

Abstract
We report the synthesis of a GDP analogue, SML-8-73-1, and a prodrug derivative, SML-10-70-1, which are selective, direct-acting covalent inhibitors of the K-Ras G12C mutant relative to wild-type Ras. Biochemical and biophysical measurements suggest that modification of K-Ras with SML-8-73-1 renders the protein in an inactive state. These first-in-class covalent K-Ras inhibitors demonstrate that irreversible targeting of the K-Ras guanine-nucleotide binding site is potentially a viable therapeutic strategy for inhibition of Ras signaling.
Keywords: Cancer, Covalent inhibitor, Drug design, K-Ras
Ras proteins are members of a large family of GTPase enzymes that are essential to transducing extracellular signals into diverse cellular responses such as proliferation, apoptosis and differentiation.[1] Ras operates as a molecular switch that is activated when growth factors bind to extracellular receptors which induce nucleotide exchange from GDP to GTP.[2–4] Wild-type Ras proteins possess a slow intrinsic GTPase activity for hydrolysis of GTP to GDP, a reaction enhanced by GTPase activating proteins (GAPs), which halt Ras signaling by switching Ras into an inactive GDP-bound signaling state. Mutations which diminish the GTPase activity or induce GAP insensitivity result in constitutively activated signaling pathways,[5] leading to deregulated cell growth, inhibition of cell death, invasiveness, and induction of angiogenesis. About 30% of all human cancers harbor activating Ras mutations making them one of the most common known genetic causes of cancer.[6,7] Moreover, cancers with high prevalence of K-Ras mutations such as lung cancer and pancreatic cancer are difficult to treat and clinical outcomes are poor even with aggressive and toxic medical interventions. Despite more than 20 years of effort in industry and academia, Ras has proven highly difficult to drug and no effective targeted therapy currently exists.[8–10] Small molecules targeting the guanine nucleotide (GN) binding site of GTPases like Ras have been largely ignored because both GTP and GDP bind to Ras with subnanomolar affinity and their intracellular concentrations are very high, leading to the widely-held conclusion that development of GN binding site-directed inhibitors is not possible.
In light of the difficulties with GTPase inhibitor development, we reasoned that a covalent approach targeting one of the more potent Ras oncogenic mutants may be feasible. Of the oncogenic Ras family members (H, K, N), K-Ras is frequently mutated with most cancer causing mutations at codons 12, 13 and 61.[6,7] G12C is a naturally occurring activating K-Ras mutation present in roughly 10–20% of all Ras-driven cancers, and roughly 50% of Ras-driven lung adenocarcinomas.[11–13] This mutation places a solvent-accessible cysteine adjacent to the active site, near the usual position of the gamma-phosphate of the native GTP and constitutively activates K-Ras (Figure 1). We hypothesized that small molecules could be developed to selectively target this activating mutation. Our group has successfully developed selective covalent kinase inhibitors,[14–16] which irreversibly occupy the ATP binding site and prevent ATP loading.[17] We envisioned that a GN based molecule which could covalently modify the GN binding site of Ras would overcome challenges in targeting K-Ras for the following reasons: (A) the guanine nucleotide scaffold would preserve many of the non-covalent interactions with Ras and (B) covalent bond formation in the GN binding site could overcome the problem of high competing nucleotide concentration by preventing further exchange upon covalent addition. As a proof of concept, we present our efforts to develop a selective, covalent Ras inhibitor that targets the GN binding site directly, and overcomes the high intracellular concentrations of GDP and GTP that would preclude binding of a non-covalent inhibitor.
Figure 1.
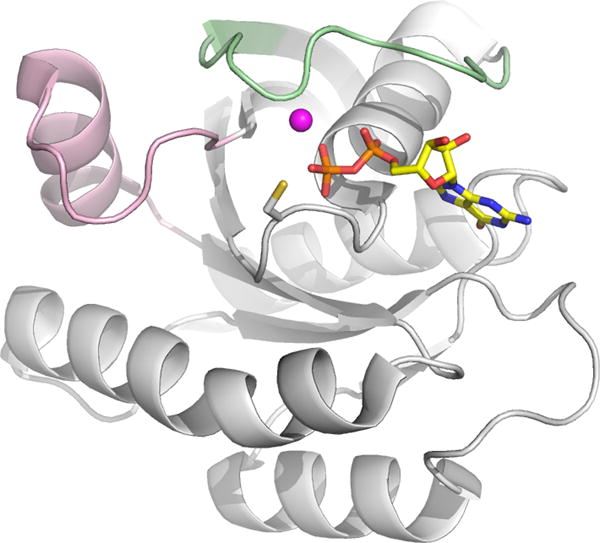
Model of K-Ras G12C bound to GDP (sticks). Cysteine 12 is located in the P-loop opposite the catalytic magnesium (magenta ball) and switches 1 (green) and 2 (pink). Model based on PDB: 4EPR.
