

Abstract
Primary open-angle glaucoma (POAG) is a leading cause of irreversible and preventable blindness and ocular hypertension is the strongest known risk factor. With current classes of drugs, management of the disease focuses on lowering intraocular pressure (IOP). Despite of their use to modify the course of the disease, none of the current medications for POAG is able to reduce the IOP by more than 25%–30%. Also, some glaucoma patients show disease progression despite of the therapeutics. This paper examines the new described physiological targets for reducing the IOP. The main cause of elevated IOP in POAG is thought to be an increased outflow resistance via the pressure-dependent trabecular outflow system, so there is a crescent interest in increasing trabecular meshwork outflow by extracellular matrix remodeling and/or by modulation of contractility/TM cytoskeleton disruption. Modulation of new agents that act mainly on trabecular meshwork outflow may be the future hypotensive treatment for glaucoma patients. There are also other agents in which modulation may decrease aqueous humour production or increase uveoscleral outflow by different mechanisms from those drugs available for glaucoma treatment. Recently, a role for the ghrelin-GHSR system in the pathophysiology modulation of the anterior segment, particularly regarding glaucoma, has been proposed.
1. Introduction
Glaucoma is a progressive optic neuropathy caused by death of the retinal ganglion cells (RGCs) and is the leading cause of irreversible blindness worldwide. The mechanism by which this progressive RGC death occurs is not fully understood. It is clear that multiple causes may give rise to the common effect of ganglion cell death.
Clinically, it is well accepted that the major risk factor for glaucoma is elevated intraocular pressure (IOP) [1, 2]. In open angle glaucoma (OAG), elevated IOP occurs from an imbalance between production and outflow of aqueous humor (AH).
The mechanical theory argues the importance of direct compression of the axonal fibers and support structures of the anterior optic nerve by elevated IOP resulting in the death of the RGCs. Lowering the IOP (baroprotection) remains the only current therapeutic approach for preserving visual function in glaucoma patients. The six classes of drugs available for glaucoma treatment (miotics, beta-blockers, alfa-agonists, epinephrine derivatives, carbonic anhydrase inhibitors, and prostaglandin analogues) act by decreasing aqueous humor production and/or by improving trabecular meshwork-Schlemm's canal or uveoscleral outflow.
Better knowledge about cellular and molecular glaucomatous changes in the aqueous production and outflow pathways opened a new horizon for new hypotensive class agents. The main cause of elevated IOP in primary open angle glaucoma (POAG) is thought to be an increased outflow resistance via the pressure-dependent trabecular outflow system by an increased accumulation of extracellular matrix (ECM) material in the trabecular meshwork, due to a disturbed balance between ECM deposition and degradation.
From all of the glaucoma medications, approved for clinical use, that decrease the intraocular pressure, only analogues prostaglandin (PG) may have a role on modulation of the molecular changes that occurred in glaucoma patients. Some studies showed that PG's analogues may induce stimulation of collagenases and other matrix metalloproteinases [3, 4] which is thought to result in dilated spaces between ciliary muscle bundles. However, other studies, using both light and electron microscopy, have found no evidence of dilated spaces between ciliary muscle bundles or other alterations in the ciliary muscle or other ocular tissues in monkeys treated with PGF2a [5].
There are 2 main new therapeutic approaches to increase outflow facility in TM. The first one includes the alteration in activities or behavior of TM cells [6]. Some agents affect cell volume and shape and loosen cell-to-cell junction and/or cell-to-extracellular matrix adhesion within the TM and inner wall of Schlemm's canal. Considerable evidence has shown that TM cells are highly contractile and play an active role in aqueous humor dynamics. Modulation of contractility of TM represents the second possible therapeutic concept within the TM. It has been shown that TM tissues possess smooth muscle cell-like properties. The contractile and relaxation properties of TM cells are regulated by several enzymes, which have become experimental therapeutic targets for lowering IOP [7–12].
There are also new therapeutic approaches to decrease aqueous humour production or to improve increased uveoscleral outflow by different subcellular pathways from those that already exist [13–16].
Moreover, a significant number of patients presenting with glaucoma continue to lose vision despite responding well to therapies that lower eye pressure. “Non-IOP-dependent” risk factors appear to be responsible for approximately 30–70% of glaucoma cases [17–20]. Enhancement of optic nerve blood supply and neuroprotection are potential treatment strategies for glaucoma, but are beyond the scope of this paper.
In this paper, we discuss potential concepts for the future treatment of OAG based on hypotensive effect by mechanisms of action that are different from current therapeutic approach that is available for clinical use (see Figure 1 and Table 1).
Figure 1.
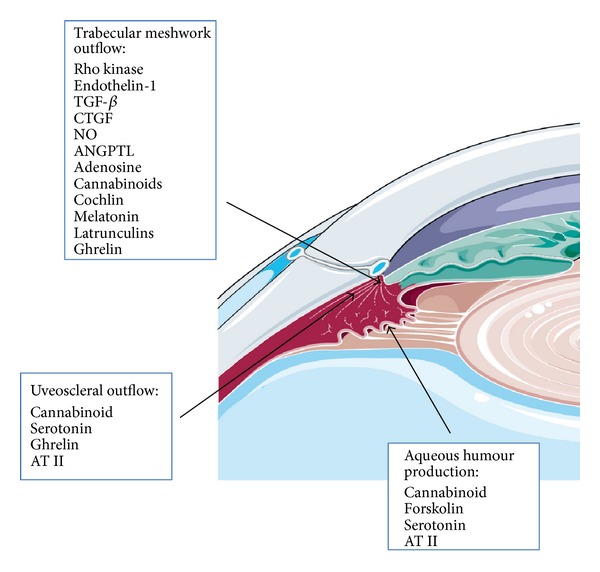
New therapeutic targets that lower intraocular pressure. Mechanisms of action are indicated in the figure.
Table 1.
New therapeutics targets for IOP lowering and possible mechanisms of action.
| Pathways | Mechanisms of action |
|---|---|
| Rho kinase | Modulation may increase TM outflow by modulation of contractility/TM cytoskeleton disruption |
| Endothelin-1 | Modulation by may increase TM outflow by modulation of contractility/TM cytoskeleton disruption |
| Transforming growth factor-β | Modulation may increase TM outflow by remodeling extracellular matrix and/or by modulation of contractility/TM cytoskeleton disruption. |
| Connective Tissue Growth Factor | Modulation may increase TM outflow by remodeling extracellular matrix |
| Nitric Oxide | Modulation by may increase TM outflow by modulation of contractility TM cells |
| Angiopoietin-like molecules | Modulation may increase TM outflow by remodeling extracellular matrix |
| Adenosine | Modulation may increase TM outflow by remodeling extracellular matrix |
| Latrunculins | Modulation may increase TM outflow by modulation of contractility/TM cytoskeleton disruption |
| Cochlin | Modulation may increase TM outflow by potential mechanosensing mechanism |
| Cannabinoids | Modulation may decrease AH production, and/or may increase TM outflow (by increase in the dimensions of Schlemm's canal and/or by remodeling extracellular matrix), and/or may increase uveoscleral outflow |
| Melatonin | Modulation may increase TM outflow trough cholinergic and noradrenergic systems |
| Ghrelin | Modulation may increase TM outflow and/or uveoscleral outflow. |
| Angiotensin II | Modulation may increase uveoscleral outflow and/or decrease AH production |
| Serotonin | Modulation may increase uveoscleral outflow and decrease AH production |
| Forskolin | Modulation may decrease AH production |
2. Materials and Methods
The focus of this paper is new therapeutics targets for IOP lowering. Excluded compounds are those with no proved intraocular hypotensive effect and those that act by same mechanism of current therapeutic approach. A Systematic revision of studies published between 2000 and 2013 in English, Spanish and Portuguese in MEDLINE, EMBASE, and Scopus was done. Search words used included glaucoma therapy, intraocular pressure, trabecular meshwork, baroprotection, novel drugs, and open angle glaucoma.
3. Results and Discussion
3.1. Rho Kinase
Originally identified as a downstream effector of the small GTPase Rho [21], Rho-associated kinase, also known as ROCK, belongs to the AGC (PKA/PKG/PKC) family of serine/threonine kinases [22].
There are two Rho-kinase isoforms, ubiquitously expressed as Rho-kinase α/ROCK2/ROKα and Rho-kinase β/ROCK1/ROKβ/p160ROCK, collectively referred to as Rho-kinase. They are highly homologous in their molecular structure (65% of the same amino acid sequence and 92% homology in kinase domains) and also in their function. Knockout null homozygous mice developed a similar phenotype between the two isoforms (eyes open at birth and omphalocele), demonstrating that the biological functions of both isoforms are redundant and cannot be separated. Also, they both act on the same major downstream substrates [22]. The Rho signaling pathway, mediated through ROCK, plays a major role in regulation of smooth muscle contraction, through the regulation of the actin-myosin filament bundles [16, 23]. Activated RhoA/ROCK leads to the phosphorylation of myosin light chain (MLC) [24], in which combined with actin induces a calcium-independent smooth muscle contraction, with formation of actin stress fibers and focal adhesions, allowing cytoskeletal rearrangement, cell motility, and proliferation [25].
Expression of RhoA GTPase, ROCKs (both ROCK 1 and ROCK2), and MLC in TM and ciliary muscle (CM) cells from humans and other species has been confirmed [16]. Physiological agonists that activate Rho kinase signaling pathway, such as endothelin-1, transforming growth factor (TGF), thrombin, and lysophospholipids, reduce aqueous humor outflow in animals [26]. Inhibition of ROCK was demonstrated to contribute to effects such as smooth muscle relaxation and alteration of intercellular junctions in the TM of the eye [27], by decreasing MLC phosphorylation [28]. Inhibitors of ROCK and Rho GTPase lower IOP in both animal models [3] and humans; a phase II clinical trial showed that K-115 has the ability to lower intraocular pressure in humans with POAG or OH [29].
Apart from this, Rho GTPase and ROCK inhibitors have potential neuroprotective effects by increasing ocular blood flow and enhancing survival of retinal ganglionary cells and axon regeneration in animal models [30]. The first Rho-kinase inhibitor was Y-27632. It is not specific for ROCK (neither for any of its isoforms) since it affects other protein kinases in higher concentrations, such as PKA and PKC. Fasudil (HA-1077) is also not specific and is the only clinically approved ROCK inhibitor so far, being used for cerebral vasospasm after subarachnoid hemorrhage in Japan [21, 22].
