 2011; Meurens et al., 2012).
In the present study, we tested the hypothesis that peripheral infection with porcine reproductive and respiratory syndrome virus (PRRSV) in the neonatal period activates hippocampal microglia and induces deficits in spatial learning in piglets. In young pigs, PRRSV preferentially infects mononuclear myeloid cells in the lungs inducing interstitial pneumonia, reduced appetite and growth, and leucopenia (Escobar et al., 2004; Liu et al., 2009; Miguel et al., 2010). PRRSV was selected over other infectious agents or mimetics (e.g., Escherichia coli or LPS) due to its ability to cause chronic respiratory tract infection.
Materials and Methods
Animals, housing, handling, and feeding.
Naturally farrowed crossbred piglets (16 females and 17 males) were obtained from nine separate litters from the University of Illinois swine herd. Piglets were killed at two different time points after inoculation (see details below), creating a 2 × 2 factorial design with the variables of treatment and time after inoculation (control, d 13: n = 6; PRRSV, d 13: n = 8; control, d 20: n = 8; PRRSV, d 20: n = 11; Fig. 1). Piglets were brought to the biomedical animal facility 48 h after birth (to allow for colostrum consumption from the dam) and housed individually in cages (0.87 m × 0.87 m × 0.49 m; length × width × height). Control and PRRSV piglets were housed in separate biomedical containment chambers, and a strict protocol of handling and cleaning was followed to prevent cross-contamination. Each cage was positioned in a rack, with stainless steel perforated wall partitions and clear acrylic front and rear doors. In addition, each cage was fitted with flooring designed for neonatal animals (Tenderfoot/NSR, Tandem Products). A toy (plastic Jingle Ball, Bio-Serv) was provided to each piglet. Room temperature was maintained at 27°C, and each cage was equipped with an electric heat pad (K&H Lectro-Kennel Heat Pad, K&H Manufacturing). Piglets were maintained on a 12 h light/dark cycle; however, during the dark cycle, minimal lighting was provided.
Figure 1.

Experimental timeline.
Piglets were handled by the experimenters multiple times per day during cleaning/feeding and daily observations. In addition, for several days before inoculation (d −4 to d −1), each piglet was permitted an exploration/exercise period in an adjacent hallway for 5 min daily. In a previous experiment (Elmore et al., 2012), we found that this habituation period reduced noncompliance (i.e., piglets failing to choose a reward arm in the given time) during cognitive behavioral testing.
Piglets were fed a nutritionally complete commercial piglet milk replacer (Advance Liqui-Wean, Milk Specialties). Milk was reconstituted fresh each morning to a final concentration of 206 g/L using tap water and supplied at a rate of 285 ml/kg body weight (BW; based on daily recorded weights). This level of feeding allowed for maintenance and growth but prevented complete satiation to ensure that the piglets remained motivated for food rewards in the behavioral task. Water was not provided separately from that used in the milk replacer. Milk replacer was delivered from a reservoir to a stainless-steel bowl (secured to the side of each cage) via a peristaltic pump (Control). Using this automated delivery system similar to that described previously (Dilger and Johnson, 2008), piglets received their daily allotted milk over 18 meals (once per hour), followed by a 6 h fasting period before behavioral testing where no milk was provided. All animal care and experimental procedures were in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals and approved by the University of Illinois Institutional Animal Care and Use Committee.
Inoculation with PRRSV and sickness measures.
At 7 d of age, piglets were inoculated intranasally with either 1 ml of 1 × 105 50% tissue culture infected dose (TCID 50) of live PRRSV (strain P129-BV), obtained from the School of Veterinary Medicine at Purdue University (West Lafayette, Indiana) or sterile PBS. Daily recorded body weights acted as an important indicator of health status throughout the study. In addition, before inoculation (d −1) and throughout the remainder of the study, daily rectal temperatures were obtained each morning. Last, the willingness of the piglets to consume their first daily meal after behavioral testing was determined using a feeding score (0 = no attempt to consume the milk; 1 = attempted to consume the milk, but did not finish within 1 min; 2 = consumed all of the milk within 1 min), which was collected from d −1 to d 12 or 19 after inoculation.
Cognitive testing using a spatial T-maze task.
Piglet spatial learning and memory were assessed using a clear plastic plus-shaped maze (essentially a double T-maze) with extra maze cues, which has been previously described and validated (Elmore et al., 2012). The maze consisted of two start arms (north and south) and two reward arms (east and west). Start arm location was alternated throughout testing, which ensured that the piglet did not solve the task using an egocentric mechanism (i.e., turn body left or right, striatum-dependent), and instead was forced to adopt an allocentric mechanism (i.e., used extra-maze visual cues to create a spatial map of the room, hippocampus-dependent) for solving the task (Fitz et al., 2008). This task is similar to that used in rodents to assess “place” and/or “direction” learning (Tolman et al., 1946; Stringer et al., 2005; Zurkovsky et al., 2006; Walsh et al., 2008). Starting on d 7 after inoculation (d 14 of age), piglets were tested daily between 08:00 h and 12:00 h by one trained experimenter. Piglets completed 10 trials per day (60 s per trial), for a total of 11 d. The first 8 d of testing constituted the acquisition phase, where piglets learned to locate the chocolate milk reward (3 ml of the same milk replacer used for regular feedings with the addition of Nesquik cocoa powder, supplied according to the manufacturer's directions) in a constant place in space, as well as direction (e.g., west reward arm), using the extra-maze visual cues. Chocolate milk was provided in both reward arms to balance for olfactory cues but was only accessible in the correct reward arm. Although PRRSV infection can reduce food intake, the combined effect of a 6 h deprivation period before testing, as well as small portions of a desired food source (i.e., 3 ml of chocolate milk) ensured that the piglets were motivated to perform the behavioral task. A performance criterion of 80% correct was applied, which, when reached, would indicate that the piglets had successfully acquired the task. Acquisition was followed by a reversal phase of testing, where the previously incorrect arm (e.g., east), was now rewarded. A video camera was mounted from the ceiling above the arena and used to record piglet movement within the maze. Piglet movement was tracked live using commercially available software (EthoVision 3.1; Noldus Information Technology), providing data on latency to reward arm choice (seconds) and total distance moved (centimeters).
Death and tissue collection.
The first subset of piglets was killed 13 d after inoculation (d 20 of age) after completing 5 d of acquisition training. This time point was determined from previous pilot work (data not shown), which showed peak performance differences between control and PRRSV piglets in the spatial T-maze task. The remaining piglets were killed after the completion of cognitive testing (20 d after inoculation, d 27 of age). All piglets were anesthetized using a telazol:ketamine:xylazine solution (50.0 mg of tiletamine, plus 50.0 mg of zolazepam, reconstituted with 2.50 ml ketamine [100 g/L] and 2.50 ml xylazine [100 g/L]; Fort Dodge Animal Health); the anesthetic combination was administered intramuscularly (i.m.) at 0.03 ml/kg BW. After verifying anesthetic induction, piglets were killed via intracardiac (i.c.) administration of sodium pentobarbital (86 mg/kg BW; Fatal Plus, Vortech Pharmaceuticals). Blood serum (∼3 ml) was collected and stored frozen (−80°C). Peripheral tissues (lung tissue and tracheobronchial lymph nodes) and brain tissue (hippocampus, prefrontal cortex, and striatum) were collected, rinsed in PBS, and stored at −80°C in RNA later (Ambion) until processing.
Peripheral PRRSV detection and cytokines.
The presence of PRRSV antibodies in the serum of all piglets at the end of the study was analyzed by the Veterinary Diagnostic Laboratory (University of Illinois, Urbana, Illinois) using a PRRSV-specific ELISA kit (IDEXX Laboratories). This assay has 98.8% sensitivity and 99.9% specificity, with an S/P ratio of >0.4 indicating a positive sample. Serum proinflammatory cytokine (IL-1β, IL-6, and TNF-α) levels at the end of the study were analyzed using porcine-specific sandwich enzyme immunoassays (R&D Systems). Samples were assayed according to the manufacturer's instructions, and mean minimum detectable concentrations were as follows: IL-1β, 6.72 pg/ml; IL-6, 2.03 pg/ml; TNF-α, 3.7 pg/ml. Interassay and intra-assay coefficients of variation were <7.2%, 5.1%, and 6.9%, respectively.