We presumed that structures utilizing a GDP scaffold would induce an inactive K-Ras conformation and designed several diphosphate compounds varying the identity of electrophile and linker length between the β-phosphate and electrophile. Molecular docking studies using a homology model of K-Ras G12C which was adapted from a K-Ras crystal structure (PDB ID: 3GFT) allowed design of promising candidates which were prioritized for synthesis. SML-8-73-1 (1) appeared to have favorable geometric properties for reaction with the cysteine in position 12 (Figure S1). The synthesis of SML-8-73-1 was achieved by reacting mono-phosphate intermediate 2 with guanosine mono-phosphate morpholidate 3 followed by incorporation of an electrophile (Scheme 1). Monophosphate 2 was prepared by tetrazole facilitated coupling of N-boc ethanolamine with dibenzyl N,N-diisopropylphosphoramidite at ambient temperature followed by oxidation with m-CPBA. The resulting dibenzyl phosphate ester was subjected to hydrogenolysis in the presence of triethylamine (TEA) to provide monophosphate 2 as a TEA salt. Di-phosphate 4 was prepared by reaction of 2 and 3 in the presence of 5-(ethylthio)-1H-tetrazole. We noticed that the free acid form of 2 is insoluble in pyridine and did not react with 3 to produce 4 suggesting that utilization of the TEA salt of 2 is critical to achieve good solubility of the mono-phosphate. Deprotection of Boc group and subsequent formation of α-chloroacetamide completed the synthesis of SML-8-73-1 (1), which was purified by preparative reverse-phase HPLC.
Scheme 1.
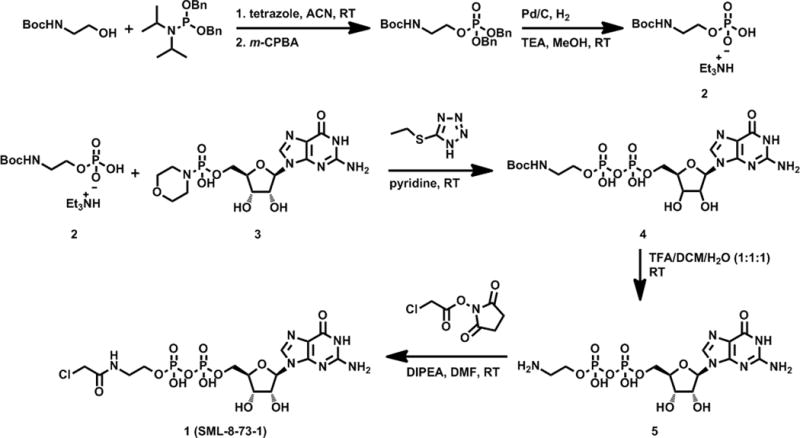
Synthesis of SML-8-73-1.
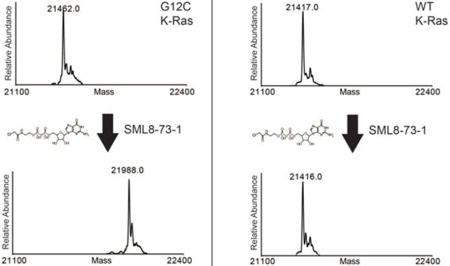
To determine if SML-8-73-1 could covalently modify K-Ras G12C, we incubated purified recombinant K-Ras at a concentration of 50 μM for 2 hours at 37°C in PBS with the inhibitor at a concentration of 2.5 mM. Electrospray ionization mass spectrometry revealed no addition of SML-8-73-1 to the wild type protein (Figures 2A,B). In contrast, a single adduct was detected when K-Ras G12C was incubated with SML-8-73-1 (Figures 2C,D). Under these conditions a molar ratio of 1:10 (K-Ras G12C:SML-8-73-1) was required for complete incorporation of compound (Figure S2). Proteolytic digestion and analysis of resulting peptides by nano LC/MS demonstrated exclusive covalent labeling of the GN-site of K-Ras G12C (cysteine residue 12; Figure 2E).
Figure 2.
