

Abstract
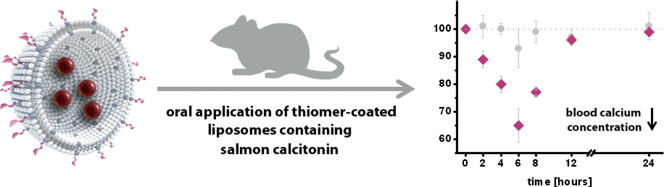
The aim of the present study was the in vivo evaluation of thiomer-coated liposomes for an oral application of peptides. For this purpose, salmon calcitonin was chosen as a model drug and encapsulated within liposomes. Subsequently, the drug loaded liposomes were coated with either chitosan–thioglycolic acid (CS–TGA) or an S-protected version of the same polymer (CS–TGA–MNA), leading to an increase in the particle size of about 500 nm and an increase in the zeta potential from approximately − 40 mV to a maximum value of about + 44 mV, depending on the polymer. Coated liposomes were demonstrated to effectively penetrate the intestinal mucus layer where they came in close contact with the underlying epithelium. To investigate the permeation enhancing properties of the coated liposomes ex vivo, we monitored the transport of fluoresceinisothiocyanate-labeled salmon calcitonin (FITC-sCT) through rat small intestine. Liposomes coated with CS–TGA–MNA showed the highest effect, leading to a 3.8-fold increase in the uptake of FITC-sCT versus the buffer control. In vivo evaluation of the different formulations was carried out by the oral application of 40 μg of sCT per rat, either encapsulated within uncoated liposomes, CS–TGA-coated liposomes or CS–TGA–MNA-coated liposomes, or given as a solution serving as negative control. The blood calcium level was monitored over a time period of 24 h. The highest reduction in the blood calcium level, to a minimum of 65% of the initial value after 6 h, was achieved for CS–TGA–MNA-coated liposomes. Comparing the areas above curves (AAC) of the blood calcium levels, CS–TGA–MNA-coated liposomes led to an 8.2-fold increase compared to the free sCT solution if applied orally in the same concentration. According to these results, liposomes coated with S-protected thiomers have demonstrated to be highly valuable carriers for enhancing the oral bioavailability of salmon calcitonin.
Keywords: Liposome, Thiomer, Chitosan, Permeation enhancement, Salmon calcitonin
Graphical abstract
1. Introduction
Biopharmaceuticals, especially peptide- and protein drugs, have become increasingly important in modern pharmacotherapy, and the market for these products is expected to increase further in the years to come [1]. In particular, their oral delivery holds enormous potential as it allows for easy and painless administration, reasonably low cost production and generally high patient compliance. Different delivery systems have been investigated in order to improve oral peptide delivery. Among them are nanoparticulate systems including liposomes, self-emulsifying systems, solid lipid nanoparticles and polymeric nanoparticles [2–5]. Other approaches to enhance the oral bioavailability of peptides are focussing on the use of permeation enhancers [6,7], enzyme inhibitors [8] or the targeting of apical membrane receptors [9]. Despite all these efforts, however, oral peptide delivery systems have not yet reached their full potential. The main obstacles which need to be overcome are the enzymatic degradation of peptides before reaching the systemic circulation, and the low permeability of such large molecules across the intestinal mucosa [8]. Both barriers have been addressed by a recently developed delivery system consisting of liposomes coated with thiolated chitosans. In different in vitro- and ex vivo studies we could clearly show that this delivery system has the potential to protect the drug towards the hostile gastric environment while prolonging the residence time on intestinal mucosal membranes additionally acting as an absorption enhancer [10,11]. So far, however, an in vivo proof-of-principle for this promising concept is missing.
It is therefore the aim of this study to evaluate these thiomer-coated liposomes in vivo utilizing salmon calcitonin (sCT), a well characterized 32 amino acid peptide [12,13], as a model drug. In order to reach this goal, sCT was encapsulated in liposomes that were coated with the mucoadhesive polymer chitosan–thioglycolic acid (CS–TGA). Furthermore, CS–TGA was optionally S-protected via conjugation with 6,6′-dithionicotinamide, resulting in a chitosan–thioglycolic acid 6-mercaptonicotinamide-conjugate (CS–TGA–MNA) being less prone to oxidation and exhibiting higher potential to form disulphide bonds with the mucus. As these coated liposomes have to penetrate into the intestinal mucus layer in order to remain there for a prolonged time and to provide a permeation enhancing effect on the mucosa, their mucus-penetration and sCT permeation-enhancing properties were evaluated on intestinal mucus and intestinal mucosa, respectively. Thereafter, thiomer-coated liposomes containing sCT were orally administered to rats, and the blood calcium levels were monitored over 24 h.
2. Materials and methods
2.1. Materials
1,2-Dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[4-(p-maleimidomethyl) cyclohexane-carboxamide] (DPPE-MCC) and the fluorescence-labeled phospholipid 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (DOPE-Liss.Rhod.) were purchased from Avanti Polar Lipids (Alabaster, AL). Chitosan–thioglycolic acid (CS–TGA; molecular weight: 150 kDa, 660 μmol SH-groups/g polymer) and a chitosan–thioglycolic acid 6-mercaptonicotinamide-conjugate (CS–TGA–MNA; based on the 150 kDa CS–TGA, 380 μmol S-protected thiol groups and 280 μmol free SH-groups/g polymer) were synthesized and provided by the Department of Pharmaceutical Technology, University of Innsbruck (Austria). Salmon calcitonin (sCT; 32 amino acids, molecular weight: 3431.89 g/mol) and fluoresceinisothiocyanate-labeled salmon calcitonin (FITC-sCT; 32 amino acids, molecular weight: 3803.29 g/mol) were synthesized and provided by piCHEM (Graz, Austria). Porcine intestinal mucus was purified and provided by Prof. Jeffrey Pearson, Medical School, University of Newcastle upon Tyne (United Kingdom). The BCA protein assay kit was purchased from Thermo Scientific (Waltham, MA, USA) and the QuantiChrom™ Calcium Assay Kit was purchased from BioAssay Systems (Hayward, CA, USA). All other chemicals were of reagent grade or of the best grade available and purchased from Sigma-Aldrich (Vienna, Austria).
