

Single, large-scale deletions of mitochondrial DNA are an important cause of mitochondrial disease, with a broad phenotypic spectrum. Grady et al. report that disease severity and progression are correlated with the size of the deletion, its location within the genome, and the deletion heteroplasmy level in skeletal muscle.
Keywords: mitochondrial diseases, mitochondrial DNA deletion, disease progression
Abstract
Single, large-scale deletions of mitochondrial DNA are a common cause of mitochondrial disease and cause a broad phenotypic spectrum ranging from mild myopathy to devastating multi-system syndromes such as Kearns-Sayre syndrome. Studies to date have been inconsistent on the value of putative predictors of clinical phenotype and disease progression such as mutation load and the size or location of the deletion. Using a cohort of 87 patients with single, large-scale mitochondrial DNA deletions we demonstrate that a variety of outcome measures such as COX-deficient fibre density, age-at-onset of symptoms and progression of disease burden, as measured by the Newcastle Mitochondrial Disease Adult Scale, are significantly (P < 0.05) correlated with the size of the deletion, the deletion heteroplasmy level in skeletal muscle, and the location of the deletion within the genome. We validate these findings with re-analysis of 256 cases from published data and clarify the previously conflicting information of the value of these predictors, identifying that multiple regression analysis is necessary to understand the effect of these interrelated predictors. Furthermore, we have used mixed modelling techniques to model the progression of disease according to these predictors, allowing a better understanding of the progression over time of this strikingly variable disease. In this way we have developed a new paradigm in clinical mitochondrial disease assessment and management that sidesteps the perennial difficulty of ascribing a discrete clinical phenotype to a broad multi-dimensional and progressive spectrum of disease, establishing a framework to allow better understanding of disease progression.
Introduction
Mitochondrial genetic defects are an important cause of neurological disease and are a result of mutations of either the nuclear or mitochondrial genome (McFarland et al., 2010; Schon et al., 2012). The ability to understand and predict the progression of mitochondrial disease is of great clinical importance to give patients guidance about their risk of developing symptoms and enabling clinicians to provide optimum patient care. However, to date, fundamental understanding of the factors governing mitochondrial disease progression is limited and studies are often contradictory in their findings. We therefore chose to concentrate on one of the most common mitochondrial diseases: patients with single, large-scale mitochondrial DNA deletions.
Characterization of the phenotypic presentation of single, large-scale mitochondrial DNA disease is traditionally divided into three main presentations (Schon et al., 2012): Kearns-Sayre syndrome (Kearns and Sayre, 1958), a multi-system childhood or teenage-onset syndrome; chronic progressive external ophthalmoplegia (CPEO; Moraes et al., 1989), a milder presentation involving principally weakness of the extraocular muscles with ptosis, but often associated with more widespread muscular weakness; and Pearson Syndrome (Pearson et al., 1979), an often fatal infantile-onset syndrome characterized by sideroblastic anaemia and exocrine pancreatic dysfunction, which may develop into Kearns-Sayre syndrome in those who survive infancy. However, as many studies attempting to unpick phenotypic presentation have noted, these divisions are somewhat imprecise and misrepresent the true spectrum of single mitochondrial DNA deletion-related disease; patients denoted CPEO are often subdivided into groups such as ‘classic CPEO’ at the mild end of the spectrum, ‘severe CPEO’ or ‘CPEO+’, and ‘partial Kearns-Sayre syndrome’ or ‘CPEO + multisystem’ (Auré et al., 2007).
