

Abstract
The corpus callosum (CC) connects the left and right cerebral hemispheres in mammals and its development requires intercellular communication at the telencephalic midline mediated by signaling proteins. Heparan sulfate (HS) is a sulfated polysaccharide that decorates cell surface and extracellular matrix proteins and regulates the biological activity of numerous signaling proteins via sugar–protein interactions. HS is subject to regulated enzymatic sulfation and desulfation and an attractive, although not proven, hypothesis is that the biological activity of HS is regulated by a sugar sulfate code. Mutant mouse embryos lacking the heparan sulfotransferases Hs2st or Hs6st1 have severe CC phenotypes and form Probst bundles of noncrossing axons flanking large tangles of midline glial processes. Here, we identify a precocious accumulation of Sox9-expressing glial cells in the indusium griseum region and a corresponding depletion at the glial wedge associated with the formation of Probst bundles along the rostrocaudal axis in both mutants. Molecularly, we found a surprising hyperactivation of Erk signaling in Hs2st−/− (2-fold) and Hs6st1−/− (6-fold) embryonic telencephalon that was most striking at the midline, where Erk signaling is lowest in wild-types, and a 2-fold increase in Fgf8 protein levels in Hs6st1−/− embryos that could underpin Erk hyperactivation and excessive glial movement to the indusium griseum. The tightly linked Hs6st1−/− CC glial and axonal phenotypes can be rescued by genetic or pharmacological suppression of Fgf8/Erk axis components. Overall, our data fit a model in which Hs2st and Hs6st1 normally generate conditions conducive to CC development by generating an HS-containing environment that keeps Erk signaling in check.
Keywords: axons, Fgf8, glia, heparan sulfate, mapk, Sox9
Introduction
The corpus callosum (CC) is the largest mammalian axon tract and mounting evidence links abnormal CC development with neuropsychological effects (Richards, 2002; Bedeschi et al., 2006; Paul et al., 2007; Donahoo and Richards, 2009; Fame et al., 2011). Secreted signaling proteins of the Fgf, Bmp, and Wnt families act through the Erk, Smad, and Tcf/Lef pathways at the corticoseptal boundary (CSB) to generate neuronal and glial structures, a key event being glial translocation from the glial wedge (GW) to the indusium griseum (IG) (Smith et al., 2006; Tole et al., 2006; Niquille et al., 2009; Moldrich et al., 2010; Sánchez-Camacho et al., 2011; Benadiba et al., 2012; Magnani et al., 2013; Amaniti et al., 2013; Niquille et al., 2013). These structures secrete Wnt, Slit, Sema, Bmp, Draxin, and Netrin proteins, which guide CC axons (CCAs) across the midline from embryonic day 16.5 (E16.5) (Serafini et al., 1996; Bagri et al., 2002; Wang et al., 2002; Shu et al., 2003b; Keeble et al., 2006; López-Bendito et al., 2007; Islam et al., 2009; Niquille et al., 2009; Choe et al., 2012; Fothergill et al., 2013).
Heparan sulfate proteoglycans are cell surface and secreted glycoproteins with heparan sulfate (HS) sidechains (polysaccharides) that are enzymatically sulfated (by specific N-, 2O-, 3O- and 6O-sulfotransferases [STs]) and 6-O-desulfated (by 6-O-sulfatases, Sulfs). The “sugar code” hypothesis proposes that the biological activity of HS is encoded in its sulfation patterns (Allen et al., 2001; Merry et al., 2001; Turnbull et al., 2001; Powell et al., 2002; Allen and Rapraeger, 2003; McLaughlin et al., 2003; Bülow and Hobert, 2004; Lee and Chien, 2004; Grobe et al., 2005; Holt and Dickson, 2005; Kinnunen et al., 2005; Kreuger et al., 2006; Pan et al., 2006; Pratt et al., 2006; Bülow et al., 2008; Pan et al., 2008; Conway et al., 2011a; Conway et al., 2011b; Qu et al., 2011).
Mutations affecting HS synthesis or sulfation cause CC phenotypes (Inatani et al., 2003; Grobe et al., 2005; Kalus et al., 2009; Conway et al., 2011a). The heparan sulfotransferases Hs2st (which place O-sulfates at the 2-position of uronic acid) and Hs6st1 (which place O-sulfates at the 6-position of N-acetylated/N-sulfated glucosamines) are expressed widely and at varying levels in the developing telencephalon, including strong expression in the GW and IG at the CSB (Pratt et al., 2006; Conway et al., 2011a). Hs2st−/− and Hs6st1−/− embryos have severe CC phenotypes in which large tangles of glial processes block the path of CCAs at the telencephalic midline, causing Probst bundle (PB) formation with a more penetrant phenotype in Hs6st1−/− embryos (Conway et al., 2011a). Here, we reveal how mutations in Hs2st or Hs6st1 affect signaling pathways critical for CC development. We observe increased GW→IG glial movement in Hs2st−/− and Hs6st1−/− embryos, increased Fgf8 protein in Hs6st1−/− embryos, and hyperactive Erk signaling in both mutants, with greater hyperactivation in Hs6st1−/− embryos. We develop the hypothesis that Hs6st1 and Hs2st normally suppress Erk signaling in the GW and that desuppression of Erk in Hs2st−/− and Hs6st1−/− embryos causes the phenotype. We test this by suppressing Fgf8/Erk axis components genetically or pharmacologically in Hs6st1−/− embryos and, as predicted, ameliorate phenotype severity.
Materials and Methods
Animals.
All mice were bred in-house in line with Home Office UK legislation. The licenses authorizing this work were approved by the University of Edinburgh and King's College London Ethical Review Committees and by the Home Office. Animal husbandry was in accordance with the UK Animals (Scientific Procedures) Act 1986 regulations.
The Hs2stLacZ (Hs2st−) mutant allele was obtained by insertion of a LacZ gene trap vector into the Hs2st locus (Bullock et al., 1998) and the Hs6st1LacZ-ires-hPLAP (Hs6st1−) mutant allele by insertion of a LacZires-hPLAP gene trap vector into intron 1 of the Hs6st1 locus (Mitchell et al., 2001). The Hs2st− and Hs6st1− alleles were maintained on an inbred CBA background; Hs2st−/− and Hs6st1−/− embryos were obtained by crossing heterozygous (Hs2st+/− or Hs6st1+/−) animals. Genotyping of Hs2st− and Hs6st1− animals was performed by PCR as described previously (Conway et al., 2011a). The Fgf8LacZ (Fgf8−) mutant allele (Ilagan et al., 2006) was maintained on a C57BL/6J inbred background. For the Hs6st1/Fgf8 rescue experiments, we crossed Hs6st1+/− (CBA inbred genetic background) with Fgf8+/− (C57BL/6J inbred genetic background) to generate Hs6st1+/−;Fgf8+/− compound heterozygotes (F1: CBAxC57BL/6J). To obtain Hs6st−/−;Fgf8+/− embryos, Hs6st+/−;Fgf8+/− animals were crossed and the resultant embryos (F2) were of a mixed CBAxC57BL/6J genetic background. Because variable penetrance is a common property of CC phenotypes (Keeble et al., 2006; Smith et al., 2006; Conway et al., 2011a; Bohlen et al., 2012), we confirmed that the Hs6st1−/− phenotype is indeed fully penetrant (13/13 embryos; Fig. 7A) in Hs6st1−/−;Fgf8+/+ F2:CBAxC57BL/6J genetic background embryos.
Figure 7.

Rescue of the Hs6st1−/− CC phenotype by repressing the Fgf8/Erk axis. A–D, Reducing Fgf8 gene dosage ameliorates the Hs6st1−/− CC phenotype. A–C, Hs6st1−/−;Fgf8+/+ (A) and Hs6st1−/−;Fgf8+/− (B,C) embryos in which B shows a completely unrescued and C a completely rescued Hs6st1−/− phenotype. D, WT (Hs6st1+/+;Fgf8+/+) control embryo. χ2 = 4.13, df = 1; p < 0.05. E–N, MEKi treatment ameliorates the Hs6st1−/− CC phenotype. Hs6st1−/− embryos from Hs6st1+/− × Hs6st1+/− crosses from uninjected (E), vehicle-injected (F), and MEKi-injected dams (G, H), in which H shows a completely unrescued and G a completely rescued Hs6st1−/− phenotype. χ2 = 6.55, df = 1; p < 0.05. A–H show immunofluorescence for the axonal marker L1 (red) and the glial marker GFAP (green) in coronal sections at E18.5. Numbers at the bottom left indicate the proportions of embryos with phenotype shown in that panel. I–N, Sox9 immunofluorescence at E18.5 on wild-type uninjected embryos (I), Hs6st1−/− vehicle-injected embryos with severe Hs6st1−/− CC phenotype (J), and Hs6st1−/− MEKi-injected embryos showing a rescue of the CC phenotype (K). Box in I shows positioning of 100-μm-wide radial strip used for quantification of numbers of Sox9+ cells in the GW and IG compartments in L–N. L–N, Sox9+ cell counts in the whole strip (GW+IG; L), the GW compartment (M), and the IG compartment (N) taken from serial sections along the rostrocaudal axis binned into rostral, medial, and caudal segments (mean for three sections for each bin). Plots shows mean ± SEM, n = 3 for all conditions. *ANOVA p < 0.05 followed by a post hoc Student's t test for MEKi (rescue) versus vehicle comparison. The total number of Sox9+-stained cells along the whole strip is similar in the WT, vehicle, and MEKi groups (L). Sox9+ cell number at the GW region is reduced in the vehicle-injected Hs6st1−/− embryos, but is returned to WT level in rescued MEKi-treated Hs6st1−/− embryos (M). Sox9+ cell number at the IG is increased in vehicle-injected Hs6st1−/− embryos, but is returned to WT level in rescued MEKi treated Hs6st1−/− embryos (N). Scale bars, 200 μm.
BrdU injection.
Bromodeoxyuridine (BrdU; Sigma) was dissolved in PBS at a concentration of 10 mg/ml. Pregnant females were injected intraperitoneally with 200 μl of BrdU at E14.5. Females were then culled at E16.5 and embryonic brains were dissected and processed for immunohistochemistry.
qRT-PCR.
RNA was extracted from medial telencephalon tissue at E16.5 using the RNAeasy Mini Kit (Qiagen). First-strand cDNA synthesis was performed using oligo (dT) primers (Promega) with a Sensiscript Reverse Transcriptase kit (Qiagen). qRT-PCR was performed on cDNA using the following primer pairs: FGF8 5′-TGGAAGCAGAGTCCGAGTTC-3′ and 5′-TGTGAATACGCAGTCCTTGC-3′ and GAPDH 5′-GGGTGTGAACCACGAGAAAT-'3 and 5′-CCTTCCACAATGCCAAAGTT-3′. qRT-PCR was performed using a Quantitect Sybr Green PCR kit (Qiagen). PCR was performed using an MJ Research Opticon Light Cycler and the abundance of each transcript (relative to GAPDH) was calculated using Opticon software and Microsoft Excel.
MEK inhibitor treatment.
The MEK inhibitor PD0325901 (Sigma) was dissolved in DMSO at a concentration of 25 mg/ml and suspended in 0.5% hydroxypropylmethyl-cellulose (Sigma) plus 0.2% Tween 80 (Sigma) to give a final inhibitor concentration of 0.5 mg/ml. MEK inhibitor was administered to pregnant females by intraperitoneal injection at a concentration of 5 mg/kg body weight daily from 14.5 to 17.5 d after fertilization. Embryos were then dissected at E18.5 and MEK-inhibitor-treated Hs6st−/− embryos were compared with vehicle-injected controls. This protocol was adapted from one used previously (Wang et al., 2012).