During the analysis of ROCK inhibitors as a potential glaucoma topical pharmacological treatment some problems arose. First of all, the lack of specificity of the inhibitors which in high concentrations also inhibit other kinases. Also, little tolerability was described, since ROCK inhibitors result in vasodilation, resulting in ocular hyperemia, which is transient, but nonetheless the main ocular side effect described so far. Regarding these last two problems, one possible solution for many authors is the combination with other IOP lowering drugs, allowing for the use of low ROCK inhibitor concentrations. Another possible solution might be the use of a prodrug, converted into a more active compound once it passes through the cornea and is present in the anterior chamber (ATS907 is an example and is currently in phase II clinical trials) [21, 22].
In conclusion, ROCK inhibitors have the ability to lower IOP in patients with POAG and/or ocular hypertension (OH) by increasing the outflow through the trabecular meshwork.
3.2. Endothelin-1
Endothelin-1 (ET-1) is a vasoconstrictive peptide that originates in the endothelium [31, 32]. Elevated concentration of endothelin-1 has been documented in the aqueous humor of patients with glaucoma. A highly significant correlation has been found between IOP and ET-1. However, there is yet to be proven a causative relation [33].
Previous studies have stated that ET-1 is capable of inducing the contraction of both trabecular meshwork cells and the cellular matrix [31, 32]. The contraction seems to be triggered by activation of the RhoA/ROCK pathway [24] which leads to increase in the outflow resistance and consequently to an IOP elevation [34, 35]. An ETA receptor antagonist would be ideal to lower IOP. Some experiments, however, suggest a correlation between ET-1 and NO: ET-1 is capable to activate NOS isoform III [34] which will induce TM relaxation if high levels of NO are present [36].
ETA receptor antagonists constitute a potential new treatment modality to manage glaucoma through IOP reduction. ETB receptor antagonists also seem to play a neuroprotective role by impeding RGCs apoptosis.
3.3. Transforming Growth Factor-β
The transforming growth factor-β (TGF-β) superfamily is a group of structurally related multifunctional regulatory proteins. There are at least three isoforms: TGF-β1, TGF-β2, and TGF-β3 [37]. In normal physiology, cell processes influenced by TGF-β are proliferation, recognition, differentiation, and apoptosis [38]. TGF-β was found to be elevated in the AH of normal [39] as well as glaucomatous human eyes, but in higher levels in the latter [40], more specifically, higher levels of TGF-β2 in POAG, being the other isoforms elevated in other forms of glaucoma [38]. Additionally, studies showed that TGF-β2 is produced mainly in the eye, with minimum serum concentration in patients with high ocular levels and no correlation between AH and serum levels [41]. Several studies proved TGF-β to be a cause of elevated IOP. A TGF-β2 infusion in an animal's anterior eye segment perfusion culture significantly increased IOP [38]. Additionally, Shepard et al. [42] observed an increased IOP and reduced AH outflow in rodents following adenoviral gene transfer of active human TGF-β2. We can then assume that elevated levels of TGF-β2 in the AH lead to an increased risk of developing elevated IOP, and consequently placing the patients at risk for optic nerve damage and vision loss.
Trivedi et al. postulate that the increased risk for developing glaucoma with increasing age might be related to the rise of TGF-β2 [43].
As for the changes that take place in the TM, TGF-β2 leads to a decrease in its cellularity, by inhibition of proliferation, stimulation of apoptosis, and phagocytosis. At the same time, it changes the number of cells, also cell phenotype is affected, shifting to a secretory type, leading to an increase in the collagen content of the ECM, which may lead to TM obstruction and decrease of AH outflow [44].
TGF-β1 and TGF-β2 not only have shown to increase ECM production, also inhibit its degradation; both isoforms β1 and β2 lead to overexpression of genes encoding ECM proteins hence increasing its production [38]. Gottanka et al. showed how in vitro treatment with TGF-β2 led to diminished outflow facility, thanks to the accumulation of fibrillar matrix materials under the inner wall of the canal of Schlemm, and also by diminished length of the canal [39]. Apart from acting directly, TGF-β2 isomer also induces expression of other effectors, such as connective tissue growth factor, thrombospondin-1, fibronectin, cochlin [45], collagen types IV and VI, and plasminogen activator inhibitor-1 (PAI-1), both in cultured human and animal TM cells [38]. PAI-1 increased levels result in decreased activity of MMPs, since it acts as a potent inhibitor of the plasminogen/plasmin system required to activate MMPs. This is another pathway through which TGF-β2 increases ECM in the TM of glaucomatous eyes [46].
Thrombospondin-1 is expressed intensely throughout the TM in glaucomatous eyes; it acts as a potent activator of TGF-β and also has its levels increased by it, making it a self-amplifying mechanism for TGF-β2 [47].
Several molecules exist to counteract or terminate TGF-β's effects in the eye. Bone morphogenetic proteins 4 and 7 (BMP-4 and BMP-7, resp.), members of the TGF-β superfamily, prevent ECM deposition and antagonize the fibrogenic actions of TGF-β2 on human TM cells, respectively [38]. In addition to this, there are molecules that bind to BMPs and inhibit them hence releasing TGF-β's effects. Some of these BMP-binding molecules, such as follistatin, gremlin, and chordin, are expressed in TM cells, and higher levels of gremlin have been found in TM cells from POAG eyes compared with nonglaucomatous eyes [45]. Treatment of TM cells with recombinant gremlin induces expression of typical TGF-β target genes, such as those for fibronectin, collagen type I, elastin, or PAI-1 [48]. Although TGF-β1 is not the main isoform expressed in POAG, we will devote the next paragraphs to its function. The transfer of active TGF-β1 gene into rat eyes led to anatomical changes in the anterior segment that resulted in increased IOP [38]. TGF-β1 is also proven to induce a myofibroblast-like phenotype in human TM cells in vitro hence leading to an increase in contractility of TM cells and decrease in outflow facility [49]. Formation of actin stress fibres in TM cells triggered by TGF-β1 is mediated by protein kinase C and Rho GTPase [50]. Both TGF-β1 and TGF-β2 increase fibronectin, but also tissue transglutaminase, in turn leads to an irreversible cross-linking of fibronectin [51]. In vitro, fibronectin enhances TM cell-mediated contraction, thus, facilitating the connection of TM cells to the surrounding matrix and the formation of stress fibres [38]. Also, the lysyl oxidase family of proteins (LOX, LOXL 1–4) has the ability to cross-link ECM proteins turning them insoluble and stiffening the ECM, and their expression is induced by treatment with all three isoforms of TGF-β [45]. There is still no report of dysregulated LOX expression in POAG. Moreover, TGF-β1 increases connective tissue growth factor and elastin production in human TM cells in vitro, which could contribute to outflow resistance [39]. Also TM cell myocilin was increased following TGF-β1 exposure in vitro [52]. Myocilin also contributes to TM outflow resistance and elevation of IOP by interacting with the TM. Additionally, accumulation of myocilin in the TM occurs in glaucomatous eyes [53].
As shown above, TGF-β plays a central role in the activity of several of the physiological pathways leading to elevated IOP in glaucomatous eyes. Evidence supports the fact that the main target of TGF-β in POAG involves the TM. TGF-β1 and TGF-β2 have been identified as potential modulators of aqueous outflow facility through ECM remodeling and also TM cellularity and contraction, making these proteins a possible target for glaucoma treatment [38].
3.4. Connective Tissue Growth Factor
Connective tissue growth factor (CTGF) is a four-module protein that performs as a signal mediator in pathways activated by TGF-β which on the other hand increases CTGF levels [54, 55]. Different types of glaucoma express different types of TGF in the aqueous humor: TGF-β2 is highest in the aqueous humor of POAG patients [41, 56] while patients with PXG present increased expression of TGF-β1 [49]. Previous studies have documented an evident effect on ECM production on cultured Tenon's capsule of PXG patients with elevated TGF-β1, while cultures from POAG patients, with high TGF-β2, experienced cell migration and collagen contraction [57]. As far as CTGF is concerned, its levels were higher in the aqueous humor of PXG patients and the magnitude of elevation seemed to be related to the severity of the disease [58].
Studying the mechanism through which CTGF contributes to the development and progression of PXG has proven to be difficult mostly due to the ability of this protein to bind receptors that activate divergent signaling pathways such as p42/44 MAPK [59], PI3J [60], RhoGTPase [61], and p38 MAPK [62]. These pathways are linked and authors have previously suggested the importance of these links in understanding the CTGF-controlled gene transcription [63]. In POAG, outflow resistance at the juxtacanalicular regions causes aqueous humor outflow resistance and increases IOP [64, 65]. The quantity and quality of TM extracellular matrix have been suggested as two of the contributing factors as POAG demonstrates increased ECM at the juxtacanalicular region outflow [66, 67]. Increased expression of CTGF induces ECM protein expression especially fibrillar and basement membrane collagens [68]. In fact, CTGF appears to directly correlate with accumulation and deposition of collagen I [69]. Browne et al. showed that CTGF increased expression of fibrillin-1 [63], corroborating previous studies that had elucidated the role of high levels of fibrillin or abnormal aggregation of fibrillin-containing microfibrils in the pathogenesis of PX syndrome [70]. In fact, in PX syndrome, there are higher levels of fibrillin containing fibrils in the extracellular matrix and fibrillin is one of the components present in the elastic fibrils in PX material.
Some studies have emphasized the role of TGF-β2 in enhancing expression and deposition of ECM molecules such as fibronectin and collagens IV and VI, which are all components of the fibrillar ECM in the JCT [51, 71, 72]. Furthermore, these experiments also revealed decreased activity of matrix metalloproteinases (MMPs), which normally degrade ECM compounds [46]. Recent study demonstrated high IOP and optic nerve damage in mice eyes in response to overexpressed CTGF [73]. This action was counteracted by Rho kinase inhibitors. The authors also confirmed that CTGF induces fibronectin as well as actin stress fibers and contractility in trabecular meshwork cells, consequently elevating IOP. Iyer and colleagues corroborated these findings by stating that Rho GTPase and Rho kinase regulate CTGF expression in TM cells, possibly through controlling actomyosin-based contraction and the levels of free G-actin [74]. Also, the changes in the actomyosin organization and myosin II activity induced by CTGF were associated with higher levels of fibronectin and laminin in TM cells. Neuromedin U is a neuropeptide which is induced by CTGF in TM cells. It facilitates AH outflow through activating actomyosin contraction of TM cells [74].
These data suggest the existence of an interaction between Rho/Rho kinase pathway activity, CTGF expression, and the contractile properties of TM cells. These interactions regulate the homeostasis of ECM synthesis and AH drainage through the trabecular pathway. Interfering with CTGF expression will therefore have effects on the ECM synthesis and AH outflow and might prove beneficial in the treatment of glaucoma.
3.5. Nitric Oxide
The maxi-K channel is a calcium-dependent potassium channel with a dense distribution in the trabecular meshwork [75, 76]. Its function is to regulate smooth muscle tone and, in fact, it acts as target for several mediating factors whose ultimate goal is smooth muscle relaxation [36].
