

Abstract
Elevated high mobility group A (HMGA) protein expression in pancreatic cancer cells is correlated with resistance to the chemotherapy agent gemcitabine. Here, we demonstrate use of HMGA-targeted AT-rich phosphorothioate DNA (AT-sDNA) aptamers to suppress HMGA carcinogenic activity. Cell growth of human pancreatic cancer cells (AsPC-1 and Miapaca-2) transfected with AT-sDNA were monitored after treatment with gemcitabine. Significant increases in cell death in AT-sDNA transfected cells compared to non-AT-rich sDNA treated cells were observed in both cell lines. The data indicate the potential use of HMGA targeted DNA aptamers to enhance chemotherapy efficacy in pancreatic cancer treatment.
Keywords: HMGA, DNA aptamer, Pancreatic cancer, Gemcitabine, AsPC-1
1. Introduction
Pancreatic adenocarcinoma was the fourth leading cause of cancer related deaths in the United States in 2010 with an estimated 43,140 new cases and 36,800 deaths [1]. A primary cause for the high mortality rates associated with pancreatic cancer is the lack of biomarkers for early detection and diagnosis. Since pancreatic cancer has no noticeable signs or symptoms in the early stages, most diagnoses occur at an advanced stage when surgical resection is not feasible. Current treatments for pancreatic cancer include chemotherapy (gemcitabine) [2,3], targeted therapy by small molecules like tarceva [4,5], radiation therapy and surgery [6]. Despite such treatments, the post-diagnosis 5-year survival rate for pancreatic cancer patients remains less than 5%.
Elevated levels of high-mobility group A (HMGA) protein expression have been reported in almost every type of human cancer [7–9]. There are two forms of HMGA proteins, HMGA1 and HMGA2, which are encoded from two different genes [10]. Both forms of HMGA are non-histone chromatin architectural transcription factors found broadly in eukaryotes [11]. HMGA proteins are expressed at high levels in embryonic tissues during early development and at very low levels in normal differentiated somatic adult cells [10,12]. Regulation of gene expression is a primary function of HMGA in these cells [10] with HMGA proteins involved in both positive and negative regulation of genes responsible for apoptosis, cell proliferation, immune response and DNA repair [13,14]. Overexpression of HMGA has been shown to increase cell proliferation contributing to tumor growth. In addition, HMGA1 interacts with the p53 tumor suppressor protein and inhibits its apoptotic activity [15]. Liau and Whang have shown that high expression levels of HMGA1 are responsible for chemotherapy resistance in pancreatic cancer cell lines [16], and suppression of HMGA1 expression by siRNA restored the cells sensitivity to gemcitabine. HMGA2 is responsible for maintaining Ras-induced epithelial–mesenchymal transition that promotes tissue invasion and metastasis. Down regulation of overexpressed HMGA2 has been shown to inhibit cell proliferation in human pancreatic cancer cell lines [17]. While the precise role that HMGA plays in cancer is not yet understood, HMGA has been suggested as a potential biomarker for tumor progression and is a drug target for cancer therapy development [18].
An early structural study showed that HMGA does not adopt a conventional protein structure composed of α helices or β sheets but rather binds in the minor groove of AT-rich double-stranded DNA through crescent-shaped DNA binding motifs referred to as “AT-hooks” [19]. In contrast to classical transcription factors that bind specific DNA sequences, HMGA acts as an architectural transcription factor [10] that binds a specific type of DNA structure, i.e. the minor groove of A:T tract DNA [19]. Due to this unique DNA binding property of HMGA, several cancer therapy drugs, such as FR900482 and FL317, have been designed as competitive HMGA1 inhibitors [20] that bind to the minor groove of AT-rich DNA; however, these drugs have shown high toxicity in humans. Recently, Maasch et al. showed that Spiegelmer NOX-A50 is a potent inhibitor of HMGA1 activity and proposed the use of artificial HMGA1 substrates that block HMGA1 binding to its natural DNA substrate [21]. In principle, decreasing all HMGA protein activity could result in inhibition of unwanted cell proliferation and reestablishment of apoptosis, reducing cancer progression.
Nucleic acid ligands designed or selected to inhibit the activity of pathogenic proteins are referred to as aptamers or “decoys”. Nucleic acid aptamers contain variable sequences and/or modified chemical structures to facilitate binding to their protein targets with high specificity and an equal to, or higher, affinity compared to their unmodified oligomer counterparts (reviewed in [22]). They are widely studied for biotechnological and therapeutic applications due to their little or no immunogenicity compared to antibodies (reviewed in [23]) and several applications have been reported. For example, overexpression of a 60-nucleotide RNA decoy used as a antiviral treatment showed inhibition of Tat-mediated HIV replication in vitro by 90% [24]. In another study, a 2′-fluoropyrimidine RNA was designed as a vascular endothelial growth factor inhibitor that reduced lung metastasis in mice [25]. A DNA aptamer targeting transcription factor E2F, which is essential in cellular proliferation regulation, was shown to decrease cell proliferation in vascular smooth muscle cells [26].
In addition to engineered specificity, an important property of DNA aptamers is that they are sometimes designed to be resistant to endogenous nuclease activity in vivo. For example, both phosphorothioate DNA (sDNA), which contains sulfur substituted for one oxygen atom in the phosphodiester backbone, and phosphorodithioate DNA, which contains sulfur substitution of two oxygen atoms in the phosphodiester backbone, have been shown to have increased resistance to nuclease S1 and DNase I activity as the number of sulfur substitutions increases [27]. The number of phosphodiester linkages in the oligonucleotide was also shown to directly affect the resistance to Bal31 endonuclease [28].
In this study we designed phosphorothioate substituted sDNA aptamers against HMGA proteins that contained multiple HMGA AT-hook binding sites (AT-sDNA) to compete for HMGA protein binding to genomic DNA and directly inhibit HMGA protein activity in pancreatic cancer cells. Since both HMGA1 and HMGA2 bind AT-rich DNA, the sDNA aptamer designed in this study is intended to inhibit the activity of both forms of HMGA. Since down regulation of both HMGA1 and HMGA2 proteins contributes to the inhibition of tumor growth, the strategy of targeting both HMGA1 and HMGA2 may result in a potentially more potent therapeutic strategy. Here we demonstrate the nuclease resistance of these DNA aptamers and increased sensitivity of pancreatic cancer cells to gemcitabine after transfection with AT-sDNA aptamers designed to inhibit the activity of HMGA proteins.
2. Materials and methods
2.1. Expression and purification of HMGA1 protein
The cDNA for full length HMGA1b was cloned into pET-30b and over-expressed in Escherichia coli BL21 (DE3). E. coli expressing HMGA1b was cultured at 37 °C to an OD600 measurement of 0.8–1.0. Protein expression was induced by the addition of 1 mM IPTG and shaking at 37 °C for 4–6 h. HMGA1 was purified by trichloroacetic acid precipitation as described previously [29]. Overexpressed HMGA1b was further purified with a Sephadex G-25 column in H2O and lyophilized. The samples were resolublized in the appropriate buffer for analysis (see below).
2.2. Electrophoretic mobility shift assays (EMSAs)
The following 28-mer oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA, USA): ATf10 5′-(56FAM)-CGCGGGGCCGCCGCGAAAAAAAAAAACCC-3′, ATs10 5′-GGGT*T*T*T*T*T*T*T*T*T*CGCGGCGGCCCCGCG-3′ (* indicates the location of phosphorothioate linkage). These oligonucleotide samples were resuspended and annealed with complementary strands in annealing solution (100 mM NaCl, 10 mM MgCl2 in H2O). AT10f was annealed with its complementary strand without a fluorescence tag, and ATs10 was annealed with a strand with no sulfur substitution. CG10 5′-GGGCCCCCCCCCCCGCGGCGGCCCCGCG-3′ and Mix10 5′-GGGCGTGACTGAGCGCGGCGGCCCCGCG-3′ were used as negative controls to demonstrate that the presence of these DNA molecules did not affect HMGA binding to AT-rich DNA. Protein samples were prepared in 25 mM Tris–HCl (pH 6.5), 50 mM NaCl, and the concentrations determined based on UV 220 nm absorbance (ε=38,200 mol−1 cm−1) [30]. Protein was mixed with DNA according to the ratio indicated and incubated at 4 °C for 15 min prior to gel analysis. The protein–DNA complexes were resolved on a 7.5% polyacrylmide gel and run with TAE buffer at 20 mA for 2–3 h at 4 °C The DNA was detected at 495 nm using the BIO-RAD VersaDoc Imaging System Model 3000 (Bio-Rad, Hercules, CA). The gels were further stained with coomassie blue and visualized by an AlphaImager (Alpha Innotech, San Leandro, CA).