Immunohistochemistry.
Embryonic mouse brains were dissected at E16.5, E17.5, or E18.5 and fixed overnight in 4% paraformaldahyde in PBS at 4°C. Brains were then either dehydrated and embedded in paraffin wax or cryoprotected using 30% sucrose in PBS and embedded in OCT for cryosectioning. Paraffin-embedded brains were sectioned coronally at 10 μm using a microtome (Leica) and frozen brains were sectioned coronally using a cryostat (Leica) at 10 μm. Cut sections were mounted serially on Superfrost Plus slides (Fisher Scientific) and allowed to dry.
Paraffin-embedded sections were dewaxed using xylene and rehydrated through an ethanol series. Sections were then boiled in 10 mm sodium citrate to aid epitope recovery. After a wash in PBS, sections were treated with blocking solution (20% goat serum in 0.1% Triton X-100/PBS) and incubated with the primary antibody diluted in blocking solution overnight at 4°C. Primary antibodies used were rabbit anti-LEF1 (1/100; Cell Signaling Technology), mouse anti-phospho-histone 3 (1/100; Cell Signaling Technology), mouse anti-phospho-MAPK1/2 (pErk1/2, 1/200; Cell Signaling Technology) and rabbit anti-phospho-SMAD1/5 (1/50; Cell Signaling Technology). Sections were then washed with PBS and incubated with a biotinylated secondary antibody (goat anti-mouse biotin or goat anti-rabbit biotin, both Vector Laboratories) diluted 1/200 in blocking solution. After washing with PBS, antibody staining was visualized using a standard avidin-biotin diaminobenzidine (DAB) detection procedure (Vector Laboratories). After DAB staining, sections were dehydrated and mounted in DPX. Bright-field images of DAB-stained sections were captured using a Leica DLMB microscope coupled to a DFC480 color digital camera.
Frozen sections were washed with 0.1% Triton X-100 in PBS and treated with the blocking solution (20% goat serum, 1% BSA in 0.1% Triton X-100/PBS). Sections were incubated with the primary antibody diluted in blocking solution overnight at 4°C. Primary antibodies used were mouse anti-NeuN (1/500; Millipore), rabbit anti-Sox9 (1/200; Millipore), rabbit anti-Tbr1 (1/200, gift from Robert Hevner), mouse anti-Fgf8 (1/5000; R&D Systems), rat anti-L1 (1/500; Millipore), rabbit anti-GFAP (1/100; Dako), and mouse anti-BrdU (1/100; BD Biosciences). Sections were washed with PBS and incubated with the appropriate secondary antibody diluted in blocking solution for 1 h at room temperature. Secondary antibodies used were goat anti-mouse Alexa Fluor 568, goat anti-rabbit Alexa Fluor 488, goat anti-rabbit Alexa Fluor 568, and goat anti-rat Alexa Fluor 568 (all used 1/200 and all from Invitrogen). Sections were counterstained with DAPI (Invitrogen) diluted 1/1000 in PBS and mounted using Vectashield Hardset H1400 (Vector Laboratories). To detect Fgf8, the above procedure was used but a TSA Plus Fluorescence System kit (PerkinElmer) was used in place of an Alexa Fluor secondary antibody. Fluorescent images were captured with a Leica AF6000 epifluorescence microscope connected to a DFC360 digital camera.
In situ hybridization. In situ hybridization was performed on frozen sections as described previously (Wallace and Raff, 1999) using a digoxigenin-labeled antisense riboprobe for Fgf8 (kindly provided by J. Rubenstein).
Quantification of cell number.
To quantify the number of Sox9- and/or BrdU-immunofluorescent positive cells at the IG region of wild-type, Hs2st−/−, and Hs6st1−/− brains (Figs. 2, 3), a counting box measuring 250 μm × 250 μm was placed on images of sections at the midline directly ventral to the ventral-most extent of the Tbr1 expression domain that marks the dorsal boundary of the IG (Fig. 1). The presence or absence of the corpus callosum or PBs within the section was noted in each section. To quantify the number of Sox9-immunofluorescence-positive cells within a strip of cells from the GW to the IG (Figs. 2, 8), a 100-μm-wide rectangle was placed on images extending from the ventricular zone of the GW to the pial surface adjacent to the IG. The rectangle was placed directly ventral to the ventral-most extent of the Tbr1 expression domain as above. The counting box was then divided into a GW and IG compartment. For each genotype, three animals were analyzed and, for each animal, three sections were analyzed (a rostral, middle, and caudal section). The rostrocaudal position of sections was determined as above.
Figure 2.
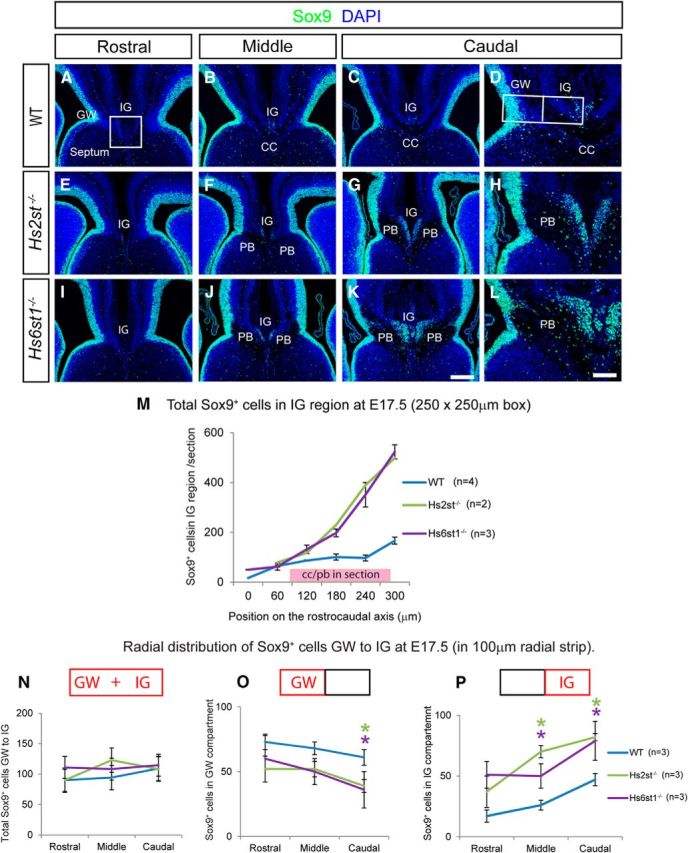
Variations in the distribution of glial cells at the telencephalic midline in wild-type, Hs2st−/−, and Hs6st1−/− embryos at E17.5. A–L, Sox9 immunofluorescence (green) at the telencephalic midline in wild-type (A–D), Hs2st−/− (E–H), and Hs6st1−/− (I–L) embryos in rostral (A,E,I), middle (B,F,J), and caudal (C,D,G,H,K,L) coronal sections through the developing CC region. Boxed region in A indicates the 250 μm × 250 μm counting area encompassing the IG region used to generate data presented in M. M, Graph showing numbers of Sox9+ nuclei in the IG region counted in serial 10 μm sections spaced at 60 μm intervals along the rostrocaudal axis (mean ± SEM for WT, n = 4 and Hs6st1−/−, n = 3; mean only for Hs2st−/−, n = 2). The pink box indicates the position of the CC (in wild-types) or PBs (in the mutants). Note that whereas midline Sox9+ cell numbers increase moving caudally in all genotypes, the rate of increase is dramatically greater in both mutants in association with PBs. D, H, L, Higher-magnification images of GW and IG region of C, G, and K, respectively. Note that in both mutants, there is a thinning of the Sox9+ area at the GW and more Sox9+ cells at the IG compared with the wild-type. Box in D shows positioning of 100 μm wide radial strip used for quantification of numbers of Sox9+ cells in the GW and IG compartments in N–P. (N–P) Sox9+ cell counts (mean ± SEM, n = 3 for all genotypes) in the whole strip (GW+IG; N), the GW compartment (O), and the IG compartment (P) taken from serial sections along the rostrocaudal axis binned into rostral, medial, and caudal segments (mean for three sections for each bin). *ANOVA p < 0.05 followed by a post hoc Student's t test for mutant versus wild-type comparison. The trend, most apparent caudally, is for more Sox9+ cells at the IG and fewer Sox9+ cells in the GW in both mutants compared with the wild-type. Scale bars: A–C, E–G, and I–K, 200 μm; D, H, and L, 100 μm.
Figure 3.

Increased migration of glial cells from the GW to the IG in Hs2st−/− and Hs6st1−/− embryos. (A–C, E–G) BrdU (red) and Sox9 (green) immunofluorescence at the telencephalic midline of WT (A, E), Hs2st−/− (B, F), and Hs6st1−/− (C, G) embryos at E16.5 after a single BrdU administration at E14.5. E–G, Higher-magnification images of the IG region in A–C, respectively. D, Quantification of the number of Sox9+BrdU+ double-labeled cells (yellow) at the IG within a 250 μm × 250 μm counting area indicated in A. Histogram shows mean ± SEM: WT, n = 4; Hs2st−/−, n = 3; Hs6st1−/−, n = 4. *ANOVA p < 0.05 followed by a post hoc Student's t test for mutant versus wild-type comparison. The number of double-labeled cells at the IG is significantly increased in both Hs2st−/− and Hs6st1−/− embryos compared with WT. H–J, PH3 immunohistochemistry shows similar location of mitotic cells at the VZ of the GW in wild-type (H), Hs2st−/− (I), and Hs6st1−/− (J) embryos and cell counts (K) confirm a similar PH3+ cell density in the GW region of the VZ between genotypes at E16.5. Histogram shows mean ± SEM, n = 3 for all genotypes. Scale bars: A–C and H–J, 100 μm; E–G, 50 μm.
Figure 1.

Similar gross organization of neuronal and glial cell bodies at the telencephalic midline in wild-type (A–C), Hs2st−/− (D–F), and Hs6st1−/− (G–I) embryos at E17.5. Shown is double immunofluorescence for Sox9 (green), which is expressed in RGC and glial cell nuclei, and the postmitotic neuronal nuclear markers NeuN (red; A, B, D, E, G, H) or Tbr1 (red; C, F, I). Nuclei in A, D, and G are counterstained with DAPI (blue). In all three genotypes, most Sox9+ cells are located in the VZ and at the midline, where they form a cluster ventral to NeuN+ or Tbr1+ neurons in the IG. In wild-types, the IG Sox9+ cell population forms above the CC axon bundle, whereas in Hs2st−/− and Hs6st1−/− embryos, it is sandwiched between the PBs. Box in A indicates the region shown at higher magnification in B, C, E, F, H, and I. A, D, and G are to same scale; bar in G is 200 μm. B, C, E, F, H, and I are to same scale, bar in I is 100 μm.
Figure 8.