2.2. Preparation of liposomes
Liposomes were prepared by dry film rehydration method as described previously [10]. Briefly, DPPC and DPPE-MCC were dissolved in chloroform and mixed in a molar ratio of 3:0.3, respectively. Subsequently, the organic solvent was evaporated in order to obtain a lipid film. This was dried overnight in a vacuum chamber, rehydrated with PBS buffer (10 mM phosphate, containing 150 mM NaCl, pH 7.4) to obtain a final lipid concentration of 30 mg/ml and incubated at 50 °C for 1 h with repeated vortexing. Sizing of so-formed multilamellar liposomes was performed by 6 freeze and thaw-cycles followed by extrusion through 200 nm polycarbonate membranes (Whatman Inc., Clifton, NJ) with a Mini-Extruder (Avanti Polar Lipids, Alabaster, AL). These empty liposomes were used for ex vivo permeation studies of FITC-sCT. For in vivo studies, sCT dissolved in PBS was used for the rehydration at a concentration of 0.4 mg/ml. Incubation and sizing were carried out as described for empty liposomes. Free sCT was separated from sCT loaded liposomes by size exclusion chromatography using a Sephadex G75 column (Amersham Biosciences, Uppsala, Sweden). The concentration of sCT within the liposomes was determined by a colorimetric assay (BCA protein assay kit, Thermo Scientific, Waltham, MA, USA). For in vitro mucus diffusion studies, empty liposomes were used, but with a fluorescence-labeled phospholipid incorporated within the bilayer, in a final molar ratio of 3.3:0.01 lipid to label. All other steps were carried out as described for empty, unlabeled DPPC/DPPE-MCC liposomes.
2.3. Coupling of thiolated chitosan
For thiomer-coated liposomes, two different polymers were used: CS–TGA and the S-protected form of the same polymer, CS–TGA–MNA (see Fig. 1). Both polymers were dissolved in bidistilled water in a concentration of 2 mg/ml and added to the liposomal suspension, which was previously diluted with PBS to a concentration of 10 mg/ml. A final lipid to polymer weight ratio of 1:1 was maintained throughout the study corresponding to a coupling rate of free SH-groups to maleimide groups of approximately 4:1. The mixture was incubated overnight at room temperature under agitation.
Fig. 1.

Reaction of CS–TGA with 6,6′-dinicotinamide leads to the S-protected thiomer CS–TGA–MNA. Both polymers were used for the coating of preformed liposomes.
2.4. Particle characterization
sCT loaded liposomes were characterized by measuring their size and zeta potential before and after polymer coupling. Size was measured using a Zetasizer 3000HS (Malvern Instruments, Herrenberg, Germany) after diluting the samples to a lipid concentration of about 0.03 mg/ml with bidistilled water. The polydispersity index of the liposomal suspension is given by the width of the size distribution. Zeta potential measurements were performed with a Zetasizer nano ZS (Malvern Instruments, Herrenberg, Germany) after dilution of the liposomes to a lipid concentration of 0.3 mg/ml with bidistilled water. All measurements were carried out at room temperature.
2.5. In vitro mucus diffusion studies
For mucus diffusion studies about 100 μl of porcine intestinal mucus were pipetted into plastic dishes with a cube-shaped bulge (5 × 5 × 5 mm). 16 μl of a 100 μg/ml solution of either uncoated liposomes, CS–TGA–coated liposomes, CS–TGA–MNA-coated liposomes or H2O, the latter serving as a control, was carefully applied on top of the mucus layer. Dishes were incubated at 37 °C and 30 rpm for 4 h and afterwards immediately frozen at − 20 °C in a cryotom (Microm, Walldorf, Germany). The frozen samples were embedded in Tissue Tek O.C.T T COMPOUND (VWR International, Vienna, Austria) and slices of 8 μm thickness were cut, transferred to SuperFrost® Plus glass slides (Thermo Scientific, Germany) and visualized immediately using the Olympus BX51 Basic Fluorescence Microscope (Hamburg, Germany), operating with a DP71 camera. All images were collected using an Uplanfl 10 × objective. For the visualization of the rhodamine-labeled liposomes a MWIG2 fluorescence filter with 520–550 nm excitation range and an emission band pass of 580IF was used (red fluorescence). A MWIBA2 filter with 460–490 nm excitation- and 510–550 nm emission range was used to visualize the autofluorescence at the same position of the samples (green fluorescence). Overlapping fluorescence images were generated with the Olympus cell^D software.