2.3. Nuclease resistance assays
The following 21 base oligonucleotides, all containing a run of either 15 consecutive adenines or thymines, were purchased from Integrated DNA Technologies, Inc. (Coralville, IA, USA): A15 5′-CCCAAAAAAAAAAAAAAACCC-3′, T15 5′-GGGTTTTTTTTTTTTTTTGGG-3′, As10 5′- CC*CA*AA*AA*AA*AA*AA*AA*AA*CC*C-5′, Ts10 5′-G*GG*TT*TT*TT*TT*TT*TT*TT*TG*GG-3′, Ts20 5′-G*G*G*T*T*T*T*T*T*T*T* T*T*T*T*T*T*T*G*G*G-3′ As20 5′-C*C*C*A*A*A*A*A*A*A*A*A*A*A*A*A*A*A*C*C*C-3′. The asterisks indicate positions of sulfur substitutions and the number following the “s” indicates the number of sulfur substitutions in each sequence. The samples were resuspended and annealed in annealing solution in the following combinations: AT15 (A15 and T15), As10Ts10, As10Ts20 and As20Ts20. Each DNA (0.25 nmol) was incubated with 0.5 unit of Deoxyri-bonuclease I (DNaseI) (Fermentas Life Sciences) at 37 °C up to 12 h. The samples were resolved on a 7% polyacrylmide gel and run with TAE buffer at 40 mA for 1–2 h at 4 °C. The gels were stained with ethidium bromide and visualized by an AlphaImager (Alpha Innotech, San Leandro, CA).
2.4. Cell cultures
Human pancreatic adenocarcinoma cell lines, Panc-1, Miapaca-2 and AsPC-1, were obtained from the American Type Culture Collection (Manassas, VA). Miapaca-2 and Panc-1 cells were grown in high glucose DMEM medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Cells were maintained in 5% CO2 humidified atmosphere at 37 °C. AsPC-1 cells were grown in Roswell Park Memorial Institute medium supplemented with 10% FBS, and 1% penicillin-streptomycin. Cells were maintained in 5% CO2 humidified atmosphere at 37 °C. Confluent cells were trypsinized using trypsin-EDTA solution and seeded into 100 mm dishes and maintained. When cells were grown to 75–85% confluence in 100 mm culture dishes, the media was aspirated and cells were trypsinized. Fresh growth media containing 10% FBS was added to the trypsinized cells (10:1 ratio) and centrifuged for 5 min at 1500 rpm at 4 °C. After resuspending the cells in the appropriate medium, the cell count was measured using a hemocytometer.
2.5. Cellular protein isolation and Western blot analysis
Cytoplasmic and nuclear protein extracts were prepared from pancreatic cancer cells using NE-PER extraction kit (Thermo Scientific, Rockford, IL). Protein concentrations were determined using a bicinchoninic acid assay with bovine serum albumin as a standard. Nuclear proteins that contained 25 or 15 μg total protein were separated by SDS-PAGE with a 4–20% Criterion gradient gel (Bio-Rad). The proteins were transferred to immunoblot PVDF membrane (0.2 μm) (Bio-Rad) and blocked in 5% dry milk in PBS supplemented with 0.2% Tween 20 (PBST). Membranes were then probed with 1:1000 dilution of rabbit anti-HMGA1 antibody (Santa Cruz Biotechnology Inc, Santa Cruz, CA) and 1:100 dilution of rabbit anti-HMGA2 antibody (Abcam), in 3% BSA in PBST at 4 °C overnight. After three washes with PBST, the membrane was blotted with secondary antibody, ant-rabbit IgG-HRP (cell signaling) 1:5000 dilution in 1% dry milk in PBST at room temperature for 1 h. An enhanced chemiluminescence detection (ECL, GE Health Care, IL) system was used to detect target proteins. To ensure equal loading of the proteins between groups, membranes were re-probed with anti-TATA binding protein antibody (Abcam).
2.6. sDNA Transfections
Cells were transfected with AT-rich phosphorothioate DNA (AT-sDNA): 5′- G*G*G*A*A*A*A*A*A*T*T*T*T*T* T*A*A*A*A*A*A*C*C*C-3′ (Integrated DNA Technologies), CG-rich phosphorothioate DNA (CG-sDNA): 5′-C*C*C*C*G*G*G*C*C*C*C*G* G*C*C*G*G*G*C*G*C*C*G* C-3′, and random phosphorothioate DNA (Mix-sDNA): 5′-C*C*C* A*C*T* G*C*A* G*T*C* G*G*A* C*T*C* A*C*T* C*G*C-3′ with the latter being used as a control that lacked a HMGA-specific binding sequence. After annealing with complementary strands, DNA was purified with a Sephadex G-25 column (GE healthcare) in H2O. Both complementary strands also contained sulfur substitution at every position. Transfections using sDNA at concentrations of 0.1 and 0.25 μg per well were conducted with 5 × 104 and 1 × 105 Miapaca-2 and AsPC-1 cells, respectively, in 24-well plates and all data were collected in quadruplicate. Miapaca-2 and AsPC-1 cells were transfected with sDNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol.
2.7. Cell growth assays in the presence of gemcitabine treatment
Cell growth was monitored using a modified crystal violet assay as previously described [31]. Briefly, AsPC-1 cells were grown in 10% FBS containing RPIM medium were trypsinized and seeded (105 cells per well) in a 24-well plate. Due to the shorter doubling time, 5000 cells per well of Miapaca-2 cells were seeded in a 24-well plate in 10% FBS containing DMEM medium. After 48 h, cells were transfected with the selected sDNA using Lipofectamine 2000 according to the manufacturer's protocol. The following day, media was changed to fresh media containing different concentrations of gemcitabine (0, 1, 10, 30, 100, 1000 nM). Cells were fixed 48 and 96 h after gemcitabine treatment. Cells were fixed with 4% paraformaldehyde in PBS for 20 min and stained in 0.1% crystal violet for 30 min. The cells were washed under running tap water, air dried, and extracted with 0.2% Triton X-100 for 30 min. The absorbance of the Triton X-100 was measured at 570 nm using a microplate reader. All results were analyzed for statistical significance using a student's unpaired t-test. The half maximal inhibitory concentration (IC50) values for gemcitabine treatment were determined using Prism software program (Graphpad software, San Diego, CA). A p-value of <0.05 was considered statistically significant.
3. Results
3.1. DNA binding assays
In order to determine if HMGA1 DNA binding activity was affected by sulfur substitution in the DNA phosphodiester backbone, fluorescence labeled DNA (ATf10 DNA) was used in competitive binding assays against various sulfur substituted DNAs where different ratios of ATf10 DNA and ATs10 DNA were added to a constant amount of purified HMGA1 (Fig. 1A). The amount of free (unshifted) ATf10 DNA was determined by monitoring the absorbance intensity at 495 nm. The ratio of ATf10 DNA to HMGA1 was held constant at 1:1.5, which resulted in nearly completely shifted ATf10 DNA in the absence of competitive sulfur-substituted DNA (Fig. 1A, lane2). As the ratio of ATs10 DNA to ATf10 DNA increased, the intensity of the free ATf10 DNA band increased (Fig. 1A, lanes 2–8), indicating that the ATs10 DNA was effectively competing for HMGA1 binding. Since ATs10 DNA has no fluorescence label, no signal could be detected when only ATs10 DNA was added to HMGA1 (Fig. 1A, lane 9). At a 1:1 ratio of ATf10 DNA to ATs10 DNA, approximately half of the ATf10 DNA was detected in the unshifted position (Fig. 1A, lane 4) indicating that HMGA1 was able to bind ATs10 DNA with a similar affinity compared to normal DNA. Thus, the following experiments were conducted under the assumption that HMGA1 bound sDNA with a similar affinity compared to normal DNA.