Early investigations of single, large scale mitochondrial DNA deletion disease found little connection between mitochondrial genetics, biochemical defects and clinical phenotype (Zeviani et al., 1988; Holt et al., 1989; Moraes et al., 1989; Rotig et al., 1995). More recent studies are rather contradictory. Skeletal muscle heteroplasmy is variously reported as non-predictive of either phenotype or age at onset (Yamashita et al., 2008), predictive of onset but not phenotype (López-Gallardo et al., 2009), or predictive of both to a limited extent (Sadikovic et al., 2010). Mitochondrial DNA deletion size is inconsistently reported as either predictive of phenotype and disease severity (Yamashita et al., 2008) or the contrary (López-Gallardo et al., 2009). Interestingly, two recent studies found that the location of the mitochondrial DNA deletion was predictive of phenotype or age at onset to an extent, though with contrary conclusions. It has been reported that mitochondrial DNA deletions including one of the three mitochondrial DNA-encoded structural components of cytochrome c oxidase (COX) (MT-CO1, MT-CO2 or MT-CO3 genes) and complex V (MT-ATP6 or MT-ATP8 genes) had significantly earlier onset (Yamashita et al., 2008) although other studies have found that deletion of the mitochondrial cytochrome b (MT-CYB) gene was significantly associated with a severe Kearns-Sayre syndrome phenotype (López-Gallardo et al., 2009).
Researchers have attempted to better segregate patients into groups that are easier to analyse, for instance by the presence of cerebellar involvement (Auré et al., 2007) or purely myopathic symptoms (López-Gallardo et al., 2009). However, for mitochondrial genetic disorders, the disease spectrum is multidimensional, with a wide range of potential systems involved, each to varying degrees of severity. This severely hampers any attempt to discretely classify the disease. The Newcastle Mitochondrial Disease Adult Scale (NMDAS) was published as a semi-quantitative rating scale to monitor mitochondrial disease (Schaefer et al., 2006). It is a clinically validated tool that has been extensively used both at our centre (Apabhai et al., 2011; Bates et al., 2013; Lax et al., 2012) and other specialist mitochondrial centres (de Laat et al., 2012; Enns et al., 2012; Orsucci et al., 2012; Yatsuga et al., 2012; Kornblum et al., 2013; Mancuso et al., 2013). The NMDAS permits quantitative analysis of both general and system-specific disease progression and has already been used in assessment of clinical progression in patients with the m.3243A>G mitochondrial DNA mutation, although this was not a longitudinal study (Whittaker et al., 2009). Linear mixed modelling has been widely used to understand disease progression in other neurodegenerative conditions including dementia (Tornatore and Grant, 2002; Hassing et al., 2004; Galvin Je and et al., 2005; Johnson et al., 2009; Knopman et al., 2009; den Heijer et al., 2010), Parkinson’s disease (Nandhagopal et al., 2009; Dobkin et al., 2011; Johnson and Galvin, 2011; Vu et al., 2012) and multiple sclerosis (Meier et al., 2007), and NMDAS facilitates such longitudinal modelling of mitochondrial disease progression.
In this study, we used repeated measures mixed modelling with collated NMDAS data for a large patient cohort to test our hypothesis that mitochondrial DNA heteroplasmy levels and mitochondrial DNA deletion size and location are predictive of single, large-scale mitochondrial DNA deletion disease progression. We also applied multiple linear regression analyses to examine the relationship between these predictors and outcome measures such as age at onset, clinical phenotype and severity of the biochemical defect (as determined by COX-deficient fibre density) in both our cohort and a meta-analysis of previously published data.
Materials and methods
Newcastle patient cohort
All patients were investigated by the NHS Highly Specialized Service for Rare Mitochondrial Disorders in Newcastle upon Tyne. The majority of the patients (55 of 87) were regularly followed in our clinic at 6 or 12 monthly intervals, assessed using the NMDAS (Schaefer et al., 2006) and recruited to the MRC Mitochondrial Disease Patient Cohort UK. The remaining 32 patients include individuals not seen in our clinic and for whom we have limited clinical information or for whom we do not have NMDAS data available. For some of these patients we have been able to ascertain disease onset from hospital records. A full listing of the clinical and molecular characteristics of the patient cohort can be found in Supplementary Table 1.
Phenotypic analysis compared patients in two groups, those classified as ‘CPEO’ or ‘CPEO + myopathy’ (54 patients) and those classified as ‘Kearns-Sayre syndrome’ (nine patients). For other analyses, we used all the patients in our cohort that had the appropriate information available, as listed in Table 1.