Model summarizing cellular and molecular phenotypes of Hs2st−/− and Hs6st1−/− embryos at the telencephalic midline. A, In WT embryos, Hs6st1 and (to a lesser extent) Hs2st activities both repress the Erk signaling pathway to maintain a level of pErk that drives appropriate numbers of Sox9+ glia (green) to translocate (pink arrows) from the GW to the IG and generates a GW ↔ IG glial balance that guides CCAs (red arrow) across the telencephalic midline in 100% of embryos. Loss of Hs2st (B) or Hs6st1 (C) activity results in a derepression of the Erk pathway, excess levels of pErk in the GW, and excess GW → IG translocation of Sox9+ glia-blocking (red X) CCAs from crossing the telencephalic midline. The Hs2st−/− and Hs6st1−/− phenotypes differ quantitatively, with Hs2st−/− embryos exhibiting 2-fold Erk hyperactivation and ∼50% of embryos developing a normal CC and Hs6st1−/− embryos exhibiting 6-fold Erk hyperactivation and 0% developing a normal CC. D, Repressing the hyperactive Erk pathway in Hs6st1−/− embryos restores the GW ↔ IG glial balance and ameliorates the severity and penetrance of the CC phenotype, with ∼33% of embryos developing a normal CC using the treatments used in this study. Thickness of “T” symbol indicates repressor strength and size of pErk text indicates pErk levels in each genotype. Numbers at bottom indicate proportion (%) of embryos of each condition that produce a normal CC.
Western blotting.
Whole telencephali were dissected at E16.5 and lysed using RIPA lysis buffer (Sigma) on ice for 30 min. The lysate was then resolved on a 4–12% bis-tris gel (Invitrogen) and transferred to a nitrocellulose membrane (Bio-Rad). The membrane was blocked using blocking solution (Li-Cor) and blotted overnight using the following primary antibodies: mouse anti-phosho-MAPK1/2 (1/1000; Cell Signaling Technology), mouse anti-MAPK1/2 (1/2000; Cell Signaling Technology), mouse anti-Fgf8 (1/1000, R&D Systems), rabbit anti-LEF1 (1/1000; Cell Signaling Technology), and rabbit anti-phospho-SMAD1/5 (1/2000; Cell Signaling Technology). For each blot performed, the membrane was also probed using an antibody for β-actin (mouse anti-β-actin or rabbit anti-β-actin, both used 1/5000 and both from Abcam) that acted as a loading control. The membrane was incubated for 2 h with the secondary antibodies, goat anti-mouse Alexa Fluor 680 (Invitrogen), and goat anti-rabbit 800 (Li-Cor). Blots were quantified using the Li-Cor scanning system. In each case, protein levels were normalized to β-actin level detected on the same membrane.
Transfection of Cos7 cells.
Cos7 cells were transfected with a cDNA expression construct expressing full-length mouse Fgf8 protein (nt201–845, GenBank accession NM_001166362) with a C-terminal myc epitope tag using a pSecTag2 vector (Invitrogen) that had been modified to remove the Ig secretory signal.
Results
Sox9+ glial cells overpopulate the IG at the expense of the GW in Hs2st−/− and Hs6st1−/− embryonic telencephalon
We previously reported striking embryonic corpus callosum agenesis and PB formation in Hs2st−/− and Hs6st1−/− mice that appeared to be caused by abnormal organization of glial cells during development (Conway et al., 2011a). To shed a more quantitative light on the glial Hs2st−/− and Hs6st1−/− phenotypes, we accurately quantified glial cells using Sox9, a transcription factor expressed both in radial glial cell (RGC) progenitors and in mature glia but switched off during neuronal differentiation, as a glial cell nuclear marker (Pompolo and Harley, 2001; Kordes et al., 2005; Nowakowski et al., 2013). At the telencephalic midline, Sox9 marks RGCs in the ventricular zone (VZ) and glia in the GW and IG (Fig. 1). Double labeling E17.5 wild-type embryos with Sox9 and the neuronal nuclear transcription factors NeuN (Fig. 1A,B) or Tbr1 (Fig. 1C) showed large numbers of Sox9-expressing (hereafter referred to as Sox9+) cells in the VZ/GW and a small tight cluster in the IG region ventral to Tbr1/NeuN-positive cells (Fig. 1A–C). Both Hs2st−/− (Fig. 1D–F) and Hs6st1−/− (Fig. 1G–I) embryos have a gross neuronal/glial organization similar to wild-types, with Sox9+ cells both in the VZ/GW and in a cluster ventral to Tbr1/NeuN-expressing neuronal cells in the IG region. However, in both mutants, the area occupied by Sox9+ glial nuclei in the IG is wider and extends further ventrally, suggesting that the ectopic GFAP-expressing glial processes we observed previously at the midline in these mutants stem from increased numbers of glia in the IG region (more on this in Results, below; Conway et al., 2011a). There is no overlap between Sox9 and NeuN/Tbr1 expression in any genotype, indicating that the normal segregation between neuronal and glial cells in the IG region is not disrupted in the mutants.
CCAs start to cross the midline at the CSB starting from E16.5, forming the U-shaped CC bundle by E17.5 (Richards, 2002). In a series of coronal sections along the rostrocaudal axis spanning the septum of E17.5 wild-type embryos, the U-shaped CC is absent in the most rostral sections (Fig. 2A) and present in the more caudal sections (Fig. 2B,C; visible in sections stained with the nuclear counterstain DAPI by virtue of low cell body density in the axon bundle), whereas Hs2st−/− and Hs6st1−/− embryos display PBs in the caudal set of sections (Fig. 2F,G,J,K; Conway et al., 2011a). We next determined how the distribution of Sox9+ cells in the IG region related to the normal formation of the CC in wild-type embryos or the abnormal formation of PBs in Hs2st−/− or Hs6st1−/− embryos along the rostralCC/PB absent → caudalCC/PB present axis. In wild-type embryos, there are very few Sox9+ cells in the IG region of the most rostral sections (Fig. 2A), with more in caudal sections dorsal to the CC (Fig. 2B,C). In both Hs2st−/− (Fig. 2E) and Hs6st1−/− (Fig. 2I) embryos, there are few Sox9+ cells rostrally, as in the wild-type, but many more in caudal sections where PBs have formed (Fig. 2F,G,J,K). Sox9+ cell counts in evenly spaced serial sections from wild-type, Hs2st−/−, and Hs6st1−/− embryos allowed us to quantify the relationship between glial cell numbers in the IG and axon phenotype along the rostrocaudal axis (Fig. 2M). In wild-type embryos (blue line in Fig. 2M), the number of Sox9+ cells was very low in the most rostral section, where the CC has not crossed the midline, and rose to a plateau of ∼80 cells/section starting at the rostral limit of the CC (left end of the pink box on the x-axis in Fig. 2M) and to ∼150 cells/section toward the caudal end of the CC. In both Hs2st−/− and Hs6st1−/− embryos (green and purple lines in Fig. 2M), there are very few Sox9+ cells in the IG region rostrally and numbers increase rapidly in successively more caudal sections, where Sox9+ cells in the IG region sit between the PBs (indicated by pink box on x-axis in Fig. 2M), increasing to a maximum of 500 cells/section caudally. In summary, the numbers of glial cells in the IG region follow a rostrallow → caudalhigh trend in all genotypes, coinciding with the rostralno CC or PB → caudalCC or PB present transition, which is much more pronounced in association with PBs in Hs2st−/− and Hs6st1−/− embryos (compare blue line with green and purple lines in Fig. 2M). We conclude that Hs2st and Hs6st1 normally participate in a mechanism that restricts the number of IG Sox9+/glial cells and that the loss of Hs2st or Hs6st1 function results in an increased number of IG glia. Careful comparison of Sox9 expression at higher magnification in the GW and IG of wild-type and mutant embryos showed that a thick Sox9+ area at the GW faced a much thinner Sox9+ area at the IG in wild-type embryos (Fig. 2D), whereas in both mutants, there was a thinner Sox9+ area at the GW coinciding with a complementary enlarged Sox9+ area at the IG (Fig. 2H,L). These tectonic features suggested that the overpopulated IG in the mutants (Fig. 2M) was at the expense of depleting the GW pool. Because Sox9 is expressed by both RGC and glial nuclei in the GW and IG, we first tested this hypothesis by counting Sox9+ cells in a radial strip (Fig. 2D) spanning the GW → IG. We found that the total number of Sox9+ cells in GW + IG compartments was approximately constant with ∼100 cells/section along the rostrocaudal axis in all three genotypes (Fig. 2N). As predicted, Hs2st−/− and Hs6st1−/− embryos exhibited fewer Sox9+ cells remaining in the GW compartment in the mutants compared with the wild-type (Fig. 2O), balancing more Sox9+ cells in the IG compartment of the mutants (Fig. 2P). This trend was apparent along the rostrocaudal axis and was most pronounced caudally, where the mutants had approximately half the number of Sox9+ cells in the GW compartment and double the number of Sox9+ cells in the IG compartment compared with wild-types.
Mechanistically, because the IG is populated with glia by a process of RGCs detaching their apical foot from the GW and translocating their nuclei toward their basal foot in the IG, we hypothesized that the change in the IG ↔ GW distribution of Sox9+ cells in the mutants (Fig. 2) is caused by excessive movement of Sox9+ cells from the GW to the IG. Cells destined to become IG glia are born in the GW around E14.5 and subsequently translocate to the IG (Shu et al., 2003a; Smith et al., 2006). We labeled cells born at E14.5 using the thymidine analog BrdU, which is incorporated into newly synthesized DNA during the S-phase, and identified cells in this BrdU+ E14.5 birthdated cohort (red signal in Fig 3A–G), which contributed to the IG glial population at E16.5 by virtue of nuclear colabeling with Sox9 (green signal in Fig. 3A–G), with colabeled BrdU+Sox9+ nuclei appearing yellow (Fig. 3A–C with IG region shown at higher magnification in Fig 3E–G). In all genotypes, there are examples of single-labeled Sox9+, BrdU− (green signal-glia not in birthdated cohort), Sox9−, BrdU+ (red signal-birthdated cohort which are not glia), and double-labeled Sox9+Brdu+ (yellow signal-birthdated cohort which are glia). Similar to E17.5 (Figs. 1, 2), there is a larger glial population at the IG in both mutants than in wild-type embryos (compare Fig. 3F,G, 3E). Quantification confirmed that, in both mutants, there was an ∼2-fold increase in numbers of colabeled nuclei compared with wild-type embryos (compare green and purple with blue bars in Fig. 3D), supporting the hypothesis that there is excessive GW → IG translocation of glial cells in Hs2st−/− and Hs6st1−/−embryos. Whereas, strictly speaking, this conclusion only applies to the subpopulation of cells that were labeled by our BrdU pulse protocol, we think it is reasonable to extend it to the larger population of BrdU−Sox9+ glial cells that reached the IG by E16.5 but were not undergoing S-phase during the BrdU pulse at E14.5.
The Sox9+ cell counts already indicate that alterations to cell proliferation or death are unlikely to account for the increased numbers of Sox9+ cells in the IG because there is no change in total Sox9+ cell numbers (Fig. 2N). Furthermore, we were unable to find any evidence for either (1) increased cell proliferation before E17.5 contributing to the increase in midline glial cells in the mutants because mitotic cells identified by phospho-histone3 (PH3) staining were restricted to the medial VZ encompassing the GW in very similar numbers in all three genotypes at E16.5 (Fig. 3H–K) or (2) decreased apoptosis, as judged by cleaved caspase-3 expression, which was similarly rare in the GW/IG of all genotypes at E16.5 (data not shown).
Together, our data support a model where increased numbers of glia in the IG region, and the corresponding depletion of the GW pool, of Hs2st−/− and Hs6st1−/− embryos compared with wild-types stem from excessive GW → IG glial cell movements. These results supply an explanation for our previous observation that GFAP expression is reduced in the GW and enhanced in the IG in these mutants and suggest that the large tangles of glial processes at the mutant midline originate from an enlarged population of glial cell bodies located in an expanded IG (Conway et al., 2011a).