Microglia isolation and cell sorting using flow cytometry.
Microglia were isolated using a method adapted from Henry et al. (2009). Approximately 500 mg of hippocampal tissue from each hemisphere (i.e., the majority of each hemisphere was collected; however, the ventral region of the hippocampus was removed for additional analyses) was collected from each piglet and rinsed in PBS. Each hemisphere was treated as its own sample for the first several steps of the protocol to maximize the number of isolated cells. Tissue was pushed through a 70 μm nylon mesh cell strainer (BD Biosciences) for homogenization using Dulbecco's PBS (Sigma) with 0.2% glucose and spun at 600 × g for 6 min to form a pellet. The supernatant was removed, and the pellet was resuspended in a 70% isotonic Percoll (GE Healthcare) solution. Layers of 50%, 35%, and 0% isotonic Percoll were overlaid to form a discontinuous gradient, which was then spun for 20 min at 2000 × g. The microglia layer, located at the interface of the 50% and 70% layers, was removed (Frank et al., 2006). At this point, isolated microglial cells from both hemispheres of each piglet were combined and spun at 600 × g for 6 min to form a pellet. This pellet was resuspended in flow buffer (PBS with 1% BSA; Fisher Scientific), 0.1% sodium azide (Sigma), and 20 mm glucose (Sigma). Purified CD16/CD32 antibodies (eBioscience) were used to block competitive binding of the Fc receptor on microglia. The cells were incubated with antibodies for receptors used to identify microglia cells: CD11b (BioLegend) and CD45 (AbD Serotec), as well as MHC II (Antibodies Online), as a marker of cell activation. Using the FACS Aria II flow cytometer (BD Biosciences), the viable microglia cell population from a massed unstained control sample (consisting of a subsample of isolated cells from each piglet) was gated according to size and granularity based on forward and side scatter properties, as well as autofluorescence. Similar to mice, cells were identified by the expression of CD11b and CD45 surface receptors. However, the expression pattern of these receptors differs from mouse data in that porcine microglia express lower levels of CD11b and higher levels of CD45 (Gregerson and Yang, 2003), identified as CD11b+/CD45int (Gregerson and Yang, 2003) versus CD11b+high/CD45low in mice (Ford et al., 1995). Cells with high CD45 expression were excluded from the analysis, as this distinct population is likely other myeloid cell types (e.g., peripheral macrophages) (Gregerson and Yang, 2003). Cells were then quantified according to MHC II positivity, resulting in a proportion of MHC II− and MHC II+ cells for each piglet.
Real-time PCR.
Total RNA from ∼75 mg of the lungs, tracheobronchial lymph nodes, hippocampus, prefrontal cortex, and striatum of each piglet was isolated using the Tri Reagent protocol (Sigma). A QuantiTect Reverse Transcription Kit (QIAGEN) was used for cDNA synthesis with integrated removal of genomic DNA contamination according to the manufacturer's protocol. Quantitative real-time PCR was performed using the Applied Biosystems TaqMan Gene Expression Assay protocol. A custom TaqMan Low Density Array card was designed, which contained a reference gene (ribosomal protein L19, RPL19) and 22 genes of interest (Table 1), including PRRSV (Chen et al., 2009), which were run in duplicate. In brief, cDNA was amplified by PCR where a target cDNA (Table 1) and reference cDNA (RPL19) were amplified simultaneously using an oligonucleotide probe with a 5′ fluorescent reporter dye (6-FAM) and a 3′ quencher dye (NFQ). Conditions for the PCR procedure included: 50°C for 2 min, 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. Fluorescence was determined on an ABI PRISM 7900HT-sequence detection system (PerkinElmer). Data were excluded from analysis for the following reasons: (1) both wells did not run; or (2) high SE between the wells (>0.5). Data were analyzed using the comparative threshold cycle (Ct) method (Livak and Schmittgen, 2001), and results are expressed as fold change to the standardized relative quantification (RQ) baseline (RQ = 1), whereby a value >1 would represent a fold increase in mRNA expression compared with control for a particular gene and a value between 0 and 1 would represent a fold decrease in mRNA expression compared with controls.
Table 1.
Quantitative real-time PCR primer information
| Gene | Classification | Accession numbera | Assay identificationb |
|---|---|---|---|
| RPL19, ribosomal protein L19 | Reference | AF435591 | Ss03375624_g1 |
| IL-10 | Anti-inflammatory | NM_214041 | Ss03382372_u1 |
| IL-13 | Anti-inflammatory | NM_213803 | Ss03392351_m1 |
| TGF-β1 | Anti-inflammatory | NM_214015 | Ss03382325_u1 |
| CD200r, cluster of differentiation 200 receptorc | Microglial | NA | NA |
| CX3CL1, fraktalkine | Neuronal | DQ991100 | Ss03377157_u1 |
| BDNF, brain-derived neurotrophic factor | Neuronal | NM_214259 | Ss03822335_s1 |
| CD200, cluster of differentiation 200 | Neuronal | AJ584612 | Ss03375826_u1 |
| MBP | Neuronal | NM_001001546 | Ss03385047_u1 |
| NGFd | Neuronal | NA | NA |
| SOD1, superoxide dismutase 1 | Neuronal | NM_001190422 | Ss03375614_u1 |
| SYP, synaptophysine | Neuronal | NA | NA |
| HMOX1, heme oxygenase (decycling) 1 | Oxidative Stress | NM_001004027 | Ss03378516_u1 |
| NOS2, nitrous oxide synthase | Oxidative Stress | NM_001143690 | Ss03374608_u1 |
| NQ01, NAD(P)H dehydrogenase, quinone 1 | Oxidative Stress | NM_001159613 | Ss03226763_g1 |
| IFN-γ | Pro-inflammatory | NM_213948 | Ss03391054_m1 |
| IL-1β | Pro-inflammatory | NM_214055 | Ss03393804_m1 |
| IL-6 | Pro-inflammatory | NM_214399 | Ss03384604_u1 |
| TNF-α | Pro-inflammatory | NM_214022 | Ss03391318_g1 |
| PRRSVf | Viral | NA | NA |
| TLR-3 | Viral | NM_001097444 | Ss03388861_m1 |
| TLR-4 | Viral/Bacterial | NM_001113039 | Ss03389780_m1 |
| TLR-7 | Viral | NM_001097434 | Ss03385429_u1 |
aNCBI GenBank accession number.
bApplied Biosystems TaqMan Gene Expression Assay identification number.
cCD200r custom probe: forward primer, TGTTCCAAGTTACTAATCAGGCTGAA; reverse primer, AGCCCATTAGCAACATGATACTCTTT; probe, ACATAGAATTGAAGGAAGGG.
dNGF custom probe: forward primer, TCAACAGGACTCACAGGAGCAA; reverse primer, ACTCCCCCCGGTGGAAA; probe, CGGTCGTCATCCC.
eSYP custom probe: forward primer, GGCCAAGGACGGCTCAT; reverse primer, TTTTCCGCCCTTAGCATGTAG; probe, CAAGATTAAATGGTACGTAGGAC.
fPRRSV custom probe (Chen et al., 2009): forward primer, CGCACCAGATGGGACCTACTT; reverse primer, ACGGTGTTCAGTGAGGGCTTT; probe, CGCTGCGTTGACTGG.
Statistical analysis.