2.6. Ex vivo permeation of salmon calcitonin
To obtain information about the permeation ability of calcitonin through the intestinal tissue, FITC-labeled sCT was used. 200–300 g non-fasting male Sprague–Dawley rats were sacrificed, their small intestines (jejunum, ileum) removed immediately and preserved in 0.9% NaCl solution (w/v). Intestines were cut into strips of about 1.5 cm, opened longitudinally and rinsed free of luminal contents with freshly prepared medium (138 mM NaCl, 1 mM MgSO4, 5 mM KCl, 10 mM glucose and 2 mM CaCl2 buffered with 10 mM Hepes; pH 6.8). Subsequently, the tissues were mounted in Ussing-type chambers with a permeation area of 0.64 cm2. The chambers were placed in a water bath preheated to 37 °C and immediately filled with 1 ml medium in the acceptor compartment and 900 μl empty liposomes (lipid concentration: 1.7 mg/ml; polymer concentration in case of coated liposomes: 1.7 mg/ml) mixed with 100 μl of a 160 μg/ml FITC-sCT solution in the donor compartment. To determine the permeation in the absence of any formulation, a FITC-sCT solution in a final concentration of 16 μg/ml was used as control. Over an incubation period of 3 h, 100 μl aliquots were withdrawn every 30 min from the acceptor chamber and replaced by 100 μl of preheated medium. The amounts of permeated drug were analyzed by fluorescence spectroscopy at an excitation wavelength of 490 nm and an emission wavelength of 525 nm using a plate reader (Tecan Austria GmbH, Austria). Cumulative corrections were made for previously removed samples. Apparent permeability coefficients (Papp; cm/s) for FITC-sCT were calculated according to the following equation,
where Q is the amount of marker (μg) permeated within 3 h, A is the diffusion area of the Ussing-type chamber (cm2), c is the initial concentration of sCT in the donor compartment (μg/cm3) and t is the time of the experiment (s). Transport enhancement ratios (ER) were calculated by:
Transepithelial electric resistance (TEER) of the intestinal tissue was measured using an epithelial voltohmmeter (EVOM®, World Precision Instruments, Germany) connected to a pair of adjacent electrodes. Measurements were performed at the beginning of the study to guarantee the integrity of intestinal tissue, and after 1, 2 and 3 h to observe the effects of the different liposomal formulations. The TEER measured prior to each experiment was set as 100%, and all other values were calculated in relation to this.
2.7. Cytotoxicity
Caco-2 (human colorectal adenocarcinoma) cells were used for cytotoxicity testing. 2 × 104 cells/well was seeded 24 h before treatment in 96 well plates and exposed to uncoated liposomes, CS–TGA-coated liposomes and CS–TGA–MNA-coated liposomes suspended in DMEM in concentrations of 0–1000 μg/ml. Exposures were performed at 37 °C in a 95% air/5% CO2 atmosphere and two time points (4 h and 24 h) were evaluated. Cytotoxicity was studied by formazan bioreduction (CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (Promega, Vienna, Austria)), ATP content (CellTiter-Glo Luminescent Cell Viability Assay (Promega)) and membrane integrity (CytoTox-ONE™ Homogeneous Membrane Integrity Assay (Promega)) as previously described [14].
2.8. In vivo studies
The protocol for in vivo studies was approved by the Animal Ethical Committee of Vienna, Austria and adheres to the Principles of Laboratory Animal Care. In vivo studies were performed with male Sprague–Dawley rats with an average body weight of 250 g. Rats were randomly divided into 6 cohorts of three animals per group and treated with different formulations as described in Section 3.5. Rats were fasted overnight before administration and for the duration of the experiment, but had free access to water. Before dosing the animals, blood samples were taken from the tail vein to determine the reference blood calcium concentration at time point zero.
In case of an oral application, aliquots (1 ml) of different formulations, or the same volume of a sCT solution in PBS, were administered through a flexible plastic stomach tube with a round tip in order to minimize trauma. Liposomal formulations as well as the sCT solution were standardized to a sCT concentration of 40 μg/ml before being applied. Apart from an oral application, the sCT solution was administered intravenously (i.v.) into the tail vein of the rat and subcutaneously (s.c.) with a dose of 1 μg and 2 μg per rat, respectively.
Blood samples were collected via the tail vein after 2, 4, 6, 8, 12 and 24 h of administration. After separating the blood cells from plasma, the latter one was stored at − 80 °C until further analysis. Blood calcium concentrations were determined by using the QuantiChrom™ Calcium Assay Kit (BioAssay Systems, Hayward, CA, USA) according to the manufacturer's instructions.
2.9. Statistical analysis
Data are presented as mean ± S.D. Data sets were compared using Student's t-test and differences were considered significant at p < 0.05, very significant at p < 0.01, and highly significant at p < 0.001. All tests were performed using the statistical and process management software MINITAB 13.0.
3. Results and discussion
3.1. Particle characterization
Thiolated chitosan was linked to functionalized liposomes by establishing a covalent thioether bond between maleimide groups present on the head-group of DPPE-MCC and free SH-groups of the polymer. This procedure has been intensively investigated using different amounts of polymer [10]. At a molar ratio of 4:1 (free SH-groups:maleimide groups) the surface of liposomes was found to be covered with polymer, and therefore, this ratio was used in the present study for coupling CS–TGA to the liposome resulting in a 1:1 weight ratio of lipid to polymer. For CS–TGA–MNA-coated liposomes the same weight ratio was used, although the amount of free SH-groups was different, as some of the free SH-groups (380 of the originally 660 μmol SH-groups/g polymer) were protected from oxidation by coupling them to mercaptonicotinamide. The results of size and zeta potential measurements for all formulations used in the study were summarized in Table 1. Coupling of the polymer to the liposome led to an increase in size from about 175 nm to 600–700 nm, depending on the polymer used. Furthermore, the zeta potential of the particles increased from highly negative to highly positive values. The reason for the smaller increase in the potential caused by CS–TGA–MNA-coating versus CS–TGA-coating was the lower amount of free SH-groups present on the surface of liposomes. The positive zeta potential is regarded to be advantageous for particles intended for oral drug delivery, as positively-charged particles were shown to adhere to the mucus, driven by electrostatic interactions with negatively charged sialic- and sulfonic residues of different mucins [15,16]. These electrostatic interactions, together with the covalent bonds, formed between thiolated chitosans and the mucus, enhanced the mucoadhesion of coated liposomes compared to uncoated ones.
Table 1.