NO is produced from L-arginine by NOS in the trabecular meshwork and cellular matrix [77]. It binds the maxi-K channel and triggers increased expression of cGMP [36, 78]. The cGMP-dependent kinase is then activated and phosphorylates the channel, prolonging the amount of time it remains open [79]. As a consequence, the efflux of potassium is greater and culminates in repolarization. At the same time, voltage-dependent calcium L-type channels close and muscle relaxes [80]. NO agonists induce relaxation of trabecular meshwork cells [77] and they appear to do so through changing the conformation of cytoskeletal proteins such as actin, myosin, and tubulin [81].
3.6. Angiopoietin-Like Molecules
Angiopoietin-like (ANGPTL) molecules are a family of glycoproteins which resemble angiopoietins structurally [82] but fail to bind the angiopoietin receptors Tie1 and Tie2 [83–85]. Despite these, some of the members in this family are potent regulators of angiogenesis [86, 87] and function as mediators in the induction of inflammation and regulation of lipid and glucose metabolism [88–90].
Angiopoietin-like 7, previously named cornea-derived transcript 6 (CDT6) [91], is expressed in the human trabecular meshwork and may be induced by treatment with dexamethasone [92–94] as well as by TGF-β2 [95], an overexpressed growth factor in the aqueous humor of glaucoma patients [41]. A beagle model of POAG demonstrated ANGPTL7 elevation in the dogs' aqueous humor which was consistent with the elevation of the same protein in the aqueous humor of glaucoma patients. Using the same animal model, the authors detected increased ANGPTL7 with disease progression. Increased expression of ANGPTL-7 is thought to arise due to elevated IOP [95].
The gene for ANGPTL-7 is located near TCF/LEF transcription factors which are key for the activation of the WNT/b-catenin signaling pathway [96]. This pathway has been previously linked with elevated IOP and glaucoma [97, 98]. The WNT secreted glycoproteins transmit signals by binding to frizzled transmembrane receptors. This results in dephosphorylation and consequent nuclear translocation of b-catenin. B-catenin will then activate expression of genes through interaction with transcription factors of the TCF/LEF family. It is therefore possible that the activation of ANGPTL7 could be mediated by the activation of the WNT pathway regulatory elements [98].
It has been studied on how ANGPTL-7 influences and modifies the trabecular meshwork. Peek et al. have postulated that ANGPTL-7 increases expression of proteoglycans and collagens types I and V [99]. Gabelt and Kaufmanwent further, stating that changes in collagen could alter the trabecular meshwork in a way compatible with increased resistance and consequent decreased aqueous humor outflow and, henceforth, cause ocular hypertension [100]. The increased expression of collagen in the sclera could also limit the aqueous humor outflow through the uveal-scleral pathway which would result in elevated IOP [101]. Studies in transgenic mice have shown that collagen I overexpression was associated with increased IOP and optic nerve damage, as observed in human POAG [102, 103]. Comes et al., on the other hand, verified decreased expression of collagen IA1, fibronectin collagen type VA1, versican, and to a lesser extent myocilin. Furthermore, induction of MMP1 was noted [104]. When ANGPTL-7 was silenced, the reverse protein expression occurred. The authors speculate that the different results concerning collagen I may be due to different cells used (transformed versus primary trabecular meshwork cells) or due to a posttranscription regulation of collagen I in these cells. Previous studies have revealed how fibronectin may be implicated in increasing outflow resistance [105]. Since overexpression of ANGPTL-7 reduces levels of fibronectin, Comes and colleagues suggest that this protein may actually facilitate rather that restrict aqueous humor outflow. This effect is supported by the increased expression of MMP1, a collagenase which has been linked to increased outflow in organ cultures [106].
Further studies are needed in order to confirm the benefit of a ANGPTL-7 agonist in the IOP lowering.
3.7. Adenosine
Adenosine modulates several physiological and pathophysiological pathways through its G protein-coupled receptors [107], and increased levels are found in retinal ischemia and elevated IOP [16]. There are four adenosine receptor subtypes known so far (A1, A2a, A2b, and A3), and A1, A2a, and A3 agonists and A3 antagonists have been considered potential IOP lowering drugs. The mechanism proposed is the secretion of MMPs in the TM, which lead to remodeling of the ECM and consequently an increase in TM outflow [16]. As for A1 agonists, preclinical studies with topical treatment showed statistically significant IOP decrease in primates [108], rabbits [109], and mice [14]. A2a agonists are currently in clinical trial phase with the expectation of lowering IOP, but interestingly, Avila et al. (2001) stated that agonists for these receptors given to monkeys increased IOP [14]. Regarding A3 receptor subtype, agonists are under clinical trial, since an unexpected finding in a previous study showed a decrease in IOP [110], but also antagonists are proposed to lower IOP, since the activation of A3 receptors leads to the activation of chloride channels in human nonpigmented ciliary epithelial cells, which potentially may lead to the increase in AH production [107, 111].
3.8. Latrunculins
Latrunculins, macrolides discovered in marine sponges, have the ability to inhibit actin polymerization [29], disrupting the TM's cytoskeleton and consequently its integrity, which results in increased outflow [112]. This increase in outflow may result from expansion of the space between SC and the TM's collagen beams and/or by increased openings between SC's inner wall cells [107]. Latrunculin-A and latrunculin-B intracamerally or topically induce a high increase in the TM's outflow and reduce IOP in monkeys [5]. Clinical trials have been undertaken, but so far no significant results were found. Several side effects were described with the highest doses used (0,02% and 0,05%), namely, mild ocular redness, irritation, and transient increase in central corneal thickness of <2,5% [113].
3.9. Cochlin
Cochlin is the product of the coagulation factor C homology gene (COCH) and is an ECM protein of unknown function. Evidence suggests association between cochlin and glaucomatous trabecular meshwork tissue [114] and that cochlin acts by cellular mechanosensitive mechanism [115–117].
Cochlin was identified in glaucomatous but not normal human TM [114]. Early cochlin protein expression was found in the TM of the glaucomatous DBA/2J mouse model as early as 3 weeks and the level of cochlin reaches a plateau around 6–8 months of age when elevation in IOP is observed [118].
Stretch activated channels (SACs), such as TREK-1, function as mechanotransducers involved in pressure regulation [115, 116], and cochlin expression has been previously shown to result in coexpression of TREK-1 [119]. Recent studies show that cochlin is involved in regulation of intraocular pressure in DBA/2J potentially through mechanosensing of the shear stress by demonstrating that overexpression and downregulation of cochlin increase and decrease intraocular pressure (IOP), respectively. Reintroduction of cochlin in cochlin-null mice increases IOP; injection of exogenous cochlin also increased IOP and increasing perfusion rates increased cochlin multimerization, which reduced the rate of cochlin proteolysis by trypsin, and proteinase K. The cochlin multimerization in response to shear stress suggests its potential mechanosensing [117].
3.10. Cannabinoids
There are two different receptor subtypes for cannabinoids, CB1 and CB2, and several compounds can activate them. Human endoligands of the cannabinoid receptor system are arachidonic acid-like substances and are called endocannabinoids. Although the expression of CB1 and CB2 has been described in ocular tissues, the main cannabinoid receptors expressed in the eye are CB1. The distribution of these receptors in the human eye has been studied, with high presence in corneal epithelium and endothelium, trabecular mesh, nonpigmented ciliary epithelium, ciliary muscle, and photoreceptor outer segments, with lesser intense presence in several other eye structures [120, 121]. Since CB1 receptor subtype is present in both structures where AH is produced and removed, it might induce an IOP reduction through a decrease in humor production and also an increase in its outflow, both through the TM and the uveoscleral routes [122]. Cannabinoids can also produce vasodilation of anterior uvea efferent blood vessels, hence, improving aqueous outflow [123]. Currently, the main hypotensive mechanism is considered to be increased outflow, by producing an increase in the dimensions of Schlemm's canal [124]. Noladin ether (endocannabinoid agonist) can induce MMP activation, which leads to remodeling of the TM and also reduces the production of actin stress fibers and focal adhesions [125]. The ocular hypotensive properties of cannabinoids are dependent on the β-adrenergic receptors stimulation. The cannabinoids reduce IOP by acting as indirect sympatholytics [126]. Cannabinoids can induce COX-2 expression, resulting in higher levels of PGE2 and MMPs, which modulates aqueous outflow resistance [127].
Marijuana (Cannabis sativa) is known to reduce IOP via systemic exposure; there are reports of IOP decrease from 30 mmHg to 15 mmHg [15]. The topical route was studied using WIN 55212-2, an agonist of CB1 and CB2 receptor subtypes. In normotensive cynomolgus monkeys, topically administered WIN 55212-2 by 0.5%, reduced IOP levels by 19%. Reduced aqueous humor flow was identified, but no changes in outflow facility were noted [123]. Also, the sublingual route was studied. Δ-9-THC (delta-9-tetrahydrocannabinol), which is the main psychoactive element, was able to reduce IOP over placebo, for only 4 hours (23.5 mmHg versus 27.3 mmHg; P = 0.026). This effect was not observed with topical application [26]. The inhalatory and intravenous routes have also proven to be successful in the reduction of IOP [122].
There is currently a proof that cannabinoids can lower IOP in humans. However, the development of tolerance and significant systemic toxicity appears to limit the usefulness of this potential treatment, making it an end-line treatment when available.
3.11. Melatonin
Melatonin is a neurohormone secreted into the blood mainly from the pineal gland. It is responsible for the regulation of the circadian rhythms of several biological functions and is regulated by the light/dark cycle [16, 128]. Three subtypes of melatonin receptors have been identified, MT1 and MT2 receptor subtypes, negatively coupled with adenylate cyclase, and MT3, coupled with phospholipase C [129].
Changes in melatonin levels in AH in light or dark, associated with IOP changes, suggest a melatonergic regulation of the circadian rhythm of IOP. A study revealed that knock-out mice for MT1 receptor subtype had higher IOP levels during the nocturnal hours than controls or knock-out mice for MT2 receptor subtype at 3 and 12 months of age. Additionally, the administration of exogenous melatonin significantly reduced IOP levels in wild-type mice, but not in the MT1 knock-out mice [130]. 5-MCA-NAT, an MT3 receptor agonist, has shown to reduce IOP by 19% in glaucomatous monkey eyes and by 40% in rabbits. Apparently, part of the mechanism of action is related to the cholinergic and noradrenergic systems, since antagonists of such systems could reverse the effect [131].
3.12. Ghrelin
Ghrelin is a 28-amino acid acylated peptide and is the endogenous ligand for the growth hormone secretagogues receptor (GHSR-1a), promoting the release of growth hormone from the pituitary independently from the growth-hormone-releasing hormone receptor [132]. Another variant of ghrelin is its unacylated form, desacyl ghrelin. This variant is identical to ghrelin except for the acyl group in the serine 3, thus, being unable to bind GHSR-1a [133].