Fig. 1.
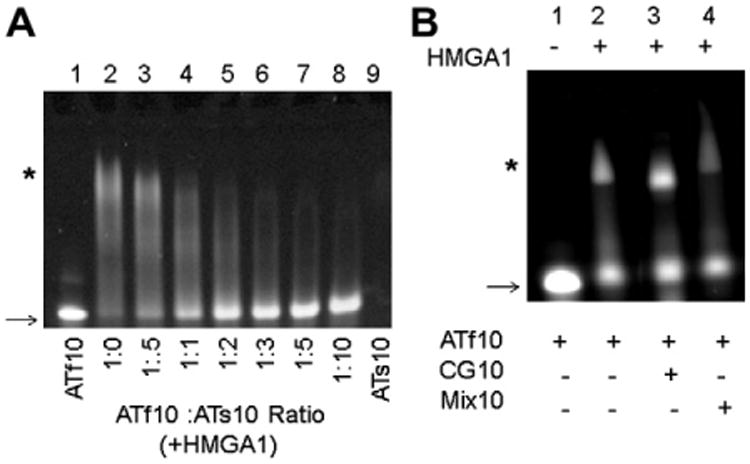
(A) EMSA of HMGA1 competitive binding assay using sDNA and unmodified DNA. Lanes 1 and 9 are controls, ATf10 and ATs10 respectively, that do not contain HMGA1. The ratios between HMGA1 and ATf10 were kept constant at 1.5 (excess HMGA1, lane 2) and an increased amount of ATs10 was added to the reactions in lanes 3–8. (B) EMSA of HMGA1 competitive binding assay using CG10 and Mix10 DNA. Lanes 2–4 contain equal amounts of ATf10 and CG10 or Mix10. ATf10 DNA: HMGA1 in lanes 2–4 was kept constant at a 1:1 ratio. The arrow indicates free (unshifted) DNA and the * indicates the complex (shifted bands) with ATf10 DNA. The image was taken at 495 nm using a BIO-RAD VersaDoc Imaging System Model 3000 in order to visualize the ATf10.
In order to demonstrate preferential HMGA binding to AT-rich DNA compared to CG-rich or mixed sequence DNA lacking AT tracks, gel shift assays were conducted using two controls, CG10 and Mix10, (Fig. 1B). The CG10 sequence only contained a mixture of C and G, and the Mix10 sequence contained a random sequence of A, T, C, G with no AT-stretch. The formation of an HMGA/ATf10 DNA complex (Fig. 1B, lane 2*) was still observed when CG10 or Mix10 DNA was present (Fig. 1B, lanes 3 and 4). These results not only demonstrated the preference of HMGA for binding AT-rich DNA, but also demonstrated that the presence of non-AT-rich DNA did not interfere with HMGA binding to AT-rich DNA.
3.2. Nuclease resistance assays
In order to have any potential clinical value, DNA-based aptamers targeting HMGA1 must have a long half-life against endogenous nuclease activity. To explore how nuclease resistance was affected by sulfur substitution in the HMGA1 DNA aptamers, sDNAs containing different numbers and positions of sulfur substitutions were subjected to nuclease resistance assays. Each sDNA was incubated with DNase I for up to 12 h (Fig. 2). The sDNA stability and nuclease activity was confirmed prior to experiments (data not shown). No DNA was detected after 30 min incubation when DNA with no sulfur substitutions or with sulfur substitutions in alternate nucleotide positions was used (Fig. 2A and B). The majority of DNA containing one strand with alternating sulfur substitution and the other strand with contiguous sulfur substitution was digested after 6 h (Fig. 2C). Only DNA with complete and contiguous sulfur substitutions on both strands remained undigested after 12 h of incubation with DNaseI (Fig. 2D).
Fig. 2.
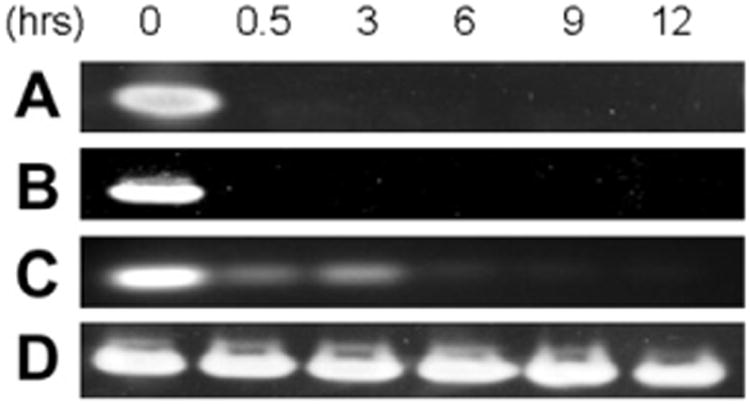
Nuclease resistance assay for sDNA with DNase I. 0.25 nmol of (A) AT15, (B) As10Ts10, (C) As10Ts20 and (D) As20Ts20 were incubated for 0–12 h and run on a 7% native gel. The gel was stained with ethidium bromide and visualized by AlphaImager.
3.3. HMGA1 expression levels in pancreatic cancer cell lines
HMGA1 expression in three human pancreatic cancer cell lines, AsPC-1, Miapaca-2 and Panc-1, was examined by Western blot analysis. AsPC-1 had the highest HMGA1 expression compared to Miapaca-2 and Panc-1 (Fig. 3A). The expression levels of HMGA2 are shown in Fig. 3B. While AsPC-1 express both HMGA1 and HMGA2, no HMGA2 protein expression was detected in Miapaca-2 cells. These results were consistent with previous findings [17]. AsPC-1 cells treated with 100 nM gemcitabine exhibited no change in HMGA1 expression levels (Fig. 3C). HMGA1 expression levels were also examined after transfection with CG-sDNA or AT-sDNA to determine if the transfection procedure affected HMGA1 expression levels. The AT-sDNA used for transfection contained an 18 adenine/thymine stretch enabling HMGA1 to bind the DNA with all three AT-hooks. As a negative control, a non-HMGA1-targetted sDNA containing an equal length of DNA but with no AT-stretches, i.e. CG-sDNA, was used. Western blot analyses indicated no changes in HMGA1 expression levels after AT-sDNA or CG-sDNA transfection (Fig. 3C).
Fig. 3.
