

Abstract
Immunoglobulin light chain (LC) amyloidosis (AL) results from overproduction of circulating amyloidogenic LC proteins and subsequent amyloid fibril deposition in organs. Mortality in AL amyloidosis patients is highly associated with a rapidly progressive AL cardiomyopathy, marked by profound impairment of diastolic and systolic cardiac function and significant early mortality. While myocardial fibril deposition contributes to the severe diastolic dysfunction seen in AL cardiomyopathy patients, the degree of fibril deposition has not been found to correlate with prognosis. Previously, we and others showed a direct cardiotoxic effect of amyloidogenic LC proteins (AL-LC), which may contribute to the pathophysiology and mortality observed in AL cardiomyopathy patients. However, the mechanisms underlying AL-LC related cardiotoxicity remain unknown. Mammalian stanniocalcin1 (STC1) is associated with a number of cellular processes including oxidative stress and cell death. Herein, we find that STC1 expression is elevated in cardiac tissue from AL cardiomyopathy patients, and is induced in isolated cardiomyocytes in response to AL-LC, but not non-amyloidogenic LC. STC1 overexpression in vitro recapitulates the pathophysiology of AL-LC mediated cardiotoxicity, with increased ROS production, contractile dysfunction and cell death. Overexpression of STC1 in vivo results in significant cardiac dysfunction and cell death. Genetic silencing of STC1 prevents AL-LC induced cardiotoxicity in cardiomyocytes and protects against AL-LC induced cell death and early mortality in zebrafish. The cardiotoxic effects of STC1 appears to be mediated via mitochondrial dysfunction as indicated by loss of mitochondrial membrane potential, ROS production and increased mitochondrial calcium levels. Collectively, this work identifies STC1 as a critical determinant of AL-LC cardiotoxicity.
Keywords: Amyloidosis, Cardiomyopathy, cardiac toxicity
‘Amyloidoses’ represent a group of diseases characterized by the extracellular deposition of amyloid fibrils in various tissues in the body [25]. Light chain (AL) amyloidosis is the most common systemic amyloidosis in the US and Europe and is the consequence of a plasma cell dyscrasia with monoclonal overproduction of amyloidogenic immunoglobulin light chain (LC) proteins and the associated deposition of amyloid fibrils in multiple tissues [26]. More than half of AL amyloidosis patients have cardiac involvement, presenting with a rapidly progressive amyloid cardiomyopathy [10, 14]. Currently, there are limited therapies available for AL amyloid cardiomyopathy, often requiring cardiac transplantation, without which the median survival is comparable to the most malignant of cancers [7]. Little is known regarding the mechanisms that underlie the pathogenesis of AL cardiomyopathy. Patients with AL cardiomyopathy are unresponsive to traditional heart failure regimens suggesting distinct, yet unknown mechanisms contribute to the pathogenesis AL cardiomyopathy. Thus, there is great need to uncover essential modulators of AL cardiomyopathy and identify novel therapeutic targets.
Interstitial amyloid fibril deposition in the heart has long been thought to be the basis for the pathogenesis of AL cardiomyopathy. Clinical observations, however, suggested an important discrepancy in survival amongst cardiac amyloidosis patients with similar degrees of cardiac fibril infiltration, but varying amyloid precursor proteins, suggesting alternative biological mechanisms for disease progression. In addition, reduction in circulating light chain levels, but not cardiac amyloid fibril deposition, is associated with improved patient outcomes [8]. The notion that amyloid precursor proteins contribute to disease pathology to as equal an extent as amyloid fibril deposition has been suggested for other amyloid disease states [43]. Moreover, our previous studies provided the first evidence for a direct, intrinsic cardiotoxic effect of amyloidogenic light chain (AL-LC) proteins in both isolated cardiomyocytes and intact mouse hearts, suggesting that AL-LC cardiotoxicity may contribute to the cardiac dysfunction observed with AL cardiomyopathy in the absence of fibril deposition [2, 24]. Our recent work further implicated non-canonical TAB1 (TAK1 binding protein 1)-mediated p38MAPK autophosphorylation as a key modulator of AL-LC cardiac toxicity [34].
Unbiased gene expression analysis of human hearts explanted from patients with AL cardiomyopathy indicated an increased differential expression of Stanniocalcin1 (STC1) (personal communication, F. del Monte). STC1 was originally identified in the corpuscles of Stannius of fish as a secreted Ca2+ regulatory hormone [15] and mammalian STC1 has been associated with a number of cellular processes including oxidative stress, inflammation, cell death and impaired calcium homeostasis, all of which have been implicated in the pathogenesis of AL-LC induced cardiotoxicity [20, 21, 30, 33]. We therefore set out to determine the role of STC1 in the pathophysiology of AL-LC cardiotoxicity as well as its potential relationship to p38MAPK activation.
Here, we demonstrate upregulation of STC1 in heart tissue from AL amyloid cardiomyopathy patients, and in isolated rat cardiomyocytes exposed to human AL-LC proteins, via a p38MAPK dependent manner. STC1 provokes mitochondrial dysfunction leading to excessive ROS production and is important for mediating AL amyloid cardiotoxicity in both cellular and in vivo models, with overexpression of STC1 recapitulating the cellular dysfunction associated with human AL-LC exposure both in vitro and in vivo. Moreover, genetic silencing of STC1 protected against AL-LC cardiotoxicity and early death in both cellular and zebrafish models.
Methods
Human tissues
All procedures related to human tissue were reviewed and approved by the Institution Review Board (IRB) at Boston University School of Medicine and Massachusetts General Hospital. Bence-Jones proteins, (AL-LC, amyloidogenic LC isolated from AL amyloid patients and Con-LC, non-amyloidogenic LC isolated from non-amyloidosis multiple myeloma patients), were purified from urine collection in collaboration with Boston University Amyloid Treatment and Research Program [5]. The purity of fractionated LC proteins was assessed by SDS-PAGE and immunoblotting as described previously [2]. Additional information regarding human LC protein is included in Table S1. Human heart tissues were collected from explanted hearts of patients with AL amyloid cardiomyopathy at the Massachusetts General Hospital. Non-disease control human hearts were obtained from the National Disease Research Interchange. Additional information regarding human samples is included in Table S2.
Animal care
All animal (mouse, rat and zebrafish) procedures and usages were reviewed and approved by the Institutional Animal Care and Use Committee at Harvard Medical School. Animals were housed in Association for Assessment and Accreditation of Laboratory Animal Care (AAALC) accredited animal care facilities under a 12 hour light-dark cycle and were fed with laboratory chow ad libidum. Rats for cardiomyocytes isolation were purchased from Charles River Laboratory (male Wistar rats, 150-200g, catalog #003). Mice were purchased from Jackson Laboratory (female FVB/NJ/mice 10weeks catalog #001800). Wild type zebrafish were purchased from Ekkwill Waterlife Resources (Ruskin). Care and breeding of zebrafish were performed as described previously [32, 41].
Chemicals and reagents
General chemicals and reagents were obtained from Sigma unless specified. Low glucose DMEM, TMRE, Rhod-2, Mitotracker-Green and Laminin were acquired from Invitrogen. Mito-tempo was from Santa Cruz Biotechnology. Trypsin and Collagenase were purchased from Willington. SB203580 compound was obtained from Calbiochem. Antibodies were obtained from Santa Cruz Biotechnology (STC1, Bax, and Bcl-2), Cell signaling Technology (p38MAPK and phosphorylated p38MAPK), Sigma (ACTININ), R&D Systems (STC1, GAPDH) or Developmental Studies Hybridoma Bank (MF20). Secondary antibodies for immunohistochemistry (Donkey anti-goat antibody-Alexa Fluo 555, Goat anti-rabbit antibody- Alexa Fluo 488, Goat anti-mouse antibody- Alexa Fluo 647) were from Invitrogen. Adenovirus expressing kit, adenovirus purification kit, and STC1 recombinant protein were purchased from Stratagene, Adenopure, and Biovendor, respectively.
Adenovirus preparation for STC1 overexpression and knockdown in isolated cardiomyocytes
Full-length murine STC1 cDNA (Open Biosystems), was subcloned into pShuttle-IRES-GFP vector (Adeasy adenovirus system, Stratagene), and later used to generate adenovirus from HEK cells according to the manufacturer's instruction. Control GFP adenovirus was generated using the same approach. To prepare STC1 shRNA adenovirus, an expression cassette containing a shRNA cloning site, U6 promoter and pCMV promoter driven GFP was obtained from pSicoR-GFP vector (Addgene) and put into pShuttle vector. shRNA oligonucleotides (GATTC TCCCT CACAC ATCA) targeting STC1 were designed using Invitrogen online software. The scrambled shRNA oligonucleotides (GACCC ACTAT CTAAC CTCT) for STC1 shRNA were designed using siRNA Wizard software (InvivoGen). Adenovirus was purified with Adenopure kit from Puresyn Inc.