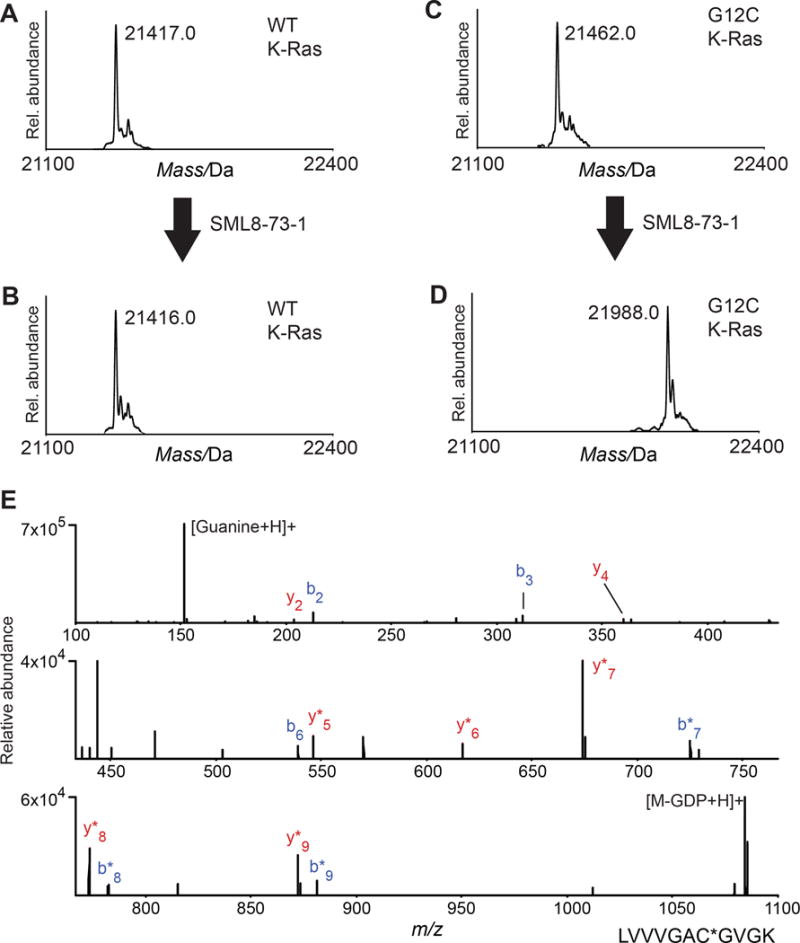
SML-8-73-1 reacts quantitatively with K-Ras G12C, but does not label WT K-Ras. Deconvoluted electrospray mass spectra obtained for (A,B) WT K-Ras and (C,D) K-Ras G12C (A,C) before and (B,D) after incubation with 2.5 mM SML-8-73-1. A mass increase of 526 Da (compound-HCl) was observed only after treatment of K-Ras G12C, indicating covalent labeling of the protein. (E) HCD MS/MS spectrum of SML-8-73-1 modified K-Ras G12C peptide (residues 6–16). Ions of type b and y are shown in blue and red, respectively, and localize the modification to C12. GDP, guanosine diphosphate; *, loss of GDP.
It is possible that the high concentrations of GTP and GDP within cells may prevent compounds like SML-8-73-1 from making productive interactions with K-Ras G12C. To investigate this we simulated cellular conditions by repeating the above experiment with the addition of 1 mM GDP and 1 mM GTP. We found that under identical incubation conditions K-Ras G12C is more than 95% labelled within 2 hours, similar to what we observe in the absence of competing nucleotides (Figure S3).
To investigate whether covalent labeling of K-Ras G12C by SML-8-73-1 stabilizes the active or inactive conformation of the protein, we used hydrogen exchange (HX) mass spectrometry (MS). HX MS provides a means to assess protein conformational dynamics by measuring the rate of hydrogen exchange of backbone amide hydrogens.[18] We performed HX MS on K-Ras G12C bound to a non-hydrolyzable GTP mimic (GMPPNP), GDP, or SML-8-73-1. Hydrogen exchange into each species was compared by monitoring the deuteration of over 40 peptides that were produced after labeling and proteolytic digestion. In most of the protein, there was no difference in deuterium incorporation between all three states (Figure S4). The regions that did show differences are summarized in Figure 3.
Figure 3.