Data analysis was conducted using the MIXED procedure of the Statistical Analysis Systems software (SAS Institute). Data were transformed as necessary to meet the assumptions of the test (e.g., homogeneity of variance). Transformations include weight (log), latency to choice (log), and total distance moved (log). For sickness measures, data were analyzed as a three-way (treatment × sex × day) repeated-measures ANOVA, where the variable pig was treated as a random effect and nested within treatment and sex. Cognitive performance data were analyzed similarly, with the addition of day nested within phase to account for both testing phases of the spatial T-maze task (i.e., acquisition and reversal). Performance during the reversal phase of cognitive testing was analyzed in two ways: (1) with all animals or (2) with only those animals that reached learning criterion (80% correct) during acquisition. As the statistical analysis revealed similar outcomes for both approaches (data not shown), cognitive performance data for both testing phases are shown with all animals included in the dataset. Gene expression data were first analyzed as treatment × sex × time. However, because very few interactions showed statistical significance, both time points were combined and the effect of treatment alone was also determined. For all other measures, which were only sampled once at the end of the study, data were analyzed as a three-way (treatment × sex × time after inoculation) ANOVA. The parameter sex was removed from the statistical model if not significant as a main effect or as part of an interaction. Post hoc paired contrasts were used to further examine significant effects. Correlations were determined using Proc Corr in SAS, and the best fit line was found using the Linear Regression function in GraphPad Prism 5 (GraphPad Software). Statistical significance was accepted at p < 0.05, statistical trends at p ≤ 0.10. Unless otherwise stated, data are presented as untransformed Least Square Means (LSM) ± SEM.
Results
PRRSV infection and measures of sickness
Serum ELISA results confirmed that all control piglets tested negative for PRRSV antibodies (S/P ratio range: 0.00–0.02) at the end of the study, whereas all PRRSV-infected piglets tested positive (S/P ratio range: 0.88–2.36). Changes in body weight, temperature, and willingness to consume the first daily meal were used to measure the sickness response of piglets infected with PRRSV. Body weight data showed a significant effect of treatment (F(1,585) = 26.55), day (F(25,585) = 915.18), and treatment × day (F(25,585) = 3.24; all effects, p < 0.001), indicating that, although all piglets gained weight throughout the course of the study, PRRSV piglets had lower average body weights at the end of the study compared with control piglets (Fig. 2A). In addition, there was an effect of sex (F(1,585) = 7.75; p = 0.006) on body weight, as female piglets (3.42 ± 0.03) weighed less than males (3.50 ± 0.03) overall. There was also a treatment × sex (F(1,585) = 5.46; p = 0.02) and treatment × sex × day interaction (F(25,585) = 1.86; p = 0.007), where PRRSV females (3.39 ± 0.04) weighed less than control females (3.45 ± 0.05; p = 0.051) and control males (3.66 ± 0.05; p < 0.001), but not PRRSV males (3.34 ± 0.04; p = 0.714), which was exacerbated over time (data not shown). Rectal temperature data showed similar results, with significant effects of treatment (F(1,529) = 156.10), day (F(21,529) = 4.28), and treatment × day (F(21,529) = 2.77; all effects, p < 0.001), revealing that PRRSV piglets had elevated temperatures after infection, which increased at ∼2 d after inoculation and returned to baseline by the end of the study (Fig. 2B). Last, the same pattern seen for body temperature was also observed for the feeding score (Fig. 2C), with treatment (F(1,508) = 57.46), day (F(20,508) = 3.39), and the interaction of these two variables (F(20,508) = 3.39) all showing statistical significance (all, p < 0.001). Interestingly, although the piglets showed reduced motivation to consume their standard milk replacer in the home cage after behavioral testing, many of the piglets willingly drank the chocolate milk in the behavioral task, which is either due to the 6 h deprivation period before testing or a preference for the chocolate milk compared with the standard milk replacer. Combined, these data reveal that sickness behavior in PRRSV piglets was evident from 2 to 14 d after inoculation, which then returned to baseline levels by the end of the study (d 20). However, body weight in PRRSV piglets remained lower than control from d 8 through d 20 after inoculation.
Figure 2.
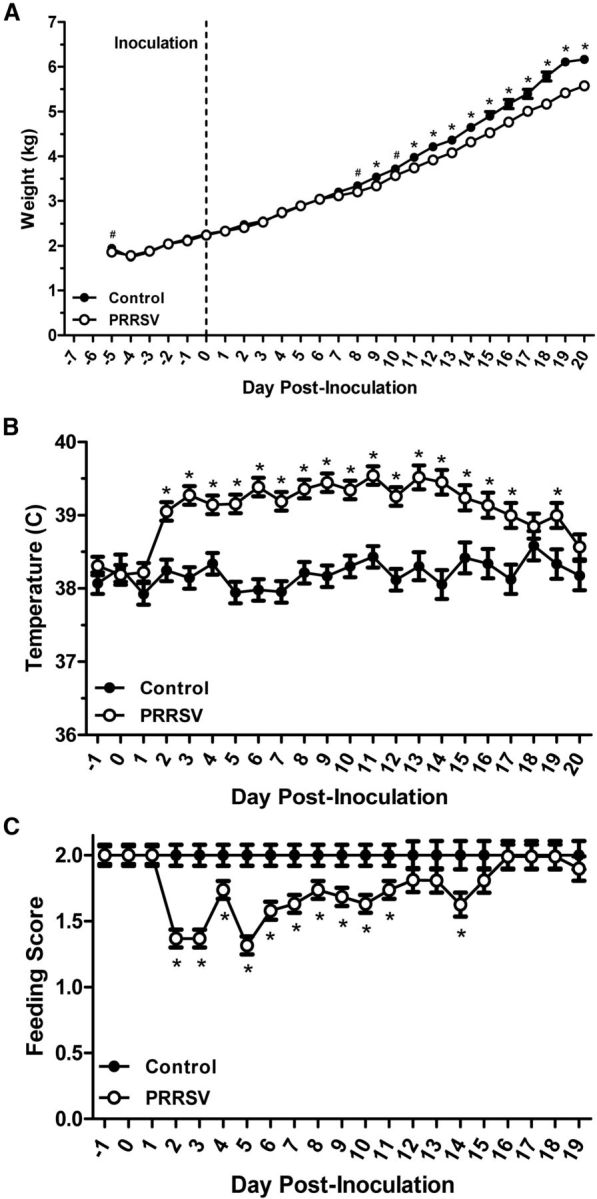
Daily recorded sickness measures (A, body weight; B, rectal temperature; C, feeding score) for control (13 and 20 d after inoculation combined, n = 14 total) and PRRSV (13 and 20 d after inoculation combined, n = 19 total) piglets throughout the course of the study. Body weight was measured from the start of the experiment, and rectal temperature and feeding score were measured beginning 1 d before inoculation. Data are LSM ± SEM. Contrasts were used to separate treatment means on each day *p < 0.05. #p < 0.10.
Serum proinflammatory cytokines
Data in Table 2 show that serum proinflammatory cytokines IL-1β (F(1,29) = 11.04; p = 0.002), IL-6 (F(1,29) = 38.93; p < 0.001), and TNF-α (F(1,29) = 28.79; p < 0.001) were elevated at the end of the study in piglets infected with PRRSV. Interestingly, there was no effect of time after inoculation (IL-1β: F(1,29) = 0.00; p = 0.987; IL-6: F(1,29) = 0.54; p = 0.468; and TNF-α: F(1,29) = 0.82; p = 0.373) or treatment × time after inoculation (IL-1β: F(1,29) = 0.11; p = 0.747; IL-6: F(1,29) = 0.57; p = 0.456; and TNF-α: F(1,29) = 0.82; p = 0.374) on any of these measures. These data demonstrate that PRRSV-infected piglets exhibited similar serum proinflammatory cytokine expression at 13 and 20 d after inoculation.
Table 2.
Serum proinflammatory cytokine concentrationsa
| CON (pg/ml) | PRRS (pg/ml) | p | |
|---|---|---|---|
| IL-1β | 1045 ± 13.6 | 1105 ± 11.7 | 0.0024 |
| IL-6 | 362 ± 1.50 | 375 ± 1.30 | < 0.001 |
| TNF-α | 827 ± 495 | 4335 ± 426 | < 0.001 |
aData from CON (13 and 20 d after inoculation combined, n = 14 total) and PRRS (13 and 20 d after inoculation combined, n = 19 total) piglets at the end of the study. Data are LSM ± SEM.