Table 1.
Summary of Newcastle patient cohort and available data in literature
| With NMDAS | Without NMDAS | Our cohort (Total) | Meta-analysis | |
|---|---|---|---|---|
| Number of patients with mitochondrial DNA deletion size and muscle mitochondrial DNA heteroplasmy data | 55 | 32 | 87 | 256 |
| Number of patients with mitochondrial DNA breakpoints identified | 52 | 31 | 83 | 184 |
| Number of patient with age at onset data | 52 | 8 | 60 | 117 |
| Number of patients with age at onset data and mitochondrial DNA breakpoints identified | 49 | 7 | 56 | 83 |
| Number of patients with COX-deficient fibre density data and mitochondrial DNA breakpoints identified | 49 | 23 | 72 | 40 |
| Number of patients presenting with a CPEO, CPEO + myopathy or KSS phenotype | 36 | 27 | 63 | 205 |
| Number of patients presenting with a CPEO, CPEO + myopathy or KSS phenotype and mitochondrial DNA breakpoints identified | 35 | 26 | 61 | 149 |
All patients have skeletal muscle heteroplasmy and deletion size data available. Where gene location is under investigation the sub-cohort of patients with identified mitochondrial DNA breakpoints is used; where location is not under consideration, the larger cohort with mitochondrial DNA deletion size is used. The small cohort of patients with phenotype data reflects the fact that patients with a multisystem phenotype are excluded from the phenotype analysis, so this number is restricted to patients with CPEO, CPEO + myopathy, or Kearns-Sayre (KSS) syndrome. The patients with NMDAS data available had a median of five assessments (interquartile range 5).
Meta-analysis
The foundation of our meta-analysis is a previous meta-analysis by López-Gallardo et al. (2009), which includes data on patient age at disease-onset, patient age at biopsy, clinical phenotype, mitochondrial DNA deletion load (heteroplasmy) in muscle, mitochondrial DNA deletion size and specific mitochondrial DNA deletion breakpoints. We used the data as published by López-Gallardo et al. (2009) except where review of the literature identified differences between the published data and that used by this group (three cases) or if there was inconsistency between the reported mitochondrial DNA deletion size and location of the breakpoints (one case). Additionally, we eliminated those cases where the reported mitochondrial DNA deletion was characterized by restriction endonuclease digests and as such there was uncertainty as to whether specific genes under scrutiny (MT-CO or MT-CYB) were deleted (seven cases) or there were other reported mutations in the same patient (nine cases). We also excluded one patient with a highly unusual mitochondrial DNA deletion in the minor arc of the mitochondrial genome. Additional cases were obtained from other publications identified through PubMed not included in the study by López-Gallardo et al. (2009) (Johns et al., 1989; Mita et al., 1989; Larsson and Holme, 1992; Oldfors et al., 1992; Ishikawa et al., 2000; Gellerich et al., 2002; Jacobs et al., 2004; Sadikovic et al., 2010). These data are detailed in Supplementary Table 2. All amendments and exclusions are listed in Supplementary Table 3. We do not include our own cohort in the meta-analysis to ensure sample independence.
For the phenotypic analysis, patients previously classified by López-Gallardo and colleagues (2009) as ‘CPEO without non-muscular signs’ were combined with patients classified ‘CPEO’ or ‘CPEO + myopathy’ from the other literature (101 cases in total) and compared with patients classified as ‘Kearns-Sayre syndrome’ from all sources (104 cases in total). For other analyses, we used all the patients in the meta-analysis that had the appropriate information available, as listed in Table 1.
Muscle biopsy histochemistry data
Sequential COX/succinate dehydrogenase histochemical reactions were performed on skeletal muscle sections following standardized protocols (Old and Johnson, 1989). The extent of the COX deficiency (percentage of COX-deficient fibres) was determined by counting all fibres in a section (minimum 200 fibres) and performed by a single researcher to ensure data were recorded consistently across the entire cohort.