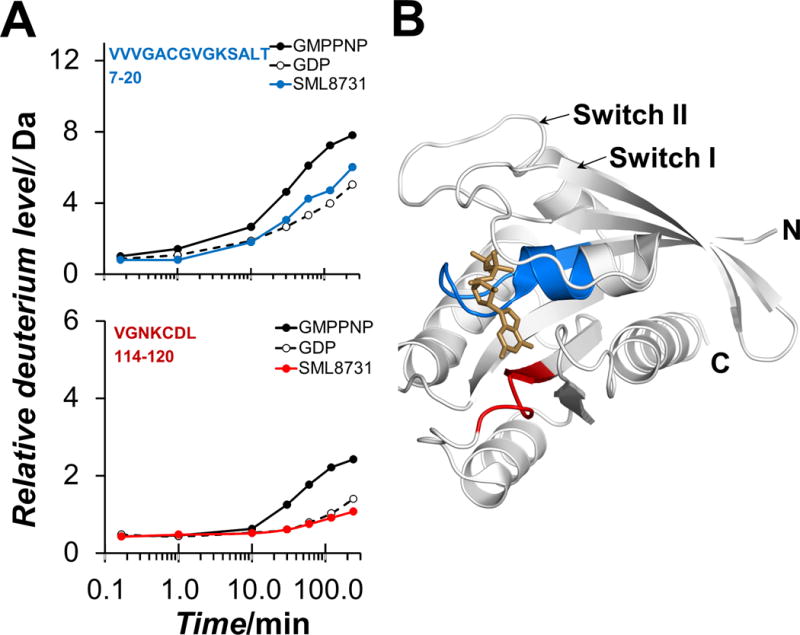
Summary of differences in deuterium exchange in K-Ras G12C bound to GMPPNP, GDP, and SML-8-73-1. Hydrogen deuterium exchange suggests that SML-8-73-1 places K-Ras G12C in the inactive state. (A) Relative deuterium uptake curves for two key peptides (top: residues 7–20, VVVGACGVGKSALT; bottom: residues 114–120, VGNKCDL) showing differences in deuterium incorporation. These are representative data: each data point is the average of duplicate mass determinations from one of three triplicates of the entire experimental measurement (see supporting information). (B) Location of the two regions from panel A, on PDB file 4Q21. GDP is shown in brown, residues 7–20 in blue, and residues 114–120 in red.
Residues 7–20 showed significantly more deuterium incorporation in the active conformation (GMPPNP-bound) compared to the other conformation (Figure 3A, top) while residues 114–120 showed slightly higher deuterium incorporation in the active state compared to the other states (Figure 3A, bottom). Both of the regions with significant differences in deuteration comprise portions of the nucleotide binding pocket (Figure 3B), with residues 7–20 in close proximity to the phosphate groups and residues 114–120 adjacent to the guanosine moiety. When K-Ras G12C was bound to covalent inhibitor SML-8-73-1, the deuterium incorporation in residues 7–20 and 114–120 mimicked that observed in the GDP-bound state (Figure 3A) suggesting that the compound likely stabilizes an inactive form of K-Ras G12C.
To corroborate that SML-8-73-1 stabilizes K-Ras G12C in an inactive state we designed a functional biochemical assay using AlphaScreen® (Perkin Elmer) technology to measure the affinity of K-Ras:nucleotide complexes for the Ras Binding Domain (RBD) of the Raf protein kinase. K-Ras preferentially binds the Raf RBD in its active, GTP-bound state.[19–21] Examination of a homology model of the RBD:K-Ras complex (based on PDB ID 1C1Y) reveals that a solvent exposed cysteine (cys 95) sits on the face of RBD opposite the K-Ras binding side. We coupled biotin to this cysteine in purified RBD.[22,23] using maleimide chemistry (Thermo Scientific) resulting in covalent addition of one biotin per RBD molecule, as verified by HABA/Avidin assay (Figure S5). N-terminally FLAG tagged K-Ras G12C preloaded with the GTP analogue GNPPNP was incubated with biotinylated Raf-RBD to form complexes. We performed a competition assay by adding untagged K-Ras G12C, which had been preloaded with various test molecules, over a range of concentrations. The assay was developed with Alpha donator and acceptor beads directed against the FLAG tag or biotin respectively. We tested multiple nucleotides for their ability to induce K-Ras G12C:Raf binding. The GDP:K-Ras G12C complex had a lower affinity for RBD (EC50 of 22.6 nM; 95% CI, 17.5–32.0 nM), than the GTP:K-Ras G12C complex (EC50 of 6.6 nM; 95% CI, 5.1–8.5 nM). Labeling of K-Ras G12C with SML-8-73-1 at a 1:10 molar ratio to K-Ras G12C prior to the assay yields a curve which is superimposable with that obtained when K-Ras G12C is treated with GDP, consistent with the notion that the compound stabilizes an inactive conformation of K-Ras G12C (Figure 4).
Figure 4.