Performance in a spatial T-maze task
PRRSV infection altered piglet performance in a spatial T-maze task (F(1,226) = 11.18; p = 0.001), which varied according to phase of testing (F(1,2926) = 64.22; p < 0.001), where PRRSV piglets performed more poorly, as indicated by the proportion of correct choices in the maze, than control piglets during acquisition (control: 0.78 ± 0.02 vs PRRSV: 0.61 ± 0.02; p < 0.001), but not reversal testing (control: 0.47 ± 0.04 vs PRRSV: 0.46 ± 0.04; p = 0.77). However, treatment × day (phase), which accounts for the effects of treatment on each day of testing within a certain phase (i.e., acquisition or reversal), was not significant (F(9,226) = 0.89; p = 0.532; Fig. 3A). In addition, piglets infected with PRRSV exhibited a longer latency to make a reward arm choice in the maze (control: 2.18 ± 0.78 s vs PRRSV: 10.42 ± 0.66 s; F(1,226) = 166.10; p < 0.001), but infection status did not alter performance in either phase of testing (F(1,226) = 2.65; p = 0.105) or the interactions of treatment × phase (F(1,226) = 0.00; p = 0.992) and treatment × day (phase) (F(9,226) = 1.37; p = 0.201; Fig. 3B). Total distance moved in the maze was greater in PRRSV piglets (control: 100.05 ± 5.16 cm vs PRRSV: 132.23 ± 4.42 cm; F(1,206) = 31.07; p < 0.001) and greater in males compared with females regardless of treatment (males: 123.92 ± 4.70 cm vs females: 108.37 ± 4.91 cm; F(1,206) = 7.07; p = 0.009). In addition, there was a trend for a treatment × phase effect (F(1,206) = 3.32; p = 0.07), where PRRSV piglets traveled a greater distance in the maze than control piglets during both acquisition (control: 94.70 ± 4.34 cm vs PRRSV: 133.96 ± 3.72 cm; p < 0.001) and reversal testing (control: 105.40 ± 8.73 cm vs PRRSV: 130.50 ± 7.47 cm; p = 0.035). There was a sex × phase effect (F(1,206) = 4.08; p = 0.045) as well, where in reversal testing, females traveled a shorter total distance (103.34 ± 8.29 cm) than males (132.57 ± 7.95 cm; p = 0.009). Last, there was a treatment × sex × phase effect (F(1,206) = 5.65; p = 0.018), where PRRSV females moved a greater distance during acquisition than control females (control: 91.46 ± 6.14 cm vs PRRSV: 135.33 ± 5.47 cm; p < 0.001), but PRRSV females moved a shorter distance during reversal than PRRSV males (females: 105.37 ± 11.08 cm vs males: 155.63 ± 10.01 cm; p < 0.001). However, there was not a treatment × sex (F(1,206) = 1.24; p = 0.266), treatment × day (phase) (F(9,206) = 0.60; p = 0.798; Fig. 3C), or a treatment × sex × day (phase) effect (F(9,206) = 1.13; p = 0.343) for total distance moved in the maze. Combined, these data reveal that PRRSV piglets showed cognitive deficits in learning a spatial T-maze task, as evidenced by a reduction in the proportion of correct reward arm choices, longer latency to make a choice, and a greater total distance moved in the maze. The finding that PRRSV piglets moved a greater total distance in the maze eliminates two potential explanations for reduced cognitive performance in the maze: decreased motor ability or lethargy resulting from infection.
Figure 3.
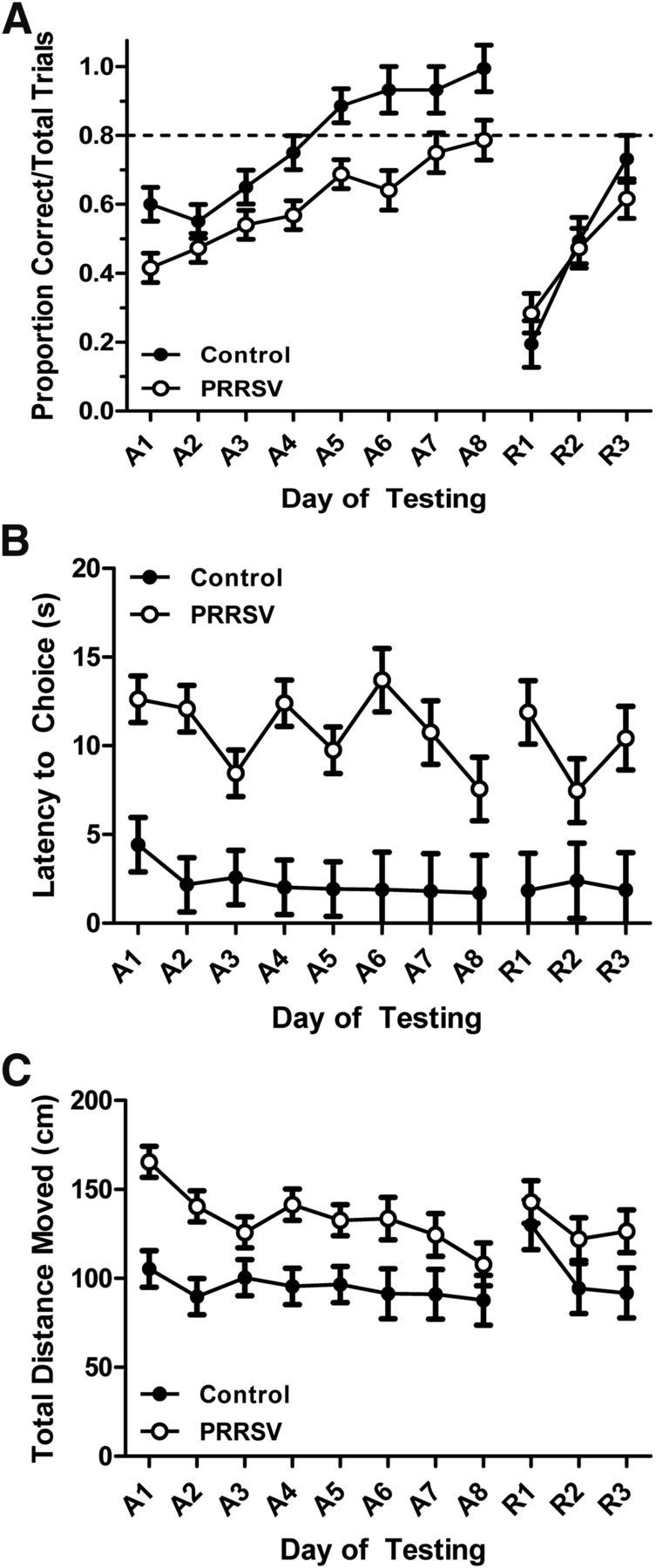
Performance of control (13 and 20 d after inoculation combined, n = 14 total) and PRRSV (13 and 20 d after inoculation combined, n = 19 total) piglets during the acquisition phase (A1–A8) and reversal phase (R1–R3) of cognitive testing in a spatial T-maze task (A, proportion of correct reward arm choices/total trials) and locomotor readouts (B, latency to choice; C, total distance moved). Data are LSM ± SEM.
Microglial activation
Isolated cells from the hippocampus were incubated with antibodies for receptors used to identify microglia cells (CD11b and CD45), as well as a marker of cell activation (MHC II; Fig. 4A,B), and sorted using flow cytometry. The presence of peripheral macrophages (characterized as CD11b+/CD45high) in the samples of isolated microglia cells was very low (control: 1.47 ± 0.47%; PRRSV: 1.68 ± 0.38%) and did not differ according to treatment (F(1,24) = 0.11; p = 0.738), time after inoculation (data not shown; F(1,24) = 1.03; p = 0.32), or sex (data not shown; F(1,24) = 0.62; p = 0.439). The proportion of activated microglia cells showed a significant effect of treatment (F(1,29) = 73.68; p < 0.001), time after inoculation (F(1,29) = 7.45; p = 0.012), and treatment × time after inoculation (F(1,29) = 7.76; p = 0.009), demonstrating that PRRSV infection resulted in a dramatic increase in activated microglial cells in the hippocampus, which was more pronounced at 13 d after inoculation than 20 d after inoculation (Fig. 4C). The proportion of activated microglia correlated with the S/P ratio obtained in the PRRSV ELISA at both 13 d (R2 = 0.35; p = 0.027) and 20 d after inoculation (R2 = 0.67; p < 0.001), demonstrating that piglets with a higher proportion of activated microglia had higher levels of PRRSV antigen in their serum. In addition, the proportion of activated microglia correlated strongly with average daily temperature in piglets killed at 13 d after inoculation (Fig. 4D) and 20 d after inoculation (R2 = 0.59; p < 0.001) and with average feeding score for piglets at these time points (d 13: Fig. 4E; d 20: R2 = 0.67; p < 0.001). Last, the proportion of activated microglia showed a trend to correlate with performance in the cognitive behavioral task at 13 d after inoculation (acquisition only; Fig. 4F) and 20 d after inoculation (acquisition and reversal combined; R2 = 0.15; p = 0.10), demonstrating that piglets with increased activated microglia in the hippocampus performed more poorly in the spatial T-maze task. Overall, PRRSV infection caused neuroinflammation in piglets, as indicated by levels of microglial activation, which correlated with markers of peripheral inflammation, sickness behavior, and reduced cognitive performance.