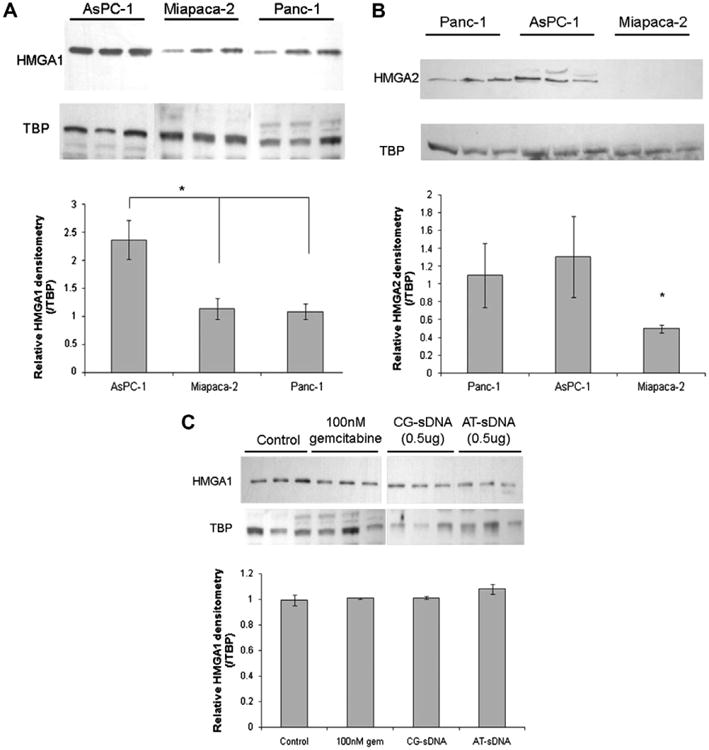
Western blot analysis of (A) HMGA1 and (B) HMGA2 expression levels in pancreatic cancer cell lines. Nuclear extracts of AsPC-1, Miapaca-2, and Panc-1 cell lines were run on a 4–12% gradient gel. In each lane, 25 μg of total nuclear protein was loaded. (C) 15 μg of total nuclear protein was loaded in each lane after AsPC-1 cells treated with 100 nM gemcitabine for 48 h and transfected with 0.5 μg of CG-sDNA, or AT-sDNA in six well plates for 48 h. The control is nuclear extract from untreated cells. Triplicates of each sample were run on the gel and analyzed with the Alpha Imager. The relative HMGA1 densities were obtained by dividing HMGA1 density by TBP density. The values were analyzed pair wise using a student t-test, with p values <0.05 considered a significant change (*p < 0.05). TBP–TATA binding protein was used as a loading control.
3.4. Effect of sDNA on cell growth
The dependence of Miapaca-2 and AsPC-1 cell viability on transfection with Mix-sDNA, CG-sDNA and AT-sDNA was assessed after 48 and 96 h (Fig. 4 and Table 1). The effect of transfection alone on cell viability was assessed to establish a transfection control (TC) baseline for comparison with combinations of sDNA transfections and gemcitabine treatment. The TC cells were transfected with Lipofectamine 200 carrying only a vehicle and cell growth was compared to the untransfected (UT) control cells. In Miapaca-2, TC cells showed a 37.7% (p < 0.005) and 37.6% (p < 0.01) decrease in cell growth compared to UT cells after 48 and 96 h, respectively (Fig. 4A), whereas in AsPC-1, TC cells showed 25.3% (p < 0.005) and 7.9% (p > 0.05) decrease in cell growth after 48 and 96 h, respectively, compared to UT cells (Fig. 4B). Similar decreases in cell growth were observed after 0.1 μg of transfection with two sDNA controls, Mix-sDNA and CG-sDNA (Fig. 4A and B). Specifically, in AsPC-1 cells, 28.0% (p < 0.005) and 33.8% (p < 0.0005) of cells were dead with Mix-sDNA and CG-sDNA transfection after 48 h, and 19.4% (p < 0.01) and 14.3% (p < 0.05) of cells were dead after 96 h, respectively. In Miapaca-2, 30.4% (p < 0.001) and 36.3% (p < 0.005) of cells were dead with Mix-sDNA and CG-sDNA transfection after 48 h, and 33.3% (p < 0.01) and 32.0% (p < 0.001) of cells were dead after 96 h, respectively. These results showed that both transfection with non-AT rich sDNA and TC cells had significantly decreased cell viability compared to UT control cells. However, comparisons between TC cells and cells transfected with Mix-sDNA, and CG-sDNA showed no significant differences between these treatments (p > 0.05) at both 48 and 96 h.
Fig. 4.
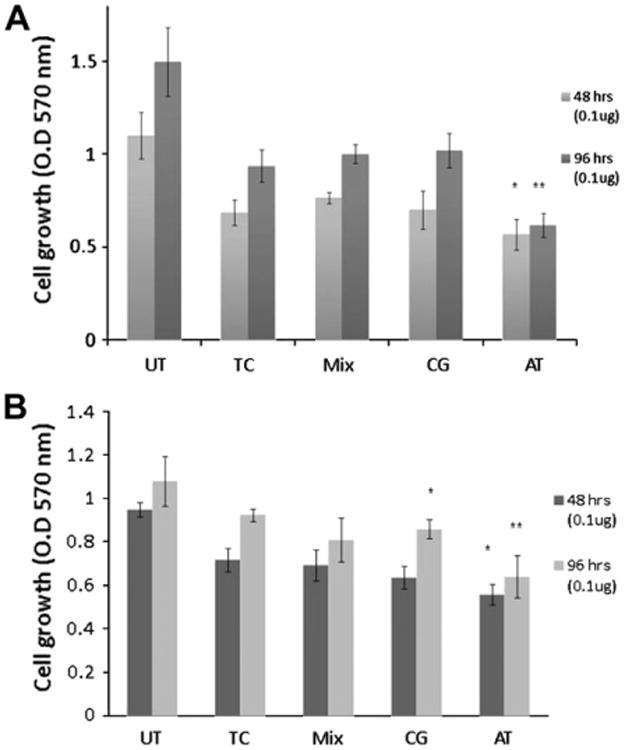
Cell viability assay after sDNA transfection. (A) Miapaca-2 and (B) AsPC-1 cells were transfected with two doses of sDNA. Cells were fixed 48 and 96 h after transfection and analyzed with the crystal violet assay as described in the methods. The absorbance was measured at 570 nm in quadruplicate. All data from control cells were combined and averaged. The values were analyzed pair wise with TC using a student t-test, with p values <0.05 considered a significant change (*p < 0.05, **p < 0.01).
Table 1.
Fold changes in numbers of viable cells between UT and transfected cells 96 h after transfection. Fold changes in numbers of viable cells between TC and sDNA transfected cells 96 h after transfection are indicated in the parentheses.
| UT | TC | Mix | CG | AT | |
|---|---|---|---|---|---|
| Miapaca-2 (0.1 μg) | 1.00 | − 1.55 (1.00) | −1.45 (1.07) | −1.43 (1.09) | −2.36 (−1.52) |
| Miapaca-2 (0.25 μg) | 1.00 | − 1.65 (1.00) | −2.15 (−1.30) | −6.07 (−3.68) | |
| AsPC-1 (0.1 μg) | 1.00 | − 1.09 (1.00) | −1.24 −1.14) | −1.20 (−1.11) | −1.57 (−1.44) |
| AsPC-1 (0.25 μg) | 1.00 | −1.09 (1.00) | −1.19 (−1.10) | −1.79 (−1.65) |
When cells were transfected with AT-sDNA, however, significant decreases in cell viability were observed compared to UT and TC controls. In Miapaca-2, 48.4% (p < 0.005) and 17.2% (p < 0.05) of the cells were dead after 48 h compared to UT and TC controls, respectively, and after 96 h, 58.9% (p < 0.001) and 34.1% (p < 0.01) of cells were dead, respectively. In AsPC-1 cells, 42.0% (p < 0.005) and 22.3% (p < 0.05) of cells were dead compared to UT and TC controls after 48 h, and 36.1% (p < 0.001) and 30.7% (p < 0.01) of cells were dead after 96 h, respectively.