Cardiomyocytes isolation and culture
Adult rat ventricular cardiomyocytes were isolated from male Wistar rats using a collagenase-based enzymatic digestion method as previously described [16]. Cardiomyocytes were treated with vehicle (ultrapure water), 20 μg/mL of Con-LC or AL-LC at designated time points as described in the Results section. Neonatal rat cardiomyocytes were isolated from 1-2 day olds Wistar rats (Charles River Laboratory #003) as previous described [13].
Cell contractility and intracellular Ca2+ measurements
Cellular function and Ca2+ homeostasis were measured in cultured adult ventricular cardiomyocytes using video edge detection and fluorescence measurements of the calcium sensitive dye, Fura-2, respectively as described previously [2]. Briefly, cardiomyocytes were infected with Adeno-STC1 or Adeno-GFP virus, and treated with either vehicle, Con-LC or AL-LC for 24 hours. Cardiomyocytes were then perfused with 1.2 mmol/L Ca2+ Tyrode's buffer at 37°C under pacing at 5 Hz. Percentage cellular shortening was calculated as the ratio of the difference between systolic and diastolic cell length over diastolic cell length. Calcium amplitude was calculated as the difference between systolic and diastolic cellular calcium levels (ratio of F360/380). 4-6 cells were measured per biological replicate and 3 biological replicates performed and grouped for statistical analysis.
Cell death assay
At the designed time points, cultured cardiomyocytes were washed with PBS, and immediately fixed in 4% paraformaldehyde. Cells were then permeablized in methanol at -20°C for 30 minutes, and incubated with TUNEL reaction mixture (Roche) in a moisture chamber for one hour at 37°C. Slides were washed with PBS, and mounted with mounting medium containing DAPI (Vectorlabs). For detection of cell death in zebrafish hearts, hearts were dissected from zebrafish in Tyrode's solution containing 3% BSA. Hearts were transferred to a microwell plate and fixed in 4% paraformaldehyde for 20 minutes. Hearts were rinsed in PBS and permeablized overnight in PBS with 0.1% Tween. Following incubation with anti- Myosin Heavy Chain (MF-20) antibody overnight at 4°C, hearts were washed 3 times in PBS, followed by incubation with secondary antibody for one hour at 37°C. Cell death was detected using TUNEL reaction mixture (Roche) in a moisture chamber for one hour at 37°C. Hearts were washed with PBS and placed directly into mounting media containing DAPI. For both cells and whole hearts, images were taken with excitation wavelengths of 405 nm and 555 nm, 5-7 pictures were taken from each slides using LSM700 confocal microscopy (Zeiss). Percent of apoptotic cell death was calculated as TUNEL positive nuclei divided by total nuclei. TUNEL positive nuclei were manually counted and the total nuclei were counted using Image J software (NIH). All the counting was performed in a blinded fashion. Caspase 3/7 activity was determined in freshly isolated homogenized extracts using a Caspase 3/7 activity kits as per manufacturer's instructions (Promega, Caspase-Glo 3/7 Assay). Briefly, 30 μg of protein was diluted to a final volume of 50 μL in PBS. 50 μL of Caspase 3/7 reaction buffer was added to each protein sample, incubated for 10 minutes in the dark, and luciferase activity was detected using a plate reader (Spectra Max).
RNA isolation and quantitative PCR
To assess gene expression, total RNA was isolated using Trizol (Invitrogen) extraction. DNAase treatment was performed subsequently to remove the residual DNA contamination (Turbo DNAase, Ambion). iScript™ cDNA Synthesis Kit (Bio-Rad) was used for first strand cDNA synthesis. Quantitative PCR was performed using standard curve method. For human tissue, STC1 forward primer: ATTCCCACCAACAAAATCCA; reverse primer: GGAAAAACATGGCAGAGGAA. 18S forward primer: TCATGTGGTGTTGAGGAAGC; reverse primer: GGCGTGGATTCTGCATAATG. For rat tissue, STC1 forward primer: CACTTCCAGATATCCGCGTT; reverse primer: CACCTGGGGTCCTTCAGATA. GAPDH forward primer: GGTGATGCTGGTGCTGAGTA; reverse primer: TTGCTGACAATCTTGAGGGA. For fish tissue, STC1 forward primer: GTGCTTAAACAGTGCGCTC; reverse primer: GCTCCTGGGTTTGATTTGGC. EF1α forward primer: CTGGAGGCCAGCTCAAACATGG; EF1α reverse primer: ACTCGTGGTGCATCTCAACAGACT.
Immunohistochemistry staining
Following cell isolation, adult cardiomyocytes were cultured overnight and fixed with 4% paraformaldehyde at room temperature. Cells were permeabilized using 2% Triton-X100 in glycerol/PBS (1:1 volume). Incubation with 3% BSA solution for 1 hour was performed to minimize non-specific binding. Cells were then incubated with cocktail of three primary antibodies (anti-STC1 [1:100], COX4 [1:1000] and ACTININ [1:500]) at 4°C for 18 hours followed by incubating with secondary antibodies in two steps at 37°C. First, donkey anti-goat Alexa Fluo 555 (1:300) was used to detect STC1. After 1xBPS wash out, goat anti-rabbit Alexa Fluo 488 (1:300) and goat anti-mouse Alexa Fluo 647 (1:300) were then added for 1 hour at 37°C to detect COX4 and ACTININ, respectively. After final washing, the slides were mounted with VECTASHIELD® mounting media (Vector Lab). Zeiss LSM700 fluorescence confocal microscope was used to visualize STC1 (Ex/Em: 550/600 nm), COX4 (Ex/Em: 488/525 nm), and ACTININ (Ex/Em: 648/680). DAPI was used to stain for the nuclei.
Immunoblotting analysis
For cultured cardiomyocytes, protein was harvested using cell lysis buffer (Cell Signaling). For mouse heart tissue, hearts were pulverized in liquid nitrogen and resuspended in cell lysis buffer. For zebrafish, 15-20 zebrafish were suspended directly in 50 μL SDS loading buffer and homogenized using a tissue homogenizer (TissueLyser II, Qiagen). Following homogenization, the samples were centrifuged and total protein homogenates were obtained. 30 μg of total protein or 18 μl of fish protein lysate was loaded onto Criterion XT bis-tris precast gels (4-12%) (Invitrogen) or PAGEr Gold precast gels (4-20%) (Lonza) for electrophoresis. Protein was electrotransferred to a PVDF membrane (Millipore) at 30 volts for 16-18 hours at 4°C. After blocking in 5% BSA in PBS, proteins of interest were detected by incubation with appropriate primary antibodies overnight at 4°C. After washing, blots were incubated with corresponding secondary antibodies. Odyssey infrared scanner (Li-Cor) was used to determine the infrared fluorescent signal and GAPDH was used as a reference gene for normalization.
Mitochondrial membrane potential measurement
Following STC1 (200ng/mL) or vehicle administration for 24 hours, cultured cardiomyocytes were incubated with cell permeable, mitochondrial membrane potential-sensitive fluorophore TMRE (Invitrogen) at the concentration of 10 nM for 30 minutes. Cardiomyocytes were then washed with warm PBS for 2 times. TMRE fluorescence was measured using SpectraMax M5 Microplate Reader (Molecular Device) with the excitation wavelength of 555 nm.
Mitochondrial calcium measurement
Following 24 hour exposure to STC1 (200ng/mL) or vehicle, cultured neonatal cardiomyocytes were incubated with cell permeable, mitochondrial specific calcium fluorophore Rhod-2 [6] (Invitrogen) at a concentration of 40 μM for 30 minutes at 37°C. Rhod-2 was reduced to a colorless dye using sodium borate. Hoechst dye (40 μM) and Mitotracker Green (250 nM) were used to visualize the nuclei and mitochondria, respectively. Cardiomyocytes were washed twice with warm PBS. Cell images were acquired using LSM700 confocal microscopy. To specifically analyze mitochondrial calcium, only Rhod-2 signal co-localized with Mitotracker Green was quantified. Analysis was performed using Cell Profiler software. For each biological replicate, 80-100 cells were analyzed per condition. Data averaged from a total of 3 biological replicates.
ROS measurement
Following STC1 (200ng/mL) or vehicle administration for 24 hours, cultured cardiomyocytes were incubated with cell permeable, redox-sensitive fluorophore DCFDA (Invitrogen) at the concentration of 20 μM for 30 minutes. Cardiomyocytes were then washed with warm PBS for 2 times. Cell images were acquired using LSM700 confocal microscopy (excitation wavelength at 488nm) and analyzed with SigmaScan Pro. For determination of mitochondrial-derived ROS, Mitotempo (Santa Cruz Biotech), a mitochondrial specific ROS scavenger, was used to pre-treat cardiomyocytes at the concentration of 100 nM for 45 minutes prior to STC1 administration.