In recent studies, ghrelin has been proposed to play important roles in the ocular tissue, both in the anterior and posterior segments. Regarding the anterior segment, ghrelin's mRNA was identified in the posterior surface of the iris and in the nonpigmented ciliary epithelium. This peptide was also shown to induce the relaxation of the iris sphincter and dilator muscles [134]. Ghrelin has also been implicated in glaucoma, being its levels significantly decreased in the aqueous humour of patients suffering from POAG and pseudoexfoliation glaucoma when compared to the control group [135, 136]. When comparing ghrelin AH levels from individuals with primary open angle glaucoma and pseudoexfoliation glaucoma, there was no significant difference [136]. The correlation between ghrelin plasma and AH levels remains controversial.
Ghrelin and desacyl ghrelin effect in the modulation of the intraocular pressure was recently studied in two animal models of osmotically induced intraocular hypertension. Ghrelin promoted a marked and sustained decrease of ocular tension when compared to control, presenting a maximal percentual decrease of 43.8 ± 12.05% in the rabbit and of 33.28 ± 6.47% in the rat. Desacyl ghrelin action did not affect the intraocular hypertension in the rabbit model, thus, indicating a role for GHSR-1a in the hypotensive effect induced by ghrelin [137]. It was also shown that ghrelin's influence on ocular hypertension is related to prostaglandins and nitric oxide production [137].
Recent studies show that ghrelin is produced by the ciliary processes and ghrelin receptor is also expressed in the trabecular meshwork and ciliary processes stroma. These data increase the already existing evidence for the ghrelin-GHSR system role in the ocular physiology. The endogenous production of ghrelin by the ciliary body together with the presence of the ghrelin receptor in the ocular tissue, namely, in the components responsible for aqueous humour dynamics, strengthens previous data that associated ghrelin with the pathophysiology of glaucoma. The hypotensive effect of ghrelin, its local production, and the GHSR-1a localization points to a possible therapeutic role in glaucoma but further studies are needed to clarify the mechanisms underlying ghrelin's influence in the ocular tension regulation.
3.13. Angiotensin II
It is already well known the role played by the renin-angiotensin system (RAS) in controlling systemic arterial pressure and ionic homeostasis. Angiotensin II (Ang II) is an endogenous potent vasoconstrictor that modulates systemic blood pressure by activating the G protein-coupled angiotensin II receptor, type I (AT1) [107]. Many of the RAS system components have been identified in human and animal eyes. Just like angiotensin I, angiotensin II and angiotensinogen are not able to cross the blood-brain barrier, and they also cannot cross the intact blood-retina barrier [138], creating a tissue-localized RAS system. RAS expression and secretory function have been shown in cultured human and rabbit ciliary body nonpigmented epithelia [139]. It was also proved that Ang II is able to induce cell proliferation in bovine TM cells and to increase the synthesis of collagen in vitro [140]. After angiotensin II administered intracamerally was shown to decrease uveo-scleral outflow, despite no changes in IOP, in rabbits [9], Wang et al. used olmesartan, an antagonist of AT1 receptors, topically in monkeys with laser-induced unilateral glaucoma and observed a dosage-dependent reduction of the IOP [141]. Topical olmesartan was already in early phase II glaucoma clinical trials when the study was terminated; even though it produced some IOP reductions, the efficacy was insufficient with no clear dose-response relationship.
A recently discovered RAS component, angiotensin-converting enzyme 2 (ACE2), can degrade Ang I to Ang (1–9) and Ang II to Ang (1–7), which in turn acts oppositely to Ang II [142], since it is an active vasodilator and antiproliferative molecule. Ang (1–7) mainly acts through a new angiotensin receptor type, Mas receptor, which is exclusive of the angiotensin (1–7) ligand [13]. Vaajanen et al. [143] assessed the effect of angiotensin II and its angiotensin (1–7) metabolite in IOP levels and aqueous humor flow in rabbits with normal ocular tension. Angiotensin (1–7) reduced IOP, probably through the Mas receptors, without any changes in the aqueous humor release flow. Topical as well as intravitreal applications of angiotensin II led to a significant increased resistance to the drainage of aqueous humor. While this effect was reverted with an AT1 antagonist, Ang (1–7) had no effect. This suggests that Ang (1–7) reduces IOP via reduced AH formation [139].
Apart from this, also ACE1 inhibitors or AT2 receptor blockers have human studies with proven effect on IOP when administered orally. The authors described a reduction in both normotensive and glaucomatous persons' IOP, although blood pressure was lowered only in patients with arterial hypertension [139]. The mechanism proposed for the hypotensive effect of ACE1 inhibitors is the decrease in the production of AH by reducing blood flow in the ciliary body [144]. Additionally, ACE1 inhibitors also promote the synthesis of prostaglandins by preventing the breakdown of bradykinin, which could in turn lower IOP by increasing the uveoscleral outflow [139].
New antiglaucomatous drug therapies can be directed towards increasing ACE2 activity and consequently increasing the formation of Ang (1–7) or towards the activation of Mas receptors directly.
3.14. Serotonin
Serotonin (5-hydroxytryptamine, 5-HT) is a biogenic monoamine neurotransmitter, a result of the hydroxylation of tryptophan and decarboxylation of tryptophan hydroxylase [107]. Seven different receptor families have been identified (5-HT1 to 5-HT7).
The interest in the serotonin pathway for glaucoma therapy started with the report of elevated IOP or angle-closure glaucoma in patients under treatment with serotonin reuptake inhibitors (SSRIs) [145]. Since these drugs enhance serotoninergic neurotransmission, a possible role for serotonin antagonists arose. Opposite from what was thought, 5-HT2 agonists, but not 5-HT1A agonists or 5-HT2 antagonists, lowered IOP when applied topically in normotensive or cynomolgus monkeys' hypertensive eyes [107]. Studies with R-2,5-dimethoxy-4-iodoamphetamine, (R-DOI; a 5-HT2A,2B,2C receptor partial agonist) showed a decreased IOP in cynomolgus monkeys, mainly through an increased uveoscleral outflow, but also led to a small increase in AH formation [146]. A differential expression of the mRNAs of several receptor subtypes has been identified in human ocular tissue, with a high density of mRNAs for 5-HT2A and 5-HT2B in the TM, with much lower levels of 5-HT2C, 5-HT5, and 5-HT7 [147]. Interestingly, in the rabbit, topical application of 5-HT can either lower or elevate IOP. This is probably due to the presence of 5-HT receptors with opposing effects, such as 5-HT1 that reduces and 5-HT7 that elevates IOP [16]. A phase II clinical trial with BVT.28949, a 5-HT2A receptor antagonist, reduced IOP by 10% with a 4-week treatment, in patients with POAG or OH [148].
On the other hand, AL-34662, a 5-HT2 receptor agonist, was able to reduce IOP by 33%, with minimal local side effects, in cynomolgus monkeys [147]. Further studies are being undertaken regarding this compound.
Serotonin receptors have proven to be an effective target for glaucoma therapies, since they can lower IOP. It remains to be understood the true function of each of its receptors, since antagonic results can occur.
3.15. Forskolin
Forskolin is a flavonoid that can lower IOP in rabbits, monkeys, and humans, through an increase in cAMP (cyclic adenosine monophosphate) levels, when applied topically in the eye [15, 149].
It consists in a lipid-soluble compound that stimulates the enzyme adenylate cyclase, increasing cAMP levels, which, in turn, act upon the ciliary epithelium, reducing aqueous humor inflow [150]. Apart from topical usage, also oral forskolin has proven to be effective. Forskolin as a food supplement decreased IOP by 20% of the initial value, reversible upon suspension of the treatment [151]. Forskolin has a different molecular mechanism from any previously used antiglaucoma drug, which makes it promising for combination therapies.
4. Conclusion
No current medication for POAG is able to reduce the IOP by more than 25%–30%. Future strategies for treatment of glaucoma must therefore take into account different mechanisms to lower IOP. Better knowledge about trabecular outflow pathway, namely, mechanisms associated with phagocytosis, cytoskeletal reorganization, cell adhesion, and matrix deposition opened a new horizon for new hypotensive class agents. The main cause of elevated IOP in POAG is thought to be an increased outflow resistance through the pressure-dependent trabecular outflow system, so there is crescent interest in increasing TM outflow by extracellular matrix remodeling and/or by modulation of contractility/TM cytoskeleton disruption. There are also other agents that act by decreasing aqueous humour production or increasing uveoscleral outflow by different mechanisms from those drugs available for glaucoma treatment. We described some physiological targets that are being currently evaluated for their role in the IOP modulation. From those, some could generate a hypotensive drug with possible clinical use.
Acknowledgment
This study was funded by Portuguese Foundation for Science and Technology, Grant PTDC/SAU-ORG/110683/2009.