The effect of sDNA transfection dose was examined using AsPC-1 cells. Cells were transfected with four different CG-sDNA and AT-sDNA doses, 0.025, 0.1, 0.25 and 1 μg, and cell growth compared to TC cells (Suppl. Fig. 1). When cells were transfected with 1.0 μg of sDNA, more than half of the cells were dead after 96 h for both AT-sDNA (53.6%, p < 0.0005) and CG-sDNA (56.4%, p < 0.001). This result showed that 1.0 μg of sDNA transfection was highly toxic to the cells independent of DNA sequence. Therefore, lower dosages of sDNA were used to characterize sensitivity to DNA transfection. When 0.25 μg sDNA was used for transfection, both cells lines were more viable compared to the 1.0 μg sDNA transfected cells, however, cell death was greater in cells transfected with AT-sDNA (39.4%, p < 0.01) compared to CG-sDNA (9.0%, p < 0.05). As the amount of sDNA transfected was decreased to 0.1 μg, the cell death rates for the two cell lines transfected with CG-sDNA and AT-sDNA were similar to 0.25 μg transfection; 9.7% (p < 0.05) CG-sDNA, and 30.7% (p < 0.01) AT-sDNA. Transfection with 0.025 μg of sDNA had very little affect on cell growth, with only an 8.5% (p < 0.05) decrease in viability observed for AT-sDNA transfected cells whereas the reduction in CG-sDNA transfected cells was not statistically significant compared to TC cells (p > 0.05). Only 0.25 μg and 0.1 μg sDNA treatments showed statistically significant differences between AT-sDNA and CG-sDNA transfected cells with p = 0.007 and p = 0.047, respectively. These results indicated that inhibition of HMGA1 activity caused increased cell death or apoptosis even in the absence of chemotherapy treatment.
3.5. Gemcitabine treatment and cell growth in the presence of sDNA aptamers
Cells transfected with 0.1 or 0.25 μg of sDNA were treated with six different concentrations of gemcitabine: 0, 1, 10, 30, 100, 1000 nM. Cells were fixed after 96 h and numbers of viable cells counted by measuring the absorbance at 570 nm. Significant decreases in gemcitabine IC50 values were observed in AsPC-1 cells with AT-sDNA treatment (Fig. 5A and B and Table 2). For example, at 0.1 and 0.25 μg AT-sDNA treatments, IC50 values were 3.8 ± 0.2 nM and 1.8 ± 0.13 nM, respectively compared to TC values of 27.98 ± 1.4 nM and 24.8 ± 1.45 nM, respectively. The IC50 values for the AT-sDNA treatments were also substantially smaller compared to the two control sDNA transfections with the Mix-sDNA having values of 69.4 ± 1.1 nM for 0.1 μg, and the CG-sDNA having values of 24.2 ± 2.2 nM and 46.5 ±1.6 nM, respectively. These results indicated that AspC-1 cells transfected with AT-sDNA were more sensitive to gemcitabine chemotherapy treatment compared to cells that did not receive this treatment. On the other hand, no significant changes in IC50 values were found in Miapaca-2 cells (Fig. 5C and D). However, there was a significant overall drop (∼10-fold, Table 3) in the number of viable Miapaca-2 cells after either 0.1 or 0.25 μg AT-sDNA transfection at doses of 100 nm or greater gemcitabine. While the AsPC-1 cells experienced a significant drop in gemcitabine IC50, the overall drop in the number of viable cells after gemcitabine treatment was considerably smaller (2-4-fold, Table 3) compared to that observed for Miapaca-2 cells (Table 3). These results indicated that cell death due to AT-sDNA transfection was substantially greater in Miapaca-2 cells compared to AsPC-1 cells. The increased cell death due to AT-sDNA transfection in comparison to controls (Table 1) presumably was a result of inhibition of HMGA proteins. The greater sensitivity of Miapaca-2 cells to AT-sDNA compared to AsPC-1 cells probably has to do with the fact that Miapaca-2 cells only express HMGA1 whereas AsPC-1 cells express both HMGA1 and HMGA2. Therefore, the effective dose of AT-sDNA towards HMGA proteins is higher to Miapaca-2 cells in comparison to AsPC-1 cells. Neither cell line treated with gemcitabine for 48 h showed a dose dependent response (Suppl. Fig. 2). This was probably due to the fact that the cell doubling times of both Miapaca-2 and AsPC-1 cells are longer than 48 h.
Fig. 5.
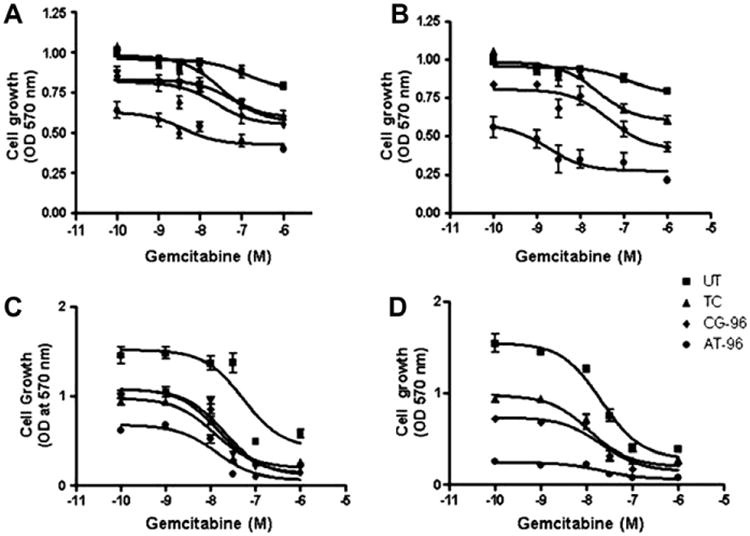
IC50 determination of three doses of sDNA transfection with gemcitabine treatment. AsPC-1: (A) 0.1 μg transfection, (B) 0.25 μg transfection, and Miapaca-2: (C) 0.1 μg transfection, (D) 0.25 μg transfection.
Table 2.
Gemcitabine IC50 values of AsPC-1 and Miapaca-2 cells after 0.025, 0.1, 0.25 μg sDNA transfections.
| Cell line | Transfection | IC50 values (nM) | ||
|---|---|---|---|---|
|
|
||||
| 0.025 μg | 0.1 μg | 0.25 μg | ||
| AsPC-1 | TC | 27.98 ± 1.4 | 24.8 ± 1.45 | |
| Mix-sDNA | 69.4 ± 1.1 | |||
| CG-sDNA | 171.53 ± 26.6 | 24.2 ± 2.2 | 46.5 ± 1.6 | |
| AT-sDNA | 107.49 ± 39.6 | 3.8 ± 0.2 | 1.8 ± .13 | |
| 0.1 μg | 0.25 μg | |||
|
|
||||
| Miapaca-2 | TC | 11.91 ± 1.2 | 21.5 ± 0.6 | |
| Mix-sDNA | 20.43 ± 1.25 | |||
| CG-sDNA | 15.11 ± 1.30 | 21.4 ± 1.3 | ||
| AT-sDNA | 13.63 ± 1.52 | 21.6 ± 1.3 | ||
Table 3.
Fold changes in numbers of viable cells normalized relative to TC for various sDNA transfections as a function of gemcitabine treatment.
| sDNA used for transfection | Gemcitabine (nM) | ||||||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| 0 | 1 | 10 | 30 | 100 | 1000 | ||
| Miapaca-2 (0.1 μg) | TC | 1.00 | 1.00 | −1.30 | −3.21 | −3.59 | −3.47 |
| Mix | 1.07 | 1.12 | 1.01 | −2.65 | −4.41 | −4.48 | |
| CG | 1.09 | 1.12 | 1.09 | 3.29 | 4.06 | 4.24 | |
| AT | −1.52 | −1.38 | −1.79 | −7.07 | −9.58 | −6.82 | |
| Miapaca-2 (0.25 μg) | TC | 1.00 | 1.00 | −1.30 | −3.21 | −3.59 | −3.47 |
| CG | −1.30 | −1.38 | −1.37 | −2.97 | −5.48 | −4.11 | |
| AT | −3.68 | −4.45 | −4.14 | −8.32 | −11.81 | −11.31 | |
| AsPC-1 (0.1 μg) | TC | 1.00 | 1.14 | −1.02 | −1.04 | −1.36 | −1.52 |
| Mix | −1.14 | −1.06 | −1.16 | −1.17 | −1.36 | −1.59 | |
| CG | −1.11 | −1.09 | −1.18 | −1.34 | −1.52 | −1.67 | |
| AT | −1.44 | −1.58 | −1.71 | −1.86 | −2.04 | −2.33 | |
| AsPC-1 (0.25 μg) | TC | 1.00 | 1.14 | −1.02 | −1.04 | −1.36 | −1.52 |
| CG | −1.10 | −1.10 | −1.21 | −1.69 | −1.36 | −2.16 | |
| AT | −1.65 | −1.92 | −2.63 | −2.66 | −2.83 | −4.32 | |
After 96 h of gemcitabine treatment, Miapaca-2 cells showed increased dose dependent responses at 0.1 μg AT-sDNA transfection compared to AsPC-1 cells at either 0.1 μg or 0.25 μg AT-sDNA transfections (Fig. 6). Responses to various concentrations of gemcitabine were similar among the three controls, TC, Mix and CG. A significant increase in sensitivity to 10 nM gemcitabine was observed in Miapaca-2 cells transfected with AT-sDNA with only 51.2% (p < 0.05) of cells surviving compared to 77.1% (p<0.05) in TC, 94.5% (p>0.05) in Mix-sDNA, and 83.9% (p>0.05) in CG-sDNA. In addition, at 1 nM gemcitabine treatment, 33.6% (p < 0.05), cells were dead with AT-sDNA transfection whereas no significant change in cell viabilities were observed in TC, Mix-sDNA, and CG-sDNA transfected cells. At higher gemcitabine treatments, 30, 100 and 1000 nM, significant decreases in cell viabilities were found in all the transfected cells (9.6–35.2%, p < 0.001) after 96 h.