Determination of level of mitochondrial DNA deletion
All molecular analyses were performed using total skeletal muscle DNA extracted using standard protocols. A validated, multiplex real-time PCR (quantitative PCR) MTND1/MTND4 assay was used to quantify mitochondrial DNA deletion levels in muscle homogenates (He et al., 2002; Krishnan et al., 2007).
Determination of mitochondrial DNA deletion size and location
Long-range PCR was used to amplify ∼9.5 kb region of the mitochondrial genome across the major arc using a single primer set corresponding to nucleotides 6378–15896 (GenBank Accession number: NC_012920.1). PCR reactions used ∼100 ng of DNA that was added to PCR mastermix [distilled H2O, LA Taq buffer (TaKaRa), 10 mM dNTPs, 20 mM forward and reverse primers and 2.5 units of LA Taq enzyme (TaKaRa)] to a total volume of 50 μl and subjected to the following cycling conditions: 94°C for 1 min; 35 cycles of 94°C for 30 s, 58°C for 30 s and 68°C for 11 min; final extension of 72°C for 10 min. Amplified products were separated through a 0.7% agarose gel, using a 1 kb DNA ladder to estimate product size and determine mitochondrial DNA deletion sizes.
Long-range PCR products were further assessed by restriction digests to map the precise location of the mitochondrial DNA deletion breakpoints (Khrapko et al., 1999). PCR amplimers (5 µl) were digested to a series of DNA fragments of known length and position within the mitochondrial genome using restriction enzymes XhoI, BamHI, XcmI and DraI (New England Biolabs) before separation through a 0.7% agarose gel. The size of restriction products allows the location of the mitochondrial DNA deletion within the genome to be estimated, guiding the choice of appropriate sequencing primers (Supplementary Table 4) to characterize mitochondrial DNA deletion breakpoints by Sanger sequencing (BigDye® Terminator chemistries using an ABI 3130xl Genetic Analyser; Applied Biosystems).
Statistical analyses
To investigate the relationship between the variables being tested, we used Box-Cox to stabilize the variance of the dependent variables (Box and Cox, 1964) and to identify optimal transformations of the variables that satisfy normality assumptions, enabling use of parametric tests. For basic analyses, a single summary data point was determined for each patient using the mean NMDAS score and mean age at assessment. Longitudinal modelling was conducted using SAS PROC MIXED in accordance with published guidelines (Singer, 1998; Diggle et al., 2002). Data covariance structure was modelled using a spatial power structure (Moser, 2004). Polynomial terms of time up to cubed (time3) were included. Models were compared using the Akaike Information Criterion (Akaike, 1974; Kuha, 2004). Residual and influence diagnostics were conducted to validate model assumptions and verify model stability. SAS version 9.2 was used throughout. For predetermined hypothesis testing, significance was determined at P < 0.05, high significance at P < 0.0001. For multiple regression, we report the standardized coefficient (b) (standardized to have unit variance) and significance value (P-value) for each parameter estimate, together with the number of subjects (n) and the coefficient of determination (R2) for the overall regression. For simple linear regression we report n, the correlation coefficient (r) and the P-value. For multiple logistic regression we report n, and the standardized coefficient and P-value for each parameter estimate. For simple logistic regression we report n, the odds ratio for the effect of the dependent variable, and the P-value.
Results
Putative predictors of disease burden and progression are intercorrelated
We began our investigations by looking at the relationships between the putative predictors of disease progression. In our patient cohort we observed a strong negative linear correlation between the level of deleted mitochondrial DNA compared with wild-type (i.e. mitochondrial DNA heteroplasmy) and mitochondrial DNA deletion size (n = 87, r = −0.49, P < 0.0001) (Fig. 1A), an observation confirmed in the meta-analysis (n = 256, r = −0.18, P = 0.0032). Mitochondrial DNA deletion size and location were also highly significantly correlated in our cohort (n = 83, r = −0.48, P < 0.0001; Fig. 1B), again confirmed by the meta-analysis (n = 184, r = −0.29, P < 0.0001). Highly significant correlations were also found between mitochondrial DNA heteroplasmy, mitochondrial DNA deletion size, and the two proposed genetic loci of interest (MT-CO and MT-CYB genes) that were identified in previous literature (Yamashita et al., 2008; López-Gallardo et al., 2009) (Table 2).