Assessment of cardiac function in zebrafish
Individual fish embryos at 3 or 4 dpf (days post fertilization) were transiently anesthetized in Tricaine methanesulfonate (Sigma) solution in E3 embryo water. Fish embryos were positioned laterally for image acquisition [35]. Axioplan (Zeiss) upright microscopy equipped with a FastCam-PCI high-speed camera (Photron) was used for image acquisition. Sequential images of heart contraction were acquired at 250 frames per second. 8-10 cardiac cycles were recorded for each fish. Long and short axes during diastole and systole were obtained using Image J (NIH) tracing during cardiac cycles. Ventricular volume was calculated using the equation Ventricular volume= (4/3) π / s2 and the stroke volume were calculated based on the change between diastole and systole. 10-12 fish were examined per condition.
Transient STC1 overexpression zebrafish model
Plasmid containing full length zebrafish STC1 was purchased from Open Biosystems (MDR1734-99237559). mMESSAGE mMACHINE Kit (Invitrogen) was used to synthesize zebrafish STC1 mRNA. STC1 mRNA in the reverse order was synthesized for control. The quality/quantity of mRNA was determined by Nanodrop spectrometer (Thermo Scientific). 20 pg of mRNA were injected into zebrafish embryos at single cell stage. Cardiac function was measured by high-speed video camera at 3 dpf (days post fertilization) and cardiac cell death as well as cardiac morphology was determined at 4dpf.
STC1 morpholino knockdown in zebrafish model of AL-LC cardiac toxicity
Morpholinos (Gene-Tools, LLC) were designed to target the boundary region between exon 2 and intron 2 of fish STC1 mRNA. Sequence used for construct development was GCCTTGCCCTGTAAAATCAACAGAA. Morpholinos were resuspended in 1X Danieau's buffer (58 mmol/L NaCl, 0.7 mmol/L KCl, 0.4 mmol/L MgSO4, 0.6 mmol/L Ca(NO3)2, 0.5 mmol/L Hepes, pH 7.6) to achieve a final concentration of 500 μmol/L. At the single cell stage, 1 nL of morpholino or control (Danieau's buffer) solution was injected. Veh (E3 water) or AL-LC (1 nL at 1 mg/mL) was delivered to the circulation of 2 dpf anaesthetized control or STC1 MO injected embryos via venous injection as described previously [29]. Cardiac function was assessed using a high-speed camera, as described above, two days post LC injections (4dpf). Three days following LC delivery (5dpf), protein expression and cell death were determined using immunoblotting for STC1 and TUNEL staining, respectively. In addition, zebrafish survival was determined and hearts or embryos were harvested for TUNEL staining or immunoblotting, respectively. Zebrafish survival was measured by counting zebrafish number in STC1 MO and control injected embryos immediately (2dpf) and 3 days (5dpf) following AL-LC or vehicle injection and the percent survival was calculated accordingly. The percent survival was averaged from 5 independent repeats.
Statistical analysis
All data are shown as mean ± standard error. Statistical differences between mean values for two groups were evaluated by Student t test using GraphPad Prism software. Statistical differences between mean values for multiple groups were evaluated by one-way analysis of variance. When comparisons were significant, post-hoc Bonferroni's test was used to compare individual groups. p<0.05 was considered as significant.
Results
STC1 is upregulated in patients with AL amyloid cardiomyopathy and in response to human AL-LC
To validate microarray gene profiling data (del Monte laboratory unpublished data), human heart samples (left ventricle) were obtained from AL amyloid cardiomyopathy patients (n=6) or control patients without heart disease (n=5) collected in cold oxygenated Wisconsin cardioplegic solution immediately following harvest. STC1 mRNA expression was quantified using real-time PCR, and found to be upregulated in hearts from patients with AL amyloid cardiomyopathy relative to control non-failing heart samples (Fig. 1a). STC1 protein expression was quantified via immunoblotting analysis of human heart samples, and similarly found to be selectively upregulated in patients with AL amyloid cardiomyopathy (Fig. 1b). To determine if STC1 expression was induced by AL-LC proteins in the absence of fibril formation, adult rat ventricular cardiomyocytes were exposed to either human AL-LC (20 μg/mL), Con-LC (20 μg/mL), or vehicle control. AL-LC but not Con-LC or vehicle, induced STC1 mRNA expression within one hour (Fig 1c), with subsequent increases in STC1 protein expression at early (2 hours) and late (24 hours) time points (Fig 1d, e). Importantly, upregulation of STC1 protein expression was observed in response to AL-LC proteins isolated from 5 different AL amyloidosis patients (Fig. 1f). These data demonstrate that STC1 expression is specifically upregulated by AL-LC proteins in both patients with AL amyloid cardiomyopathy and in isolated cardiomyocytes.
Fig. 1.
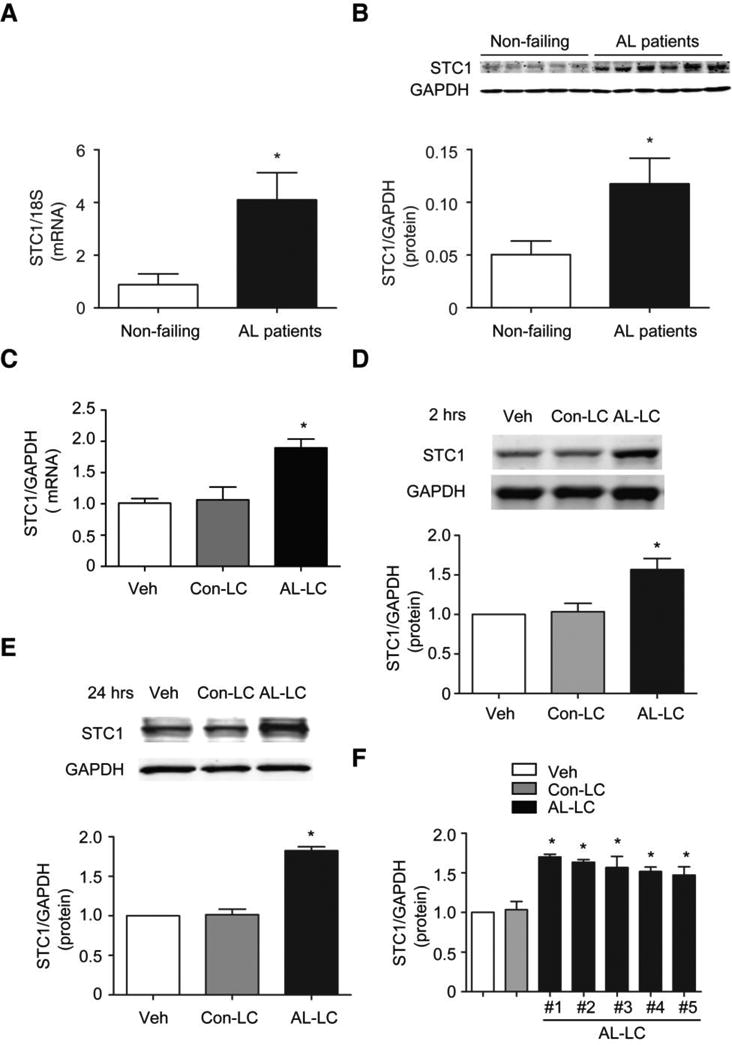
STC1 is upregulated in patients with AL cardiomyopathy and in response to AL-LC stimulation. a STC1 gene expression (normalized to 18S) and b STC1 protein expression relative to GAPDH were upregulated in AL cardiomyopathy patients (N=6) as compared to non-failing control patients (N=5). AL-LC triggers c STC1 gene and d, e protein expression in isolated cardiomyocytes exposed to AL-LC for 2 hour and 24 hours, respectively, relative to those exposed to Con-LC or Veh. f STC1 was upregulated early with AL-LC obtained from 5 independent patients with amyloid cardiomyopathy as compared to Veh or Con-LC. * p<0.05 between indicated groups.
AL-LC induces STC1 upregulation in a p38MAPK dependent manner
Previously, we have shown that p38MAPK activation occurs within minutes of AL-LC exposure and is required for AL-LC induced cardiotoxicity [34]. To determine whether p38MAPK regulates STC1 induction, isolated cardiomyocytes were treated with AL-LC, Con-LC, or vehicle in the presence or absence of SB203580, a selective p38MAPK inhibitor [34]. Inhibition of p38MAPK completely prevented STC1 upregulation in response to AL-LC (Fig. 2a). In an established mouse model of AL-LC cardiotoxicity, generated by parenteral injection of AL-LC proteins [34], human AL-LC induced cardiac STC1 protein expression in mice, compared to Con-LC treated mice (Fig. 2b). AL-LC induction of cardiac STC1 expression, however, was abolished in transgenic mice with cardiac expression of dominant negative p38MAPK (DN-TG) (Fig. 2b). Collectively, these in vitro and in vivo data demonstrate that AL-LC induction of STC1 is downstream of p38MAPK activation, a necessary early signaling event upstream of AL-LC induced cardiac toxicity.