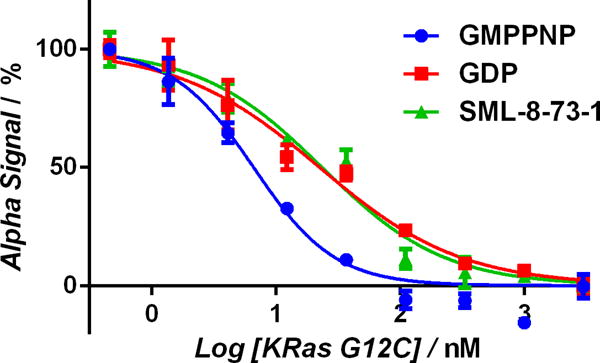
Alpha assay confirms that modification with SML-8-73-1 renders K-Ras G12C biochemically inactive. Untagged GMPPNP-loaded K-Ras G12C was incubated with a 10 fold molar excess GDP (red), SML-8-73-1 (green) or buffer (blue). Under these conditions K-Ras G12C becomes completely labelled by SML-8-73-1 or loaded with GDP or GMPPNP. These K-Ras G12C preparations were then added at the indicated concentrations to compete for binding with Alpha-tagged K-Ras G12C (donor):Raf-RBD (acceptor) complexes which had been pre-formed. SML-8-73-1-bound K-Ras G12C (green) resembles GDP-bound K-Ras G12C (red) with respect to affinity for Raf-RBD.
We further confirmed the results of the Alpha assay using an FP variant of the K-Ras-RBD binding assay wherein the RBD was labelled with the fluorescent dye Oregon Green (Figure S6) and the protein binding-dependent FP signal was measured. GDP-bound K-Ras G12C mimicked the SML-8-73-1 labeled protein, and both possessed lower affinity for K-Ras G12C bound to the GTP analogue GMPPNP (Figures S7). Wild type K-Ras behaved similarly in this assay suggesting that the G12C mutation does not significantly affect the interaction between K-Ras and RBD or between K-Ras and the nucleotides GTP and GDP (Figure S8).
SML-8-73-1 cannot pass through the cell membrane because it contains two ne gatively charged phosphate groups. “Caging” is a method whereby charged ions can be chemically modified to mask the charged group and thereby allow for passive cellular uptake.[24] The caging moiety is typically designed to be removed intracellularly as consequence of enzymatic cleavage. Caged bisphosphates, where both phosphates are modified, have not been reported and indeed we discovered that these compounds were unstable as a consequence of hydrolysis of the phosphate ester. However, we were able to modify the beta phosphate as an alanine ester phosphoramidate, resulting in a partially-caged version of SML-8-73-1, SML-10-70-1 (Scheme S2, Figure 5). This phosphoramidate has been previously used to ‘cage’ phosphates and intracellular hydrolysis of the lipophilic ester is reported to result in regeneration of the phosphate group.[25]
Figure 5.
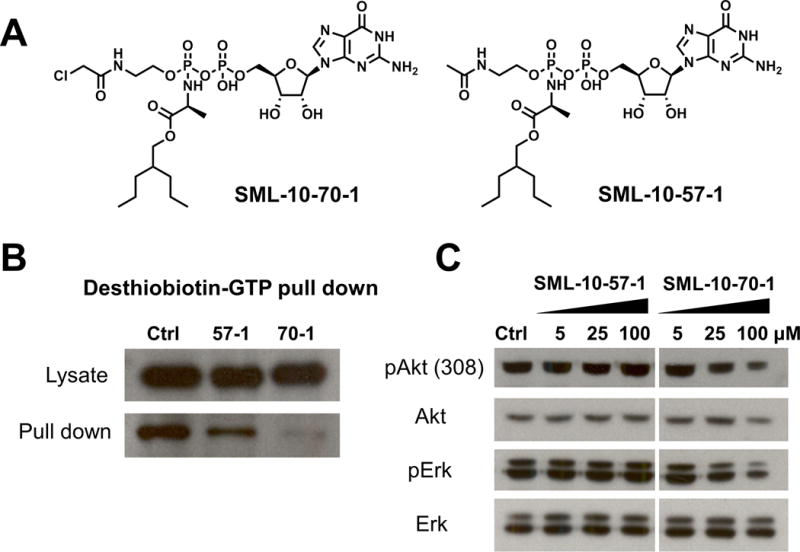
The caged analogue SML-10-70-1 is cell permeable and disrupts Ras signaling. (A) Chemical structrues of caged SML-10-70-1 and the non-reactive negative control, SML-10-57-1. (B) Treatment of H358 cells with SML-10-70-1 at 100 μM prior to probing with desthiobiotin-GTP decreases the amount of K-Ras which can be pulled down with streptavidin as compared to cells treated with non-reactive control suggeting that SML-10-70-1 penetrates into cells and accesses the active site of K-Ras-G12C. (C) Treatment of H358 cells with SML-10-70-1 decreases levels of pErk and pAkt as compared to treatment with negative control (SML-10-57-1) suggesting a compound-dependent effect on K-Ras signaling.