Figure 1.

Putative predictors of disease progression are intercorrelated. (A) Skeletal muscle heteroplasmy is negatively correlated with mitochondrial DNA deletion size in our cohort. n = 87, r = −0.49, P < 0.0001. 95% CI is shown. The dense cluster of points around 5.0kb represents the cohort of patients with the common 4977 bp mitochondrial DNA deletion. (B) Mitochondrial DNA deletion size is negatively correlated with the location of the mitochondrial DNA deletion midpoint in our cohort. n = 83, r = −0.48, P < 0.0001. 95% CI is shown.
Table 2.
Intercorrelations between putative predictors of disease burden and progression
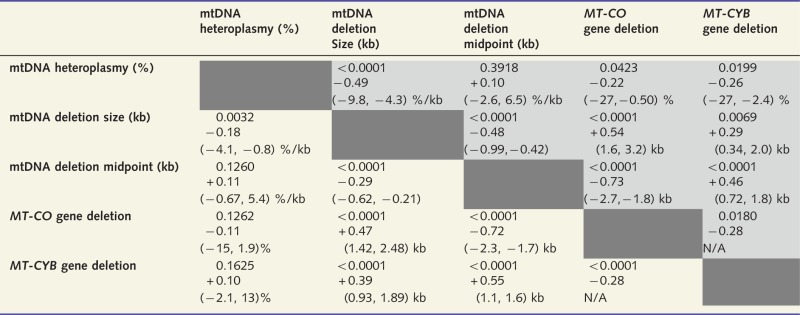 |
Shaded cells (upper right) show for our cohort P-values, correlation coefficients, and 95% confidence intervals for linear regression gradient or estimated difference due to specific mitochondrial DNA (mtDNA) gene deletion; unshaded cells (bottom left) show the same for the meta-analysis. Units for 95% confidence intervals identify y and x for linear regression, except in the case of mitochondrial DNA deletion size (kb) versus mitochondrial DNA deletion midpoint (kb). The strongest correlations in each data set are between mitochondrial DNA deletion midpoint or size and MT-CO gene deletion; larger mitochondrial DNA deletions tend to include MT-CO genes. MT-CYB deletion is also associated with larger mitochondrial DNA deletion size and mitochondrial DNA deletions. The same trends are seen in all cases in our cohort and the meta-analysis, excepting that in our cohort MT-CYB gene deletion is significantly associated with lower mitochondrial DNA heteroplasmy, whereas in the meta-analysis there is a non-significant trend relating MT-CYB gene deletion with higher mitochondrial DNA heteroplasmy levels.
Age at onset, clinical phenotype and NMDAS progression are correlated with muscle heteroplasmy and mitochondrial DNA deletion size
The square root of age at onset was used in all analyses, which was identified by Box-Cox as the optimal transform. For the subjects in our patient cohort with known age at onset (n = 60), age at onset was significantly correlated with both mitochondrial DNA deletion size (b = −0.41, P = 0.0039) and muscle mitochondrial DNA heteroplasmy (b = −0.42, P = 0.0027) using multiple linear regression (R2 = 0.18) (Fig. 2A). Similarly, in the meta-analysis (n = 117), both mitochondrial DNA deletion size (b = −0.30, P = 0.0008) and muscle mitochondrial DNA heteroplasmy (b = −0.30, P = 0.0010) were significantly correlated with age at onset (R2 = 0.15).
Figure 2.