Fig. 2.
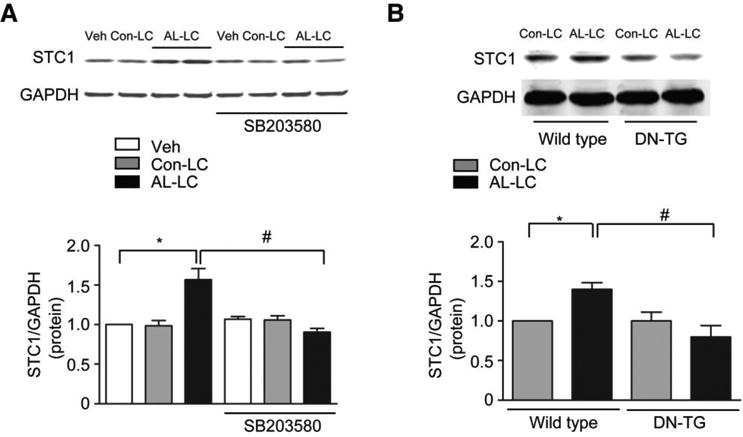
AL-LC induction of STC1 is p38MAPK dependent in vitro and in vivo. a AL-LC induced upregulation of STC1 protein expression in cardiomyocytes was abolished in the presence of p38MAPK inhibitor, SB203580 (5 μmol/L). b AL-LC triggered upregulation of cardiac STC1 protein expression was prevented in p38MAPK dominate negative (DN-TG) mice. * p<0.05, AL-LC vs. vehicle or Con-LC, # p<0.05 AL-LC with vs. without SB203580 or AL-LC infused wild type mice vs. DN-TG mice. N=3 for each group.
STC1 induces mitochondrial dysfunction and provokes ROS generation
To further investigate the function of STC1 in cardiomyocytes, we sought to delineate the cellular location of STC1. Using immunohistochemical staining, we found that STC1 is mostly expressed in nuclei and mitochondria (Fig. 3a). Mitochondrial localization was confirmed through co-staining for STC1 and the mitochondrial protein, COX4, as shown in Fig. 3b and Fig. 3c. Given its mitochondrial localization, we then examined whether STC1 alters mitochondrial function by measuring mitochondrial membrane potential using the cell-permeable mitochondrial membrane potential-sensitive dye tetramethylrhodamine, ethyl ester (TMRE). Our data showed that STC1 treatment significantly reduced mitochondrial membrane potential (Fig. 3d). Moreover, we found that STC1 induced ROS production can be attenuated by pretreatment with MitoTempo, a mitochondrial specific ROS scavenger (Fig. 3e, f). To determine whether STC1 affects cardiomyocyte mitochondrial calcium levels, mitochondrial calcium concentration was measured using the mitochondrial calcium probe, Rhod2. We found that STC1 significantly elevates mitochondrial calcium levels (Fig 3g, h), suggesting a role for STC1 in the dysregulation of calcium homeostasis and mitochondrial function.
Fig. 3.
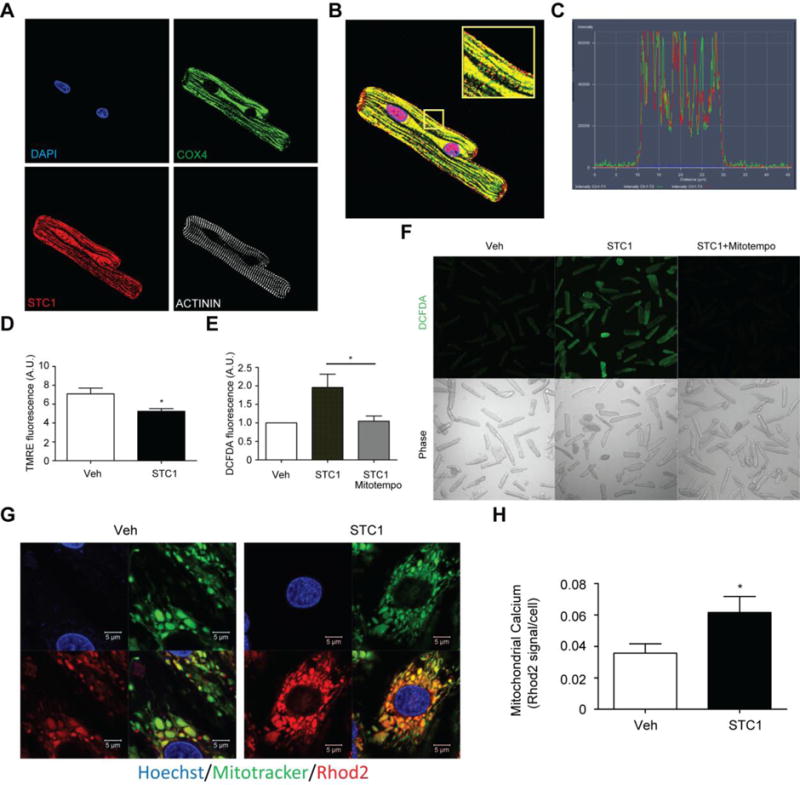
STC1 affects mitochondrial calcium homeostasis and function. a Subcellular localization of STC1 in cardiomyocytes was determined using immunohistochemical staining against DAPI (blue), STC1 (red), mitochondrial COX4 (green), and the myofilament protein, ACTININ (white). b Merged images of immunohistochemical staining indicates that STC1 is co-localized to the mitochondria and nucleus. c Line scanning analysis demonstrates close overlap of spectral profiles for the red fluorophore (STC1) and green fluorophore (COX4), confirming the mitochondrial localization of STC1. d Mitochondrial membrane potential is decreased in cardiomyocytes exposed to 200 ng/mL STC1 for 24 hours as determined by TMRE, a mitochondrial membrane potential sensitive fluorescence dye. e AL-LC induced ROS was prevented in cardiomyocytes treated with a mitochondrial targeted ROS scavenger, MitoTempo. f Representative DCFDA fluorescence images with corresponding bright field images on right. g Representative fluorescent images of Rhod2 (red) and mitotracker (green), suggesting increased mitochondrial calcium in response to 200 ng/mL STC1 stimulation for 24 hours. h Summarized data of mitochondrial calcium levels. * p<0.05 between indicated groups.
STC1 recapitulates AL-LC cardiac pathology in vitro and in vivo
To define the effects of STC1 upregulation on cardiomyocytes, STC1 was overexpressed via adenoviral mediated gene transfer in isolated adult rat cardiomyocytes. Expression of STC1 (Adeno-STC1) or control GFP (Adeno-GFP) was titrated to levels comparable to that induced by human AL-LC exposure, ∼1.5-2 fold increase (Fig. S1), to avoid non-specific toxic effects associated with excessive protein expression. STC1 overexpression decreased cell shortening (Fig 4a) and diminished calcium transient amplitude (Fig 4b) concomitantly, as measured in single cardiomyocytes using video edge detection techniques and a fluorescent calcium sensitive dye. Furthermore, STC1 overexpression increased cell death, as assessed by Bax/Bcl-2 ratio (Fig. 4c) and quantification of TUNEL positive cells (Fig. 4d). In summary, STC1 overexpression caused cardiomyocyte dysfunction and cell death, recapitulating AL-LC induced cardiac toxicity in cardiomyocytes [34].
Fig. 4.