Fig. 6.
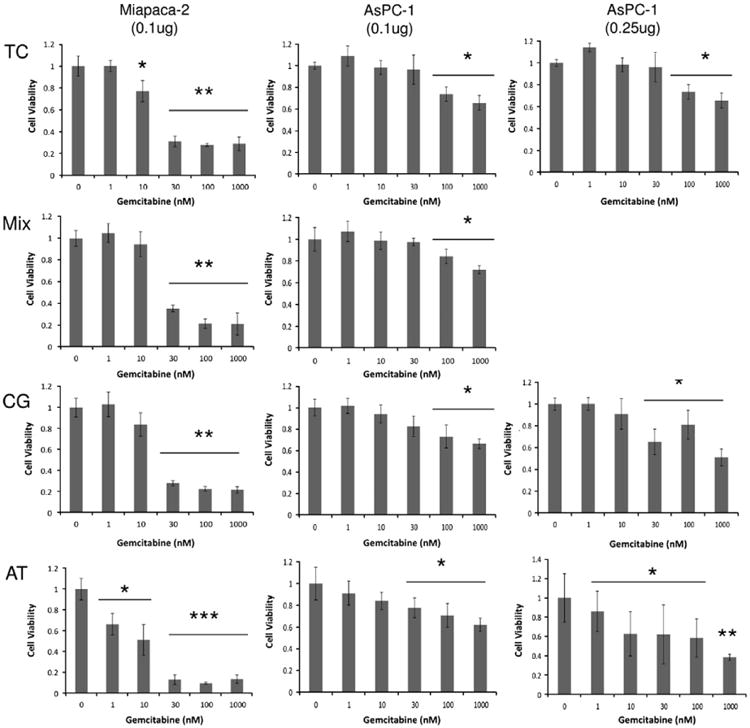
(Left) Dose dependent response to gemcitabine treatment after sDNA transfection in Miapaca-2 at 96 h. Cells were transfected with 0.1 μg of Mix-sDNA, CG-sDNA, and AT-sDNA. Dose dependent responses to gemcitabine treatment in AsPC-1 at 96 h and 0.1 μg (middle), and 0.25 μg (right) transfection. Data was normalized with the 0 nM gemcitabine treated cells and the resulting value defined as a value of 1. The values were analyzed pair wise using a student t-test, with p values <0.05 considered a significant change (*p < 0.05, **p < 0.01, ***p < 0.001).
AsPC-1 cell sensitivity to gemcitabine treatment was assayed for 0.1 and 0.25 μg sDNA transfection doses at six different gemcitabine concentrations (Fig. 6). At 0.025 μg AT-sDNA transfection, though not significant, a slight increase in sensitivity to 30 nM gemcitabine was observed with the number of cells surviving at 92.9% (p >0.05) in TC, 92.8% (p > 0.05) in CG-sDNA transfection, and 89.5% (p < 0.05) in AT-sDNA transfection (data not shown). In contrast to Miapaca-2 cells, the increase in gemcitabine sensitivity was less notable in AsPC-1 cells at 0.1 μg AT-sDNA transfection where AsPC-1 cell survivals were 98.3% (p>0.05) in TC, 94.0% (p > 0.05) in CG-sDNA transfection, and 84.1% (p < 0.05) in AT-sDNA transfection for the 10 nM gemcitabine dose. At the 0.25 μg transfection, the increase in AsPC-1 gemcitabine sensitivity became more pronounced. AsPC-1 cell viability was significantly decreased with 10 nM gemcitabine and AT-sDNA transfection (62.7%, p < 0.05) compared to TC (98.3%, p > 0.05) and CG-sDNA transfection (91.9%, p > 0.05). Within the AT-sDNA transfection set of experiments, the dose dependent responses to gemcitabine were stronger at the 0.25 μg transfection compared to the 0.1 μg transfection. For example, 14.9% (p < 0.05) and 41.7% (p < 0.05) of the cells died in the 0.25 μg AT-sDNA transfections at 1 and 100 nM gemcitabine doses, respectively, compared to 9.0% (p > 0.05) and 29.4% (p < 0.05) of cells dying at 1 and 100 nM gemcitabine treatments with 0.1 μg AT-sDNA transfection, respectively. At the lowest AT-sDNA transfection dose of 0.025 μg, an insignificant effect on cell viability was observed with only 8.5% cell death (p > 0.05) in the absence gemcitabine treatment (Fig. 4B), whereas cell death increased significantly to 31% (p < 0.05) when 1000 nM gem-citabine treatment was administered (Fig. 6).
4. Discussion
Overexpression of HMGA protein is a hallmark of many human cancers, and, consequently, HMGA has been suggested as a therapeutic target for cancer treatment. HMGA, a DNA binding architectural transcription factor, is involved in both positive and negative regulation of many genes that could play a role in carcinogenesis, including those responsible for apoptosis, cell proliferation, immune response and DNA repair. It is well established that both HMGA1 and HMGA2 bind to the minor groove of short stretches of AT-rich DNA [32–35]. Motivated by the fact that HMGA activity is mediated through DNA binding, numerous studies have demonstrated different methods that take advantage of HMGA's DNA binding properties to inhibit HMGA activity. For example, several studies have focused on HMGA inactivation either using small molecules that competitively bind to HMGA1 DNA binding sites or cross-link HMGA1 to DNA [36,37]. Others have developed drugs such as FR900482, Hoechst 33258 and netropsin that compete for binding with HMGA1 to the minor groove of AT-rich DNA sequences, which results in competitive inhibition of HMGA1 DNA binding and diminished HMGA1 activity [20,38,39], however, these molecules have been shown to have high toxicity. An alternative approach to counteract carcinogenic effects of HMGA protein in cancer cells has been to knock down HMGA1 or HMGA2 expression levels using either anti-sense RNA or RNA interference. In the former example, the use of antisense RNA was accomplished by infecting the tumor cells with an adenovirus carrying the HMGA1 gene in the antisense orientation, and transcription of the antisense HMGA1 gene resulted in production of anti-sense mRNA, which was complementary to the mRNA produced for HMGA1 in the sense orientation, resulting in the double stranded mRNA which then blocks subsequent translation and synthesis of HMGA1. This use of antisense RNA was shown to reduce cell proliferation in pancreatic cancer cell lines and inhibited tumor growth after transfected cells were transplantated into nude mice [40]. RNA interference has also been used to down regulate HMGA1 expression in pancreatic cancer cell lines, resulting in recovery of sensitivity to the chemotherapy reagent, gemcitabine, and the cells showed an increase in gemcitabine-induced apoptosis [16]. Recently, direct HMGA1 inhibition was demonstrated using L-RNA oligo-nucleotides selected to bind specifically to HMGA1, i.e. Spiegelmer NOX-A50 [21] suggesting the potential use of nucleotide analogs as HMGA1 inhibitors. Collectively, these studies have further demonstrated that HMGA over-expression plays a key role in regulating cell proliferation in carcinogenesis in many type of cancers, and the potential value of disrupting HMGA activity to increase the efficacy of chemotherapy agents such as gemcitabine in treating cancers. Nonetheless, none of these methods has yet achieved successful clinical use and there is need for continued development of novel and alternative approaches to disrupt the carcinogenic activity of elevated HMGA levels in human cancer cells.