Heteroplasmy and deletion size are linearly correlated with age at onset and NMDAS score progression. (A) Age at onset is predicted by both mitochondrial DNA heteroplasmy and deletion size. The y-axis shows the square root of age at onset. Data are from our cohort (n = 60, R2 = 0.18). Both mitochondrial DNA heteroplasmy (P = 0.0027) and deletion size (P = 0.0039) are significantly correlated with age at onset in our cohort using multiple regression. P-values are for both predictors as continuous variables, mitochondrial DNA deletion size is dichotomous for visualization only. (B) Phenotype and average NMDAS score are highly significantly correlated (P < 0.0001). Individual comparison P-values are shown. (C) NMDAS progression (scaled NMDAS points per year) is highly significantly correlated with both mitochondrial DNA deletion size (P < 0.0001) and heteroplasmy (P < 0.0001) (n = 55, R2 = 0.49). The y-axis shows scaled NMDAS score per year. P-values are for both predictors as continuous variables, deletion size is dichotomous for visualization only. KSS = Kearns-Sayre syndrome.
In a phenotypic analysis, patients identified with Kearns-Sayre syndrome were directly compared to those with CPEO or CPEO + myopathy; intermediate phenotypes (in particular non-Kearns-Sayre syndrome, multisystem disease phenotype) were excluded from this analysis. For such patients from our cohort (n = 64) we found that both mitochondrial DNA heteroplasmy (b = −1.4, P = 0.0020) and mitochondrial DNA deletion size (b = −0.74, P = 0.0318) were significantly correlated with phenotype using multiple logistic regression. Similarly, in the meta-analysis (n = 192), both mitochondrial DNA heteroplasmy (b = −0.56, P < 0.0001) and mitochondrial DNA deletion size (b = −0.18, P = 0.0453) were significantly correlated with phenotype.
Average NMDAS score correlated with traditional phenotype classification (Fig. 2B). Box-Cox identified the fourth root of the NMDAS score (NMDAS0.25) as the optimal transformation for linear regression; this was divided by the age at assessment to measure NMDAS progression. In our cohort (n = 55), we found that both mitochondrial DNA deletion size (b = 0.49, P < 0.0001) and mitochondrial DNA heteroplasmy (b = 0.70, P < 0.0001) were significant predictors of NMDAS progression using multiple regression (R2 = 0.49) (Fig. 2C).
Disease burden and progression of patients with a common mitochondrial DNA deletion are correlated with heteroplasmy
We also investigated the cohort of patients whose mitochondrial DNA disease is associated with a common 4977 bp single, large-scale mitochondrial DNA deletion (Schon et al., 1989). Using a combined data set of the meta-analysis and our own patient cohort, we observed that muscle mitochondrial DNA heteroplasmy was a significant predictor of both clinical phenotype [n = 85, odds ratio for 10% change in heteroplasmy 1.43, 95% confidence interval (CI) 1.13–1.81, P = 0.0030] and also age of disease onset (n = 37, r = −0.44, P = 0.0063).
COX-deficient fibre density is dependent on muscle heteroplasmy and deletion location but not deletion size
We next studied the relationship between the proportion of COX-deficient muscle fibres in patient biopsies, levels of muscle mitochondrial DNA heteroplasmy and MT-CO gene deletion (namely, deletion of part or all of at least one of the MT-CO1, MT-CO2 or MT-CO3 genes within the deleted mitochondrial DNA molecule). The square root of COX-deficient fibre density was used in all analyses, which was identified by Box-Cox as the optimal transform.
In our patient cohort (n = 72), we observed that both muscle mitochondrial DNA heteroplasmy (b = 0.68, P < 0.0001) and MT-CO gene deletion (b = 0.31, P = 0.0018) were significantly correlated with COX-deficient fibre density using multiple linear regression (R2 = 0.43) (Fig. 3). Similarly, in the meta-analysis (n = 39), both mitochondrial DNA heteroplasmy (b = 0.49, P = 0.0012) and MT-CO gene deletion (b = 0.34, P = 0.0192) were significantly correlated with COX-deficient fibre density (R2 = 0.31).
Figure 3.

COX-deficient fibre density is dependent on skeletal muscle mitochondrial DNA heteroplasmy and deletion of MT-CO genes. The y-axis shows the square root of the COX-deficient fibre density %. Data are from our cohort, n = 72, R2 = 0.43. Heteroplasmy (P < 0.0001) and deletion of MT-CO genes (P = 0.0018) are both significant predictors. Separate regression lines are shown for those that delete one or more MT-CO genes (n = 63, 95% CI for regression line shown) and those that do not (n = 9, gradient of regression line is not significantly non-zero, CI not shown).