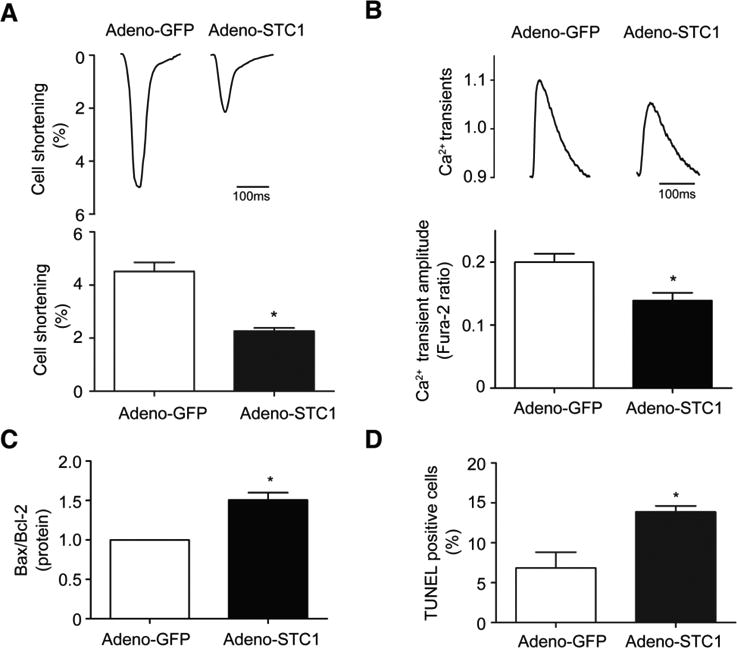
STC1 induces contractile dysfunction and cell death in cardiomyocytes. Adenoviral mediated overexpression of STC1 in cardiomyocytes (Adeno-STC1) decreased a cellular contractile function (% cell shortening) and b intracellular calcium transient amplitude as well as increased cell death determined using c Bax/Bcl2 ratio and d TUNEL assay relative to control (Adeno-GFP). Representative tracing of cell shortening and calcium transient are shown in the upper panels of a and b, respectively. N=3 for each group. * p<0.05 between indicated groups
To determine whether STC1 adversely affects cardiac function and cell viability in vivo, STC1 was transiently overexpressed in zebrafish via delivery of STC1 mRNA at the one-cell stage of development followed by measurement of cardiac function, TUNEL assay and morphological assessment as illustrated in Fig. 5a. The size and purity of the mRNA was confirmed as showed in Fig. S2a. STC1 mRNA (Fig. 5b) and protein expression levels (Fig. 5c) were confirmed to be upregulated in zebrafish 4dpf (4 days following STC1 mRNA injection). Equal protein loading was confirmed with Ponceau staining (Fig. S2b). Using high-speed camera imaging, a reduction in stroke volume (Fig. 5d) and cardiac output (Fig. 5e) were observed as early as 3 days post STC1 mRNA injection (3dpf). No difference in heart rate was found between STC1 overexpressing fish and controls (Fig. 5f). Strikingly, only STC1-overexpressing zebrafish displayed pericardial edema, a characteristic landmark for cardiac dysfunction in zebrafish (Fig. 5g). Additionally, we observed increased cardiac cell death in cardiomyocytes in STC1-overexpressing zebrafish compared with controls, as assessed by TUNEL and cardiac marker (MF20) co-staining of isolated hearts at 4 dpf (Fig. 5h, i). A similar increase in cardiac cell death was observed in STC1-overexpressing zebrafish at 5 dpf, as compared with controls (Fig. S3a, b). In accordance with the phenotype observed in the STC1 overexpressing zebrafish, transient overexpression of STC1 in mouse hearts via in vivo adenoviral-mediated gene transfer (Fig. S4a) resulted in a similar depression of cardiac function (Fig. S4b). Additionally, cardiac cell death was increased in STC1-overexpressing mouse hearts, assessed by increased Bax/Bcl-2 ratio (Fig. S4c) and caspase 3/7 activity (Fig. S4d).
Fig. 5.
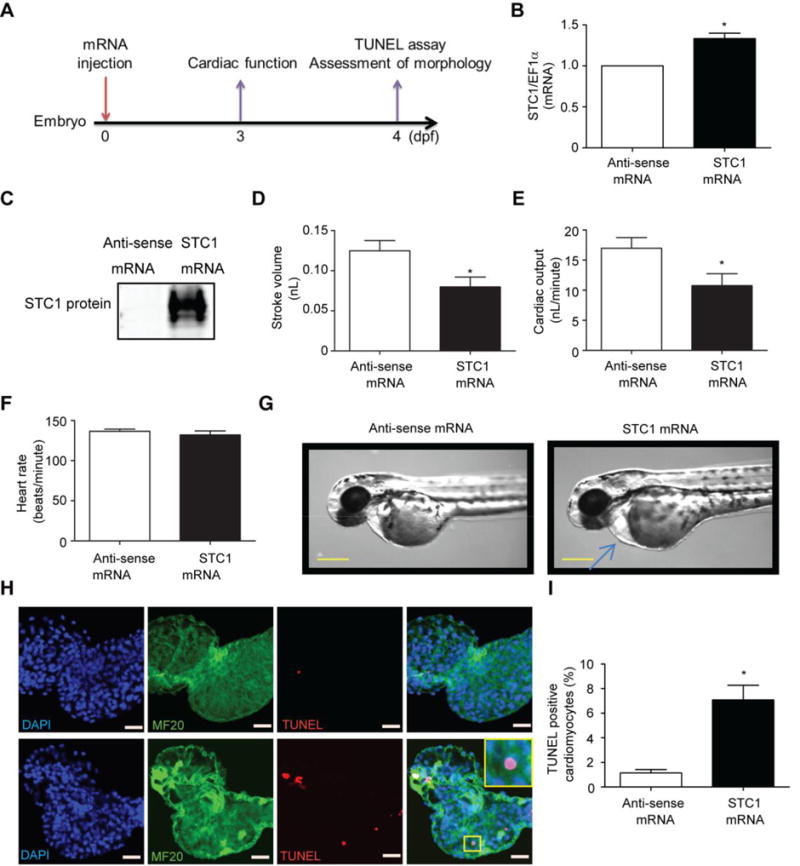
Overexpression of STC1 induces contractile dysfunction and cell death in vivo. a Time course of the experiments. STC1 or anti-sense control mRNA was injected into zebrafish at day 0 (one-cell stage). STC1 b gene and c protein expression measured at day 3 post mRNA injection were increased in STC1 mRNA injected zebrafish. STC1mRNA injection in zebrafish also altered cardiac function as demonstrated by the decreased d stroke volume and e cardiac output without change in f heart rate. g Representative images demonstrating pericardial edema with STC1 expression in zebrafish. Scale bar represents 400 μm. h Representative images of isolated zebrafish hearts stained with DAPI (blue) for nuclei, the cardiac specific marker, MF-20 (green) and TUNEL (red). i Quantification of TUNEL positive nuclei in cardiomyocytes. Scale bar=20 μm. * p<0.05 between indicated groups. (N=12 for cardiac function analysis and N=6 for TUNEL assay)
STC1 is required for AL-LC induced cardiac toxicity in vitro and in vivo
To determine whether STC1 plays a causal role in AL-LC induced cardiomyocyte dysfunction and death, STC1 expression was genetically silenced using adenoviral-mediated expression of shRNA targeting STC1 (Adeno-STC1-shRNA) in adult cardiomyocytes. Adeno-STC1-shRNA attenuated STC1 expression induced by AL-LC in adult cardiomyocytes compared to scrambled controls (Fig. 6a), and prevented AL-LC induced contractile dysfunction (Fig. 6b) and impaired calcium homeostasis (Fig. 6c). AL-LC-induced cell death was attenuated in Adeno-STC1-shRNA treated cardiomyocytes (Fig. 6d). These data demonstrate a requirement for STC1 in mediating AL-LC induced cellular dysfunction and death.
Fig. 6.
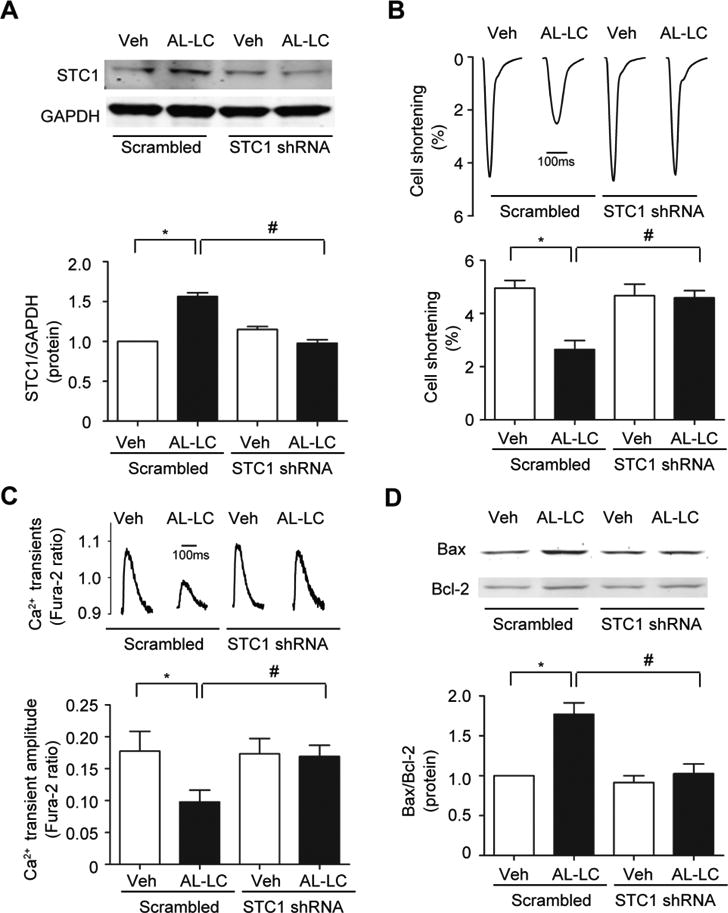
STC1 is required for AL-LC induced dysfunction and cell death. Isolated cardiomyocytes were infected with STC1 shRNA adenovirus or scrambled shRNA adenovirus 24 hours prior to AL-LC or vehicle exposure. a Adenoviral mediated STC1 shRNA in cardiomyocytes effectively prevent the upregulation of STC1 in response to AL-LC. AL-LC triggered b cellular dysfunction, c impaired calcium homeostasis and d cell death were attenuated in Adeno-STC1-shRNA infected cardiomyocytes. N=3 per condition. *, # p<0.05 between indicated groups.