In the study reported here, we have tested the ability of our AT-sDNA aptamer to increase the gemcitabine sensitivity of two human pancreatic cancer cell lines: AsPC-1, which has been shown to express high level of both HMGA1 and HMGA2, and Miapaca-2 which has high HMGA1 expression but no HMGA2 expression [17]. Increased sensitivity to gemcitabine treatment was observed in both pancreatic cancer cell lines following HMGA1-tar-geted AT-sDNA aptamer transfection. We believe that HMGA-targeted AT-sDNA aptamers transfected into pancreatic cancer cells restore gemcitabine sensitivity by attenuating HMGA function by competitive sequestration of excess HMGA, thereby preventing excess HMGA1 from binding to chromosomal DNA transcription factors and other proteins involved in regulating apoptosis and cell proliferation in tumor progression. Furthermore, the data suggest that HMGA-targeted AT-sDNA aptamer inhibition of HMGA results in rescue of cellular apoptosis induced by gemcitabine treatment. Since these HMGA-targeted DNA aptamers have no direct effect on HMGA gene expression levels, do not bind directly to genomic DNA, and should not stimulate immunogenic responses, HMGA-targeted AT-sDNA aptamer treatment, potentially in localized combination therapy with gemcitabine, could render cancer cells more sensitive to existing chemotherapy reagents and result in fewer side effects. Collectively, the results reported in this study demonstrate a potential clinical value of using specifically targeted sulfur substituted DNA aptamers for cancer treatment specifically targeting the carcinogenic activity of HMGA protein.
Supplementary Material
Acknowledgments
The authors would like to acknowledge support of Miami University and the Ohio Board of Regents for funding to establish the Ohio Eminent Scholar Laboratory where the work was performed. The authors would also like to acknowledge support from Bruker Biospin, Inc. that enabled development of the statistical significance analysis software used in the analysis of the data reported in this paper. M.W. would like to acknowledge support of a Miami University Cell, Molecular, and Structural Biology Fellowship. The work was funded by the National Institutes of Health National Cancer Institutes; Grant Number: 1R15CA152985.
Abbreviations
- ATfDNA
fluorescence labeled DNA
- AT-sDNA
AT-rich phosphorothioate DNA
- CG-sDNA
CG-rich phosphorothioate DNA
- DMEM
Dulbecco's Modified Eagle Medium
- DNaseI
Deoxyribonuclease I
- EMSA
electrophoretic mobility shift assay
- FBS
fetal bovine serum
- HMGA
high mobility group A
- IC50
half maximal inhibitory concentration
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- Mix-sDNA
random sequence phosphorothioate DNA
- sDNA
phosphorothioate DNA
- TAE
Tris base, acetic acid and EDTA buffer
Footnotes
Appendix A. Supplementary material: Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.canlet.2011.10.005.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Bianchi V, Borella S, Calderazzo F, Ferraro P, Chieco Bianchi L, Reichard P. Inhibition of ribonucleotide reductase by 2′-substituted deoxycytidine analogs: possible application in AIDS treatment. Proc Natl Acad Sci USA. 1994;91:8403–8407. doi: 10.1073/pnas.91.18.8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szekeres T, Fritzer-Szekeres M, Elford HL. The enzyme ribonucleotide reductase: target for antitumor and anti-HIV therapy. Crit Rev Clin Lab Sci. 1997;34:503–528. doi: 10.3109/10408369709006424. [DOI] [PubMed] [Google Scholar]
- 4.Rocha-Lima CM, Raez LE. Erlotinib (tarceva) for the treatment of non-small-cell lung cancer and pancreatic cancer. P&T: Peer-Rev J Formul Manage. 2009;34:554–564. [PMC free article] [PubMed] [Google Scholar]
- 5.Laurent-Puig P, Taieb J. Lessons from tarceva in pancreatic cancer: where are we now, and how should future trials be designed in pancreatic cancer? Curr Opin Oncol. 2008;20:454–458. doi: 10.1097/CCO.0b013e32830218d6. [DOI] [PubMed] [Google Scholar]
- 6.Sharma C, Eltawil KM, Renfrew PD, Walsh MJ, Molinari M. Advances in diagnosis, treatment and palliation of pancreatic carcinoma: 1990–2010. World J Gastroenterol: WJG. 2011;17:867–897. doi: 10.3748/wjg.v17.i7.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nat Rev Cancer. 2007;7:899–910. doi: 10.1038/nrc2271. [DOI] [PubMed] [Google Scholar]
- 8.Fedele M, Bandiera A, Chiappetta G, Battista S, Viglietto G, Manfioletti G, Casamassimi A, Santoro M, Giancotti V, Fusco A. Human colorectal carcinomas express high levels of high mobility group HMGI(Y) proteins. Cancer Res. 1996;56:1896–1901. [PubMed] [Google Scholar]
- 9.Abe N, Watanabe T, Suzuki Y, Matsumoto N, Masaki T, Mori T, Sugiyama M, Chiappetta G, Fusco A, Atomi Y. An increased high-mobility group A2 expression level is associated with malignant phenotype in pancreatic exocrine tissue. Br J Cancer. 2003;89:2104–2109. doi: 10.1038/sj.bjc.6601391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grosschedl R, Giese K, Pagel J. HMG domain proteins: architectural elements in the assembly of nucleoprotein structures. Trends Genet: TIG. 1994;10:94–100. doi: 10.1016/0168-9525(94)90232-1. [DOI] [PubMed] [Google Scholar]
- 11.Murua Escobar H, Soller JT, Richter A, Meyer B, Winkler S, Bullerdiek J, Nolte I. “Best friends” sharing the HMGA1 gene: comparison of the human and canine HMGA1 to orthologous other species. J Heredity. 2005;96:777–781. doi: 10.1093/jhered/esi083. [DOI] [PubMed] [Google Scholar]
- 12.Abe N, Watanabe T, Masaki T, Mori T, Sugiyama M, Uchimura H, Fujioka Y, Chiappetta G, Fusco A, Atomi Y. Pancreatic duct cell carcinomas express high levels of high mobility group I(Y) proteins. Cancer Res. 2000;60:3117–3122. [PubMed] [Google Scholar]
- 13.Bustin M, Reeves R. High-mobility-group chromosomal proteins: architectural components that facilitate chromatin function. Prog Nucleic Acid Res Mol Biol. 1996;54:35–100. doi: 10.1016/s0079-6603(08)60360-8. [DOI] [PubMed] [Google Scholar]
- 14.Chiappetta G, Avantaggiato V, Visconti R, Fedele M, Battista S, Trapasso F, Merciai BM, Fidanza V, Giancotti V, Santoro M, Simeone A, Fusco A. High level expression of the HMGI (Y) gene during embryonic development. Oncogene. 1996;13:2439–2446. [PubMed] [Google Scholar]
- 15.Pierantoni GM, Rinaldo C, Esposito F, Mottolese M, Soddu S, Fusco A. High Mobility Group A1 (HMGA1) proteins interact with p53 and inhibit its apoptotic activity. Cell Death Differ. 2006;13:1554–1563. doi: 10.1038/sj.cdd.4401839. [DOI] [PubMed] [Google Scholar]