As the inclusion of MT-CO genes within the deleted mitochondrial DNA region was significantly correlated with a larger mitochondrial DNA deletion size in both our cohort and the meta-analysis, we also examined whether there was a correlation between mitochondrial DNA deletion size and COX-deficient fibre density. Using multiple regression with all three predictors, in our patient cohort (n = 72) mitochondrial DNA deletion size (b = −0.081, P = 0.5104) was not a significant predictor, although both mitochondrial DNA heteroplasmy (b = 0.65, P < 0.0001) and MT-CO gene deletion (b = 0.35, P = 0.0028) remained significant (R2 = 0.44). Similarly, in the meta-analysis (n = 39), mitochondrial DNA deletion size (b = −0.00010, P = 0.5586) was not a significant predictor; however, both mitochondrial DNA heteroplasmy (b = 0.48, P = 0.0016) and MT-CO gene deletion (b = 0.43, P = 0.0445) remained significant (R2 = 0.32).
Longitudinal mixed modelling shows that mitochondrial DNA heteroplasmy, mitochondrial DNA deletion size and location are predictors of disease progression
Using longitudinal mixed modelling we showed that muscle mitochondrial DNA heteroplasmy (P < 0.0001) and mitochondrial DNA deletion size (P < 0.0001) were both highly significantly correlated with NMDAS progression in our patient cohort (n = 55) (Fig. 4). The interaction between mitochondrial DNA deletion size and mitochondrial DNA heteroplasmy was also significant (P = 0.0046), which is exemplified by comparing Fig. 4A with Fig. 4B, where mitochondrial DNA deletion size has a stronger effect at high mitochondrial DNA heteroplasmy levels than low mitochondrial DNA heteroplasmy levels; and Fig. 4C with Fig. 4E, where mitochondrial DNA heteroplasmy has a strong effect with large mitochondrial DNA deletions but negligible effect with small mitochondrial DNA deletions.
Figure 4.

The effect of mitochondrial DNA deletion size and heteroplasmy on NMDAS progression. All panels show 95% CI. (A and B) The effect of mitochondrial DNA deletion size at 80% and 40% heteroplasmy, respectively. Deletion size is shown to have a greater impact at high heteroplasmy than at low heteroplasmy. (C–E) The effect of mitochondrial DNA heteroplasmy for a 2.0 kb, 5.0 kb and 8.0 kb mitochondrial DNA deletion, respectively. For small deletions the effect of heteroplasmy on NMDAS progression is negligible, but for larger deletions the effect is substantial. (F) The effect of deletion location for a 5.0 kb deletion present at 80% heteroplasmy; progression is faster when MT-CYB is included in the deleted region. A–E are generated from a model using time, deletion size and heteroplasmy as predictors. The model used for F has an additional deletion location predictor (MT-CYB gene inclusion).
The location of the mitochondrial DNA deletion within the mitochondrial genome was also shown to affect disease progression. With mitochondrial DNA heteroplasmy and mitochondrial DNA deletion size in the model, deletion of the MT-CYB gene is significantly predictive of faster progression (P = 0.0085, n = 52) (Fig. 4F).
Individual patient progression can be modelled longitudinally
As mixed modelling allows incorporation of random effects to model unknown variance, we were also able to use the clinical and molecular genetic data obtained to model the progress of individual patients and predict their expected course of disease development.