The necessity of STC1 in mediating AL-LC induced cardiac dysfunction and mortality was examined in vivo using our zebrafish model of AL-LC toxicity [29]. Consistent with in vitro experiments, injection of human AL-LC induced STC1 expression (Fig. 7a). The time course of morpholino injection and various functional and morphological readouts are outlined in Fig. 7b. Similar to in vitro observations, AL-LC injection decreased cardiac function (Fig. 7c, d) and increased cardiac cell death (Fig. 7e, f) as well as increased mortality (Fig. 7g), all of which were prevented in STC1 morphants (Fig. 7c-g). It is notable that morpholino mediated knockdown successfully attenuated AL-LC induced STC1 upregulation over the entire time frame of study as showed in Fig. 7h. Collectively, these results further established STC1 as a critical mediator of AL-LC induced cardiac pathology and suggest that antagonism of STC1 may serve as a novel therapeutic treatment approach.
Fig. 7.
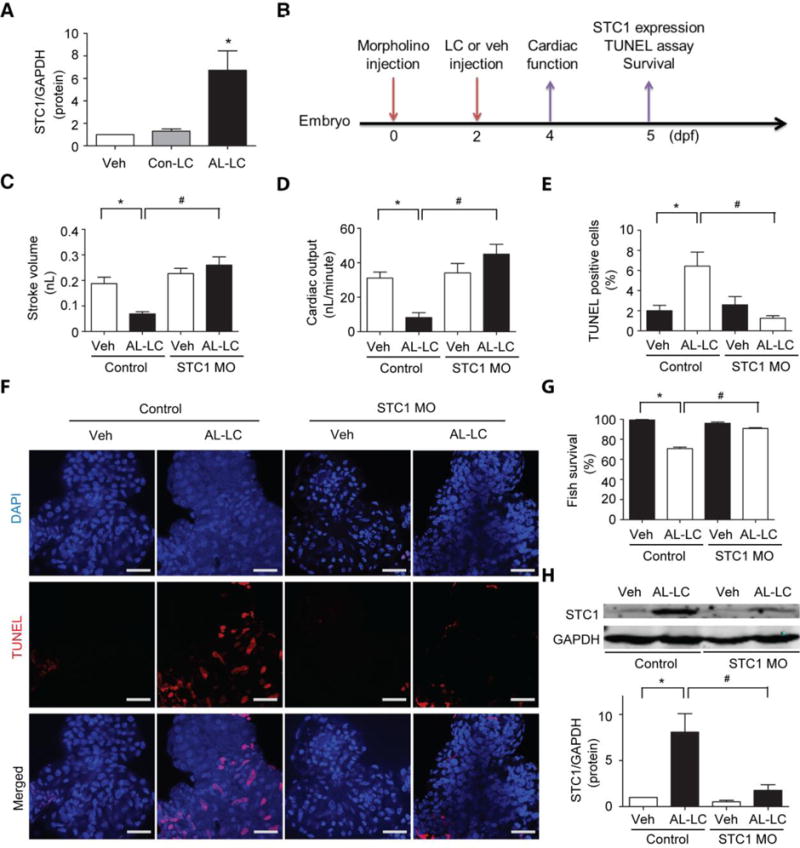
Antagonism of STC1 protects against AL-LC induced toxicity in a zebrafish model. AL-LC or Con-LC was introduced via venous injection at 48 hpf, at a final concentration of 100 μg/mL. E3 water injection served as Veh control. a Immunoblotting analysis for STC1 protein expression in zebrafish following venous injection of AL-LC but not Con-LC or Veh 3 days post-injection. b Time line of the experiments conducted. Cardiac contractile function decreased in AL-LC injected zebrafish as shown by the decreased c stroke volume and d cardiac output. AL-LC injection also increased e. cell death as determined by TUNEL assay. f Representative images with nuclei stained with DAPI (blue) and TUNEL positive cells (red). AL-LC induced cardiac dysfunction, cell death and mortality were prevented in STC1 MO injected zebrafish (c-g). h STC1 protein knockdown via STC1 MO, as assessed using immunoblotting at 5 days post MO injection Scale bar=20 μm. (N=12 per group for cardiac function analysis, N=6 hearts per group for TUNEL analysis, N=55-70 per group for survival analysis. *, # p<0.05 between indicated groups.
Discussion
While less common than other forms of cardiomyopathy, the prevalence of AL cardiomyopathy is comparable to that of Hodgkin's lymphoma or chronic myelogenous leukemia [11]. Importantly, current statistics may provide an underestimation of the prevalence of AL cardiomyopathy due to under-diagnosis resulting from insidious onset of disease, rapid progression, and insufficient clinical awareness. Unlike other forms of cardiomyopathy, AL amyloidosis cardiomyopathy is largely unresponsive to standard treatment regimens, resulting in early mortality [10]. In this study, we provide critical molecular insight into the pathogenesis of this fatal disease and identify STC1 as a key mediator of AL-LC cardiotoxicity both in vitro and in vivo, highlighting the potential of STC1 as a viable therapeutic target.
It was initially hypothesized that AL amyloid cardiomyopathy was the direct result of an infiltrative disease process associated with extracellular deposition of amyloid fibrils within the heart coupled with passive restriction of cardiac function. However, clinical observations were discordant with this proposed mechanism, as little correlation between the degree of cardiac fibril infiltration and cardiac dysfunction had been found amongst patients with AL amyloid cardiomyopathy [8]. Instead, mortality was been found to correlate with the levels of circulating LC [8]. Moreover, reduction of circulating AL-LC was shown to have a correlative improvement in cardiac performance and prolonged patient survival, despite the persistence of cardiac amyloid deposits [31]. In line with these clinical observations, our laboratory and via subsequent independent confirmation by others [27, 28, 36] demonstrated a direct cardiotoxic effect of AL-LC proteins in the absence of fibril formation, both in vitro and in vivo [2, 24, 34]. Thus, AL amyloidosis results from two intermingling pathogenic mechanisms involving both amyloid fibril deposition and direct cardiac toxicity of AL-LC precursor protein. The finding that activation of a non-canonical p38MAPK pathway within minutes of AL-LC exposure provided the first evidence of the acute induction of specific AL-LC mediated signaling pathways in the heart [34]. In this study, we reveal STC1 as a novel downstream molecule of AL-LC mediated p38MAPK activation. Importantly, we show that STC1 expression was induced by AL-LC proteins isolated from five separate AL patients, but not by non-amyloidogenic Con-LC proteins from multiple myeloma patients, suggesting that the observed STC1 upregulation is not due to a potential artifact associated with any one human AL-LC sample, but rather may be generalizable to AL amyloid cardiomyopathy.
STC1 is a glycoprotein identified in bony fish as a calcium regulatory hormone [38]. The mammalian STC1 homolog shares sequence homology with its fish counterpart, and has been implicated in a multitude of cellular processes, including ischemic injury, apoptosis, angiogenesis, inflammation, cancer biology, bone and muscle development, cellular metabolism, calcium regulation, differentiation and endothelial permeability [4, 9, 12, 17, 18, 20, 30, 33, 38-40, 42, 44, 45]. Numerous reports have shown a role for STC1 as a mediator of apoptosis [20, 22, 42], demonstrating both pro-apoptotic and anti-apoptotic roles depending upon cell type and stimulus. A recent report by Nguyen et al suggested a pathologic role for STC1 in promoting apoptosis under conditions of oxidative stress [30]. Additionally, STC1 has been identified as a mediator of apoptotic cell death in amyloidogenic Aβ protein-treated endothelial cells and radiated fibroblasts [1, 23]. Our study suggests a critical role for STC1 extending beyond localized amyloidosis, to systemic amyloid diseases, specifically AL amyloid cardiomyopathy. Importantly, cardiac STC1 expression is differentially increased in patients with AL amyloid cardiomyopathy but not idiopathic dilated cardiomyopathy (del Monte Laboratory unpublished data). We were able to simulate the increased STC1 observed in AL cardiomyopathy patients in fish, mouse and isolated rat cardiomyocytes, upon exposure to human AL-LC, suggesting that upregulation of STC1 occurs in a cardiomyocyte autonomous process independent of cardiac endothelial or fibroblast cells (Fig. S5). Moreover, STC1 activation was highly dependent upon p38MAPK activation, determined by both in vitro pharmacologic inhibition and in vivo using mice overexpressing dominant negative p38MAPK.