Hs6st1 suppresses Fgf8 protein levels at the telencephalic midline
The translocation of glia from the GW to the IG is regulated by Fgf signaling. Blocking the Fgf pathway by abrogating Fgf receptor function prevents glial cells from reaching the IG and, conversely, implanting Fgf8-soaked beads into the cerebral cortex induces excessive translocation of RGCs to the pial surface, where they differentiate into mature glia (Smith et al., 2006; Tole et al., 2006). Therefore, the possibility of Fgf8 overexpression as a plausible mechanism to explain the Hs2st−/− and/or Hs6st1−/− glial phenotype was examined in more detail. Western blotting of protein extracted from whole telencephalon of E16.5 wild-type, Hs2st−/−, and Hs6st1−/− embryos showed that although there was no difference in global Fgf8 protein levels between wild-type and Hs2st−/− embryos, there was a marked 2-fold increase in Hs6st1−/− embryos (Fig. 4A). Immunofluorescence showed that, at this stage, Fgf8 protein was mainly located in the extreme ventral region of the medial telencephalon and either absent or below detection threshold elsewhere in wild-type and Hs2st−/− telencephalon (Fig. 4B,E,C,F). In contrast, Fgf8 protein was detected much more dorsally adjacent to the GW/IG in Hs6st1−/− embryos (Fig. 4D,G). The use of this Fgf8 antibody in mice has been widely published (Dobrowolski et al., 2009; Lania et al., 2009; Toyoda et al., 2010; Crespo-Enriquez et al., 2012) and we confirmed its specificity in our hands by demonstrating specific staining of Cos7 cells transfected with an Fgf8 cDNA expression construct (compare Fgf8 transfected cells in Fig. 4H with untransfected controls in Fig. 4I) and by taking advantage of the severely hypomorphic Fgf8neo allele (Meyers et al., 1998) to confirm that the immunofluorescence signal was undetectable in sections taken from Fgf8neo/neo embryos (Fig. 4J). We next investigated whether the changes in Fgf8 protein levels and distribution in Hs6st1−/− embryos stem from changes in Fgf8 mRNA levels or distribution. qRT-PCR analysis comparing RNA extracted from wild-type and Hs6st1−/− medial telencephalon (encompassing the septum and medial cerebral cortex) revealed no significant differences in Fgf8 mRNA levels (Fig. 4K) and in situ hybridization shows that Fgf8 mRNA expression is most prominent in the GW and IG of both wild-type and Hs6st1−/− embryos, with no obvious difference in its distribution (compare Fig. 4L, 4M). Together, these data indicate that Hs6st1 acts on Fgf8 via a posttranscriptional mechanism. In conclusion, the GW/IG region is exposed to higher levels of Fgf8 protein in Hs6st1−/− embryos than in the other gentoypes, revealing an unexpectedly specific role for Hs6st1 in suppressing levels of Fgf8 protein and suggesting a molecular mechanism for the glial translocation phenotype in Hs6st1−/− embryos. In contrast, Fgf8 protein levels are unaltered in Hs2st−/− embryos, indicating that Hs2st and Hs6st1 have distinct molecular roles.
Figure 4.
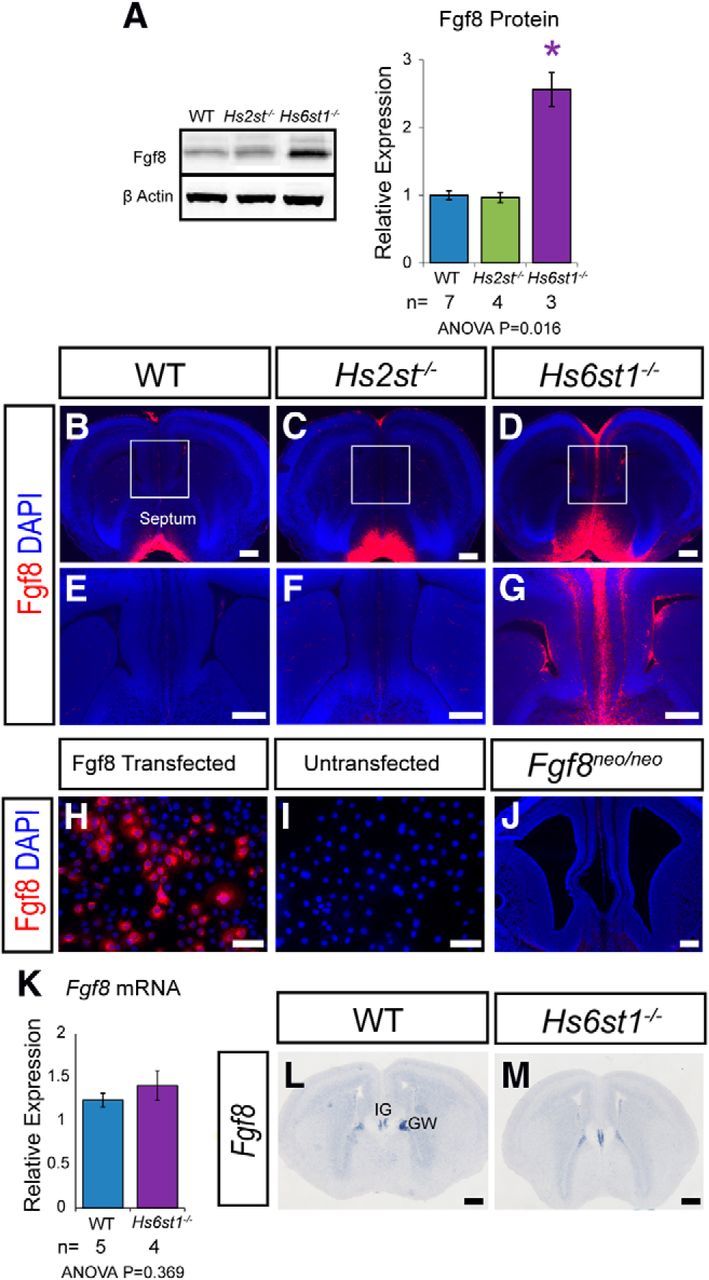
Fgf8 protein and mRNA expression in the telencephalon at E16.5 in wild-type, Hs2st−/−, and Hs6st1−/− embryos. A, Western blot of telencephalic protein and quantification for Fgf8 protein with β-actin protein used for normalization. Histograms shows mean ± SEM for WT, n = 7; Hs2st−/−, n = 4, Hs6st1−/−, n = 3. *ANOVA p < 0.05 followed by a post hoc Student's t test for mutant versus wild-type comparison. B–G, Immunofluorescence for Fgf8 (red) on coronal sections. Boxed areas in B–D shown at higher magnification in E–G. Fgf8 protein levels are much higher at the telencephalic midline of Hs6st1−/− embryos compared with the other genotypes. H, I, COS7 cells transfected with a mouse Fgf8 cDNA expression construct show specific immunofluorescent staining (H), whereas untransfected cells do not (I). J, Fgf8 immunofluorescence is below detection limit in Fgf8neo/neo-matched telencephalic tissue homozygous for a severely hypomorphic Fgf8 allele. K, qRT-PCR analysis of RNA extracted from the medial telencephalon shows no significant difference in Fgf8 mRNA expression between WT and Hs6st1−/− embryos. Histograms shows mean ± SEM for WT, n = 5; Hs6st1−/−, n = 4. L, M, In situ hybridization shows that in both WT (L) and Hs6st1−/− (M) embryos Fgf8 mRNA is similarly localized to the IG and GW and expressed below detection threshold elsewhere in the telencephalon. Scale bars: B–G and J–M, 200 μm; I and J, 100 μm.
Hyperactive Erk signaling coincides with the CC phenotype in Hs2st−/− and Hs6st1−/− embryos
Fgf8 signals through the Erk pathway. The final step of this signaling cascade involves the phosphorylation of Erk by Mek1/2 kinases to generate the transcriptionally active phospho-Erk (pErk). We therefore examined the possibility that elevated Fgf8 levels in Hs6st1−/− embryos in the developing telencephalon (Fig. 4) might overstimulate the Erk pathway (Thisse and Thisse, 2005; Borello et al., 2008; Iwata and Hevner, 2009; Guillemot and Zimmer, 2011; Toyoda et al., 2010). First, we used Western blotting to quantify the levels of pErk (normalized to levels of β-actin in the same blot) in the telencephalon of E16.5 wild-type, Hs2st−/−, and Hs6st1−/− embryos and discovered a robust trend: wild-type < Hs2st−/− ≪ Hs6st1−/− with a 2-fold increase in Hs2st−/− and a 6-fold increase in Hs6st1−/− genotypes, respectively (Fig. 5A). We confirmed that this was not a consequence of a general increase in the levels of Erk protein in the mutants by quantifying levels of total Erk (normalized to levels of β-actin in the same blot), which showed that Erk levels were similar between genotypes, although there was a small but significant decrease in Hs2st−/− embryos (Fig. 5B). Consistent with previous reports, immunohistochemistry showed that, in wild-type embryos, pErk is most abundant in cells in the VZ of the lateral telencephalon at the pallial-subpallial boundary (PSPB) and forms a PSPBhigh → CSBlow gradient in the cerebral cortex from the PSPB (open arrowhead in Fig. 5C) to the GW at the CSB (filled arrowhead in Fig. 5C; Faedo et al., 2010). The overall pattern is rather similar in Hs2st−/− embryos, although, consistent with the Western blot data, the staining is stronger (Fig. 5D). In Hs6st1−/− embryonic telencephalon, there is a global increase in pErk compared with wild-types, which is particularly apparent medially, where pErk levels are normally lowest and the PSPBhigh → CSBlow gradient is flattened (compare relatively uniform staining in cerebral cortex between open and filled arrowheads in the Hs6st1−/− telencephalon in Fig. 5E with the wild-type in Fig. 5C). Higher magnification shows that pErk is detectable in some GW cells in all three genotypes (Fig. 5F–H; arrows indicate highest expressing cells in each panel), with many cells exhibiting nuclear localization, suggesting that pErk is regulating nuclear gene transcription. In the wild-type GW, cells expressing detectable levels of pErk make up a relatively small subpopulation of the total (Fig. 5F). In the Hs2st−/− GW (Fig. 5G) and the Hs6st1−/− GW (Fig. 5H), there are more pErk-expressing cells and individual cells express higher levels of pErk than in the wild-type (compare staining intensity in individual cells indicated by arrows in Fig. 5F with that in Fig. 5G,H). This effect is more pronounced in the Hs6st1−/− GW, which is packed with cells expressing high levels of pErk, than the Hs2st−/− GW (compare Fig. 5G,H), although the levels of pErk within cells appears similar (compare staining intensity of individual cells indicated by arrow in Fig. 5G with that in Fig. 5H). The Hs6st1−/− ≫ Hs2st−/− > wild-type pErk expression trend persists at E17.5 both globally (Fig. 5I–K) and in GW cells (Fig. 5L–N). In conclusion, there is a striking increase in pErk in GW cells in Hs6st1−/− and Hs2st−/− embryos, with the amount of deregulation correlating well with the severity of the glial phenotype.
Figure 5.