References
- 1.Sommer A, Tielsch JM, Katz J, et al. Relationship between intraocular pressure and primary open angle glaucoma among white and black Americans: the Baltimore eye survey. Archives of Ophthalmology. 1991;109(8):1090–1095. doi: 10.1001/archopht.1991.01080080050026. [DOI] [PubMed] [Google Scholar]
- 2.Kass MA, Heuer DK, Higginbotham EJ, et al. The Ocular Hypertension Treatment Study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Archives of Ophthalmology. 2002;120(6):701–713. doi: 10.1001/archopht.120.6.701. [DOI] [PubMed] [Google Scholar]
- 3.Weinreb RN, Toris CB, Gabelt BT, Lindsey JD, Kaufman PL. Effects of prostaglandins on the aqueous humor outflow pathways. Survey of Ophthalmology. 2002;47(4, supplement 1):S53–S64. doi: 10.1016/s0039-6257(02)00306-5. [DOI] [PubMed] [Google Scholar]
- 4.Weinreb RN, Lindsey JD. Metalloproteinase gene transcription in human ciliary muscle cells with latanoprost. Investigative Ophthalmology & Visual Science. 2002;43(3):716–722. [PubMed] [Google Scholar]
- 5.Camras CB, Friedman AH, Rodrigues MM, Tripathi BJ, Tripathi RC, Podos SM. Multiple dosing of prostaglandin F(2α) or epinephrine on cynomolgus monkey eyes—III. Histopathology. Investigative Ophthalmology & Visual Science. 1988;29(9):1428–1436. [PubMed] [Google Scholar]
- 6.Kaufman PL. Pharmacologic trabeculocanalotomy: facilitating aqueous outflow by assaulting the meshwork cytoskeleton, junctional complexes, and extracellular matrix. Archives of Ophthalmology. 1992;110(1):34–36. doi: 10.1001/archopht.1992.01080130036022. [DOI] [PubMed] [Google Scholar]
- 7.Fukata Y, Amano M, Kaibuchi K. Rho-Rho-kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends in Pharmacological Sciences. 2001;22(1):32–39. doi: 10.1016/s0165-6147(00)01596-0. [DOI] [PubMed] [Google Scholar]
- 8.Honjo M, Tanihara H, Inatani M, et al. Effects of Rho-associated protein kinase inhibitor Y-27632 on intraocular pressure and outflow facility. Investigative Ophthalmology & Visual Science. 2001;42(1):137–144. [PubMed] [Google Scholar]
- 9.Tian B, Geiger B, Epstein DL, Kaufman PL. Cytoskeletal involvement in the regulation of aqueous humor outflow. Investigative Ophthalmology & Visual Science. 2000;41(3):619–623. [PubMed] [Google Scholar]
- 10.Tian B, Brumback LC, Kaufman PL. ML-7, chelerythrine and phorbol ester increase outflow facility in the monkey eye. Experimental Eye Research. 2000;71(6):551–566. doi: 10.1006/exer.2000.0919. [DOI] [PubMed] [Google Scholar]
- 11.Rao PV, Deng P, Sasaki Y, Epstein DL. Regulation of myosin light chain phosphorylation in the trabecular meshwork: role in aqueous humour outflow facility. Experimental Eye Research. 2005;80(2):197–206. doi: 10.1016/j.exer.2004.08.029. [DOI] [PubMed] [Google Scholar]
- 12.Thieme H, Nuskovski M, Nass JU, Pleyer U, Strauss O, Wiederholt M. Mediation of calcium-independent contraction in trabecular meshwork through protein kinase C and rho-A. Investigative Ophthalmology & Visual Science. 2000;41(13):4240–4246. [PubMed] [Google Scholar]
- 13.Munoz-Negrete FJ, Pérez-López M, Won Kim HR, Rebolleda G. New developments in glaucoma medical treatment. Archivos de la Sociedad Española de Oftalmología. 2009;84(10):491–500. doi: 10.4321/s0365-66912009001000003. [DOI] [PubMed] [Google Scholar]
- 14.Avila MY, Stone RA, Civan MM. A1-, A2a- and A3-subtype adenosine receptors modulate intraocular pressure in the mouse. British Journal of Pharmacology. 2001;134(2):241–245. doi: 10.1038/sj.bjp.0704267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beidoe G, Mousa SA. Current primary open-angle glaucoma treatments and future directions. Clinical Ophthalmology. 2012;6:1699–1707. doi: 10.2147/OPTH.S32933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee AJ, Goldberg I. Emerging drugs for ocular hypertension. Expert Opinion on Emerging Drugs. 2011;16(1):137–161. doi: 10.1517/14728214.2011.521631. [DOI] [PubMed] [Google Scholar]
- 17.Pinazo-Duran MD, Zanon-Moreno V, Garcia-Medina JJ, Gallego-Pinazo R. Evaluation of presumptive biomarkers of oxidative stress, immune response and apoptosis in primary open-angle glaucoma. Current Opinion in Pharmacology. 2013;13(1):98–107. doi: 10.1016/j.coph.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 18.Wierzbowska J, Robaszkiewicz J, Figurska M, Stankiewicz A. Future possibilities in glaucoma therapy. Case Reports and Clinical Practice Review. 2010;16(11):RA252–RA259. [PubMed] [Google Scholar]
- 19.Armaly MF, Krueger DE, Maunder L. Biostatistical analysis of the collaborative glaucoma study—I. Summary report of the risk factors for glaucomatous visual-field defects. Archives of Ophthalmology. 1980;98(12):2163–2171. doi: 10.1001/archopht.1980.01020041015002. [DOI] [PubMed] [Google Scholar]
- 20.Shiose Y, Kitazawa Y, Tsukahara S, et al. Epidemiology of glaucoma in Japan. A nationwide glaucoma survey. Japanese Journal of Ophthalmology. 1991;35(2):133–155. [PubMed] [Google Scholar]
- 21.Amano M, Nakayama M, Kaibuchi K. Rho-kinase/ROCK: a key regulator of the cytoskeleton and cell polarity. Cytoskeleton. 2010;67(9):545–554. doi: 10.1002/cm.20472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hahmann C, Schroeter T. Rho-kinase inhibitors as therapeutics: from pan inhibition to isoform selectivity. Cellular and Molecular Life Sciences. 2010;67(2):171–177. doi: 10.1007/s00018-009-0189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Colligris B, Crooke A, Huete F, Pintor J. Potential role of Rho-associated protein kinase inhibitors for glaucoma treatment. Recent Patents on Endocrine, Metabolic & Immune Drug Discovery. 2012;6(2):89–98. doi: 10.2174/187221412800604554. [DOI] [PubMed] [Google Scholar]
- 24.Nakajima E, Nakajima T, Minagawa Y, Shearer TR, Azuma M. Contribution of ROCK in contraction of trabecular meshwork: proposed mechanism for regulating aqueous outflow in monkey and human eyes. Journal of Pharmaceutical Sciences. 2005;94(4):701–708. doi: 10.1002/jps.20285. [DOI] [PubMed] [Google Scholar]
- 25.Tokushige H, Inatani M, Nemoto S, et al. Effects of topical administration of Y-39983, a selective Rho-associated protein kinase inhibitor, on ocular tissues in rabbits and monkeys. Investigative Ophthalmology & Visual Science. 2007;48(7):3216–3222. doi: 10.1167/iovs.05-1617. [DOI] [PubMed] [Google Scholar]
- 26.Zhang M, Maddala R, Rao PV. Novel molecular insights into RhoA GTPase-induced resistance to aqueous humor outflow through the trabecular meshwork. American Journal of Physiology. 2008;295(5):C1057–C1070. doi: 10.1152/ajpcell.00481.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fang X, Chen YT, Sessions EH, et al. Synthesis and biological evaluation of 4-quinazolinones as Rho kinase inhibitors. Bioorganic & Medicinal Chemistry Letters. 2011;21(6):1844–1848. doi: 10.1016/j.bmcl.2011.01.039. [DOI] [PubMed] [Google Scholar]
- 28.Anderson DR, Drance SM, Schulzer M. Comparison of glaucomatous progression between untreated patients with normal-tension glaucoma and patients with therapeutically reduced intraocular pressures. American Journal of Ophthalmology. 1998;126(4):487–497. doi: 10.1016/s0002-9394(98)00223-2. [DOI] [PubMed] [Google Scholar]
- 29.Zhang K, Zhang L, Weinreb RN. Ophthalmic drug discovery: novel targets and mechanisms for retinal diseases and glaucoma. Nature Reviews Drug Discovery. 2012;11(7):541–559. doi: 10.1038/nrd3745. [DOI] [PubMed] [Google Scholar]
- 30.Rao VP, Epstein DL. Rho GTPase/Rho kinase inhibition as a novel target for the treatment of glaucoma. BioDrugs. 2007;21(3):167–177. doi: 10.2165/00063030-200721030-00004. [DOI] [PubMed] [Google Scholar]
- 31.Lepple-Wienhues A, Stahl F, Willner U, Schafer R, Wiederholt M. Endothelin-evoked contractions in bovine ciliary muscle and trabecular meshwork: interaction with calcium, nifedipine and nickel. Current Eye Research. 1991;10(10):983–989. doi: 10.3109/02713689109020335. [DOI] [PubMed] [Google Scholar]
- 32.Renieri G, Choritz L, Rosenthal R, Meissner S, Pfeiffer N, Thieme H. Effects of endothelin-1 on calcium-independent contraction of bovine trabecular meshwork. Graefe’s Archive for Clinical and Experimental Ophthalmology. 2008;246(8):1107–1115. doi: 10.1007/s00417-008-0817-4. [DOI] [PubMed] [Google Scholar]
- 33.Choritz L, Machert M, Thieme H. Correlation of endothelin-1 concentration in aqueous humor with intraocular pressure in primary open angle and pseudoexfoliation glaucoma. Investigative Ophthalmology & Visual Science. 2012;53(11):7336–7342. doi: 10.1167/iovs.12-10216. [DOI] [PubMed] [Google Scholar]
- 34.Cellini M, Versura P, Zamparini E, Bendo E, Campos EC. Effects of endothelin-1 and flunarizine on human trabecular meshwork cell contraction. Experimental Biology and Medicine. 2006;231(6):1081–1084. [PubMed] [Google Scholar]
- 35.Choritz L, Rosenthal R, Fromm M, Foerster MH, Thieme H. Pharmacological and functional characterization of endothelin receptors in bovine trabecular meshwork and ciliary muscle. Ophthalmic Research. 2005;37(4):179–187. doi: 10.1159/000086471. [DOI] [PubMed] [Google Scholar]
- 36.Wiederholt M, Sturm A, Lepple-Wienhues A. Relaxation of trabecular meshwork and ciliary muscle by release of nitric oxide. Investigative Ophthalmology & Visual Science. 1994;35(5):2515–2520. [PubMed] [Google Scholar]
- 37.De Caestecker M. The transforming growth factor-β superfamily of receptors. Cytokine and Growth Factor Reviews. 2004;15(1):1–11. doi: 10.1016/j.cytogfr.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 38.Prendes MA. The role of transforming growth factor beta in glaucoma and the therapeutic implications. British Journal of Ophthalmology. 2013;97(6):680–686. doi: 10.1136/bjophthalmol-2011-301132. [DOI] [PubMed] [Google Scholar]