Composition of all liposomal suspensions used within this study and characterization concerning their size, polydispersity and zeta potential after coupling the polymer to the liposomal surface (means ± SD; n ≥ 3).
| Sample description | Lipid compositiona | Encapsulated drugb | Added polymerc | Size [nm] | Polydispersity index | Zeta potential [mV] |
|---|---|---|---|---|---|---|
| Uncoated liposomes | DPPC/DPPE-MCC | sCT | – | 174.8 ± 0.9 | 0.19 ± 0.01 | − 39.8 ± 2.4 |
| CS–TGA–coated liposomes | DPPC/DPPE-MCC | sCT | CS–TGA, 150kD | 709.2 ± 36.0 | 0.34 ± 0.05 | + 43.5 ± 1.6 |
| CS–TGA–MNA-coated liposomes | DPPC/DPPE-MCC | sCT | CS–TGA–MNA, 150 kDa | 604.8 ± 29.6 | 0.91 ± 0.16 | + 27.9 ± 1.1 |
The molar ratio of DPPC to DPPE-MCC was 3:0.3 in all cases.
Encapsulation efficiency: 69 ± 12%.
A polymer to lipid weight ratio of 1:1 was used for all formulations.
3.2. In vitro mucus diffusion studies
To study the ability of coated liposomes to penetrate the mucus, mucus diffusion studies were carried out using porcine intestinal mucus. Several studies showed that the use of native mucus gives more reliable information about the in vivo interaction of particles and mucus, than a solution of commercially available purified gastric mucin would do [17,18]. Furthermore, porcine mucus was chosen since its structure and molecular weight were very similar to humans [17,19]. After incubating the mucus layer with coated- or uncoated rhodamine-labeled liposomes, or water in case of the control, cross sections of the frozen mucus were visualized using fluorescence microscopy. From every part of the mucus, an image corresponding to the red fluorescence of the rhodamine-labeled liposomes and one corresponding to the green fluorescence of the mucus was taken. All images are shown in Fig. 2. The first three pictures show the negative control, where only water was applied to the mucus. The picture in the middle (merge) illustrates that all areas, which show red fluorescence, also show the same signal when visualizing green fluorescence, corresponding to the autofluorescence of the mucus. The second three pictures display the mucus, where uncoated liposomes, being about 175 nm in size (see Table 1), were applied. Liposomes can be seen as a diffuse, red color, being located mainly in the bottom part of the mucus. This heterogeneous distribution of small particles in a viscous environment was also found by Dawson et al. [20], who worked with 100- and 200 nm particles in cystic fibrosis sputum.
Fig. 2.

Diffusion of rhodamine-labeled liposomes through porcine intestinal mucus. Mucus layers were incubated with uncoated liposomes (B), CS–TGA-coated liposomes (C), or CS–TGA–MNA coated liposomes (D) and compared with a control omitting any particles (A). Green (I) and red fluorescence (II) images were merged in order to distinguish between particles (red) and autofluorescence (yellow). The mucus layer thickness is displayed by the two dotted lines; the right side corresponds to the mucus surface, where particles have been applied.
Generally, smaller particles show comparatively higher permeation properties through gastrointestinal mucus than larger ones [21]. Apart from size, further important factors influencing mucus diffusion properties of particles are surface charge [18] and hydrophobicity [17,22]. Several viruses, for example, evolved strategies in order to penetrate the mucus barrier like (i) being small enough not to be sterically hindered by the mucin mesh, (ii) possessing surfaces without hydrophobic moieties, and (iii) exhibiting a surface densely coated with positive and negative charges resulting in a neutral net charge [4,22]. Thus, particles permeate best if they are small, neutrally charged and do not possess hydrophobic areas on their surfaces. In this context, it was questionable, whether liposomes coated with thiolated chitosan, being quite big, positively charged and highly mucoadhesive [10], would be able to penetrate into the mucus at all. Nevertheless, images for CS–TGA- and CS–TGA–MNA-coated liposomes (Fig. 2C, D) clearly demonstrated that both are capable of penetrating the mucus. This can be explained by the fact that mucoadhesion is not a spontaneous but a time dependent process. As a consequence at least two mechanisms are taking place in parallel: On the one hand, particles penetrate the mucus based on a concentration gradient towards the epithelium, and on the other hand, over time, particles get immobilized in the mucus. Hence, some thiomer-coated liposomes will stick already to the mucus surface, but others will make it to the epithelium, where they can interact with the mucosa. Additionally, mucoadhesive particles, like the thiomer-coated liposomes investigated in this study, offer in this context the advantage of a limited back diffusion when the concentration gradient is gone. Mucus penetrating properties of large, mucoadhesive particles was also observed by others, e.g. Cone [22] or Wang et al. [23], who found that high concentrations of large, mucophilic agents may alter the microstructure of the mucus, increase its average mesh spacing and cause the formation of channels. In agreement with this hypothesis, we suggest that aggregates of coated liposomes interacted with the mucus, leading to a reorganization of the mucus mesh, where smaller aggregates or single thiomer-coated liposomes were able to intrude. Bearing in mind that the mucus layer in this study was thicker than it would be in vivo, where it is estimated to be below ~ 200 μm [4], thiomer-coated liposomes clearly possessed the ability to diffuse into the mucus thereby getting in close contact with the underlying epithelium.
3.3. Ex vivo permeation of FITC-sCT
Previous investigations showed that liposomes coated with thiolated chitosans exhibit permeation enhancing and efflux pump inhibitory properties, using the model substances fluoresceinisothiocyanate-dextran (FD4) and rhodamine-123 (Rho-123) [11]. Now, permeation experiments with FITC-sCT should clarify whether the delivery system has the potential to increase the absorption of peptide drugs. To address this question, we mixed FITC-sCT with coated and uncoated liposomes and investigated the transport through rat small intestine by monitoring the fluorescence in the basolateral compartment over a time period of 3 h. This time frame was chosen as the average transit time through the small intestine is about 3–4 h [19]. To investigate only the effect on the transport of the drug, excluding any influence due to different release kinetics, FITC-sCT was not encapsulated within liposomes, but simply mixed with different formulations before being added to the apical compartment of the chamber.