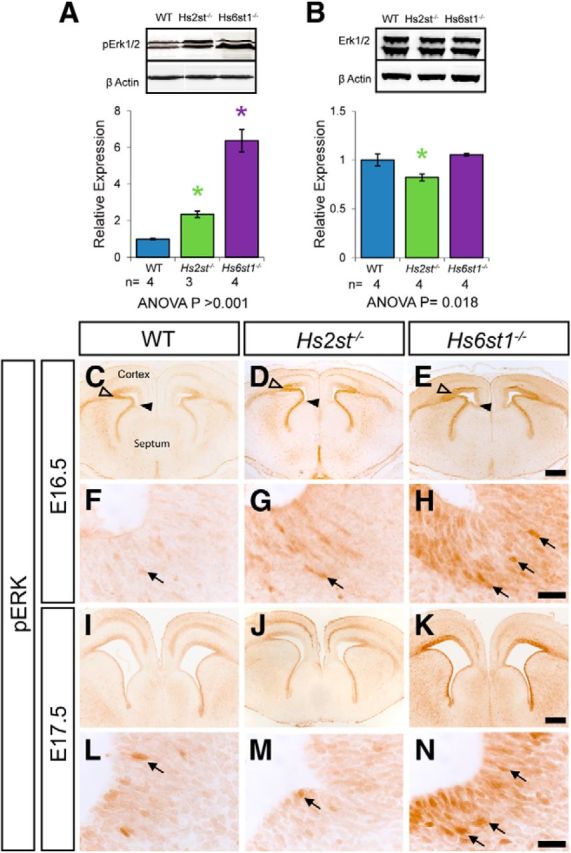
Erk signaling in the embryonic telencephalon in wild-type, Hs2st−/−, and Hs6st1−/− embryos at E16.5 (A–H) and E17.5 (I–N). A, Western blot for pErk1/2 and β-actin (loading control) in protein extracted from whole telencephalon, quantification shows pErk1/2 levels relative to β-actin. pErk1/2 expression is significantly increased in both Hs2st−/− and Hs6st1−/− embryos compared with wild-type. B, Western blot for total Erk1/2, and β-actin (loading control) shows no increase in Erk1/2 expression in either Hs2st−/− or Hs6st1−/− embryos. Histograms in A and B show mean ± SEM for WT, n = 7; Hs2st−/−, n = 4, Hs6st1−/−, n = 3. *ANOVA p < 0.05 followed by a post hoc Student's t test for mutant versus wild-type comparison. C–N pErk1/2 immunohistochemistry on (C–H) E16.5 and (I–N) E17.5 coronal section from wild-type (C, F, I, L), Hs2st−/− (D, G, J, M), and Hs6st1−/− (E, H, K, N) embryos. Arrowheads in C–E, demarcate the cerebral cortex from the GW at the CSB (filled arrowhead) to the PSPB (unfilled arrowhead). F–H and L–N are higher magnifications of the GW area in C–E and I–K. Both mutants exhibit a global increase in pErk1/2 compared with wild-type, which is particularly striking at the telencephalic midline, where levels are relatively low in wild-type embryos. Within the GW, a larger number of pErk1/2-positive cells (examples indicated by arrows) can be seen in the Hs2st−/− (G, M), which is even more pronounced in the Hs6st1−/− (H, N) compared with WT (F, L). Scale bars: C–E and I–K, 200 μm; F–H and L–N, 20 μm.
Bmp and Wnt signaling are not grossly altered in E16.5 Hs2st−/− and Hs6st1−/− telencephalon
We next investigated whether perturbation to the Wnt or Bmp pathways might contribute to the Hs2st−/− and Hs6st1−/− CC phenotypes. Western blotting of protein extracted from whole telencephalon revealed that Lef1 (a readout of Wnt/β-catenin signaling; Hovanes et al., 2001; Filali et al., 2002; Fotaki et al., 2013) protein levels did not vary between genotypes (Fig. 6A) and phosphorylated Smad (pSmad1/5, a readout of Bmp signaling) protein levels showed a weak trend of wild-type < Hs2st−/− < Hs6st1−/− with a 1.5-fold increase in Hs6st1−/− telencephalon (Fig. 6A). Immunohistochemistry showed that the distribution of Lef1 (compare Fig. 6B,D,F) and pSmad1/5 (compare Fig. 6C,E,G) were very similar between wild-type and mutant embryos at the telencephalic midline. In summary, we were unable to find evidence for major perturbation to the Wnt or Bmp pathways in Hs2st−/− or Hs6st1−/− telencephalon coinciding with the CC phenotype.
Figure 6.
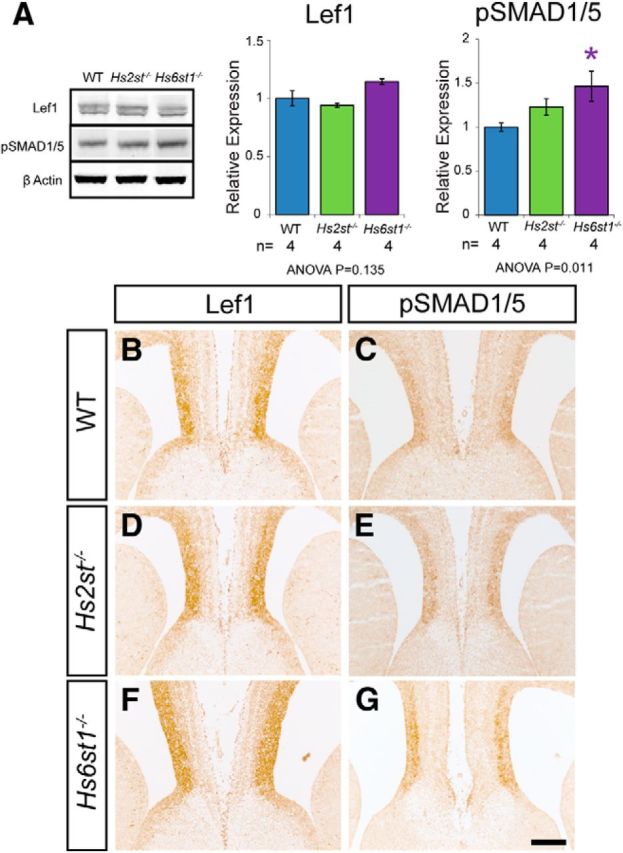
Lef1 and pSmad1/5 protein expression in the embryonic telencephalon of wild-type, Hs2st−/−, and Hs6st1−/− embryos at E16.5. A, Western blots for Lef1, pSMAD1/5, and β-actin (loading control) in protein extracted from whole telencephalon. Quantification shows relative protein expression relative to β-actin level. Note that Lef1 and pSMAD1/5 were quantified in separate blots, which were each simultaneously probed with β-actin. Lef1 expression appears unchanged in both Hs2st−/− and Hs6st1−/− embryos compared with wild-type. pSMAD1/5 expression is slightly increased in Hs2st−/− embryos compared with wild-type and is significantly increased in Hs6st1−/− embryos compared with wild-type. Histograms shows mean ± SEM for WT, n = 4; Hs2st−/−, n = 4, Hs6st1−/−, n = 4. *ANOVA p < 0.05 followed by a post hoc Student's t test for mutant versus wild-type comparison. B–G, Immunohistochemistry for Lef1 (B, D, F) and pSMAD1/5 (C, E, G) at E16.5 on coronal section of wild-type (B, C), Hs2st−/− (D, E), and Hs6st1−/− (F, G) telencephalon. The expression pattern of both Lef1 and pSMAD1/5 at the telencephalic midline appears similar in WT, Hs2st−/−, and Hs6st1−/− embryos. Scale bar, 200 μm.
Genetic or pharmacological suppression of Fgf8/Erk axis components ameliorates the Hs6st1−/− phenotype
The data suggest that the Hs6st1−/− phenotype stems from hyperactivation of the Fgf8/Erk axis causing excessive GW → IG glial cell translocation and preventing CCAs navigating the midline. We reasoned that if this is the case, then suppressing components of the Fgf8/Erk pathway would rescue both glial and axonal CC phenotypes.
First, we used a genetic approach to reduce gene dosage of Fgf8 from two (Fgf8+/+) to one (Fgf8+/−) functional alleles in Hs6st1−/− embryos. These experiments were performed on an F2 CBA × C57BL/6J genetic background and we were careful to control for effect of genetic background (see Materials and Methods). Comparison of phenotype frequencies between the Hs6st1−/−;Fgf8+/+ and Hs6st1−/−;Fgf8+/− genotypes at E18.5 showed all Hs6st−/−;Fgf8+/+ embryos (13/13 embryos; Fig. 7A) and two-thirds of Hs6st1−/−;Fgf8+/− embryos (5/7 embryos; Fig. 7B), with the severe Hs6st1−/− CC phenotype manifesting GFAP-expressing ectopic midline glia (green) and L1-expressing PBs (red), as described previously (Conway et al., 2011a). Consistent with our hypothesis, the remaining Hs6st1−/−;Fgf8+/− embryos (2/7 embryos; Fig. 7C) displayed a remarkably complete rescue with appropriately positioned GFAP expressing GW and IG glia (green) and a smooth L1-expressing U-shaped CC (red) indistinguishable from wild-type embryos (Fig. 7D).
Next, we used a pharmacological approach to target the Erk-activating kinase Mek in Hs6st1−/− embryos. These experiments were performed on the CBA background for which we originally described the Hs6st1−/− phenotype (Conway et al., 2011a). Erk signaling is hyperactive in mice harboring mutations in the Erk-inhibitor Nf1 and these mice have brain abnormalities that are rescued by treating with the potent and specific Mek1/2 inhibitor (MEKi) PD0325901 (Wang et al., 2012). We used the same MEKi dosage and treated pregnant females from Hs6st1+/− × Hs6st1+/− crosses daily with MEKi between E14.5 and E17.5 to span the period of normal and abnormal CC formation (Shu et al., 2003a; Smith et al., 2006; Conway et al., 2011a). Immunofluorescence for L1 (red) and GFAP (green) showed that all Hs6st1−/− embryos from uninjected (10/10 embryos; Fig. 7E) or mock injected (vehicle with no MEKi) dams (3/3 embryos; Fig. 7F) have severe Hs6st1−/− CC phenotype that we have consistently seen on the CBA genetic background (Conway et al., 2011a). In stark contrast, whereas approximately half the Hs6st1−/− embryos recovered from MEKi-injected dams exhibited the full CC phenotype (4/7 embryos; Fig. 7H), the remainder displayed a complete phenotype rescue (3/7 embryos; Fig. 7G) with complete restoration of GFAP expressing glia to their normal positions and the recovery of the L1-expressing U-shaped CCA bundle. We next quantitatively tested the prediction that the rescue of the Hs6st1−/− CC followed from the restoration of the GW ↔ IG glial balance. At E18.5, similar to E17.5 (Fig. 2), wild-type embryos have a thick area of Sox9+ cells at the GW facing a relatively small number of Sox9+ cells at the IG (Fig. 7I) and untreated Hs6st1−/− embryos have a much thinner zone of Sox9+ at the GW and more Sox9+ cells at the IG (Fig. 7J). MEKi treated Hs6st1−/− embryos displaying the full rescue phenotype (Fig. 7K) showed a Sox9+ cell distribution closely resembling wild-types (Fig. 7I) with a thick zone of Sox9+ cells in the GW facing fewer Sox9+ cells in the IG. Counts of Sox9+ cells in the GW and IG regions along the rostrocaudal axis (following the same protocol used in Fig. 2) revealed that although the Hs6st1 genotype and MEKi treatment did not affect the total number of Sox9+ cells along the GW → IG path (Fig. 7L), Hs6st1−/− embryos in which the CC had been rescued by MEKi treatment had approximately twice the number of Sox9+ cells in the GW (Fig. 7M) and half the number of Sox9+ cells in the IG (Fig. 7N) as Hs6st1−/− embryos that had been vehicle injected and displayed the severe CC phenotype. In fact, the GW and IG counts for MEKi-treated rescued Hs6st1−/− embryos (red lines in Fig. 7M,N) were very similar to those for wild-type embryos (blue lines in Fig. 7M,N) in contrast to vehicle-injected (unrescued) Hs6st1−/− embryos (purple lines in Fig. 7M,N). These data strongly support the hypothesis that repressing the Erk pathway in Hs6st1−/− embryos restores the wild-type GW ↔ IG glial cell balance by damping excessive GW → IG translocation and allows the development of a normal CC. The amelioration of the Hs6st1−/− CC phenotype by suppressing the Erk pathway from E14.5 on establishes that elevated Erk signaling after E14.5, as opposed to an irreversible change occurring before E14.5, is a primary cause of the Hs6st1−/− phenotype and identifies a 3 d window during which Hs6st1 activity must keep Erk signaling in check for normal CC development to proceed.