- 39.Gottanka J, Chan D, Eichhorn M, Lütjen-Drecoll E, Ethier CR. Effects of TGF-beta2 in perfused human eyes. Investigative Ophthalmology & Visual Science. 2004;45(1):153–158. doi: 10.1167/iovs.03-0796. [DOI] [PubMed] [Google Scholar]
- 40.Min SH, Lee T-I, Chung YS, Kim HK. Transforming growth factor-beta levels in human aqueous humor of glaucomatous, diabetic and uveitic eyes. Korean Journal of Ophthalmology. 2006;20(3):162–165. doi: 10.3341/kjo.2006.20.3.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tripathi RC, Li J, Chan FA, Tripathi BJ. Aqueous humor in glaucomatous eyes contains an increased level of TGF-β2. Experimental Eye Research. 1994;59(6):723–728. doi: 10.1006/exer.1994.1158. [DOI] [PubMed] [Google Scholar]
- 42.Shepard AR, Cameron Millar J, Pang I-H, Jacobson N, Wang W-H, Clark AF. Adenoviral gene transfer of active human transforming growth factor-β2 elevates intraocular pressure and reduces outflow facility in rodent eyes. Investigative Ophthalmology & Visual Science. 2010;51(4):2067–2076. doi: 10.1167/iovs.09-4567. [DOI] [PubMed] [Google Scholar]
- 43.Trivedi RH, Nutaitis M, Vroman D, Crosson CE. Influence of race and age on aqueous humor levels of transforming growth factor-beta 2 in glaucomatous and nonglaucomatous eyes. Journal of Ocular Pharmacology and Therapeutics. 2011;27(5):477–480. doi: 10.1089/jop.2010.0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Da B, Cao Y, Wei H, Chen Z, Shui Y, Li Z. Antagonistic effects of tranilast on proliferation and collagen synthesis induced by TGF-β2 in cultured human trabecular meshwork cells. Journal of Huazhong University of Science and Technology. 2004;24(5):490–496. doi: 10.1007/BF02831117. [DOI] [PubMed] [Google Scholar]
- 45.Wordinger RJ, Fleenor DL, Hellberg PE, et al. Effects of TGF-β2, BMP-4, and gremlin in the trabecular meshwork: implications for glaucoma. Investigative Ophthalmology & Visual Science. 2007;48(3):1191–1200. doi: 10.1167/iovs.06-0296. [DOI] [PubMed] [Google Scholar]
- 46.Fuchshofer R, Welge-Lussen U, Lütjen-Drecoll E. The effect of TGF-β2 on human trabecular meshwork extracellular proteolytic system. Experimental Eye Research. 2003;77(6):757–765. doi: 10.1016/s0014-4835(03)00220-3. [DOI] [PubMed] [Google Scholar]
- 47.Liton PB, Liu X, Challa P, Epstein DL, Gonzalez P. Induction of TGF-β1 in the trabecular meshwork under cyclic mechanical stress. Journal of Cellular Physiology. 2005;205(3):364–371. doi: 10.1002/jcp.20404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fuchshofer R, Tamm ER. The role of TGF-β in the pathogenesis of primary open-angle glaucoma. Cell and Tissue Research. 2012;347(1):279–290. doi: 10.1007/s00441-011-1274-7. [DOI] [PubMed] [Google Scholar]
- 49.Schlötzer-Schrehardt U, Zenkel M, Küchle M, Sakai LY, Naumann GOH. Role of transforming growth factor-β1 and its latent form binding protein in pseudoexfoliation syndrome. Experimental Eye Research. 2001;73(6):765–780. doi: 10.1006/exer.2001.1084. [DOI] [PubMed] [Google Scholar]
- 50.Nakamura Y, Hirano S, Suzuki K, Seki K, Sagara T, Nishida T. Signaling mechanism of TGF-β1-induced collagen contraction mediated by bovine trabecular meshwork cells. Investigative Ophthalmology & Visual Science. 2002;43(11):3465–3472. [PubMed] [Google Scholar]
- 51.Welge-Lüßen U, May CA, Lütjen-Drecoll E. Induction of tissue transglutaminase in the trabecular meshwork by TGF- β1 and TGF-β2. Investigative Ophthalmology & Visual Science. 2000;41(8):2229–2238. [PubMed] [Google Scholar]
- 52.Tamm ER, Russell P, Epstein DL, Johnson DH, Piatigorsky J. Modulation of myocilin/TIGR expression in human trabecular meshwork. Investigative Ophthalmology & Visual Science. 1999;40(11):2577–2582. [PubMed] [Google Scholar]
- 53.Konz DD, Flügel-Koch C, Ohlmann A, Tamm ER. Myocilin in the trabecular meshwork of eyes with primary open-angle glaucoma. Graefe’s Archive for Clinical and Experimental Ophthalmology. 2009;247(12):1643–1649. doi: 10.1007/s00417-009-1152-0. [DOI] [PubMed] [Google Scholar]
- 54.Weston BS, Wahab NA, Mason RM. CTGF mediates TGF-β-induced fibronectin matrix deposition by upregulating active α5β1 integrin in human mesangial cells. Journal of the American Society of Nephrology. 2003;14(3):601–610. doi: 10.1097/01.asn.0000051600.53134.b9. [DOI] [PubMed] [Google Scholar]
- 55.Kothapalli D, Grotendorst GR. CTGF modulates cell cycle progression in cAMP-arrested NRK fibroblasts. Journal of Cellular Physiology. 2000;182(1):119–126. doi: 10.1002/(SICI)1097-4652(200001)182:1<119::AID-JCP13>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 56.Inatani M, Tanihara H, Katsuta H, Honjo M, Kido N, Honda Y. Transforming growth factor-β2 levels in aqueous humor of glaucomatous eyes. Graefe’s Archive for Clinical and Experimental Ophthalmology. 2001;239(2):109–113. doi: 10.1007/s004170000241. [DOI] [PubMed] [Google Scholar]
- 57.Kottler UB, Jünemann AGM, Aigner T, Zenkel M, Rummelt C, Schlötzer-Schrehardt U. Comparative effects of TGF-β1 and TGF-β2 on extracellular matrix production, proliferation, migration, and collagen contraction of human Tenon’s capsule fibroblasts in pseudoexfoliation and primary open-angle glaucoma. Experimental Eye Research. 2005;80(1):121–134. doi: 10.1016/j.exer.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 58.Gottanka J, Flügel-Koch C, Martus P, Johnson DH, Lütjen-Drecoll E. Correlation of pseudoexfoliative material and optic nerve damage in pseudoexfoliation syndrome. Investigative Ophthalmology & Visual Science. 1997;38(12):2435–2446. [PubMed] [Google Scholar]
- 59.Crean JKG, Finlay D, Murphy M, et al. The role of p42/44 MAPK and protein kinase B in connective tissue growth factor induced extracellular matrix protein production, cell migration, and actin cytoskeletal rearrangement in human mesangial cells. Journal of Biological Chemistry. 2002;277(46):44187–44194. doi: 10.1074/jbc.M203715200. [DOI] [PubMed] [Google Scholar]
- 60.Crean JK, Furlong F, Finlay D, et al. Connective tissue growth factor [CTGF]/CCN2 stimulates mesangial cell migration through integrated dissolution of focal adhesion complexes and activation of cell polarization. The FASEB Journal. 2004;18(13):1541–1543. doi: 10.1096/fj.04-1546fje. [DOI] [PubMed] [Google Scholar]
- 61.Crean JK, Furlong F, Mitchell D, McArdle E, Godson C, Martin F. Connective tissue growth factor/CCN2 stimulates actin disassembly through Akt/protein kinase B-mediated phosphorylation and cytoplasmic translocation of p27(Kip-1) The FASEB Journal. 2006;20(10):1712–1714. doi: 10.1096/fj.05-5010fje. [DOI] [PubMed] [Google Scholar]
- 62.Furlong F, Crean J, Thornton L, O’Leary R, Murphy M, Martin F. Dysregulated intracellular signaling impairs CTGF-stimulated responses in human mesangial cells exposed to high extracellular glucose. American Journal of Physiology. 2007;292(6):F1691–F1700. doi: 10.1152/ajprenal.00342.2006. [DOI] [PubMed] [Google Scholar]
- 63.Browne JG, Ho SL, Kane R, et al. Connective tissue growth factor is increased in pseudoexfoliation glaucoma. Investigative Ophthalmology & Visual Science. 2011;52(6):3660–3666. doi: 10.1167/iovs.10-5209. [DOI] [PubMed] [Google Scholar]
- 64.Grant WM. Experimental aqueous perfusion in enucleated human eyes. Archives of Ophthalmology. 1963;69:783–801. doi: 10.1001/archopht.1963.00960040789022. [DOI] [PubMed] [Google Scholar]
- 65.Johnson M. What controls aqueous humour outflow resistance? Experimental Eye Research. 2006;82(4):545–557. doi: 10.1016/j.exer.2005.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lutjen-Drecoll E, Shimizu T, Rohrbach M, Rohen JW. Quantitative analysis of “plaque material” between ciliary muscle tips in normal- and glaucomatous eyes. Experimental Eye Research. 1986;42(5):457–465. doi: 10.1016/0014-4835(86)90005-9. [DOI] [PubMed] [Google Scholar]
- 67.Rohen JW, Lutjen-Drecoll E, Flugel C, Meyer M, Grierson I. Ultrastructure of the trabecular meshwork in untreated cases of primary open-angle glaucoma (POAG) Experimental Eye Research. 1993;56(6):683–692. doi: 10.1006/exer.1993.1085. [DOI] [PubMed] [Google Scholar]
- 68.Leask A, Abraham DJ. All in the CCN family: essential matricellular signaling modulators emerge from the bunker. Journal of Cell Science. 2006;119(23):4803–4810. doi: 10.1242/jcs.03270. [DOI] [PubMed] [Google Scholar]
- 69.Yokoi H, Mukoyama M, Sugawara A, et al. Role of connective tissue growth factor in fibronectin expression and tubulointerstitial fibrosis. American Journal of Physiology. 2002;282(5):F933–F942. doi: 10.1152/ajprenal.00122.2001. [DOI] [PubMed] [Google Scholar]
- 70.Schlötzer-Schrehardt U, Von der Mark K, Sakai LY, Naumann GOH. Increased extracellular deposition of fibrillin-containing fibrils in pseudoexfoliation syndrome. Investigative Ophthalmology & Visual Science. 1997;38(5):970–984. [PubMed] [Google Scholar]
- 71.Fuchshofer R, Birke M, Welge-Lussen U, Kook D, Lütjen-Drecoll E. Transforming growth factor-β2 modulated extracellular matrix component expression in cultured human optic nerve head astrocytes. Investigative Ophthalmology & Visual Science. 2005;46(2):568–578. doi: 10.1167/iovs.04-0649. [DOI] [PubMed] [Google Scholar]
- 72.Fuchshofer R, Yu AHL, Welge-Lüssen U, Tamm ER. Bone morphogenetic protein-7 is an antagonist of transforming growth factor-β2 in human trabecular meshwork cells. Investigative Ophthalmology & Visual Science. 2007;48(2):715–726. doi: 10.1167/iovs.06-0226. [DOI] [PubMed] [Google Scholar]
- 73.Junglas B, Kuespert S, Seleem AA, et al. Connective tissue growth factor causes glaucoma by modifying the actin cytoskeleton of the trabecular meshwork. American Journal of Pathology. 2012;180(6):2386–2403. doi: 10.1016/j.ajpath.2012.02.030. [DOI] [PubMed] [Google Scholar]