All results displaying the cumulative transport of FITC-sCT across the intestinal membrane, determined by increasing concentrations of fluorescent-labeled peptide in the basolateral compartment of the Ussing-type chamber, were illustrated in Fig. 3. The resulting Papp values and the enhancement ratios are listed in Table 2. Already in the presence of uncoated liposomes, a slightly increased permeation of FITC-sCT could be detected compared to the buffer control, reflected by an enhancement ratio of 1.8. This might be due to interactions of maleimide-functionalized phospholipid head-groups with glycoproteins of the mucus, leading to a re-organization of mucins causing a change in viscosity of the mucus and an easier penetration of the drug [24]. Nevertheless, the permeation enhancing effect of coated liposomes was much more pronounced given by values of 2.7 and 3.8 for CS–TGA-coated and CS–TGA–MNA-coated liposomes, respectively. As discussed previously, thiomers were shown to open tight junctions by reducing oxidized glutathione on the cell surface, which, in its reduced form, is able to inhibit the protein tyrosine phosphatase causing dephosphorylation (= closing) of tight junctions [25,26]. In addition to the opening of tight junctions, thiomers inhibit efflux pumps and thereby prevent drugs from being transported back to the luminal side of the tissue once they are absorbed from the gut [27,28]. As reported previously [29], S-protected thiomers, like the CS–TGA–MNA used for this study, provide better permeation enhancing- and efflux pump inhibitory properties than the corresponding, unprotected thiomer due to a higher reactivity of S-protected thiol groups. We could confirm this effect within our studies, reflected by a 1.4-fold improvement of FITC-sCT permeation due to the use of S-protected thiomers instead of conventional ones; a result that was also found when investigating the transport of FD4 across intestinal membranes [11].
Fig. 3.

Absorptive permeation studies of FITC-calcitonin across rat intestinal tissue. Effect of uncoated liposomes (△) and liposomes coated with the thiolated chitosans CS–TGA (■) and the S-protected version CS–TGA–MNA (♦) in comparison to the FITC-sCT control (×). Indicated values are the means ± SD of at least three experiments.
Table 2.
Comparison of Papp values of uncoated liposomes, CS–TGA-coated liposomes and CS–TGA–MNA-coated liposomes. Enhancement ratios result from the comparison of each test solution with the FITC-sCT control solution. Indicated values represent the means ± SD of at least three experiments (*p < 0.05 and **p < 0.01 compared to the buffer control). Additionally the effect of named test compounds was tested on the TEER.
| Substrate | Test compound | Papp × 10− 5 [cm/s] | Fold increase in Papp | Fold decrease in TEER |
|---|---|---|---|---|
| FITC-sCT | Buffer | 0.48 ± 0.02 | – | – |
| Uncoated liposomes | 0.86 ± 0.34 | 1.8 | 1.2 | |
| CS–TGA-coated liposomes | 1.29 ± 0.03 | 2.7** | 1.6* | |
| CS–TGA–MNA-coated liposomes | 1.80 ± 0.32 | 3.8* | 1.7* |
In addition to the permeation of FITC-sCT, the TEER was monitored, which gave direct information about the tissue integrity: If the TEER was too low from the start or it becomes too low during the experiment, the tissue was most probably damaged either by mounting it in the chamber or by taking samples with the pipette. In such a case, tissues were discarded immediately. In addition, monitoring the TEER during a permeation experiment provides information about opening and closing of tight junctions. In the absence of any test compounds, only a slight decrease in the TEER was measured over 3 h (see Fig. 4). The addition of uncoated liposomes, CS–TGA-coated liposomes and CS–TGA–MNA-coated liposomes resulted in a reduction of the TEER, whereby the slightest decrease was detected for uncoated liposomes and the highest decrease was measured for liposomes coated with S-protected thiolated chitosan. These results reflect and corroborate the findings from monitoring the drug permeation, where the highest permeation was found in the presence of CS–TGA–MNA-coated liposomes.
Fig. 4.

Decrease of the transepithelial electrical resistance (TEER) after adding uncoated liposomes (△) and liposomes coated with the thiolated chitosans CS–TGA (■) and its S-protected form CS–TGA–MNA (♦) in comparison to FITC-sCT control (×). Indicated values are the means ± SD of at least three experiments.
3.4. Cytotoxicity
For the assessment of cytotoxicity, the metabolic functions according to formazan bioreduction and cellular ATP content were determined. In addition, cell membrane integrity was assessed by the release of lactate dehydrogenase. In all assays, using up to 1 mg/ml of thiomer-coated liposomes, no indication for cellular damage was observed after incubation for 4 h and 24 h. Consistent with these results, cell membrane integrity, verified by the absence of lactate dehydrogenate release, was maintained. Taken together, these results demonstrate no cytotoxic effects in the concentration range tested.