Figure 4.
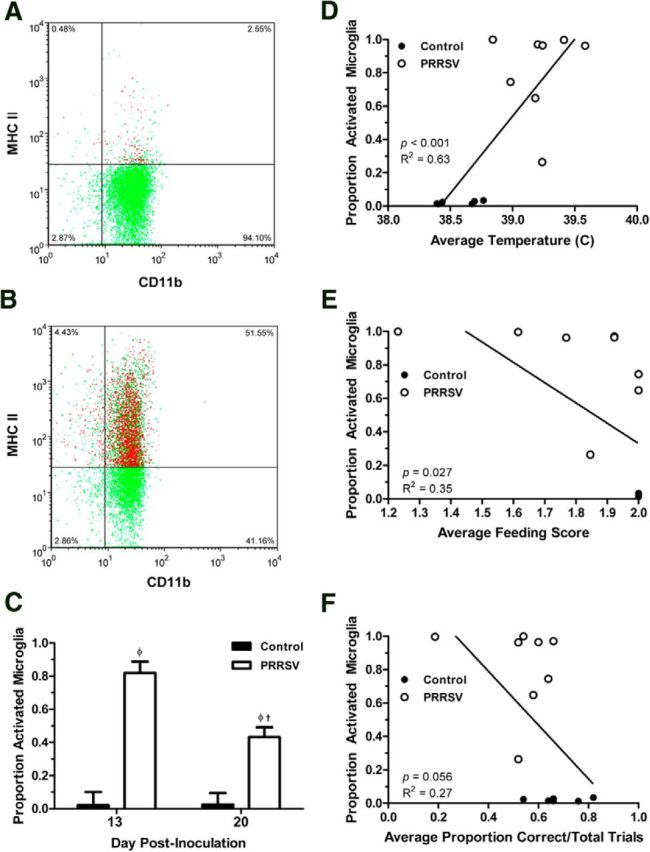
A representative two-color dot plot for a control (A) and a PRRSV (B) piglet is shown; cells were incubated with antibodies for receptors used to identify microglia cells (CD11b and CD45, data not shown), as well as a marker of cell activation (MHC II, shown). The proportion of activated microglia cells at the end of the study in piglets killed 13 d after inoculation (control 1: n = 6; PRRSV 1: n = 8) and 20 d after inoculation (control 2: n = 8; PRRSV 2: n = 11; C). In addition, the correlation of this measure to the average rectal temperature (D), the average feeding score (E), and performance in the cognitive behavioral task (F) for piglets killed at 13 d after inoculation are shown. A–C, Data are LSM ± SEM. Contrasts were used to separate treatment means within each time point (control vs PRRSV at 13 d after inoculation; control vs PRRSV at 20 d after inoculation; φp < 0.05) and between cohorts (control 13 d vs control 20 d after inoculation; PRRSV 13 d vs PRRSV 20 d after inoculation; †p < 0.05).
Gene expression
PRRSV was detected in both the lung (control: 1.00 ± 0.00 vs PRRSV: 3414.10 ± 1055.80; raw data; F(1,31) = 8.06; p = 0.008) and the tracheobronchial lymph node (control: 1.00 ± 0.00 vs PRRSV: 1437.56 ± 385.72; raw data; F(1,25) = 7.58; p = 0.011) of PRRSV-infected piglets but was not present in control piglets. In the lung (Fig. 5A), PRRSV piglets showed an increase in IL-10, but a decrease in TGF-β1, both of which are anti-inflammatory genes. PRRSV piglets showed a decrease in neuronal factors (BDNF, CD200, NGF, and SYP) and a marker of oxidative stress (NOS2), but an increase in CD200r (a microglial receptor), proinflammatory cytokines (IFN-γ, IL-1β, and TNF-α), and a viral marker (TLR-7) in the lungs. The lymph node (Fig. 5B) revealed fewer significant differences between control and PRRSV piglets than the lung, with an increase in proinflammatory genes (IFN-γ, IL-6, and TNF-α) and a decrease in a single viral marker (TLR-3).
Figure 5.

Relative abundance of mRNA in peripheral tissues (A, lung; B, tracheobronchial lymph node) and brain tissues (C, hippocampus; D, prefrontal cortex; E, striatum) for control (13 and 20 d after inoculation combined, n = 14 total) and PRRSV (13 and 20 d after inoculation combined, n = 19 total) piglets. Only significant treatment effects are shown. Data are LSM ± SEM. The gray line indicates the standardized Relative Quantification (RQ) baseline (RQ = 1); all data are expressed as relative fold change to the baseline. Contrasts were used to separate treatment means for each gene. *p < 0.05. #p < 0.10.
Within the brain tissues (Fig. 5C–E), PRRSV was detected in the hippocampus (control: 1.00 ± 0.00 vs PRRSV: 32.93 ± 10.82; raw data; F(1,24) = 6.82; p = 0.015), prefrontal cortex (control: 1.00 ± 0.00 vs PRRSV: 240.87 ± 115.02; raw data; F(1,22) = 3.17; p = 0.089), and striatum (control: 1.00 ± 0.00 vs PRRSV: 129.35 ± 61.85; raw data; F(1,24) = 3.37; p = 0.079) of PRRSV-infected piglets, but was not present in control piglets. IL-13 was not detected in the hippocampus or striatum of either PRRSV or control piglets and, therefore, could not be analyzed. PRRSV piglets showed an increase in anti-inflammatory genes (IL-10 and/or TGF-β1) in all brain tissues, but only a significant increase in proinflammatory genes (IFN-γ, IL-1β, and TNF-α) in the hippocampus and striatum, but not the prefrontal cortex. CD200r expression was increased in the prefrontal cortex and the striatum of PRRSV piglets, whereas the hippocampus and the striatum showed a decrease in neuronal factors (CD200, MBP, and/or NGF). All three brain tissues showed an increase in a marker of oxidative stress (HMOX1) and in at least one of the viral markers (TLR-3, TLR-4, and/or TLR-7). Gene expression data for the interaction of treatment × time after inoculation for peripheral and central tissues are shown in Table 3.
Table 3.
Relative abundance of mRNA in peripheral and central tissuesa
| Tissue | Gene | Control |
PRRSV |
||
|---|---|---|---|---|---|
| 13 d after inoculation | 20 d after inoculation | 13 d after inoculation | 20 d after inoculation | ||
| Lung | IL-13 | 1.10 ± 0.59 | 2.24 ± 0.68 | 3.17 ± 0.68c | 0.48 ± 0.45b,c |
| CD200r | 1.41 ± 0.59 | 1.41 ± 0.51 | 3.66 ± 0.51c | 1.31 ± 0.55b | |
| BDNF | 1.11 ± 0.58 | 3.85 ± 0.50b | 0.32 ± 0.50 | 0.33 ± 0.45c | |
| IL-6 | 1.16 ± 0.44 | 2.72 ± 0.38b | 1.69 ± 0.38 | 0.96 ± 0.32c | |
| TLR-7 | 1.19 ± 0.67 | 1.97 ± 0.58 | 4.24 ± 0.58c | 2.32 ± 0.49b | |
| Lymph node | SOD1 | 1.01 ± 0.27 | 1.55 ± 0.23 | 2.07 ± 0.23c | 1.69 ± 0.20 |
| Prefrontal cortex | TGF-β1 | 1.26 ± 0.27 | 0.78 ± 0.21 | 1.15 ± 0.21 | 1.58 ± 0.18c |
| NGF | 1.89 ± 0.44 | 2.40 ± 0.34 | 3.35 ± 0.39c | 2.24 ± 0.29b | |
| NOS2 | 1.16 ± 0.16 | 0.78 ± 0.12 | 0.68 ± 0.12c | 0.79 ± 0.11 | |
| Striatum | BDNF | 1.00 ± 276 | 26.1 ± 175 | 1004 ± 225c | 138 ± 148b |
| IFN-γ | 1.00 ± 23.5 | 1.00 ± 20.4 | 1.89 ± 33.2c | 57.6 ± 19.2b,c | |
| TLR-3 | 1.17 ± 1.28 | 1.50 ± 0.99 | 6.42 ± 1.28c | 2.14 ± 0.74b | |
aSignificant interactions of treatment × time after inoculation.
bControl 13 d versus Control 20 d after inoculation; PRRSV 13 d versus PRRSV 20 d after inoculation; p < 0.05.
cControl versus PRRSV at 13 d after inoculation; Control versus PRRSV at 20 d after inoculation; p < 0.05.