While profound diastolic dysfunction due to extensive amyloid fibril deposition is a predominant manifestation of AL cardiomyopathy, systolic dysfunction is also present. The degree of systolic dysfunction may actually be underestimated using traditional echocardiographic parameters such as ejection fraction, and recently has been detected using more sensitive approaches including strain analysis [19]. In isolated cardiomyocytes, exposure to AL-LC markedly decreases contractile dysfunction and we observed a similar impairment of systolic function in vivo in zebrafish exposed to either AL-LC, or overexpressing STC1.
Our report is the first to demonstrate a pathological role of cardiac STC1 in AL-LC induced cardiac toxicity. The adverse effect of STC1 in the heart is consistent with reports suggesting that STC1 promotes cell death in multiple cell types in response to diverse stress signals. STC1 can act as a negative regulator of fibroblast cell survival under conditions of oxidative stress [30]. STC1 has been reported to be induced at both mRNA and protein levels by H2O2 and furthermore, STC1 null mouse embryonic fibroblasts are resistant to growth inhibition and cell death induced by H2O2 or 20% O2. Reintroduction of STC1 into null mouse embryonic fibroblasts increases cell death under conditions of oxidative stress [30]. In contrast, in other cell types, such as neurons or mesenchymal stem cells, STC1 has been suggested to exert protective functions [1, 45]. Our in vivo data showing deleterious effects of STC1 are supported by two independent STC1 transgenic mouse studies in which STC1 overexpression results in decreased muscle mass and deterioration in muscle function [12, 37]. While STC1's role is highly context dependent, genetic ablation of STC1 in mice was associated with little to no basal phenotype [3], further highlighting the potential of targeted STC1 antagonism as a therapeutic strategy. Our data show that STC1 mediated mitochondrial damage, potentially via calcium overload, the resulting ROS production and loss of mitochondrial integrity, may be the underlying mechanism for STC1-induced cardiac toxicity. Consistent with our findings, using cellular models and STC1 transgenic mice, reports have indicated that STC1 directly targets the mitochondria, causing profound dysfunction and deformation of mitochondria [9, 12]. Relevant to our finding, recent studies have shown that STC1 can impair mitochondrial calcium homeostasis [9]. The observed increased mitochondria calcium in response to STC1 may, in part, contribute to the decrease in cytosolic calcium concentration. Altered cytosolic and mitochondrial calcium levels may result in impaired contractile function and elevated ROS / mitochondrial dysfunction, respectively. Antagonizing STC1 resulted in beneficial effects in both in vitro and in vivo, in isolated cardiomyocytes and zebrafish, respectively. However, it remains to be determined whether antagonizing STC1 alters the natural progression of amyloid cardiomyopathy in other animal models, including those involving chronic exposure to AL-LC, as well as whether antagonizing STC1 is a viable option for the treatment of amyloid cardiomyopathy in patients.
The current lack of understanding of the molecular mechanisms underlying AL amyloid cardiomyopathy is a critical bottleneck in the development of treatment and diagnostics for this disease. The work presented here represents efforts towards understanding the mechanisms underlying this fatal disease and while we demonstrate a critical role for STC1 in mediating AL-LC induced proteotoxicity in vitro and in vivo, further studies in both animal models and patients, are required to more definitively define the pathophysiologic basis for amyloid cardiomyopathy and the role of STC1 in this process.
Supplementary Material
Acknowledgments
The authors thank Ms. Wendy Du at Carnegie Mellon University, Mr. Soeun Ngoy at the Brigham and Women's Hospital (BWH) Cardiovascular Physiology Core, Ms. Gloria Chan at Boston University (BU) Amyloidosis Center for excellent technical assistances and Drs. Yingyi Zhang, Judith Gwathmey and Thomas E. Macgillivray for providing critical reagents. This work was supported, in part, by the National Institutes of Health (NIH), HL088533, HL086967, HL093148, HL099073 (R.L.), DK90696 (D.C.S.), AG031804 (L.H.C.), American Heart Association (C.A.M.), and the Cardiac Amyloid Center, BWH (R.H.F., R.L.) and the Gruss and Wildflower Foundations and the Amyloid Research Fund at BU (L.H.C., D.C.S.). G.M. is supported by Associazione Italiana per la Ricercasul Cancro Special Program 5x mille, Molecular Clinical Oncology. S.M. is supported by NIH T32 (T32HL007604).
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- 1.Block GJ, Ohkouchi S, Fung F, Frenkel J, Gregory C, Pochampally R, Dimattia G, Sullivan DE, Prockop DJ. Multipotent stromal cells are activated to reduce apoptosis in part by upregulation and secretion of stanniocalcin-1. Stem Cells. 2009;27:670–681. doi: 10.1002/stem.20080742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brenner DA, Jain M, Pimentel DR, Wang B, Connors LH, Skinner M, Apstein CS, Liao R. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res. 2004;94:1008–1010. doi: 10.1161/01.RES.0000126569.75419.74. [DOI] [PubMed] [Google Scholar]
- 3.Chang AC, Cha J, Koentgen F, Reddel RR. The murine stanniocalcin 1 gene is not essential for growth and development. Mol Cell Biol. 2005;25:10604–10610. doi: 10.1128/MCB.25.23.10604-10610.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang AC, Jellinek DA, Reddel RR. Mammalian stanniocalcins and cancer. Endocr Relat Cancer. 2003;10:359–373. doi: 10.1677/erc.0.0100359. [DOI] [PubMed] [Google Scholar]
- 5.Connors LH, Jiang Y, Budnik M, Theberge R, Prokaeva T, Bodi KL, Seldin DC, Costello CE, Skinner M. Heterogeneity in primary structure, post-translational modifications, and germline gene usage of nine full-length amyloidogenic kappa1 immunoglobulin light chains. Biochemistry. 2007;46:14259–14271. doi: 10.1021/bi7013773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Du J, Wang Y, Hunter R, Wei Y, Blumenthal R, Falke C, Khairova R, Zhou R, Yuan P, Machado-Vieira R, McEwen BS, Manji HK. Dynamic regulation of mitochondrial function by glucocorticoids. Proc Natl Acad Sci U S A. 2009;106:3543–3548. doi: 10.1073/pnas.0812671106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dubrey SW, Cha K, Anderson J, Chamarthi B, Reisinger J, Skinner M, Falk RH. The clinical features of immunoglobulin light-chain (AL) amyloidosis with heart involvement. QJM. 1998;91:141–157. doi: 10.1093/qjmed/91.2.141. [DOI] [PubMed] [Google Scholar]
- 8.Dubrey SW, Cha K, Skinner M, LaValley M, Falk RH. Familial and primary (AL) cardiac amyloidosis: echocardiographically similar diseases with distinctly different clinical outcomes. Heart. 1997;78:74–82. doi: 10.1136/hrt.78.1.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ellard JP, McCudden CR, Tanega C, James KA, Ratkovic S, Staples JF, Wagner GF. The respiratory effects of stanniocalcin-1 (STC-1) on intact mitochondria and cells: STC-1 uncouples oxidative phosphorylation and its actions are modulated by nucleotide triphosphates. Mol Cell Endocrinol. 2007;264:90–101. doi: 10.1016/j.mce.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 10.Falk RH. Cardiac amyloidosis: a treatable disease, often overlooked. Circulation. 2011;124:1079–1085. doi: 10.1161/CIRCULATIONAHA.110.010447. [DOI] [PubMed] [Google Scholar]
- 11.Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005;112:2047–2060. doi: 10.1161/CIRCULATIONAHA.104.489187. [DOI] [PubMed] [Google Scholar]
- 12.Filvaroff EH, Guillet S, Zlot C, Bao M, Ingle G, Steinmetz H, Hoeffel J, Bunting S, Ross J, Carano RA, Powell-Braxton L, Wagner GF, Eckert R, Gerritsen ME, French DM. Stanniocalcin 1 alters muscle and bone structure and function in transgenic mice. Endocrinology. 2002;143:3681–3690. doi: 10.1210/en.2001-211424. [DOI] [PubMed] [Google Scholar]