In conclusion, independent treatment strategies to antagonize the Fgf8/Erk pathway genetically or pharmacologically produced strikingly similar effects on both Hs6st1−/− CC phenotype severity and penetrance. Approximately one-third (5/14) of treated embryos exhibited a full rescue and the remaining two-thirds (9/14) exhibited no rescue at all compared with a fully penetrant severe phenotype in all (26/26) untreated Hs6st1−/− embryos (χ2 = 10.61, df = 1; p < 0.01). It is noteworthy that either both or neither axon and glial phenotypes were rescued and we never saw abnormal axons with normal glia or vice versa in any (0/14) of the rescue experiment embryos examined.
Discussion
Our salient findings are as follows: (1) the loss of either Hs2st or Hs6st1 function alters the telencephalic midline glial environment by allowing excess GW → IG glial cell movement; (2) Hs6st1 posttranscriptionally suppresses the levels of Fgf8 protein levels in the GW/IG region; (3) Hs6st1, and to a lesser extent, Hs2st suppress the Erk signaling pathway in the telencephalon including at the GW; and (4) repressing the Fgf8/Erk axis rescues the Hs6st1−/− CC phenotype. These data suggest a model in which, in wild-type embryos, pErk levels are tightly regulated, with Hs2st and Hs6st1 playing important repressor functions to generate the right levels of pErk to drive the right amount of GW → IG glial translocation and generate the GW ↔ IG glial cell balance needed to guide CCAs across the midline (Fig. 8A). The loss of either Hs2st (Fig. 8B) or Hs6st1 (Fig. 8C) repressor function elevates pErk above normal levels, causing excessive GW → IG glial translocation and preventing CCAs from crossing the midline. Conversely, repressing the hyperactive Erk pathway in an Hs6st1−/− embryo restores the normal GW ↔ IG glial cell balance and CCA midline crossing (Fig. 8D).
The Erk pathway is one of four mitogen-activated protein kinase (Mapk) cascades that transduce extracellular signals into the cell. At the core of Erk signaling pathway are the two Erk kinases, Erk1 and Erk2, the phosphorylation of which by Mek1/2 kinases activates Erk kinase activity and regulation of gene expression, cell proliferation, migration, differentiation, and/or death (Rubinfeld and Seger, 2005). Both gain- and loss-of-function mutations in ERK pathway components in humans have been linked to a wide spectrum of phenotypes spanning developmental processes (Samuels et al., 2009). Genetically stimulating the Erk pathway during mouse cerebral cortex development with constitutively active Mek or inactive Nf1 alleles causes precocious glial output, whereas suppressing Erk by inactivating Mek has the opposite effect, both without changes in cell proliferation or apoptosis (Li et al., 2012; Pucilowska et al., 2012; Wang et al., 2012). The increased numbers of mature glia at the telencephalic midline of both Hs2st−/− and Hs6st1−/− embryos with no obvious associated changes in cell proliferation resembles these models (Conway et al., 2011a; present study). Knock-out of the Erk inhibitors Sprouty1/2 also elevates Erk signaling and causes midline glial tangles with CC agenesis, although Sprouty1/2 −/− embryos additionally exhibit increased cell proliferation, which may contribute to the glial output (Faedo et al., 2010; Magnani et al., 2014). The spectrum of phenotypes in Hs6st1, Mek, Hs2st, Nf1, and Sprouty1/2 mutant models that share Erk hyperactivation likely stems from differences in Erk and non-Erk pathway disturbance and spatiotemporal activity.
A clear implication of our study is that the correct GW ↔ IG balance of glia must be maintained at an optimal level for normal CC development. Here, we addressed Erk hyperactivation resulting in excessive GW → IG glial translocation and CC agenesis. Fgf receptor null mutations, which would be expected to inhibit the Erk pathway, have the opposite effect and inhibit GW → IG glial translocation but also produce CC agenesis (Tole et al., 2006, Smith et al., 2006). The overall cellular picture that emerges is of a finely poised system in which the correct GW ↔ IG balance of glia, itself dependent on a narrow range of Erk signaling levels, is needed to shepherd axons across the telencephalic midline.
In humans, hypomorphic mutations in HS6ST1 have been linked to the pituitary developmental disorder idiopathic hypogonadotrophic hypogonadism (IHH), as have loss-of-function mutations in FGF8 and FGFR1 (Tornberg et al., 2011). Interestingly, FGFR1 mutations enhance the IHH phenotype in patients harboring HS6ST1 mutations, suggesting that, in the human IHH context, HS6ST1 positively regulates FGF signaling. How the HS6ST1 ↔ FGF8 interaction relates to ERK signaling in IHH is unknown, but contrasts with the antagonistic Fgf8 ↔ Hs6st1 genetic interaction that we have described here in the mouse and indicates species- and/or tissue-specific interaction.
Classically, HS acts as a coreceptor with Fgfrs for Fgfs and regulates Fgf diffusion kinetics (Schlessinger et al., 2000; Harmer, 2006; Yu et al., 2009; Guillemot and Zimmer, 2011; Duchesne et al., 2012). Fgf signaling during the development of the mouse forebrain is a complex and dynamic procedure, marshalling Fgf2, Fgf3, Fgf7, Fgf8, Fgf15, Fgf17, and Fgf18 ligands and Fgfr1, Fgfr2, and Fgfr3 receptors that signal through a combination of Erk and non-Erk pathways (Thisse and Thisse, 2005; Borello et al., 2008; Iwata and Hevner, 2009; Guillemot and Zimmer, 2011; Toyoda et al., 2010). Puzzlingly, in contrast to the Erk hyperactivation that we see in Hs2st−/− or Hs6st1−/− telencephalon, the consensus of other studies in which HS biosynthesis is perturbed genetically is hypoactive Fgf/Erk signaling attributed to excessive diffusion diluting Fgf ligands below activation threshold and/or weak Fgf/Fgfr binding, rendering cells unresponsive to Fgf (Qu et al., 2011; Shimokawa et al., 2011; Qu et al., 2012). We suspect major perturbations imposed on HS by genetic ablation of the core HS biosynthetic Ext enzymes, which abolish HS synthesis altogether, or of multiple HS-modifying enzymes simultaneously maximize gross changes in, or even complete loss of, HS sulfation and this is why we see a different class of phenotype in our single mutation Hs2st−/− and Hs6st1−/− embryos, which would be expected to have less perturbed HS (Inatani et al., 2003; Qu et al., 2011; Shimokawa et al., 2011; Qu et al., 2012). Perhaps analogously, a missense mutation in Fgf9, which changes its interaction kinetics with HS and ectopically activates Fgf9 targets, causes a completely different bone phenotype to null Fgf9 alleles which have no signaling activity (Harada et al., 2009).
We found a striking correlation between Erk hyperactivity and CC phenotype penetrance (summarized in Fig. 8A–D). Hs6st1−/− embryos have greater Erk hyperactivation and higher penetrance than Hs2st−/− embryos and treatments that suppress Fgf8/Erk axis components in Hs6st1−/− embryos reduce CC phenotype penetrance. Matched Hs6st1−/− embryonic littermates sharing the same amount of drug from the maternal bloodstream and/or the same genetic background, as well other environmental factors, were either fully rescued or not rescued at all, suggesting a more interesting explanation than suboptimal rescue treatment. Our data imply a surprisingly binary “on/off” switch property of this system in which genetic and environmental factors, channelling through Erk after E14.5, fix the probability of the CC developing normally. Mechanistically, we suspect that Erk controls gene expression programs governing an initiation event involving a small number of cells critical for the subsequent fate of many more cells during CC development. How this might work is uncertain but plausible scenarios include pioneer effects in which once one or a few axons manage to cross the midline, likely in close association with glia, this seeds the formation of the whole CC structure via cell ↔ cell interaction mechanisms such as fasciculation. Consistent with this idea, we always see an extremely tight linkage between glial and axonal phenotypes in the Fgf8/Erk Hs6st1−/− rescue experiments (14/14 embryos in which neither or both axons and glia attain normal CC configuration). This tight linkage can be broken in other situations as axon and glial phenotypes are uncoupled in Hs6st1−/−;Slit2−/− embryos in which removal of Slit2 allows axons to burrow through ectopic glia at the midline (Conway et al., 2011a). These and other studies point to glial mispositioning (caused by Erk) as the primary cause and the noncrossing axon phenotype (caused by Slit2 expressed by mispositioned glia) as a secondary consequence in the Hs6st1−/−, and likely the Hs2st−/−, phenotype (Bagri et al., 2002; Shu et al., 2003b; Smith et al., 2006; Conway et al., 2011a). However, because the full extent of Erk ↔ glia ↔ axon causal relationships during CC development are not fully understood, there are additional possibilities worth exploring in the future. CC axons may influence the glial phenotype and/or various combinations of Hs2st, Hs6st1, Fgf8, and Erk and other factors may contribute directly to both axon and glial phenotypes.
Here, we identify distinct molecular roles for Hs2st and Hs6st1 in the developing mammalian telencephalon. Hs6st1 suppresses levels of Fgf8 protein posttranscriptionally and has a large negative effect on pErk levels, whereas Hs2st does not suppress levels of Fgf8 protein and has a smaller negative effect on pErk. All of this fits the “sugar code” idea that Hs2st and Hs6st1 each instruct HS to function in a molecularly distinct manner. Formally testing this hypothesis in our system awaits the development of more sensitive glycomic technologies to reveal how the fine structure of HS, ideally at the cellular level, correlates with normal and abnormal CC development. Alternative explanations, for example, that the distinct Hs2st−/− and Hs6st1−/− phenotypes reflect different sites of action of the two enzymes, also need to be investigated because, in other systems, there is genetic evidence that Hs2st and the three mammalian Hs6st isoforms have redundant functions and/or are able to compensate for one another's function, arguing against Hs2st and Hs6st imparting unique molecular instructions onto HS in all situations (Kamimura et al., 2006; Qu et al., 2011; (Conway et al., 2011b). In light of all of these unanswered questions, the nonredundant roles we find for Hs2st and Hs6st1 during CC development provide a powerful experimental system for deciphering the roles of Hs2st and Hs6st1 in the sugar code or establishing an alternative model to account for their distinct genetic functions.
Footnotes
This work was supported by the Medical Research Council (Grant G0701460) and The Wellcome Trust (Grants 094832/Z/10/Z and 091475). We thank Kirsty Ferrier, Gwen Harrison, Erin O'Hare, Samantha Martin, and Da Mi for excellent technical assistance; Tian Yu for coordinating Fgf8 matings and embryo collections; BRR staff for animal husbandry; and Scott Guimond and Thomas Theil for discussion. The Fgf8 mutant lines were kindly provided by Gail Martin.