Discussion
Very little is known about the effects of peripheral infection in the neonatal period on neuroinflammation and cognitive behavior, especially in humans and other animals whose brain is gyrencephalic and undergoes major perinatal growth. Therefore, in the present study, we inoculated neonatal piglets with a virus that induced pneumonia and examined neuroinflammation, behavior, and spatial learning. A unique aspect of the study is that due to the size of the piglet brain, it was possible to isolate microglia from the hippocampus instead of the whole homogenized brain. We used Percoll gradient centrifugation, coupled with antibody staining and flow cytometry, to identify CD11b+/CD45int cells, which are considered to be microglia. Further staining allowed us to show substantial MHC II expression by microglia in the hippocampus due to infection, which was still evident in microglia isolated nearly 3 weeks after inoculation. Microglial MHC II expression was positively correlated with other markers of neuroinflammation, sickness behavior, and deficits in spatial learning. We believe this to be the first study to report in a neonatal gyrencephalic species a relationship between peripheral infection, microglial cell activation in the hippocampus, and deficits in spatial learning.
The dramatic effect of neonatal infection on microglial cell expression of MHC II is notable because of its role in formation, refinement, maintenance, and plasticity of the brain. The period from birth to age 2 is a critical phase of postnatal brain development in humans. Total brain volume doubles the first year and reaches 80–90% of adult volume by age 2 (Knickmeyer et al., 2008). This phase of rapid growth represents a sensitive period, wherein environmental insults may alter neurodevelopment (McEwen, 1999). Indications that neuroinflammation in the neonatal period can affect brain development and have a role in psychopathology are emerging from studies of autism spectrum disorder (ASD). A population-based cohort study of children born in Denmark from 1980 to 2002 reported that children admitted to the hospital for an infectious disease displayed a significantly higher rate of ASD diagnosis later (Atladóttir et al., 2010). The increased diagnosis of ASD was independent of the type of infectious pathogen (viral vs bacterial), indicating the involvement of a generalized immune response (e.g., neuroinflammation). In another large population-based case–control study among children born in Northern California from 1995 to 1999, children diagnosed with an infection in the first month of life were more likely to be diagnosed later with ASD (Rosen et al., 2007). An emerging hypothesis is that disruptions in MHC expression in the developing brain due to infection and immune dysregulation contribute to the altered brain connectivity and function seen in individuals with ASD (Needleman and McAllister, 2012). Although the present study did not focus on brain development per se, it did show that peripheral infection affected expression of MHC II by hippocampal microglia for a significant period of time. This was associated with reduced expression of NGF in the hippocampus, which is consistent with what others have found in rodents after peripheral respiratory infection (Jurgens et al., 2012). Expression of a gene necessary for myelination (i.e., MBP) was also reduced in the hippocampus and striatum of PRRSV piglets. The expression of MHC II was related to poorer performance in a spatial T-maze task, which is consistent with the view that neuroinflammation underlies infection-related deficits in cognition (Yirmiya and Goshen, 2011). Similar to human infants, in piglets the first month after birth (i.e., the period studied here) is characterized by a large expansion in total brain volume, including the hippocampus (Conrad et al., 2012). The present study focused on microglial activation, neuroinflammation, and hippocampal function within a narrow developmental window, but the results clearly warrant further investigation into the long-term structural and functional effects of infection on the hippocampus and other brain regions.
We think that the increased MHC II expression in microglia from PRRSV piglets was likely the result of stimulation of neural and humoral communication pathways from the peripheral immune system to the brain (Dantzer et al., 2008), which are known to lead to microglial cell activation and neuroinflammation. We cannot discount, however, the potential influence of PRRSV entering the CNS. The ability of PRRSV to enter the brain (Rossow et al., 1999; Shin and Molitor, 2002a, b; Tian et al., 2007) and microglia (Rossow et al., 1999) has been reported previously. In the present study, PRRSV was present in the lung and tracheobronchial lymph node, but to a much lesser extent in the hippocampus, prefrontal cortex, and striatum. Although uncertain, the very low levels detected by RT-PCR in brain tissues may have been due to contamination by peripheral blood because piglets were not perfused before sample collection. This seems likely because PRRSV, which preferentially infects mononuclear myeloid cells (e.g., microglia), was not detected in isolated hippocampal microglia from PRRSV-infected piglets (data not shown). In any case, consistent with prior reports (Escobar et al., 2004; Klinge et al., 2009; Liu et al., 2009; Miguel et al., 2010), in both peripheral and central tissues, the general effect of PRRSV infection was to increase expression of genes encoding for proinflammatory cytokines. For example, PRRSV piglets showed an increase in IFN-γ in nearly all tissues, including the brain. IFN-γ is an antiviral cytokine that upregulates MHC II (Kettenmann et al., 2011) and potentiates further cytokine production after infection (De Simoni et al., 1997). In brain tissues, this tended to be associated with increased expression of several receptors that recognize pathogen-associated molecular patterns (TLR-3, TLR-4, and TLR-7).
PRRSV also caused an increase in the anti-inflammatory genes IL-10 and/or TGF-β1 in all brain tissues, as well as an increase in a gene involved in oxidative stress, HMOX1. It is known that HMOX1 is antioxidative, is involved with the anti-inflammatory response, and is protective during infection (Courtney and Maxwell, 2008; Piantadosi et al., 2011). Therefore, it is not surprising that both of these subsets of genes would be upregulated after PRRSV infection. Immunomodulatory factor CD200 is broadly distributed throughout the brain (Wright et al., 2003) and is known to play an important role in downregulating inflammation and microglial activation (Hoek et al., 2000; Jurgens and Johnson, 2012). In this study, CD200 was reduced in the hippocampus and striatum of PRRSV-infected pigs (previously observed in mice with influenza infection) (Jurgens et al., 2012), whereas its receptor CD200r, which is located on myeloid cells and is involved in regulation of macrophage function (Wright et al., 2003), was increased in the prefrontal cortex and the striatum. Similar to influenza infection in mice (Rygiel et al., 2009), loss of CD200-CD200r signaling regulation could lead to excessive production of antiviral responses, leading to increased immune pathology in the PRRSV infection model. It appears that, at least within the striatum, microglia were attempting to compensate for a reduction in the CD200 ligand caused by PRRSV infection by upregulating the inhibitory immune receptor CD200r.
In response to PPRSV infection, the number of MHC II-positive microglia in the hippocampus increased dramatically to ∼80%. Differential expression of MHC II in response to a pathogen is not unique to our neonatal infection model, as this finding has been observed in other models of infection (murine acquired immunodeficiency syndrome) (Mutnal et al., 2013), neurodegenerative disease and aging (Jurgens and Johnson, 2012), as well as brain injury (Nagamoto-Combs et al., 2010). Although beyond the scope of the present study, to understand why some microglia undergo a phenotypic shift in response to insult whereas others do not is a significant issue with major clinical ramifications. However, an important point is that, although MHC II is an accepted marker of microglial activation (Saijo and Glass, 2011), it is now clear that resting microglia can transition between M1 (considered “classically activated,” proinflammatory) and M2 (considered “alternatively activated,” anti-inflammatory and neuroprotective) phenotypes (Mosser and Edwards, 2008; Kigerl et al., 2009; Michelucci et al., 2009). It is important to note that MHC II positivity cannot be categorized as entirely M1 or M2, as both classical and alternative activation involve presentation of MHC II (Gordon, 2003). In future studies, it will be critical to determine how M1 and M2 microglia are regulated, especially because our data suggest that MHC II activation is important for the expression of sickness behaviors and cognitive performance in neonates with respiratory infection.