3.5. In vivo studies
The main objective of the present work was to evaluate the potential of thiomer-coated liposomes as oral delivery system for peptide drugs. sCT was used as a model drug and its effect on the blood calcium level was monitored over 24 h. As a positive control and to evaluate the sensitivity of the analytical test method, salmon calcitonin was injected i.v. (1 μg/rat) and s.c. (2 μg/rat) and compared with results obtained for an oral application of free sCT (40 μg/rat). The mean initial serum calcium value (“0 h”) was taken as 100% and all the following concentrations were given as percentage of this value. As shown in Fig. 5, both, the i.v. as well as the s.c. injection of sCT, have significant effects on the blood calcium level, whereas almost no effect was detected for the orally administered sCT solution, even when a much higher dose was applied. The maximal decrease in the blood calcium level (cmin) was about 40% and 27% for i.v., and s.c. injected sCT, respectively, and was observed about 6 h after administration (tmin). This time correlates well with findings from other research groups who applied sCT to rats via these application routes [30,31]. All values for cmin, tmin and the AAC (area above the curve; marks the area between the 100%-line and the blood calcium level curve) are summarized in Table 3.
Fig. 5.

Effect of different application routes of sCT solution on the blood calcium level: The applied dose per rat was either 40 μg in case of an oral application (×), 2 μg in case of a s.c. application (□) or 1 μg for an i.v. injection (○). Indicated values are the means ± SD of three rats.
Table 3.
List of all formulations used for the in vivo study and summary of the results. Each group consists of three rats and all formulations were applied in liquid form. Blood samples were taken from the tail vain after 0, 2, 4, 6, 8, 12 and 24 h. Cmin, minimum serum calcium concentration (% of initial); tmin, time to cmin; AAC, area above the blood calcium level time curves; ER, enhancement ratio of different oral formulations (*p < 0.05, **p < 0.01 and ***p < 0.001 compared to the sCT solution in PBS).
| Groups | Application route | Dose [μg/rat] | cmin [%] | tmin [h] | AAC0–24h [% * h] | ER |
|---|---|---|---|---|---|---|
| sCT in PBS | i.v. | 1 | 60 ± 3 | 6 | 334 ± 46 | – |
| sCT in PBS | s.c. | 2 | 73 ± 4 | 6 | 296 ± 2 | – |
| sCT in PBS | Oral | 40 | 93 ± 7 | 6 | 29 ± 6 | – |
| sCT encapsulated liposomes, uncoated | Oral | 40 | 85 ± 2 | 6 | 121 ± 17 | 4.2* |
| sCT encapsulated liposomes, CS–TGA coated | Oral | 40 | 82 ± 4 | 4 | 164 ± 28 | 5.7* |
| sCT encapsulated liposomes, CS–TGA–MNA coated | Oral | 40 | 65 ± 4 | 6 | 239 ± 12 | 8.2*** |
In order to enhance the oral bioavailability of sCT, it was encapsulated within liposomes which were applied either with or without thiomer coating. The effects on the blood calcium level were shown in Fig. 6 and Table 3. Comparing the AAC among different formulations clearly demonstrates that the best result was obtained for liposomes coated with CS–TGA–MNA, leading to an enhancement ratio of 8.2, relative to the same amount of free sCT administered orally. Uncoated liposomes as well as liposomes coated with conventional thiomers also led to a considerable, but smaller increase in the AAC. In another study where drug loaded thiomer nanoparticles were orally administered to rats, the best results were also obtained for nanoparticles consisting of S-protected thiomers [32]. These and our results highlight once again the superiority of S-protected thiomers over conventional thiomers, which is caused by the higher reactivity of S-protected thiomers as discussed above.
Fig. 6.

Decrease in the blood calcium level as a biological response to the oral application of sCT encapsulated within different liposomal formulations or given as a solution. sCT containing liposomes were coated with either CS–TGA (■) or CS–TGA–MNA (♦) and compared to uncoated, sCT loaded liposomes (△) and free sCT in solution (×). The applied dose per rat was 40 μg sCT in all cases. Indicated values are the means ± SD of three rats.
Even though it is difficult to compare the results of different research groups due to differences in the calcitonin dose, the dosage form and/or the detection method, we aimed to assess the potential of our best formulation with respect to the outcomes of other studies [30,33,34]. Since the values for the AAC were not available, the assessment of our formulation was based on a comparison of the cmin values given. This reveals that a calcium reduction of 65%, as achieved with CS–TGA–MNA-coated liposomes, demonstrates an excellent result and confirms the high potential of our leading formulation.
A general question that arises when working with mucoadhesive formulations like thiolated polymers is whether they would not stick to the mucus within the stomach. To answer this question, we refer to a study by Takeuchi et al., who designed liposomal formulations for the oral administration of eel calcitonin based on mucoadhesive chitosan-coated liposomes [35]. Within their work, they reported that the remaining percentage of chitosan-coated liposomes in the stomach 1 h after administration was less than 10%, which should also be true for the formulations used herein. A reason for this observation might be found in the mucus turnover time, which is very short in the stomach, and so limits the diffusion of even very small mucus penetrating particles [4,22]. Concluding from this study and our ex vivo- and in vivo results, we propose the following scenario to happen in the body: Thiomer-coated liposomes are stable in the gastric environment [11] and therefore able to keep encapsulated sCT inside the core until reaching the small intestine. When the intestinal mucus is reached, covalent bonds are formed between SH-groups of the thiomers and mucus glycoproteins, which lead to a prolonged residence time of the delivery system at the site of drug absorption [10]. Our preceding study already showed that encapsulated compounds are released within the small intestine, triggered by simulated intestinal fluid [11]. By opening tight junctions and inhibiting efflux pumps, thiomer-coated liposomes, which are still present within the mucus, promote the permeation of released calcitonin through the gut wall and into the bloodstream, thus enhancing its oral bioavailability.