We next investigated whether SML-10-70-1 is capable of penetrating cell membranes and covalently engaging K-Ras G12C. We designed a cellular ‘target engagement’ assay where we measured the ability of SML-10-70-1 to protect K-Ras G12C from subsequent labelling with desthiobiotin-GTP in lysates. Desthiobiotin-GTP is able to react with a conserved lysine in the K-Ras GTP site such that K-Ras becomes covalently biotinylated.[26] The K-Ras G12C expressing H358 cells were treated with SML-10-70-1 or a negative control containing no electrophile (SML-10-57-1) for 6 hours prior to lysis. Lysates were incubated with desthiobiotin-GTP and biotinylated proteins recovered using streptavidin beads. Treatment of cells with SML-10-70-1 decreased the recovery of biotinylated-Ras suggesting that SML-10-70-1 is able to penetrate into cells and effectively competes for the GTP binding pocket (Figure 5B). We investigated the effects of SML-10-70-1 on signalling to Erk and Akt, two key K-Ras effector pathways. SML-10-70-01 but not the non-covalent SML-10-57-1 was able to attenuate Akt and Erk phosphorylation albeit only at a high concentration of 100 μM.
We further tested for anti-proliferative activity of SML-10-70-01 in H23, H358 and A549 cells. H23 and H358 cells express the K-Ras G12C mutation and are considered K-Ras dependent for growth, while A549 cells have a G12S mutation and are considered K-Ras independent.[27] Upon treatment with SML-10-70-1, antiproliferative effects were seen in all lines with an EC50 of 43.8 μM, 47.6 μM and 26.6 μM for A549, H23 and H358 cells respectively.
Efforts to directly target Ras proteins have largely proven unsuccessful. Targeting farnesyl transferase (FT) which adds an isoprenyl group to the C-terminus of Ras and is essential for Ras signaling appeared promising in preclinical studies, but did not progress beyond early phase trials because of toxicity and lack of efficacy, likely because of compensatory geranylgeranyl transferase or other isoprenyl transferase activity for K-Ras and N-Ras.[28] Peptides have been used to disrupt protein-protein interactions between Ras and downstream signaling molecules but this approach has not produced clinically useful agents because the peptides had poor ‘drug-like’ properties. Along similar lines, small molecule ligands which bind to the surface of K-Ras are also in early stages of development.[10] Antisense oligonucleotides directed against K-Ras have demonstrated efficacy in cell culture but have not been advanced to clinical investigation.[9] Notably, recent work targeting the prenyl-bindng pocket of phosphodiesterase 6 (PDE6) delta subunit (PDEδ) has shown promise as a means of disrupting oncogenic signaling by K-Ras.[29] PDEδ regulates the spatial distribution of Ras family proteins by solubilizing depalmitoylated Ras proteins and facilitating diffusion to the Golgi for palmitoylation. Downregulation of PDEδ or disruption of the Ras:PDEδ interaction by small molecule inhibitors modulates oncogenic Ras signaling and results in an anti-proliferative effect on Ras-transformed cells.[30]
As another potential approach to directly targeting oncogenic Ras, we have synthesized a GDP analogue, SML-8-73-1, containing an electrophilic chloroacetamide attached to the beta phosphate which can covalently modify cysteine 12 of K-Ras G12C. The covalent modification is specific to mutant K-Ras G12C when compared to wild-type protein, and would therefore specifically target those Ras-driven cancers in which the K-Ras G12C mutation is present (i.e., 10–20% of all Ras-driven cancers and roughly 50% of Ras-driven lung adenocarcinomas). Labeling with SML-8-73-1 stabilizes an inactive form of K-Ras G12C as assessed by biochemical and biophysical means. This work demonstrates that specific GN-directed covalent inhibitors of K-Ras G12C can be prepared and can compete with GTP and GDP for active site binding in a cellular context. Extending this paradigm to other Ras forms may be possible by specifically targeting other active site side chains such as lysine. SML-8-73-1 possesses two negative charges and appears to be cell impermeable. A caged version of SML-8-73-1 is able to penetrate into cells, but only at high concentrations where it likely shows limited target selectivity as evidenced by the general cytotoxicity independent of K-Ras G12C status in lung cancer cell lines. Future efforts will explore cellular delivery strategies including alternative pro-drugs, non-charged phosphate bioisosteres and encapsulation strategies that will be required to assess the potential of this pharmacological approach in cellular and in vivo models of K-Ras driven cancer.