- 13.Fisch S, Gray S, Heymans S, Haldar SM, Wang B, Pfister O, Cui L, Kumar A, Lin Z, Sen-Banerjee S, Das H, Petersen CA, Mende U, Burleigh BA, Zhu Y, Pinto YM, Liao R, Jain MK. Kruppel-like factor 15 is a regulator of cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2007;104:7074–7079. doi: 10.1073/pnas.0701981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guan J, Mishra S, Falk RH, Liao R. Current perspectives on cardiac amyloidosis. Am J Physiol Heart Circ Physiol. 2012;302:H544–552. doi: 10.1152/ajpheart.00815.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishibashi K, Imai M. Prospect of a stanniocalcin endocrine/paracrine system in mammals. Am J Physiol Renal Physiol. 2002;282:F367–375. doi: 10.1152/ajprenal.00364.2000. [DOI] [PubMed] [Google Scholar]
- 16.Jain M, Brenner DA, Cui L, Lim CC, Wang B, Pimentel DR, Koh S, Sawyer DB, Leopold JA, Handy DE, Loscalzo J, Apstein CS, Liao R. Glucose-6-phosphate dehydrogenase modulates cytosolic redox status and contractile phenotype in adult cardiomyocytes. Circ Res. 2003;93:e9–16. doi: 10.1161/01.RES.0000083489.83704.76. [DOI] [PubMed] [Google Scholar]
- 17.Kanellis J, Bick R, Garcia G, Truong L, Tsao CC, Etemadmoghadam D, Poindexter B, Feng L, Johnson RJ, Sheikh-Hamad D. Stanniocalcin-1, an inhibitor of macrophage chemotaxis and chemokinesis. Am J Physiol Renal Physiol. 2004;286:F356–362. doi: 10.1152/ajprenal.00138.2003. [DOI] [PubMed] [Google Scholar]
- 18.Koizumi K, Hoshiai M, Ishida H, Ohyama K, Sugiyama H, Naito A, Toda T, Nakazawa H, Nakazawa S. Stanniocalcin 1 prevents cytosolic Ca2+ overload and cell hypercontracture in cardiomyocytes. Circ J. 2007;71:796–801. doi: 10.1253/circj.71.796. [DOI] [PubMed] [Google Scholar]
- 19.Koyama J, Falk RH. Prognostic significance of strain Doppler imaging in light-chain amyloidosis. JACC Cardiovasc Imaging. 2010;3:333–342. doi: 10.1016/j.jcmg.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 20.Lai KP, Law AY, Yeung HY, Lee LS, Wagner GF, Wong CK. Induction of stanniocalcin-1 expression in apoptotic human nasopharyngeal cancer cells by p53. Biochem Biophys Res Commun. 2007;356:968–975. doi: 10.1016/j.bbrc.2007.03.074. [DOI] [PubMed] [Google Scholar]
- 21.Law AY, Ching LY, Lai KP, Wong CK. Identification and characterization of the hypoxia-responsive element in human stanniocalcin-1 gene. Mol Cell Endocrinol. 2010;314:118–127. doi: 10.1016/j.mce.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 22.Law AY, Lai KP, Lui WC, Wan HT, Wong CK. Histone deacetylase inhibitor-induced cellular apoptosis involves stanniocalcin-1 activation. Exp Cell Res. 2008;314:2975–2984. doi: 10.1016/j.yexcr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Li K, Dong D, Yao L, Dai D, Gu X, Guo L. Identification of STC1 as an beta-amyloid activated gene in human brain microvascular endothelial cells using cDNA microarray. Biochem Biophys Res Commun. 2008;376:399–403. doi: 10.1016/j.bbrc.2008.08.158. [DOI] [PubMed] [Google Scholar]
- 24.Liao R, Jain M, Teller P, Connors LH, Ngoy S, Skinner M, Falk RH, Apstein CS. Infusion of light chains from patients with cardiac amyloidosis causes diastolic dysfunction in isolated mouse hearts. Circulation. 2001;104:1594–1597. [PubMed] [Google Scholar]
- 25.Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583–596. doi: 10.1056/NEJMra023144. [DOI] [PubMed] [Google Scholar]
- 26.Merlini G, Seldin DC, Gertz MA. Amyloidosis: pathogenesis and new therapeutic options. J Clin Oncol. 2011;29:1924–1933. doi: 10.1200/JCO.2010.32.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Migrino RQ, Hari P, Gutterman DD, Bright M, Truran S, Schlundt B, Phillips SA. Systemic and microvascular oxidative stress induced by light chain amyloidosis. Int J Cardiol. 2010;145:67–68. doi: 10.1016/j.ijcard.2009.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Migrino RQ, Truran S, Gutterman DD, Franco DA, Bright M, Schlundt B, Timmons M, Motta A, Phillips SA, Hari P. Human microvascular dysfunction and apoptotic injury induced by AL amyloidosis light chain proteins. Am J Physiol Heart Circ Physiol. 2011;301:H2305–2312. doi: 10.1152/ajpheart.00503.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mishra S, Guan J, Plovie E, Seldin DC, Connors LH, Merlini G, Falk RH, Macrae CA, Liao R. Human amyloidogenic light chain proteins result in cardiac dysfunction, cell death and early mortality in zebrafish. Am J Physiol Heart Circ Physiol. 2013;305:H95–H103. doi: 10.1152/ajpheart.00186.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen A, Chang AC, Reddel RR. Stanniocalcin-1 acts in a negative feedback loop in the prosurvival ERK1/2 signaling pathway during oxidative stress. Oncogene. 2009;28:1982–1992. doi: 10.1038/onc.2009.65. [DOI] [PubMed] [Google Scholar]
- 31.Palladini G, Lavatelli F, Russo P, Perlini S, Perfetti V, Bosoni T, Obici L, Bradwell AR, D'Eril GM, Fogari R, Moratti R, Merlini G. Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood. 2006;107:3854–3858. doi: 10.1182/blood-2005-11-4385. [DOI] [PubMed] [Google Scholar]
- 32.Panakova D, Werdich AA, Macrae CA. Wnt11 patterns a myocardial electrical gradient through regulation of the L-type Ca(2+) channel. Nature. 2010;466:874–878. doi: 10.1038/nature09249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sheikh-Hamad D, Bick R, Wu GY, Christensen BM, Razeghi P, Poindexter B, Taegtmeyer H, Wamsley A, Padda R, Entman M, Nielsen S, Youker K. Stanniocalcin-1 is a naturally occurring L-channel inhibitor in cardiomyocytes: relevance to human heart failure. Am J Physiol Heart Circ Physiol. 2003;285:H442–448. doi: 10.1152/ajpheart.01071.2002. [DOI] [PubMed] [Google Scholar]
- 34.Shi J, Guan J, Jiang B, Brenner DA, Del Monte F, Ward JE, Connors LH, Sawyer DB, Semigran MJ, Macgillivray TE, Seldin DC, Falk R, Liao R. Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38alpha MAPK pathway. Proc Natl Acad Sci U S A. 2010;107:4188–4193. doi: 10.1073/pnas.0912263107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin JT, Pomerantsev EV, Mably JD, MacRae CA. High-resolution cardiovascular function confirms functional orthology of myocardial contractility pathways in zebrafish. Physiol Genomics. 2010;42:300–309. doi: 10.1152/physiolgenomics.00206.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sikkink LA, Ramirez-Alvarado M. Cytotoxicity of amyloidogenic immunoglobulin light chains in cell culture. Cell Death Dis. 2010;1:e98. doi: 10.1038/cddis.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Varghese R, Gagliardi AD, Bialek PE, Yee SP, Wagner GF, Dimattia GE. Overexpression of human stanniocalcin affects growth and reproduction in transgenic mice. Endocrinology. 2002;143:868–876. doi: 10.1210/en.143.3.868. [DOI] [PubMed] [Google Scholar]
- 38.Wagner GF, Dimattia GE. The stanniocalcin family of proteins. J Exp Zoolog A Comp Exp Biol. 2006;305:769–780. doi: 10.1002/jez.a.313. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Huang L, Abdelrahim M, Cai Q, Truong A, Bick R, Poindexter B, Sheikh-Hamad D. Stanniocalcin-1 suppresses superoxide generation in macrophages through induction of mitochondrial UCP2. J Leukoc Biol. 2009;86:981–988. doi: 10.1189/jlb.0708454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Westberg JA, Serlachius M, Lankila P, Penkowa M, Hidalgo J, Andersson LC. Hypoxic preconditioning induces neuroprotective stanniocalcin-1 in brain via IL-6 signaling. Stroke. 2007;38:1025–1030. doi: 10.1161/01.STR.0000258113.67252.fa. [DOI] [PubMed] [Google Scholar]
- 41.Westerfield M. The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio) University of Oregon Press; Eugene: 2000. [Google Scholar]
- 42.Wu S, Yoshiko Y, De Luca F. Stanniocalcin 1 acts as a paracrine regulator of growth plate chondrogenesis. J Biol Chem. 2006;281:5120–5127. doi: 10.1074/jbc.M506667200. [DOI] [PubMed] [Google Scholar]
- 43.Yankner BA, Lu T. Amyloid beta-protein toxicity and the pathogenesis of Alzheimer disease. J Biol Chem. 2009;284:4755–4759. doi: 10.1074/jbc.R800018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yeung HY, Lai KP, Chan HY, Mak NK, Wagner GF, Wong CK. Hypoxia-inducible factor-1-mediated activation of stanniocalcin-1 in human cancer cells. Endocrinology. 2005;146:4951–4960. doi: 10.1210/en.2005-0365. [DOI] [PubMed] [Google Scholar]
- 45.Zhang K, Lindsberg PJ, Tatlisumak T, Kaste M, Olsen HS, Andersson LC. Stanniocalcin: A molecular guard of neurons during cerebral ischemia. Proc Natl Acad Sci U S A. 2000;97:3637–3642. doi: 10.1073/pnas.070045897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.