- 74.Iyer P, Maddala R, Pattabiraman PP, Rao PV. Connective tissue growth factor-mediated upregulation of neuromedin U expression in trabecular meshwork cells and its role in homeostasis of aqueous humor outflow. Investigative Ophthalmology & Visual Science. 2012;53(8):4952–4962. doi: 10.1167/iovs.12-9681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lepple-Wienhues A, Rauch R, Clark AF, Grassmann A, Berweck S, Wiederholt M. Electrophysiological properties of cultured human trabecular meshwork cells. Experimental Eye Research. 1994;59(3):305–311. doi: 10.1006/exer.1994.1112. [DOI] [PubMed] [Google Scholar]
- 76.Stumpff F, Strauss O, Boxberger M, Wiederholt M. Characterization of maxi-K-channels in bovine trabecular meshwork and their activation by cyclic guanosine monophosphate. Investigative Ophthalmology & Visual Science. 1997;38(9):1883–1892. [PubMed] [Google Scholar]
- 77.Nathanson JA, McKee M. Identification of an extensive system of nitric oxide-producing cells in the ciliary muscle and outflow pathway of the human eye. Investigative Ophthalmology & Visual Science. 1995;36(9):1765–1773. [PubMed] [Google Scholar]
- 78.Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacological Reviews. 1991;43(2):109–142. [PubMed] [Google Scholar]
- 79.Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. American Journal of Physiology. 1995;268(4):C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- 80.Pattabiraman PP, Rao PV. Mechanistic basis of Rho GTPase-induced extracellular matrix synthesis in trabecular meshwork cells. American Journal of Physiology. 2010;298(3):C749–C763. doi: 10.1152/ajpcell.00317.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stumpff F, Wiederholt M. Regulation of trabecular meshwork contractility. Ophthalmologica. 2000;214(1):33–53. doi: 10.1159/000027471. [DOI] [PubMed] [Google Scholar]
- 82.Oike Y, Tabata M. Angiopoietin-like proteins—potential therapeutic targets for metabolic syndrome and cardiovascular disease. Circulation Journal. 2009;73(12):2192–2197. doi: 10.1253/circj.cj-09-0710. [DOI] [PubMed] [Google Scholar]
- 83.Kim I, Kim H-G, Kim H, et al. Hepatic expression, synthesis and secretion of a novel fibrinogen/angiopoietin-related protein that prevents endothelial-cell apoptosis. Biochemical Journal. 2000;346(3):603–610. [PMC free article] [PubMed] [Google Scholar]
- 84.Camenisch G, Pisabarro MT, Sherman D, et al. ANGPTL3 stimulates endothelial cell adhesion and migration via integrin αvβ3 and induces blood vessel formation in vivo. Journal of Biological Chemistry. 2002;277(19):17281–17290. doi: 10.1074/jbc.M109768200. [DOI] [PubMed] [Google Scholar]
- 85.Katoh Y, Katoh M. Comparative integromics on Angiopoietin family members. International Journal of Molecular Medicine. 2006;17(6):1145–1149. [PubMed] [Google Scholar]
- 86.Oike Y, Ito Y, Maekawa H, et al. Angiopoietin-related growth factor (AGF) promotes angiogenesis. Blood. 2004;103(10):3760–3765. doi: 10.1182/blood-2003-04-1272. [DOI] [PubMed] [Google Scholar]
- 87.Hato T, Tabata M, Oike Y. The role of angiopoietin-like proteins in angiogenesis and metabolism. Trends in Cardiovascular Medicine. 2008;18(1):6–14. doi: 10.1016/j.tcm.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 88.Ono M, Shimizugawa T, Shimamura M, et al. Protein region important for regulation of lipid metabolism in angiopoietin-like 3 (ANGPTL3): ANGPTL3 is cleaved and activated in vivo. Journal of Biological Chemistry. 2003;278(43):41804–41809. doi: 10.1074/jbc.M302861200. [DOI] [PubMed] [Google Scholar]
- 89.Oike Y, Akao M, Yasunaga K, et al. Angiopoietin-related growth factor antagonizes obesity and insulin resistance. Nature Medicine. 2005;11(4):400–408. doi: 10.1038/nm1214. [DOI] [PubMed] [Google Scholar]
- 90.Xu A, Lam MC, Chan KW, et al. Angiopoietin-like protein 4 decreases blood glucose and improves glucose tolerance but induces hyperlipidemia and hepatic steatosis in mice. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(17):6086–6091. doi: 10.1073/pnas.0408452102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Peek R, Van Elske Gelderen B, Bruinenberg M, Kijlstm A. Molecular cloning of a new angiopoietinlike factor from the human cornea. Investigative Ophthalmology & Visual Science. 1998;39(10):1782–1788. [PubMed] [Google Scholar]
- 92.Gonzalez P, Epstein DL, Borras T. Characterization of gene expression in human trabecular meshwork using single-pass sequencing of 1060 clones. Investigative Ophthalmology & Visual Science. 2000;41(12):3678–3693. [PubMed] [Google Scholar]
- 93.Tomarev SI, Wistow G, Raymond V, Dubois S, Malyukova I. Gene expression profile of the human trabecular meshwork: NEIBank sequence tag analysis. Investigative Ophthalmology & Visual Science. 2003;44(6):2588–2596. doi: 10.1167/iovs.02-1099. [DOI] [PubMed] [Google Scholar]
- 94.Rozsa FW, Reed DM, Scott KM, et al. Gene expression profile of human trabecular meshwork cells in response to long-term dexamethasone exposure. Molecular Vision. 2006;12:125–141. [PubMed] [Google Scholar]
- 95.Zhao X, Ramsey KE, Stephan DA, Russell P. Gene and protein expression changes in human trabecular meshwork cells treated with transforming growth factor-β . Investigative Ophthalmology & Visual Science. 2004;45(11):4023–4034. doi: 10.1167/iovs.04-0535. [DOI] [PubMed] [Google Scholar]
- 96.Cadigan KM. Wnt-β-catenin signaling. Current Biology. 2008;18(20):R943–R947. doi: 10.1016/j.cub.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 97.Wang W-H, McNatt LG, Pang I-H, et al. Increased expression of the WNT antagonist sFRP-1 in glaucoma elevates intraocular pressure. Journal of Clinical Investigation. 2008;118(3):1056–1064. doi: 10.1172/JCI33871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Comes N, Borrás T. Individual molecular response to elevated intraocular pressure in perfused postmortem human eyes. Physiological Genomics. 2009;38(2):205–225. doi: 10.1152/physiolgenomics.90261.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Peek R, Kammerer RA, Frank S, Otte-Höller I, Westphal JR. The angiopoietin-like factor cornea-derived transcript 6 is a putative morphogen for human cornea. Journal of Biological Chemistry. 2002;277(1):686–693. doi: 10.1074/jbc.M105746200. [DOI] [PubMed] [Google Scholar]
- 100.Gabelt BT, Kaufman PL. Changes in aqueous humor dynamics with age and glaucoma. Progress in Retinal and Eye Research. 2005;24(5):612–637. doi: 10.1016/j.preteyeres.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 101.Kuchtey J, Källberg ME, Gelatt KN, Rinkoski T, Komàromy AM, Kuchtey RW. Angiopoietin-like 7 secretion is induced by glaucoma stimuli and its concentration is elevated in glaucomatous aqueous humor. Investigative Ophthalmology & Visual Science. 2008;49(8):3438–3448. doi: 10.1167/iovs.07-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mabuchi F, Lindsey JD, Aihara M, Mackey MR, Weinreb RN. Optic nerve damage in mice with a targeted type I collagen mutation. Investigative Ophthalmology & Visual Science. 2004;45(6):1841–1845. doi: 10.1167/iovs.03-1008. [DOI] [PubMed] [Google Scholar]
- 103.Aihara M, Lindsey JD, Weinreb RN. Ocular hypertension in mice with a targeted type I collagen mutation. Investigative Ophthalmology & Visual Science. 2003;44(4):1581–1585. doi: 10.1167/iovs.02-0759. [DOI] [PubMed] [Google Scholar]
- 104.Comes N, Buie LK, Borrás T. Evidence for a role of angiopoietin-like 7 (ANGPTL7) in extracellular matrix formation of the human trabecular meshwork: implications for glaucoma. Genes to Cells. 2011;16(2):243–259. doi: 10.1111/j.1365-2443.2010.01483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Faralli JA, Schwinn MK, Gonzalez JM, Jr., Filla MS, Peters DM. Functional properties of fibronectin in the trabecular meshwork. Experimental Eye Research. 2009;88(4):689–693. doi: 10.1016/j.exer.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bradley JM, Vranka J, Colvis CM, et al. Effect of matrix metalloproteinases activity on outflow in perfused human organ culture. Investigative Ophthalmology & Visual Science. 1998;39(13):2649–2658. [PubMed] [Google Scholar]
- 107.Chen J, Runyan SA, Robinson MR. Novel ocular antihypertensive compounds in clinical trials. Clinical Ophthalmology. 2011;5(1):667–677. doi: 10.2147/OPTH.S15971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tian B, Gabelt BT, Crosson CE, Kaufman PL. Effects of adenosine agonists on intraocular pressure and aqueous humor dynamics in cynomolgus monkeys. Experimental Eye Research. 1997;64(6):979–989. doi: 10.1006/exer.1997.0296. [DOI] [PubMed] [Google Scholar]
- 109.Crosson CE. Adenosine receptor activation modulates intraocular pressure in rabbits. Journal of Pharmacology and Experimental Therapeutics. 1995;273(1):320–326. [PubMed] [Google Scholar]
- 110.Avni I, Garzozi HJ, Barequet IS, et al. Treatment of dry eye syndrome with orally administered CF101: data from a phase 2 clinical trial. Ophthalmology. 2010;117(7):1287–1293. doi: 10.1016/j.ophtha.2009.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Civan MM, Macknight ADC. The ins and outs of aqueous humour secretion. Experimental Eye Research. 2004;78(3):625–631. doi: 10.1016/j.exer.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 112.Peterson JA, Tian B, McLaren JW, Hubbard WC, Geiger B, Kaufman PL. Latrunculins’ effects on intraocular pressure, aqueous humor flow, and corneal endothelium. Investigative Ophthalmology & Visual Science. 2000;41(7):1749–1758. [PubMed] [Google Scholar]
- 113.Ritch R, Schiewe M, Zink RC, et al. Latrunculin B (INS115644) reduces intraocular pressure (IOP) in ocular hypertension (OHT) and primary open angle glaucoma (POAG) Investigative Ophthalmology & Visual Science. 2010;51 ARVO E-Abstract and Poster 6432. [Google Scholar]