Supplementary Material
Footnotes
This work was supported by CPRIT (R1207), The Welch Foundation (I1829), NIH P01NS047572, the Strategic Research Initiative at the Dana Farber Cancer Institute (JAM), the Dana Farber Cancer Institute/Northeastern University Joint Program in Cancer Drug Development, NIH GM101135 (J.R.E.), a research collaboration with the Waters Corp (J.R.E.), Sally Gordon Fellowship of the Damon Runyon Cancer Research Foundation (DRG 112-12) (M.E.P.), DOD Breast Cancer Research Program Postdoctoral Fellowship (BC120208) (ME.P.).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Sang Min Lim, Department of Cancer Biology, Dana-Farber Cancer Institute, 450 Brookline Avenue, Boston, MA 02215; Department of Biological Chemistry & Molecular Pharmacology, Harvard Medical School, 250 Longwood Avenue, Boston, MA 02115; Department of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA 02138.
Kenneth D. Westover, Departments of Biochemistry and Radiation Oncology, The University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd. Dallas, TX 75390.
Scott B. Ficarro, Department of Cancer Biology, Dana-Farber Cancer Institute, 450 Brookline Avenue, Boston, MA 02215 Blais Proteomics Center, Dana-Farber Cancer Institute, 450 Brookline Avenue, Boston, MA 02115.
Rane A. Harrison, Department of Chemistry and Chemical Biology, Northeastern University, 360 Huntington Avenue, Boston, MA 02115
Hwan Geun Choi, Department of Cancer Biology, Dana-Farber Cancer Institute, 450 Brookline Avenue, Boston, MA 02215; Department of Biological Chemistry & Molecular Pharmacology, Harvard Medical School, 250 Longwood Avenue, Boston, MA 02115.
Michael E. Pacold, Department of Cancer Biology, Dana-Farber Cancer Institute, 450 Brookline Avenue, Boston, MA 02215 Whitehead Institute for Biomedical Research, 9 Cambridge Center, Cambridge, MA 02139.
Martin Carrasco, Departments of Biochemistry and Radiation Oncology, The University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd. Dallas, TX 75390.
John Hunter, Departments of Biochemistry and Radiation Oncology, The University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd. Dallas, TX 75390.
Nam Doo Kim, New Drug Development Center, Daegu-Gyeongbuk Medical Innovation Foundation, Daegu, 706–010, South Korea.
Ting Xie, Department of Cancer Biology, Dana-Farber Cancer Institute, 450 Brookline Avenue, Boston, MA 02215; Department of Biological Chemistry & Molecular Pharmacology, Harvard Medical School, 250 Longwood Avenue, Boston, MA 02115; Department of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA 02138.
Taebo Sim, Chemical Kinomics Research Center, Korea Institute of Science and Technology, Seoul, 130–650, South Korea.
Pasi A. Jänne, Department of Medical Oncology, Dana-Farber Cancer Institute, 450 Brookline Avenue, Boston, MA 02115
Matthew Meyerson, Department of Medical Oncology, Dana-Farber Cancer Institute, 450 Brookline Avenue, Boston, MA 02115; Broad Institute of Harvard and MIT, 320 Charles St., Cambridge, MA 02141; Department of Pathology, Harvard Medical School, 77 Avenue Louis Pasteur, Boston, MA 02115.
Jarrod A. Marto, Department of Cancer Biology, Dana-Farber Cancer Institute, 450 Brookline Avenue, Boston, MA 02215 Blais Proteomics Center, Dana-Farber Cancer Institute, 450 Brookline Avenue, Boston, MA 02115.
John R. Engen, Department of Chemistry and Chemical Biology, Northeastern University, 360 Huntington Avenue, Boston, MA 02115
Nathanael S. Gray, Department of Cancer Biology, Dana-Farber Cancer Institute, 450 Brookline Avenue, Boston, MA 02215; Department of Biological Chemistry & Molecular Pharmacology, Harvard Medical School, 250 Longwood Avenue, Boston, MA 02115.