4. Conclusion
Within the present study, an oral delivery system based on the coating of liposomes with S-protected- and conventional thiolated chitosan was evaluated. Coated liposomes were demonstrated to effectively penetrate into the intestinal mucus layer where they come in close contact with the underlying epithelium, and can act as permeation enhancers. Indeed, our ex vivo studies showed that the addition of thiomer-coated liposomes to a solution of salmon calcitonin leads to an increased permeation of the peptide drug through rat small intestine. The results from the in vivo study, where different formulations were applied orally to rats, revealed a remarkable reduction of the blood calcium level by about 35% for liposomes coated with S-protected thiomers. In summary, our data provide a proof of concept for the use of thiomer-coated liposomes to increase the bioavailability of orally administered calcitonin, which is likely to be expandable to many other peptides as well.
Acknowledgment
This work was supported by the Austrian Science Fund (FWF project no. P 23515-B11 to A.B.) and the Austrian Nano-Initiative, who co-financed this work as part of the Nano-Health project (project no: 819721 to R.P.), financed by the Austrian Research Promotion Agency (FFG). The authors would like to thank Professor Jeffrey Pearson from the Medical School, University of Newcastle upon Tyne (United Kingdom), for providing the isolated porcine mucus and Miss Grace Hatton for carefully reading the manuscript.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
K. Gradauer, Email: kerstin1329@gmx.at.
J. Barthelmes, Email: jan.barthelmes@uibk.ac.at.
C. Vonach, Email: caroline.vonach@medunigraz.at.
G. Almer, Email: gunter.almer@medunigraz.at.
H. Mangge, Email: harald.mangge@medunigraz.at.
B. Teubl, Email: teubl_b@yahoo.com.
E. Roblegg, Email: eva.roblegg@uni-graz.at.
S. Dünnhaupt, Email: Sarah.Duennhaupt@uibk.ac.at.
E. Fröhlich, Email: eleonore.froehlich@klinikum-graz.at.
A. Bernkop-Schnürch, Email: andreas.bernkop@uibk.ac.at.
R. Prassl, Email: ruth.prassl@medunigraz.at.
References
- 1.IMARC . IMARC; 2013. Global Biopharmaceutical Market Report (2010–2015) [Google Scholar]
- 2.Fricker G., Kromp T., Wendel A., Blume A., Zirkel J., Rebmann H., Setzer C., Quinkert R.O., Martin F., Muller-Goymann C. Phospholipids and lipid-based formulations in oral drug delivery. Pharm. Res. 2010;27:1469–1486. doi: 10.1007/s11095-010-0130-x. [DOI] [PubMed] [Google Scholar]
- 3.Shukla D., Chakraborty S., Singh S., Mishra B. Lipid-based oral multiparticulate formulations — advantages, technological advances and industrial applications. Expert Opin. Drug Deliv. 2011;8:207–224. doi: 10.1517/17425247.2011.547469. [DOI] [PubMed] [Google Scholar]
- 4.Ensign L.M., Cone R., Hanes J. Oral drug delivery with polymeric nanoparticles: the gastrointestinal mucus barriers. Adv. Drug Deliv. Rev. 2012;64:557–570. doi: 10.1016/j.addr.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elsabahy M., Wooley K.L. Design of polymeric nanoparticles for biomedical delivery applications. Chem. Soc. Rev. 2012;41:2545–2561. doi: 10.1039/c2cs15327k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aungst B.J. Intestinal permeation enhancers. J. Pharm. Sci. 2000;89:429–442. doi: 10.1002/(SICI)1520-6017(200004)89:4<429::AID-JPS1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 7.Aungst B.J. Absorption enhancers: applications and advances. AAPS J. 2012;14:10–18. doi: 10.1208/s12248-011-9307-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park K., Kwon I.C., Park K. Oral protein delivery: current status and future prospect. React. Funct. Polym. 2011;71:280–287. [Google Scholar]
- 9.Brayden D.J., Mrsny R.J. Oral peptide delivery: prioritizing the leading technologies. Ther. Deliv. 2011;2:1567–1573. doi: 10.4155/tde.11.114. [DOI] [PubMed] [Google Scholar]
- 10.Gradauer K., Vonach C., Leitinger G., Kolb D., Frohlich E., Roblegg E., Bernkop-Schnurch A., Prassl R. Chemical coupling of thiolated chitosan to preformed liposomes improves mucoadhesive properties. Int. J. Nanomedicine. 2012;7:2523–2534. doi: 10.2147/IJN.S29980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gradauer K., Dunnhaupt S., Vonach C., Szollosi H., Pali-Scholl I., Mangge H., Jensen-Jarolim E., Bernkop-Schnurch A., Prassl R. Thiomer-coated liposomes harbor permeation enhancing and efflux pump inhibitory properties. J. Control. Release. 2013;165:207–215. doi: 10.1016/j.jconrel.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Azria M., Copp D.H., Zanelli J.M. 25 years of salmon calcitonin: from synthesis to therapeutic use. Calcif. Tissue Int. 1995;57:405–408. doi: 10.1007/BF00301940. [DOI] [PubMed] [Google Scholar]
- 13.Chesnut C.H., III, Azria M., Silverman S., Engelhardt M., Olson M., Mindeholm L. Salmon calcitonin: a review of current and future therapeutic indications. Osteoporos. Int. 2008;19:479–491. doi: 10.1007/s00198-007-0490-1. [DOI] [PubMed] [Google Scholar]
- 14.Frohlich E., Samberger C., Kueznik T., Absenger M., Roblegg E., Zimmer A., Pieber T.R. Cytotoxicity of nanoparticles independent from oxidative stress. J. Toxicol. Sci. 2009;34:363–375. doi: 10.2131/jts.34.363. [DOI] [PubMed] [Google Scholar]