Footnotes
This work was supported by NIH HD069899 and NIFA ILLU-538-913 (to R.W.J.). We thank the following staff and students for their assistance with this project: Ravi Amin, Brandi Burton, Kaleigh Evans, Tara Garcia, Trisha Gibbons, Tyler Kuper, Hongnan Liu, Sarah Main, Geraldine Petr, Barbara Pilas, Peter Prestia, Casey Radlowski, Brigitte Townsend, Paige Wachholz, and Sara Westergaard.
The authors declare no competing financial interests.
References
- Atladóttir HO, Thorsen P, Schendel DE, Østergaard L, Lemcke S, Parner ET. Association of hospitalization for infection in childhood with diagnosis of autism spectrum disorders: a Danish cohort study. Arch Pediatr Adolesc Med. 2010;164:470–477. doi: 10.1001/archpediatrics.2010.9. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Rudy JW, Watkins LR, Maier SF. A behavioural characterization of neonatal infection-facilitated memory impairment in adult rats. Behav Brain Res. 2006;169:39–47. doi: 10.1016/j.bbr.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Boissé L, Mouihate A, Ellis S, Pittman QJ. Long-term alterations in neuroimmune responses after neonatal exposure to lipopolysaccharide. J Neurosci. 2004;24:4928–4934. doi: 10.1523/JNEUROSCI.1077-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen NH, Chen XZ, Hu DM, Yu XL, Wang LL, Han W, Wu JJ, Cao Z, Wang CB, Zhang Q, Wang BY, Tian KG. Rapid differential detection of classical and highly pathogenic North American Porcine Reproductive and Respiratory Syndrome virus in China by a duplex real-time RT-PCR. J Virol Methods. 2009;161:192–198. doi: 10.1016/j.jviromet.2009.06.007. [DOI] [PubMed] [Google Scholar]
- Conrad MS, Dilger RN, Johnson RW. Brain growth of the domestic pig (Sus scrofa) from 2 to 24 weeks of age: a longitudinal MRI study. Dev Neurosci. 2012;34:291–298. doi: 10.1159/000339311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney AE, Maxwell AP. Heme oxygenase 1: does it have a role in renal cytoprotection? Am J Kidney Dis. 2008;51:678–690. doi: 10.1053/j.ajkd.2007.11.033. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O'Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson H. A comparative assessment of the pig, mouse, and human genomes: structural and functional analysis of genes involved in immunity and inflammation. In: McAnulty PA, editor. The Minipig in Biomedical Research. Boca Raton, FL: CRC; 2011. pp. 321–341. [Google Scholar]
- De Simoni MG, Terreni L, Chiesa R, Mangiarotti F, Forloni GL. Interferon-gamma potentiates interleukin (IL)-6 and tumor necrosis factor-alpha but not IL-1beta induced by endotoxin in the brain. Endocrinology. 1997;138:5220–5226. doi: 10.1210/en.138.12.5220. [DOI] [PubMed] [Google Scholar]
- Dickerson JWT, Dobbing J. Prenatal and postnatal growth and development of the central nervous system of the pig. Proc R Soc Lond B Biol Scis. 1967;166:384–395. doi: 10.1098/rspb.1967.0002. [DOI] [PubMed] [Google Scholar]
- Dilger RN, Johnson RW. Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. J Leukoc Biol. 2008;84:932–939. doi: 10.1189/jlb.0208108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3:79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- Elmore MR, Dilger RN, Johnson RW. Place and direction learning in a spatial T-maze task by neonatal piglets. Anim Cogn. 2012;15:667–676. doi: 10.1007/s10071-012-0495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar J, Van Alstine WG, Baker DH, Johnson RW. Decreased protein accretion in pigs with viral and bacterial pneumonia is associated with increased myostatin expression in muscle. J Nutr. 2004;134:3047–3053. doi: 10.1093/jn/134.11.3047. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Sidwell R, Akhter P, Sedgewick J, Thuras P, Bailey K, Kist D. Human influenza viral infection in utero increases nNOS expression in hippocampi of neonatal mice. Synapse. 1998;29:84–88. doi: 10.1002/(SICI)1098-2396(199805)29:1<84::AID-SYN8>3.0.CO%3B2-7. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Emamian ES, Kist D, Sidwell RW, Nakajima K, Akhter P, Shier A, Sheikh S, Bailey K. Defective corticogenesis and reduction in Reelin immunoreactivity in cortex and hippocampus of prenatally infected neonatal mice. Mol Psychiatry. 1999;4:145–154. doi: 10.1038/sj.mp.4000520. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Earle J, Kanodia R, Kist D, Emamian ES, Patterson PH, Shi L, Sidwell R. Prenatal viral infection leads to pyramidal cell atrophy and macrocephaly in adulthood: implications for genesis of autism and schizophrenia. Cell Mol Neurobiol. 2002;22:25–33. doi: 10.1023/A:1015337611258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Reutiman TJ, Folsom TD, Huang H, Oishi K, Mori S, Smee DF, Pearce DA, Winter C, Sohr R, Juckel G. Maternal infection leads to abnormal gene regulation and brain atrophy in mouse offspring: implications for genesis of neurodevelopmental disorders. Schizophr Res. 2008;99:56–70. doi: 10.1016/j.schres.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliciano DM, Bordey A. Newborn cortical neurons: only for neonates? Trends Neurosci. 2013;36:51–61. doi: 10.1016/j.tins.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, Gibbs RB, Johnson DA. Selective lesion of septal cholinergic neurons in rats impairs acquisition of a delayed matching to position T-maze task by delaying the shift from a response to a place strategy. Brain Res Bull. 2008;77:356–360. doi: 10.1016/j.brainresbull.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting: phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- Frank MG, Wieseler-Frank JL, Watkins LR, Maier SF. Rapid isolation of highly enriched and quiescent microglia from adult rat hippocampus: immunophenotypic and functional characteristics. J Neurosci Methods. 2006;151:121–130. doi: 10.1016/j.jneumeth.2005.06.026. [DOI] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Gregerson DS, Yang J. CD45-positive cells of the retina and their responsiveness to in vivo and in vitro treatment with IFN-gamma or anti-CD40. Invest Ophthalmol Vis Sci. 2003;44:3083–3093. doi: 10.1167/iovs.02-1014. [DOI] [PubMed] [Google Scholar]
- Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehling KA, Erdman D, Grijalva CG, Zhu Y, Szilagyi P. The burden of respiratory syncytial virus infection in young children. N Engl J Med. 2009;360:588–598. doi: 10.1056/NEJMoa0804877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1 beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23:309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, Blom B, Homola ME, Streit WJ, Brown MH, Barclay AN, Sedgwick JD. Downregulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290:1768–1771. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Mishima K, Aoo N, Liu AX, Egashira N, Iwasaki K, Fujiwara M, Ikenoue T. Dexamethasone prevents long-lasting learning impairment following a combination of lipopolysaccharide and hypoxia-ischemia in neonatal rats. Am J Obstet Gynecol. 2005;192:719–726. doi: 10.1016/j.ajog.2004.12.048. [DOI] [PubMed] [Google Scholar]