References
- 1.Barbacid M. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 2.Milburn MV, Tong L, deVos AM, Brunger A, Yamaizumi Z, Nishimura S, Kim S-H. Science. 1990;247:939–945. doi: 10.1126/science.2406906. [DOI] [PubMed] [Google Scholar]
- 3.Kim S-H, Privé GG, Milburn MV. In: Handbook of Experimental Pharmacology, Vol. 108/I: GTPases in Biology. Dickey BF, Birnbaumer L, editors. Springer-Verlag; Berlin Heidelberg: 1993. pp. 177–194. [Google Scholar]
- 4.Vetter IR, Wittinghofer A. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 5.Scheffzek K, Ahmadian MR, Kabsch W, Wiesmüller L, Lautwein A, Schmitz F, Wittinghofer A. Science. 1997;277:333–338. doi: 10.1126/science.277.5324.333. [DOI] [PubMed] [Google Scholar]
- 6.Bos JL. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 7.Prior IA, Lewis PD, Mattos C. Cancer Res. 2012;72:2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Downward J. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 9.Gysin S, Salt M, Young A, McCormick F. Genes Cancer. 2011;2:359–372. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang W, Fang G, Rudolph J. Bioorg Med Chem Lett. 2012;22:5766–5776. doi: 10.1016/j.bmcl.2012.07.082. [DOI] [PubMed] [Google Scholar]
- 11.Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR, Futreal PA. Nucleic Acids Res. 2011;39:D945–950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones S, Lambert S, Williams GT, Best JM, Sampson JR, Cheadle JP. Br J Cancer. 2004;90:1591–1593. doi: 10.1038/sj.bjc.6601747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greulich H, Kaplan B, Mertins P, Chen TH, Tanaka KE, Yun CH, Zhang X, Lee SH, Cho J, Ambrogio L, Liao R, Imielinski M, Banerji S, Berger AH, Lawrence MS, Zhang J, Pho NH, Walker SR, Winckler W, Getz G, Frank D, Hahn WC, Eck MJ, Mani DR, Jaffe JD, Carr SA, Wong KK, Meyerson M. Proc Natl Acad Sci U S A. 2012;109:14476–14481. doi: 10.1073/pnas.1203201109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, Cortot AB, Chirieac L, Iacob RE, Padera R, Engen JR, Wong KK, Eck MJ, Gray NS, Jänne PA. Nature. 2009;462:1070–1074. doi: 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou W, Hur W, McDermott U, Dutt A, Xian W, Ficarro SB, Zhang J, Sharma SV, Brugge J, Meyerson M, Settleman J, Gray NS. Chem Biol. 2010;17:285–295. doi: 10.1016/j.chembiol.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang T, Inesta-Vaquera F, Niepel M, Zhang J, Ficarro SB, Machleidt T, Xie T, Marto JA, Kim N, Sim T, Laughlin JD, Park H, LoGrasso PV, Patricelli M, Nomanbhoy TK, Sorger PK, Alessi DR, Gray NS. Chem Biol. 2012;19:140–154. doi: 10.1016/j.chembiol.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Yang PL, Gray NS. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marcsisin SR, Engen JR. Anal Bioanal Chem. 2010;397:967–972. doi: 10.1007/s00216-010-3556-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moodie SA, Willumsen BM, Weber MJ, Wolfman A. Science. 1993;260:1658–1661. doi: 10.1126/science.8503013. [DOI] [PubMed] [Google Scholar]
- 20.Vojtek AB, Hollenberg SM, Cooper JA. Cell. 1993;74:205–214. doi: 10.1016/0092-8674(93)90307-c. [DOI] [PubMed] [Google Scholar]
- 21.Zhang XF, Settleman J, Kyriakis JM, Takeuchi-Suzuki E, Elledge SJ, Marshall MS, Bruder JT, Rapp UR, Avruch J. Nature. 1993;364:308–313. doi: 10.1038/364308a0. [DOI] [PubMed] [Google Scholar]
- 22.Nassar N, Horn G, Herrmann C, Block C, Janknecht R, Wittinghofer A. Nat Struct Biol. 1996;3:723–729. doi: 10.1038/nsb0896-723. [DOI] [PubMed] [Google Scholar]
- 23.Nassar N, Horn G, Herrmann C, Scherer A, McCormick F, Wittinghofer A. Nature. 1995;375:554–560. doi: 10.1038/375554a0. [DOI] [PubMed] [Google Scholar]
- 24.Adams SR, Tsien RY. Annu Rev Physiol. 1993;55:755–784. doi: 10.1146/annurev.ph.55.030193.003543. [DOI] [PubMed] [Google Scholar]
- 25.Hecker SJ, Erion MD. J Med Chem. 2008;51:2328–2345. doi: 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]
- 26.Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, Kozarich JW. Biochemistry. 2007;46:350–358. doi: 10.1021/bi062142x. [DOI] [PubMed] [Google Scholar]
- 27.Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, Settleman J. Cancer Cell. 2009;15:489–500. doi: 10.1016/j.ccr.2009.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ellis CA, Clark G. Cell Signal. 2000;12:425–434. doi: 10.1016/s0898-6568(00)00084-x. [DOI] [PubMed] [Google Scholar]
- 29.Zimmermann G, Papke B, Ismail S, Vartak N, Chandra A, Hoffmann M, Hahn SA, Triola G, Wittinghofer A, Bastiaens PI, Waldmann HG. Nature. 2013;497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 30.Chandra A, Grecco HE, Pisupati V, Perera D, Cassidy L, Skoulidis F, Ismail SA, Hedberg C, Hanzal-Bayer M, Venkitaraman AR, Wittinghofer A, Bastiaens PI. Nat Cell Biol. 2012;14:148–158. doi: 10.1038/ncb2394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.