- 15.Hassan E.E., Gallo J.M. A simple rheological method for the in vitro assessment of mucin-polymer bioadhesive bond strength. Pharm. Res. 1990;7:491–495. doi: 10.1023/a:1015812615635. [DOI] [PubMed] [Google Scholar]
- 16.Khutoryanskiy V.V. Advances in mucoadhesion and mucoadhesive polymers. Macromol. Biosci. 2011;11:748–764. doi: 10.1002/mabi.201000388. [DOI] [PubMed] [Google Scholar]
- 17.Larhed A.W., Artursson P., Grasjo J., Bjork E. Diffusion of drugs in native and purified gastrointestinal mucus. J. Pharm. Sci. 1997;86:660–665. doi: 10.1021/js960503w. [DOI] [PubMed] [Google Scholar]
- 18.Crater J.S., Carrier R.L. Barrier properties of gastrointestinal mucus to nanoparticle transport. Macromol. Biosci. 2010;10:1473–1483. doi: 10.1002/mabi.201000137. [DOI] [PubMed] [Google Scholar]
- 19.Kararli T.T. Comparison of the gastrointestinal anatomy, physiology, and biochemistry of humans and commonly used laboratory animals. Biopharm. Drug Dispos. 1995;16:351–380. doi: 10.1002/bdd.2510160502. [DOI] [PubMed] [Google Scholar]
- 20.Dawson M., Wirtz D., Hanes J. Enhanced viscoelasticity of human cystic fibrotic sputum correlates with increasing microheterogeneity in particle transport. J. Biol. Chem. 2003;278:50393–50401. doi: 10.1074/jbc.M309026200. [DOI] [PubMed] [Google Scholar]
- 21.Aljayyoussi G., Abdulkarim M., Griffiths P., Gumbleton M. Pharmaceutical nanoparticles and the mucin biopolymer barrier. Bioimpacts. 2012;2:173–174. doi: 10.5681/bi.2012.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cone R.A. Barrier properties of mucus. Adv. Drug Deliv. Rev. 2009;61:75–85. doi: 10.1016/j.addr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y.Y., Lai S.K., So C., Schneider C., Cone R., Hanes J. Mucoadhesive nanoparticles may disrupt the protective human mucus barrier by altering its microstructure. PLoS One. 2011;6:e21547. doi: 10.1371/journal.pone.0021547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai S.K., Wang Y.Y., Wirtz D., Hanes J. Micro- and macrorheology of mucus. Adv. Drug Deliv. Rev. 2009;61:86–100. doi: 10.1016/j.addr.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clausen A.E., Bernkop-Schnurch A. Thiolated carboxymethylcellulose: in vitro evaluation of its permeation enhancing effect on peptide drugs. Eur. J. Pharm. Biopharm. 2001;51:25–32. doi: 10.1016/s0939-6411(00)00130-2. [DOI] [PubMed] [Google Scholar]
- 26.Clausen A.E., Kast C.E., Bernkop-Schnurch A. The role of glutathione in the permeation enhancing effect of thiolated polymers. Pharm. Res. 2002;19:602–608. doi: 10.1023/a:1015345827091. [DOI] [PubMed] [Google Scholar]
- 27.Werle M., Hoffer M. Glutathione and thiolated chitosan inhibit multidrug resistance P-glycoprotein activity in excised small intestine. J. Control. Release. 2006;111:41–46. doi: 10.1016/j.jconrel.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 28.Foger F., Kafedjiiski K., Hoyer H., Loretz B., Bernkop-Schnurch A. Enhanced transport of P-glycoprotein substrate saquinavir in presence of thiolated chitosan. J. Drug Target. 2007;15:132–139. doi: 10.1080/10611860601140798. [DOI] [PubMed] [Google Scholar]
- 29.Dunnhaupt S., Barthelmes J., Rahmat D., Leithner K., Thurner C.C., Friedl H., Bernkop-Schnurch A. S-protected thiolated chitosan for oral delivery of hydrophilic macromolecules: evaluation of permeation enhancing and efflux pump inhibitory properties. Mol. Pharm. 2012;9:1331–1341. doi: 10.1021/mp200598j. [DOI] [PubMed] [Google Scholar]
- 30.Guggi D., Kast C.E., Bernkop-Schnurch A. In vivo evaluation of an oral salmon calcitonin-delivery system based on a thiolated chitosan carrier matrix. Pharm. Res. 2003;20:1989–1994. doi: 10.1023/b:pham.0000008047.82334.7d. [DOI] [PubMed] [Google Scholar]
- 31.Cheng W., Satyanarayanajois S., Lim L.Y. Aqueous-soluble, non-reversible lipid conjugate of salmon calcitonin: synthesis, characterization and in vivo activity. Pharm. Res. 2007;24:99–110. doi: 10.1007/s11095-006-9128-9. [DOI] [PubMed] [Google Scholar]
- 32.Dunnhaupt S., Barthelmes J., Iqbal J., Perera G., Thurner C.C., Friedl H., Bernkop-Schnurch A. In vivo evaluation of an oral drug delivery system for peptides based on S-protected thiolated chitosan. J. Control. Release. 2012;160:477–485. doi: 10.1016/j.jconrel.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 33.Werle M., Makhlof A., Takeuchi H. Carbopol–lectin conjugate coated liposomes for oral peptide delivery. Chem. Pharm. Bull. (Tokyo) 2010;58:432–434. doi: 10.1248/cpb.58.432. [DOI] [PubMed] [Google Scholar]
- 34.Cetin M., Youn Y.S., Capan Y., Lee K.C. Preparation and characterization of salmon calcitonin–biotin conjugates. AAPS PharmSciTech. 2008;9:1191–1197. doi: 10.1208/s12249-008-9165-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeuchi H., Matsui Y., Yamamoto H., Kawashima Y. Mucoadhesive properties of carbopol or chitosan-coated liposomes and their effectiveness in the oral administration of calcitonin to rats. J. Control. Release. 2003;86:235–242. doi: 10.1016/s0168-3659(02)00411-x. [DOI] [PubMed] [Google Scholar]