- Jurgens HA, Johnson RW. Dysregulated neuronal-microglial cross-talk during aging, stress and inflammation. Exp Neurol. 2012;233:40–48. doi: 10.1016/j.expneurol.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgens HA, Amancherla K, Johnson RW. Influenza infection induces neuroinflammation, alters hippocampal neuron morphology, and impairs cognition in adult mice. J Neurosci. 2012;32:3958–3968. doi: 10.1523/JNEUROSCI.6389-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinge KL, Vaughn EM, Roof MB, Bautista EM, Murtaugh MP. Age-dependent resistance to porcine reproductive and respiratory syndrome virus replication in swine. Virol J. 2009;6:177. doi: 10.1186/1743-422X-6-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knickmeyer RC, Gouttard S, Kang C, Evans D, Wilber K, Smith JK, Hamer RM, Lin W, Gerig G, Gilmore JH. A structural MRI study of human brain development from birth to 2 years. J Neurosci. 2008;28:12176–12182. doi: 10.1523/JNEUROSCI.3479-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohman RA, Tarr AJ, Sparkman NL, Bogale TM, Boehm GW. Neonatal endotoxin exposure impairs avoidance learning and attenuates endotoxin-induced sickness behavior and central IL-1beta gene transcription in adulthood. Behav Brain Res. 2008;194:25–31. doi: 10.1016/j.bbr.2008.06.018. [DOI] [PubMed] [Google Scholar]
- Liu CH, Chaung HC, Chang HL, Peng YT, Chung WB. Expression of Toll-like receptor mRNA and cytokines in pigs infected with porcine reproductive and respiratory syndrome virus. Vet Microbiol. 2009;136:266–276. doi: 10.1016/j.vetmic.2008.11.016. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Stress and hippocampal plasticity. Annu Rev Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- Meurens F, Summerfield A, Nauwynck H, Saif L, Gerdts V. The pig: a model for human infectious diseases. Trends Microbiol. 2012;20:50–57. doi: 10.1016/j.tim.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Schwendener S, Feldon J, Yee BK. Prenatal and postnatal maternal contributions in the infection model of schizophrenia. Exp Brain Res. 2006;173:243–257. doi: 10.1007/s00221-006-0419-5. [DOI] [PubMed] [Google Scholar]
- Michelucci A, Heurtaux T, Grandbarbe L, Morga E, Heuschling P. Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: effects of oligomeric and fibrillar amyloid-beta. J Neuroimmunol. 2009;210:3–12. doi: 10.1016/j.jneuroim.2009.02.003. [DOI] [PubMed] [Google Scholar]
- Miguel JC, Chen J, Van Alstine WG, Johnson RW. Expression of inflammatory cytokines and Toll-like receptors in the brain and respiratory tract of pigs infected with porcine reproductive and respiratory syndrome virus. Vet Immunol Immunopathol. 2010;135:314–319. doi: 10.1016/j.vetimm.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutnal MB, Schachtele SJ, Hu S, Lokensgard JR. T-cell reconstitution during murine acquired immunodeficiency syndrome produces neuroinflammation and mortality in animals harboring opportunistic viral brain infection. J Neuroinflammation. 2013;10:98. doi: 10.1186/1742-2094-10-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagamoto-Combs K, Morecraft RJ, Darling WG, Combs CK. Long-term gliosis and molecular changes in the cervical spinal cord of the rhesus monkey after traumatic brain injury. J Neurotrauma. 2010;27:565–585. doi: 10.1089/neu.2009.0966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman LA, McAllister AK. The major histocompatibility complex and autism spectrum disorder. Dev Neurobiol. 2012;72:1288–1301. doi: 10.1002/dneu.22046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnet P, Kelley KW, Bluthé RM, Dantzer R. Expression and regulation of interleukin-1 receptors in the brain: role in cytokines-induced sickness behavior. J Neuroimmunol. 2002;125:5–14. doi: 10.1016/S0165-5728(02)00022-X. [DOI] [PubMed] [Google Scholar]
- Piantadosi CA, Withers CM, Bartz RR, MacGarvey NC, Fu P, Sweeney TE, Welty-Wolf KE, Suliman HB. Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J Biol Chem. 2011;286:16374–16385. doi: 10.1074/jbc.M110.207738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen NJ, Yoshida CK, Croen LA. Infection in the first 2 years of life and autism spectrum disorders. Pediatrics. 2007;119:e61–e69. doi: 10.1542/peds.2006-1788. [DOI] [PubMed] [Google Scholar]
- Rossow KD, Shivers JL, Yeske PE, Polson DD, Rowland RR, Lawson SR, Murtaugh MP, Nelson EA, Collins JE. Porcine reproductive and respiratory syndrome virus infection in neonatal pigs characterised by marked neurovirulence. Vet Rec. 1999;144:444–448. doi: 10.1136/vr.144.16.444. [DOI] [PubMed] [Google Scholar]
- Rygiel TP, Rijkers ES, de Ruiter T, Stolte EH, van der Valk M, Rimmelzwaan GF, Boon L, van Loon AM, Coenjaerts FE, Hoek RM, Tesselaar K, Meyaard L. Lack of CD200 enhances pathological T cell responses during influenza infection. J Immunol. 2009;183:1990–1996. doi: 10.4049/jimmunol.0900252. [DOI] [PubMed] [Google Scholar]
- Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol. 2011;11:775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- Sanai N, Nguyen T, Ihrie RA, Mirzadeh Z, Tsai HH, Wong M, Gupta N, Berger MS, Huang E, Garcia-Verdugo JM, Rowitch DH, Alvarez-Buylla A. Corridors of migrating neurons in the human brain and their decline during infancy. Nature. 2011;478:382–386. doi: 10.1038/nature10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Fatemi SH, Sidwell RW, Patterson PH. Maternal influenza infection causes marked behavioral and pharmacological changes in the offspring. J Neurosci. 2003;23:297–302. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JH, Molitor TW. Localization of porcine reproductive and respiratory syndrome virus infection in boars by in situ riboprobe hybridization. J Vet Sci. 2002a;3:87–96. [PubMed] [Google Scholar]
- Shin JH, Molitor TW. Assessment of porcine reproductive and respiratory syndrome virus RNA load in sera and tissues during acute infection. J Vet Sci. 2002b;3:75–86. [PubMed] [Google Scholar]
- Stringer KG, Martin GM, Skinner DM. The effects of hippocampal lesions on response, direction, and place learning in rats. Behav Neurosci. 2005;119:946–952. doi: 10.1037/0735-7044.119.4.946. [DOI] [PubMed] [Google Scholar]
- Thibault KL, Margulies SS. Age-dependent material properties of the porcine cerebrum: effect on pediatric inertial head injury criteria. J Biomech. 1998;31:1119–1126. doi: 10.1016/S0021-9290(98)00122-5. [DOI] [PubMed] [Google Scholar]
- Tian K, Yu X, Zhao T, Feng Y, Cao Z, Wang C, Hu Y, Chen X, Hu D, Tian X, Liu D, Zhang S, Deng X, Ding Y, Yang L, Zhang Y, Xiao H, Qiao M, Wang B, Hou L, et al. Emergence of fatal PRRSV variants: unparalleled outbreaks of atypical PRRS in China and molecular dissection of the unique hallmark. PloS One. 2007;2:e526. doi: 10.1371/journal.pone.0000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolman EC, Ritchie BF, Kalish D. Studies in spatial learning: place learning versus response learning. J Exp Psychol. 1946;36:221–229. doi: 10.1037/h0060262. [DOI] [PubMed] [Google Scholar]
- Walsh SJ, Harley CW, Corbett D, Skinner DM, Martin GM. CA1 ischemic injury does not affect the ability of Mongolian gerbils to solve response, direction, or place problems. Brain Res. 2008;1187:194–200. doi: 10.1016/j.brainres.2007.10.050. [DOI] [PubMed] [Google Scholar]
- Wright GJ, Cherwinski H, Foster-Cuevas M, Brooke G, Puklavec MJ, Bigler M, Song Y, Jenmalm M, Gorman D, McClanahan T, Liu MR, Brown MH, Sedgwick JD, Phillips JH, Barclay AN. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J Immunol. 2003;171:3034–3046. doi: 10.4049/jimmunol.171.6.3034. [DOI] [PubMed] [Google Scholar]
- Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immunity. 2011;25:181–213. doi: 10.1016/j.bbi.2010.10.015. [DOI] [PubMed] [Google Scholar]
- Zurkovsky L, Brown SL, Korol DL. Estrogen modulates place learning through estrogen receptors in the hippocampus. Neurobiol Learn Mem. 2006;86:336–343. doi: 10.1016/j.nlm.2006.07.008. [DOI] [PubMed] [Google Scholar]