- 16.Liau SS, Whang E. HMGA1 is a molecular determinant of chemoresistance to gemcitabine in pancreatic adenocarcinoma. Clin Cancer Res: Official J Am Assoc Cancer Res. 2008;14:1470–1477. doi: 10.1158/1078-0432.CCR-07-1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watanabe S, Ueda Y, Akaboshi Si, Hino Y, Sekita Y, Nakao M. HMGA2 maintains oncogenic RAS-induced epithelial–mesenchymal transition in human pancreatic cancer cells. Am J Pathol. 2009;174:854–868. doi: 10.2353/ajpath.2009.080523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liau SS, Rocha F, Matros E, Redston M, Whang E. High mobility group AT-hook 1 (HMGA1) is an independent prognostic factor and novel therapeutic target in pancreatic adenocarcinoma. Cancer. 2008;113:302–314. doi: 10.1002/cncr.23560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reeves R, Nissen MS. The A.T-DNA-binding domain of mammalian high mobility group I chromosomal proteins. A novel peptide motif for recognizing DNA structure. J Biol Chem. 1990;265:8573–8582. [PubMed] [Google Scholar]
- 20.Beckerbauer L, Tepe JJ, Cullison J, Reeves R, Williams RM. FR900482 class of anti-tumor drugs cross-links oncoprotein HMG I/Y to DNA in vivo. Chem Biol. 2000;7:805–812. doi: 10.1016/s1074-5521(00)00028-4. [DOI] [PubMed] [Google Scholar]
- 21.Maasch C, Vater A, Buchner K, Purschke WG, Eulberg D, Vonhoff S, Klussmann S. Polyetheylenimine-polyplexes of Spiegelmer NOX-A50 directed against intracellular high mobility group protein A1 (HMGA1) reduce tumor growth in vivo. J Biol Chem. 2010;285:40012–40018. doi: 10.1074/jbc.M110.178533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nielsen PE. DNA analogues with nonphosphodiester backbones. Annu Rev Biophys Biomol Struct. 1995;24:167–183. doi: 10.1146/annurev.bb.24.060195.001123. [DOI] [PubMed] [Google Scholar]
- 23.Nimjee SM, Rusconi CP, Sullenger BA. Aptamers: an emerging class of therapeutics. Annu Rev Med. 2005;56:555–583. doi: 10.1146/annurev.med.56.062904.144915. [DOI] [PubMed] [Google Scholar]
- 24.Sullenger BA, Gallardo HF, Ungers GE, Gilboa E. Overexpression of TAR sequences renders cells resistant to human immunodeficiency virus replication. Cell. 1990;63:601–608. doi: 10.1016/0092-8674(90)90455-n. [DOI] [PubMed] [Google Scholar]
- 25.Huang J, Moore J, Soffer S, Kim E, Rowe D, Manley CA, O'Toole K, Middlesworth W, Stolar C, Yamashiro D, Kandel J. Highly specific antiangiogenic therapy is effective in suppressing growth of experimental Wilms tumors. J Pediatr Surg. 2001;36:357–361. doi: 10.1053/jpsu.2001.20716. [DOI] [PubMed] [Google Scholar]
- 26.Morishita R, Gibbons GH, Horiuchi M, Ellison KE, Nakama M, Zhang L, Kaneda Y, Ogihara T, Dzau VJ. A gene therapy strategy using a transcription factor decoy of the E2F binding site inhibits smooth muscle proliferation in vivo. Proc Natl Acad Sci USA. 1995;92:5855–5859. doi: 10.1073/pnas.92.13.5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cummins L, Graff D, Beaton G, Marshall WS, Caruthers MH. Biochemical and physicochemical properties of phosphorodithioate DNA. Biochemistry. 1996;35:8734–8741. doi: 10.1021/bi960318x. [DOI] [PubMed] [Google Scholar]
- 28.Monia BP, Johnston JF, Sasmor H, Cummins LL. Nuclease resistance and antisense activity of modified oligonucleotides targeted to Ha-ras. J Biol Chem. 1996;271:14533–14540. doi: 10.1074/jbc.271.24.14533. [DOI] [PubMed] [Google Scholar]
- 29.Reeves R. HMGA proteins: isolation, biochemical modifications, and nucleosome interactions. Methods Enzymol. 2004;375:297–322. doi: 10.1016/s0076-6879(03)75020-4. [DOI] [PubMed] [Google Scholar]
- 30.Huth JR, Bewley CA, Nissen MS, Evans JN, Reeves R, Gronenborn AM, Clore GM. The solution structure of an HMG-I(Y)-DNA complex defines a new architectural minor groove binding motif. Nat Struct Biol. 1997;4:657–665. doi: 10.1038/nsb0897-657. [DOI] [PubMed] [Google Scholar]
- 31.Sheriff S, Ali M, Yahya A, Haider KH, Balasubramaniam A, Amlal H. Neuropeptide Y Y5 receptor promotes cell growth through extracellular signal-regulated kinase signaling and cyclic AMP inhibition in a human breast cancer cell line. Mol Cancer Res: MCR. 2010;8:604–614. doi: 10.1158/1541-7786.MCR-09-0301. [DOI] [PubMed] [Google Scholar]
- 32.Strauss F, Varshavsky A. A protein binds to a satellite DNA repeat at three specific sites that would be brought into mutual proximity by DNA folding in the nucleosome. Cell. 1984;37:889–901. doi: 10.1016/0092-8674(84)90424-0. [DOI] [PubMed] [Google Scholar]
- 33.Elton TS, Reeves R. Purification and postsynthetic modifications of friend erythroleukemic cell high mobility group protein HMG-I. Anal Biochem. 1986;157:53–62. doi: 10.1016/0003-2697(86)90195-8. [DOI] [PubMed] [Google Scholar]
- 34.Reeves R, Elton TS, Nissen MS, Lehn D, Johnson KR. Posttranscriptional gene regulation and specific binding of the nonhistone protein HMG-I by the 3′ untranslated region of bovine interleukin 2 cDNA. Proc Natl Acad Sci. 1987;84:6531–6535. doi: 10.1073/pnas.84.18.6531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solomon MJ, Strauss F, Varshavsky A. A mammalian high mobility group protein recognizes any stretch of six A·T base pairs in duplex DNA. Proc Natl Acad Sci USA. 1986;83:1276–1280. doi: 10.1073/pnas.83.5.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rajski SR, Williams RM. Observations on the covalent cross-linking of the binding domain (BD) of the high mobility group I/Y (HMGI/Y) proteins to DNA by FR66979. Bioorg Med Chem. 2000;8:1331–1342. doi: 10.1016/s0968-0896(00)00078-x. [DOI] [PubMed] [Google Scholar]
- 37.Rajski SR, Williams RM. DNA cross-linking agents as antitumor drugs. Chem Rev. 1998;98:2723–2796. doi: 10.1021/cr9800199. [DOI] [PubMed] [Google Scholar]
- 38.Vega MC, Garcia Saez I, Aymami J, Eritja R, Van der Marel GA, Van Boom JH, Rich A, Coll M. Three-dimensional crystal structure of the A-tract DNA dodecamer d(CGCAAATTTGCG) complexed with the minor-groove-binding drug Hoechst 33258. Eur J Biochem/FEBS. 1994;222:721–726. doi: 10.1111/j.1432-1033.1994.tb18917.x. [DOI] [PubMed] [Google Scholar]
- 39.Grant MA, Baron RM, Macias AA, Layne MD, Perrella MA, Rigby AC. Netropsin improves survival from endotoxaemia by disrupting HMGA1 binding to the NOS2 promoter. Biochem J. 2009;418:103–112. doi: 10.1042/BJ20081427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trapasso F, Sarti M, Cesari R, Yendamuri S, Dumon KR, Aqeilan RI, Pentimalli F, Infante L, Alder H, Abe N, Watanabe T, Viglietto G, Croce CM, Fusco A. Therapy of human pancreatic carcinoma based on suppression of HMGA1 protein synthesis in preclinical models. Cancer Gene Ther. 2004;11:633–641. doi: 10.1038/sj.cgt.7700745. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.