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Allen BL, Rapraeger AC. Spatial and temporal expression of heparan sulfate in mouse development regulates FGF and FGF receptor assembly. J Cell Biol. 2003;163:637–648. doi: 10.1083/jcb.200307053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen BL, Filla MS, Rapraeger AC. Role of heparan sulfate as a tissue-specific regulator of FGF-4 and FGF receptor recognition. J Cell Biol. 2001;155:845–858. doi: 10.1083/jcb.200106075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaniti EM, Hasenpusch-Theil K, Li Z, Magnani D, Kessaris N, Mason JO, Theil T. Gli3 is required in Emx1+ progenitors for the development of the corpus callosum. Dev Biol. 2013;376:113–124. doi: 10.1016/j.ydbio.2013.02.001. [DOI] [PubMed] [Google Scholar]
- Bagri A, Marín O, Plump AS, Mak J, Pleasure SJ, Rubenstein JL, Tessier-Lavigne M. Slit proteins prevent midline crossing and determine the dorsoventral position of major axonal pathways in the mammalian forebrain. Neuron. 2002;33:233–248. doi: 10.1016/S0896-6273(02)00561-5. [DOI] [PubMed] [Google Scholar]
- Bedeschi MF, Bonaglia MC, Grasso R, Pellegri A, Garghentino RR, Battaglia MA, Panarisi AM, Di Rocco M, Balottin U, Bresolin N, Bassi MT, Borgatti R. Agenesis of the corpus callosum: clinical and genetic study in 63 young patients. Pediatr Neurol. 2006;34:186–193. doi: 10.1016/j.pediatrneurol.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Benadiba C, Magnani D, Niquille M, Morlé L, Valloton D, Nawabi H, Ait-Lounis A, Otsmane B, Reith W, Theil T, Hornung JP, Lebrand C, Durand B. The ciliogenic transcription factor RFX3 regulates early midline distribution of guidepost neurons required for corpus callosum development. PLoS Genet. 2012;8:e1002606. doi: 10.1371/journal.pgen.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlen MO, Bailoo JD, Jordan RL, Wahlsten D. Hippocampal commissure defects in crosses of four inbred mouse strains with absent corpus callosum. Genes Brain Behav. 2012;11:757–766. doi: 10.1111/j.1601-183X.2012.00802.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borello U, Cobos I, Long JE, McWhirter JR, Murre C, Rubenstein JL. FGF15 promotes neurogenesis and opposes FGF8 function during neocortical development. Neural Dev. 2008;3:17. doi: 10.1186/1749-8104-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock SL, Fletcher JM, Beddington RS, Wilson VA. Renal agenesis in mice homozygous for a gene trap mutation in the gene encoding heparan sulfate 2-sulfotransferase. Genes Dev. 1998;12:1894–1906. doi: 10.1101/gad.12.12.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bülow HE, Hobert O. Differential sulfations and epimerization define heparan sulfate specificity in nervous system development. Neuron. 2004;41:723–736. doi: 10.1016/S0896-6273(04)00084-4. [DOI] [PubMed] [Google Scholar]
- Bülow HE, Tjoe N, Townley RA, Didiano D, van Kuppevelt TH, Hobert O. Extracellular sugar modifications provide instructive and cell-specific information for axon-guidance choices. Curr Biol. 2008;18:1978–1985. doi: 10.1016/j.cub.2008.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe Y, Siegenthaler JA, Pleasure SJ. A cascade of morphogenic signaling initiated by the meninges controls corpus callosum formation. Neuron. 2012;73:698–712. doi: 10.1016/j.neuron.2011.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway CD, Howe KM, Nettleton NK, Price DJ, Mason JO, Pratt T. Heparan sulfate sugar modifications mediate the functions of slits and other factors needed for mouse forebrain commissure development. J Neurosci. 2011a;31:1955–1970. doi: 10.1523/JNEUROSCI.2579-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway CD, Price DJ, Pratt T, Mason JO. Analysis of axon guidance defects at the optic chiasm in heparan sulphate sulphotransferase compound mutant mice. J Anat. 2011b;219:734–742. doi: 10.1111/j.1469-7580.2011.01432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo-Enriquez I, Partanen J, Martinez S, Echevarria D. Fgf8-related secondary organizers exert different polarizing planar instructions along the mouse anterior neural tube. PLoS One. 2012;7:e39977. doi: 10.1371/journal.pone.0039977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolski R, Hertig G, Lechner H, Wörsdörfer P, Wulf V, Dicke N, Eckert D, Bauer R, Schorle H, Willecke K. Loss of connexin43-mediated gap junctional coupling in the mesenchyme of limb buds leads to altered expression of morphogens in mice. Hum Mol Genet. 2009;18:2899–2911. doi: 10.1093/hmg/ddp227. [DOI] [PubMed] [Google Scholar]
- Donahoo AL, Richards LJ. Understanding the mechanisms of callosal development through the use of transgenic mouse models. Semin Pediatr Neurol. 2009;16:127–142. doi: 10.1016/j.spen.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Duchesne L, Octeau V, Bearon RN, Beckett A, Prior IA, Lounis B, Fernig DG. Transport of fibroblast growth factor 2 in the pericellular matrix is controlled by the spatial distribution of its binding sites in heparan sulfate. PLoS Biol. 2012;10:e1001361. doi: 10.1371/journal.pbio.1001361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faedo A, Borello U, Rubenstein JL. Repression of Fgf signaling by sprouty1–2 regulates cortical patterning in two distinct regions and times. J Neurosci. 2010;30:4015–4023. doi: 10.1523/JNEUROSCI.0307-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fame RM, MacDonald JL, Macklis JD. Development, specification, and diversity of callosal projection neurons. Trends Neurosci. 2011;34:41–50. doi: 10.1016/j.tins.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filali M, Cheng N, Abbott D, Leontiev V, Engelhardt JF. Wnt-3A/beta-catenin signaling induces transcription from the LEF-1 promoter. J Biol Chem. 2002;277:33398–33410. doi: 10.1074/jbc.M107977200. [DOI] [PubMed] [Google Scholar]
- Fotaki V, Smith R, Pratt T, Price DJ. Foxg1 is required to limit the formation of ciliary margin tissue and Wnt/beta-catenin signalling in the developing nasal retina of the mouse. Dev Biol. 2013;380:299–313. doi: 10.1016/j.ydbio.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fothergill T, Donahoo AL, Douglass A, Zalucki O, Yuan J, Shu T, Goodhill GJ, Richards LJ. Netrin-DCC signaling regulates corpus callosum formation through attraction of pioneering axons and by modulating Slit2-mediated repulsion. Cereb Cortex. 2013 doi: 10.1093/cercor/bhs395. doi: 10.1093/cercor/bhs395. Advance online publication. [DOI] [PubMed] [Google Scholar]
- Grobe K, Inatani M, Pallerla SR, Castagnola J, Yamaguchi Y, Esko JD. Cerebral hypoplasia and craniofacial defects in mice lacking heparan sulfate Ndst1 gene function. Development. 2005;132:3777–3786. doi: 10.1242/dev.01935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemot F, Zimmer C. From cradle to grave: the multiple roles of fibroblast growth factors in neural development. Neuron. 2011;71:574–588. doi: 10.1016/j.neuron.2011.08.002. [DOI] [PubMed] [Google Scholar]
- Harada M, Murakami H, Okawa A, Okimoto N, Hiraoka S, Nakahara T, Akasaka R, Shiraishi Y, Futatsugi N, Mizutani-Koseki Y, Kuroiwa A, Shirouzu M, Yokoyama S, Taiji M, Iseki S, Ornitz DM, Koseki H. FGF9 monomer-dimer equilibrium regulates extracellular matrix affinity and tissue diffusion. Nat Genet. 2009;41:289–298. doi: 10.1038/ng.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer NJ. Insights into the role of heparan sulphate in fibroblast growth factor signalling. Biochem Soc Trans. 2006;34:442–445. doi: 10.1042/BST0340442. [DOI] [PubMed] [Google Scholar]
- Holt CE, Dickson BJ. Sugar codes for axons? Neuron. 2005;46:169–172. doi: 10.1016/j.neuron.2005.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovanes K, Li TW, Munguia JE, Truong T, Milovanovic T, Lawrence Marsh J, Holcombe RF, Waterman ML. Beta-catenin-sensitive isoforms of lymphoid enhancer factor-1 are selectively expressed in colon cancer. Nat Genet. 2001;28:53–57. doi: 10.1038/ng0501-53. [DOI] [PubMed] [Google Scholar]
- Ilagan R, Abu-Issa R, Brown D, Yang YP, Jiao K, Schwartz RJ, Klingensmith J, Meyers EN. Fgf8 is required for anterior heart field development. Development. 2006;133:2435–2445. doi: 10.1242/dev.02408. [DOI] [PubMed] [Google Scholar]
- Inatani M, Irie F, Plump AS, Tessier-Lavigne M, Yamaguchi Y. Mammalian brain morphogenesis and midline axon guidance require heparan sulfate. Science. 2003;302:1044–1046. doi: 10.1126/science.1090497. [DOI] [PubMed] [Google Scholar]
- Islam SM, Shinmyo Y, Okafuji T, Su Y, Naser IB, Ahmed G, Zhang S, Chen S, Ohta K, Kiyonari H, Abe T, Tanaka S, Nishinakamura R, Terashima T, Kitamura T, Tanaka H. Draxin, a repulsive guidance protein for spinal cord and forebrain commissures. Science. 2009;323:388–393. doi: 10.1126/science.1165187. [DOI] [PubMed] [Google Scholar]
- Iwata T, Hevner RF. Fibroblast growth factor signaling in development of the cerebral cortex. Dev Growth Differ. 2009;51:299–323. doi: 10.1111/j.1440-169X.2009.01104.x. [DOI] [PubMed] [Google Scholar]
- Kalus I, Salmen B, Viebahn C, von Figura K, Schmitz D, D'Hooge R, Dierks T. Differential involvement of the extracellular 6-O-endosulfatases Sulf1 and Sulf2 in brain development and neuronal and behavioral plasticity. J Cell Mol Med. 2009;13:4505–4521. doi: 10.1111/j.1582-4934.2008.00558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamimura K, Koyama T, Habuchi H, Ueda R, Masu M, Kimata K, Nakato H. Specific and flexible roles of heparan sulfate modifications in Drosophila FGF signaling. J Cell Biol. 2006;174:773–778. doi: 10.1083/jcb.200603129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeble TR, Halford MM, Seaman C, Kee N, Macheda M, Anderson RB, Stacker SA, Cooper HM. The Wnt receptor Ryk is required for Wnt5a-mediated axon guidance on the contralateral side of the corpus callosum. J Neurosci. 2006;26:5840–5848. doi: 10.1523/JNEUROSCI.1175-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnunen T, Huang Z, Townsend J, Gatdula MM, Brown JR, Esko JD, Turnbull JE. Heparan 2-O-sulfotransferase, hst-2, is essential for normal cell migration in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2005;102:1507–1512. doi: 10.1073/pnas.0401591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordes U, Cheng YC, Scotting PJ. Sox group E gene expression distinguishes different types and maturational stages of glial cells in developing chick and mouse. Brain Res Dev Brain Res. 2005;157:209–213. doi: 10.1016/j.devbrainres.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Kreuger J, Spillmann D, Li JP, Lindahl U. Interactions between heparan sulfate and proteins: the concept of specificity. J Cell Biol. 2006;174:323–327. doi: 10.1083/jcb.200604035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lania G, Zhang Z, Huynh T, Caprio C, Moon AM, Vitelli F, Baldini A. Early thyroid development requires a Tbx1-Fgf8 pathway. Dev Biol. 2009;328:109–117. doi: 10.1016/j.ydbio.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Chien CB. When sugars guide axons: insights from heparan sulphate proteoglycan mutants. Nat Rev Genet. 2004;5:923–935. doi: 10.1038/nrg1490. [DOI] [PubMed] [Google Scholar]