- 114.Bhattacharya SK, Rockwood EJ, Smith SD, et al. Proteomics reveal cochlin deposits associated with glaucomatous trabecular meshwork. Journal of Biological Chemistry. 2005;280(7):6080–6084. doi: 10.1074/jbc.M411233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kung C. A possible unifying principle for mechanosensation. Nature. 2005;436(7051):647–654. doi: 10.1038/nature03896. [DOI] [PubMed] [Google Scholar]
- 116.Patel AJ, Lazdunski M, Honoré E. Lipid and mechano-gated 2P domain K+ channels. Current Opinion in Cell Biology. 2001;13(4):422–427. doi: 10.1016/s0955-0674(00)00231-3. [DOI] [PubMed] [Google Scholar]
- 117.Goel M, Sienkiewicz AE, Picciani R, Wang J, Lee RK, Bhattacharya SK. Cochlin, intraocular pressure regulation and mechanosensing. PLoS ONE. 2012;7(4) doi: 10.1371/journal.pone.0034309.e34309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bhattacharya SK, Annangudi SP, Salomon RG, Kuchtey RW, Peachey NS, Crabb JW. Cochlin deposits in the trabecular meshwork of the glaucomatous DBA/2J mouse. Experimental Eye Research. 2005;80(5):741–744. doi: 10.1016/j.exer.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 119.Goel M, Sienkiewicz AE, Picciani R, Lee RK, Bhattacharya SK. Cochlin induced TREK-1 co-expression and annexin A2 secretion: role in trabecular meshwork cell elongation and motility. PLoS ONE. 2011;6(8) doi: 10.1371/journal.pone.0023070.e23070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Straiker AJ, Maguire G, Mackie K, Lindsey J. Localization of cannabinoid CB1 receptors in the human anterior eye and retina. Investigative Ophthalmology & Visual Science. 1999;40(10):2442–2448. [PubMed] [Google Scholar]
- 121.Yazulla S, Studholme KM, McIntosh HH, Deutsch DG. Immunocytochemical localization of cannabinoid CB1 receptor and fatty acid amide hydrolase in rat retina. Journal of Comparative Neurology. 1999;415(1):80–90. doi: 10.1002/(sici)1096-9861(19991206)415:1<80::aid-cne6>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 122.Pinar-Sueiro S, Rodríguez-Puertas R, Vecino E. Cannabinoid applications in glaucoma. Archivos de la Sociedad Espanola de Oftalmologia. 2011;86(1):16–23. doi: 10.1016/j.oftal.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 123.Chien FY, Wang R-F, Mittag TW, Podos SM. Effect of WIN 55212-2, a cannabinoid receptor agonist, on aqueous humor dynamics in monkeys. Archives of Ophthalmology. 2003;121(1):87–90. doi: 10.1001/archopht.121.1.87. [DOI] [PubMed] [Google Scholar]
- 124.Porcella A, Maxia C, Gessa GL, Pani L. The synthetic cannabinoid WIN55212-2 decreases the intraocular pressure in human glaucoma resistant to conventional therapies. European Journal of Neuroscience. 2001;13(2):409–412. doi: 10.1046/j.0953-816x.2000.01401.x. [DOI] [PubMed] [Google Scholar]
- 125.Njie YF, Kumar A, Qiao Z, Zhong L, Song Z-H. Noladin ether acts on trabecular meshwork cannabinoid (CB1) receptors to enhance aqueous humor outflow facility. Investigative Ophthalmology & Visual Science. 2006;47(5):1999–2005. doi: 10.1167/iovs.05-0729. [DOI] [PubMed] [Google Scholar]
- 126.Hudson BD, Beazley M, Szczesniak A-M, Straiker A, Kelly MEM. Indirect sympatholytic actions at β-adrenoceptors account for the ocular hypotensive actions of cannabinoid receptor agonists. Journal of Pharmacology and Experimental Therapeutics. 2011;339(3):757–767. doi: 10.1124/jpet.111.185769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Rösch S, Ramer R, Brune K, Hinz B. R+-methanandamide and other cannabinoids induce the expression of cyclooxygenase-2 and matrix metalloproteinases in human nonpigmented ciliary epithelial cells. Journal of Pharmacology and Experimental Therapeutics. 2006;316(3):1219–1228. doi: 10.1124/jpet.105.092858. [DOI] [PubMed] [Google Scholar]
- 128.Crooke A, Colligris B, Pintor J. Update in glaucoma medicinal chemistry: emerging evidence for the importance of melatonin analogues. Current Medicinal Chemistry. 2012;19(21):3508–3522. doi: 10.2174/092986712801323234. [DOI] [PubMed] [Google Scholar]
- 129.Pintor J, Peláez T, Hoyle CHV, Peral A. Ocular hypotensive effects of melatonin receptor agonists in the rabbit: further evidence for an MT3 receptor. British Journal of Pharmacology. 2003;138(5):831–836. doi: 10.1038/sj.bjp.0705118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Alcantara-Contreras S, Baba K, Tosini G. Removal of melatonin receptor type 1 increases intraocular pressure and retinal ganglion cells death in the mouse. Neuroscience Letters. 2011;494(1):61–64. doi: 10.1016/j.neulet.2011.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Babizhayev MA, Bunin Ya. A. Lipid peroxidation in open-angle glaucoma. Acta Ophthalmologica. 1989;67(4):371–377. doi: 10.1111/j.1755-3768.1989.tb01617.x. [DOI] [PubMed] [Google Scholar]
- 132.Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402(6762):656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 133.Hosoda H, Kojima M, Matsuo H, Kangawa K. Ghrelin and des-acyl ghrelin: two major forms of rat ghrelin peptide in gastrointestinal tissue. Biochemical and Biophysical Research Communications. 2000;279(3):909–913. doi: 10.1006/bbrc.2000.4039. [DOI] [PubMed] [Google Scholar]
- 134.Rocha-Sousa A, Saraiva J, Henriques-Coelho T, Falcão-Reis F, Correia-Pinto J, Leite-Moreira AF. Ghrelin as a novel locally produced relaxing peptide of the iris sphincter and dilator muscles. Experimental Eye Research. 2006;83(5):1179–1187. doi: 10.1016/j.exer.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 135.Rocha-Sousa A, Alves-Faria P, Falcão-Pires I, Falcão-Reis F, Leite-Moreira AF. Analyses of aqueous humour ghrelin levels of eyes with and without glaucoma. British Journal of Ophthalmology. 2009;93(1):131–132. doi: 10.1136/bjo.2007.134627. [DOI] [PubMed] [Google Scholar]
- 136.Katsanos A, Dastiridou A, Georgoulias P, Cholevas P, Kotoula M, Tsironi EE. Plasma and aqueous humour levels of ghrelin in open-angle glaucoma patients. Clinical and Experimental Ophthalmology. 2011;39(4):324–329. doi: 10.1111/j.1442-9071.2010.02463.x. [DOI] [PubMed] [Google Scholar]
- 137.Rocha-Sousa A, Tavares-Silva M, Pereira-Silva P, et al. Ghrelin is expressed in the ciliary body and presents a significant and sustained hypotensive effect in acute glaucoma models. ARVO Meeting Abstracts. 2012;53 [Google Scholar]
- 138.Danser AH, Derkx FH, Admiraal PJ, Deinum J, de Jong PT, Schalekamp MA. Angiotensin levels in the eye. Investigative Ophthalmology & Visual Science. 1994;35(3):1008–1018. [PubMed] [Google Scholar]
- 139.Vaajanen A, Vapaatalo H. Local ocular renin-angiotensin system—a target for glaucoma therapy? Basic and Clinical Pharmacology and Toxicology. 2011;109(4):217–224. doi: 10.1111/j.1742-7843.2011.00729.x. [DOI] [PubMed] [Google Scholar]
- 140.Ramirez M, Davidson EA, Luttenauer L, et al. The renin-angiotensin system in the rabbit eye. Journal of Ocular Pharmacology and Therapeutics. 1996;12(3):299–312. doi: 10.1089/jop.1996.12.299. [DOI] [PubMed] [Google Scholar]
- 141.Wang R-F, Podos SM, Mittag TW, Yokoyoma T. Effect of CS-088, an angiotensin AT1 receptor antagonist, on intraocular pressure in glaucomatous monkey eyes. Experimental Eye Research. 2005;80(5):629–632. doi: 10.1016/j.exer.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 142.Tikellis C, Bernardi S, Burns WC. Angiotensin-converting enzyme 2 is a key modulator of the renin-angiotensin system in cardiovascular and renal disease. Current Opinion in Nephrology and Hypertension. 2011;20(1):62–68. doi: 10.1097/MNH.0b013e328341164a. [DOI] [PubMed] [Google Scholar]
- 143.Vaajanen A, Vapaatalo H, Kautiainen H, Oksala O. Angiotensin (1–7) reduces intraocular pressure in the normotensive rabbit eye. Investigative Ophthalmology & Visual Science. 2008;49(6):2557–2562. doi: 10.1167/iovs.07-1399. [DOI] [PubMed] [Google Scholar]
- 144.Reitsamer HA, Kiel JW. Relationship between ciliary blood flow and aqueous production in rabbits. Investigative Ophthalmology & Visual Science. 2003;44(9):3967–3971. doi: 10.1167/iovs.03-0088. [DOI] [PubMed] [Google Scholar]
- 145.Costagliola C, Parmeggiani F, Semeraro F, Sebastiani A. Selective serotonin reuptake inhibitors: a review of its effects on intraocular pressure. Current Neuropharmacology. 2008;6(4):293–310. doi: 10.2174/157015908787386104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gabelt BT, Okka M, Dean TR, Kaufman PL. Aqueous humor dynamics in monkeys after topical R-DOI. Investigative Ophthalmology & Visual Science. 2005;46(12):4691–4696. doi: 10.1167/iovs.05-0647. [DOI] [PubMed] [Google Scholar]
- 147.Sharif NA, McLaughlin MA, Kelly CR. AL-34662: a potent, selective, and efficacious ocular hypotensive serotonin-2 receptor agonist. Journal of Ocular Pharmacology and Therapeutics. 2007;23(1):1–13. doi: 10.1089/jop.2006.0093. [DOI] [PubMed] [Google Scholar]
- 148.Bunch TJ, Seeman JL, Gabelt BT, et al. BVT.28949 as a potential ocular hypotensive agent in monkeys. Investigative Ophthalmology & Visual Science. 2004;45 ARVO E-Abstract 5040. [Google Scholar]
- 149.Wagh VD, Patil PN, Surana SJ, Wagh KV. Forskolin: upcoming antiglaucoma molecule. Journal of Postgraduate Medicine. 2012;58(3):199–202. doi: 10.4103/0022-3859.101396. [DOI] [PubMed] [Google Scholar]
- 150.Metzger H, Lindner E. The positive inotropic-acting forskolin, a potent adenylatecyclase activator. Arzneimittel-Forschung. 1981;31(8):1248–1250. [PubMed] [Google Scholar]
- 151.Pescosolido N, Librando A. Oral administration of an association of forskolin, rutin and vitamins B1 and B2 potentiates the hypotonising effects of pharmacological treatments in POAG patients. La Clinica terapeutica. 2010;161(3):e81–e85. [PubMed] [Google Scholar]