- Li X, Newbern JM, Wu Y, Morgan-Smith M, Zhong J, Charron J, Snider WD. MEK is a key regulator of gliogenesis in the developing brain. Neuron. 2012;75:1035–1050. doi: 10.1016/j.neuron.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Bendito G, Flames N, Ma L, Fouquet C, Di Meglio T, Chedotal A, Tessier-Lavigne M, Marín O. Robo1 and Robo2 cooperate to control the guidance of major axonal tracts in the mammalian forebrain. J Neurosci. 2007;27:3395–3407. doi: 10.1523/JNEUROSCI.4605-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani D, Hasenpusch-Theil K, Benadiba C, Yu T, Basson MA, Price DJ, Lebrand C, Theil T. Gli3 controls corpus callosum formation by positioning midline guideposts during telencephalic patterning. Cereb Cortex. 2014;24:186–198. doi: 10.1093/cercor/bhs303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani D, Hasenpusch-Theil K, Theil T. Gli3 controls subplate formation and growth of cortical axons. Cereb Cortex. 2013;23:2542–2551. doi: 10.1093/cercor/bhs237. [DOI] [PubMed] [Google Scholar]
- McLaughlin D, Karlsson F, Tian N, Pratt T, Bullock SL, Wilson VA, Price DJ, Mason JO. Specific modification of heparan sulphate is required for normal cerebral cortical development. Mech Dev. 2003;120:1481–1488. doi: 10.1016/j.mod.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Merry CL, Bullock SL, Swan DC, Backen AC, Lyon M, Beddington RS, Wilson VA, Gallagher JT. The molecular phenotype of heparan sulfate in the Hs2st-/- mutant mouse. J Biol Chem. 2001;276:35429–35434. doi: 10.1074/jbc.M100379200. [DOI] [PubMed] [Google Scholar]
- Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat Genet. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- Mitchell KJ, Pinson KI, Kelly OG, Brennan J, Zupicich J, Scherz P, Leighton PA, Goodrich LV, Lu X, Avery BJ, Tate P, Dill K, Pangilinan E, Wakenight P, Tessier-Lavigne M, Skarnes WC. Functional analysis of secreted and transmembrane proteins critical to mouse development. Nat Genet. 2001;28:241–249. doi: 10.1038/90074. [DOI] [PubMed] [Google Scholar]
- Moldrich RX, Gobius I, Pollak T, Zhang J, Ren T, Brown L, Mori S, De Juan Romero C, Britanova O, Tarabykin V, Richards LJ. Molecular regulation of the developing commissural plate. J Comp Neurol. 2010;518:3645–3661. doi: 10.1002/cne.22445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niquille M, Garel S, Mann F, Hornung JP, Otsmane B, Chevalley S, Parras C, Guillemot F, Gaspar P, Yanagawa Y, Lebrand C. Transient neuronal populations are required to guide callosal axons: a role for semaphorin 3C. PLoS Biol. 2009;7:e1000230. doi: 10.1371/journal.pbio.1000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niquille M, Minocha S, Hornung JP, Rufer N, Valloton D, Kessaris N, Alfonsi F, Vitalis T, Yanagawa Y, Devenoges C, Dayer A, Lebrand C. Two specific populations of GABAergic neurons originating from the medial and the caudal ganglionic eminences aid in proper navigation of callosal axons. Dev Neurobiol. 2013;73:647–672. doi: 10.1002/dneu.22075. [DOI] [PubMed] [Google Scholar]
- Nowakowski TJ, Fotaki V, Pollock A, Sun T, Pratt T, Price DJ. MicroRNA-92b regulates the development of intermediate cortical progenitors in embryonic mouse brain. Proc Natl Acad Sci U S A. 2013;110:7056–7061. doi: 10.1073/pnas.1219385110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Woodbury A, Esko JD, Grobe K, Zhang X. Heparan sulfate biosynthetic gene Ndst1 is required for FGF signaling in early lens development. Development. 2006;133:4933–4944. doi: 10.1242/dev.02679. [DOI] [PubMed] [Google Scholar]
- Pan Y, Carbe C, Powers A, Zhang EE, Esko JD, Grobe K, Feng GS, Zhang X. Bud specific N-sulfation of heparan sulfate regulates Shp2-dependent FGF signaling during lacrimal gland induction. Development. 2008;135:301–310. doi: 10.1242/dev.014829. [DOI] [PubMed] [Google Scholar]
- Paul LK, Brown WS, Adolphs R, Tyszka JM, Richards LJ, Mukherjee P, Sherr EH. Agenesis of the corpus callosum: genetic, developmental and functional aspects of connectivity. Nat Rev Neurosci. 2007;8:287–299. doi: 10.1038/nrn2107. [DOI] [PubMed] [Google Scholar]
- Pompolo S, Harley VR. Localisation of the SRY-related HMG box protein, SOX9, in rodent brain. Brain Res. 2001;906:143–148. doi: 10.1016/S0006-8993(01)02574-4. [DOI] [PubMed] [Google Scholar]
- Powell AK, Fernig DG, Turnbull JE. Fibroblast growth factor receptors 1 and 2 interact differently with heparin/heparan sulfate. Implications for dynamic assembly of a ternary signaling complex. J Biol Chem. 2002;277:28554–28563. doi: 10.1074/jbc.M111754200. [DOI] [PubMed] [Google Scholar]
- Pratt T, Conway CD, Tian NM, Price DJ, Mason JO. Heparan sulphation patterns generated by specific heparan sulfotransferase enzymes direct distinct aspects of retinal axon guidance at the optic chiasm. J Neurosci. 2006;26:6911–6923. doi: 10.1523/JNEUROSCI.0505-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucilowska J, Puzerey PA, Karlo JC, Galán RF, Landreth GE. Disrupted ERK signaling during cortical development leads to abnormal progenitor proliferation, neuronal and network excitability and behavior, modeling human neuro-cardio-facial-cutaneous and related syndromes. J Neurosci. 2012;32:8663–8677. doi: 10.1523/JNEUROSCI.1107-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Carbe C, Tao C, Powers A, Lawrence R, van Kuppevelt TH, Cardoso WV, Grobe K, Esko JD, Zhang X. Lacrimal gland development and Fgf10-Fgfr2b signaling are controlled by 2-O- and 6-O-sulfated heparan sulfate. J Biol Chem. 2011;286:14435–14444. doi: 10.1074/jbc.M111.225003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X, Pan Y, Carbe C, Powers A, Grobe K, Zhang X. Glycosaminoglycan-dependent restriction of FGF diffusion is necessary for lacrimal gland development. Development. 2012;139:2730–2739. doi: 10.1242/dev.079236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards LJ. Axonal pathfinding mechanisms at the cortical midline and in the development of the corpus callosum. Braz J Med Biol Res. 2002;35:1431–1439. doi: 10.1590/S0100-879X2002001200004. [DOI] [PubMed] [Google Scholar]
- Rubinfeld H, Seger R. The ERK cascade: a prototype of MAPK signaling. Mol Biotechnol. 2005;31:151–174. doi: 10.1385/MB:31:2:151. [DOI] [PubMed] [Google Scholar]
- Samuels IS, Saitta SC, Landreth GE. MAP'ing CNS development and cognition: an ERKsome process. Neuron. 2009;61:160–167. doi: 10.1016/j.neuron.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Camacho C, Ortega JA, Ocaña I, Alcántara S, Bovolenta P. Appropriate Bmp7 levels are required for the differentiation of midline guidepost cells involved in corpus callosum formation. Dev Neurobiol. 2011;71:337–350. doi: 10.1002/dneu.20865. [DOI] [PubMed] [Google Scholar]
- Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, Linhardt RJ, Mohammadi M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. 2000;6:743–750. doi: 10.1016/S1097-2765(00)00073-3. [DOI] [PubMed] [Google Scholar]
- Serafini T, Colamarino SA, Leonardo ED, Wang H, Beddington R, Skarnes WC, Tessier-Lavigne M. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell. 1996;87:1001–1014. doi: 10.1016/S0092-8674(00)81795-X. [DOI] [PubMed] [Google Scholar]
- Shimokawa K, Kimura-Yoshida C, Nagai N, Mukai K, Matsubara K, Watanabe H, Matsuda Y, Mochida K, Matsuo I. Cell surface heparan sulfate chains regulate local reception of FGF signaling in the mouse embryo. Dev Cell. 2011;21:257–272. doi: 10.1016/j.devcel.2011.06.027. [DOI] [PubMed] [Google Scholar]
- Shu T, Puche AC, Richards LJ. Development of midline glial populations at the corticoseptal boundary. J Neurobiol. 2003a;57:81–94. doi: 10.1002/neu.10252. [DOI] [PubMed] [Google Scholar]
- Shu T, Sundaresan V, McCarthy MM, Richards LJ. Slit2 guides both precrossing and postcrossing callosal axons at the midline in vivo. J Neurosci. 2003b;23:8176–8184. doi: 10.1523/JNEUROSCI.23-22-08176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KM, Ohkubo Y, Maragnoli ME, Rasin MR, Schwartz ML, Sestan N, Vaccarino FM. Midline radial glia translocation and corpus callosum formation require FGF signaling. Nat Neurosci. 2006;9:787–797. doi: 10.1038/nn1705. [DOI] [PubMed] [Google Scholar]
- Thisse B, Thisse C. Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev Biol. 2005;287:390–402. doi: 10.1016/j.ydbio.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Tole S, Gutin G, Bhatnagar L, Remedios R, Hébert JM. Development of midline cell types and commissural axon tracts requires Fgfr1 in the cerebrum. Dev Biol. 2006;289:141–151. doi: 10.1016/j.ydbio.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Tornberg J, Sykiotis GP, Keefe K, Plummer L, Hoang X, Hall JE, Quinton R, Seminara SB, Hughes V, Van Vliet G, Van Uum S, Crowley WF, Habuchi H, Kimata K, Pitteloud N, Bülow HE. Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc Natl Acad Sci U S A. 2011;108:11524–11529. doi: 10.1073/pnas.1102284108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoda R, Assimacopoulos S, Wilcoxon J, Taylor A, Feldman P, Suzuki-Hirano A, Shimogori T, Grove EA. FGF8 acts as a classic diffusible morphogen to pattern the neocortex. Development. 2010;137:3439–3448. doi: 10.1242/dev.055392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull J, Powell A, Guimond S. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 2001;11:75–82. doi: 10.1016/S0962-8924(00)01897-3. [DOI] [PubMed] [Google Scholar]
- Wallace VA, Raff MC. A role for Sonic hedgehog in axon-to-astrocyte signalling in the rodent optic nerve. Development. 1999;126:2901–2909. doi: 10.1242/dev.126.13.2901. [DOI] [PubMed] [Google Scholar]
- Wang Y, Thekdi N, Smallwood PM, Macke JP, Nathans J. Frizzled-3 is required for the development of major fiber tracts in the rostral CNS. J Neurosci. 2002;22:8563–8573. doi: 10.1523/JNEUROSCI.22-19-08563.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kim E, Wang X, Novitch BG, Yoshikawa K, Chang LS, Zhu Y. ERK inhibition rescues defects in fate specification of Nf1-deficient neural progenitors and brain abnormalities. Cell. 2012;150:816–830. doi: 10.1016/j.cell.2012.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SR, Burkhardt M, Nowak M, Ries J, Petrásek Z, Scholpp S, Schwille P, Brand M. Fgf8 morphogen gradient forms by a source-sink mechanism with freely diffusing molecules. Nature. 2009;461:533–536. doi: 10.1038/nature08391. [DOI] [PubMed] [Google Scholar]