We studied five individual patients using a predictive model that incorporated muscle mitochondrial DNA heteroplasmy, mitochondrial DNA deletion size and involvement of MT-CYB gene deletion as predictive factors (Fig. 5). Expected progression is shown with 95% prediction intervals. The effect of mitochondrial DNA deletion size is exemplified by comparing Patients 4 and 5, as these only differ in this single parameter. The effect of mitochondrial DNA heteroplasmy is shown by comparing Patients 1 and 3, who have similarly sized mitochondrial DNA deletions (6.9 kb and 6.5 kb, respectively) but at differing mitochondrial DNA heteroplasmy levels. Patient 2 (9.1kb mitochondrial DNA deletion) shows a rapid progression despite a low level of mitochondrial DNA heteroplasmy (37% mitochondrial DNA deletion load), on account of the exceptionally large mitochondrial DNA deletion, whereas Patient 1 demonstrates that even a single NMDAS score can represent a useful prognostic input.
Figure 5.

Longitudinal modelling of five individual patients with single, large-scale mitochondrial DNA deletion disease. The chosen patients are representative of the range of rates of disease progression found in our cohort. Actual NMDAS assessment scores are depicted as crosses joined by solid lines. Each patient is shown with their predicted progression trendline with 95% prediction intervals, and is labelled with deletion size and heteroplasmy. Only Patient 2 includes part of the MT-CYB gene in their deletion.
Discussion
The aim of our study was to further understanding of the nature and progression of mitochondrial DNA disease. This remains challenging despite the fact that the first mitochondrial DNA mutations were described 25 years ago (Holt et al., 1988). The relationship between a specific mitochondrial DNA mutation and clinical phenotype and progression is complex for many common mitochondrial DNA mutations not least because of heteroplasmy and variation in mitochondrial DNA mutation load. In addition, a particular mitochondrial DNA mutation can cause several different disease phenotypes depending on specific organ involvement and these can be difficult to categorize without the benefit of a scale of disease burden. This study has focused on patients with single, large-scale mitochondrial DNA deletions as these represent a common cause of mitochondrial disease and exemplify many of the challenges that are observed with other mitochondrial DNA defects. We have identified that muscle mitochondrial DNA heteroplasmy levels, mitochondrial DNA deletion size and mitochondrial DNA deletion location are all important in understanding the expression and progression of clinical disease.
Two major factors have helped to define the relationship between single, large-scale mitochondrial DNA deletions and disease progression. The first is the use of a validated rating scale that covers the major clinical features of mitochondrial disease. Traditional phenotyping of mitochondrial disease is difficult because of the frequent involvement of multiple systems. The NMDAS, as a quantitative measure of mitochondrial disease burden, allows us to study disease progression with unprecedented power, and longitudinal modelling of successive NMDAS assessments for patients demonstrates the benefits of such an approach. Second is the use of statistical techniques, including multiple regression analyses that we have applied to our own data and those previously published in the literature, using the Box-Cox transformation to identify optimal transformations to normality. In trying to understand the nature and progression of single, large-scale mitochondrial DNA deletion disease, where there is intercorrelation between the predictive factors, these techniques are required to correctly identify significant findings; indeed, the reanalysis of previously reported data using these techniques has clarified previous uncertainties.
Our longitudinal modelling shows that disease burden and progression is predicted by muscle mitochondrial DNA heteroplasmy level, mitochondrial DNA deletion size and the location of the mitochondrial DNA deletion within the mitochondrial genome. These findings are reinforced by a series of results, from both our cohort and the meta-analysis, demonstrating that both phenotype and age at onset are predicted by mitochondrial DNA deletion size and mitochondrial DNA heteroplasmy, and also by the significance of heteroplasmy as a predictor in a cohort of patients with the common 4977 bp mitochondrial DNA deletion. Previous studies have been contradictory regarding the use of these factors as predictors, reporting that both phenotype and age at onset were dependent on mitochondrial DNA deletion size, but not significantly related to mitochondrial DNA heteroplasmy (Yamashita et al., 2008), or that the clinical phenotype was related to mitochondrial DNA heteroplasmy, but unrelated to mitochondrial DNA deletion size, although age at onset was related to both factors (López-Gallardo et al., 2009). Skeletal muscle mitochondrial DNA heteroplasmy and mitochondrial DNA deletion size have also been reported as not being useful in disease progression prediction (Auré et al., 2007). More seminal studies also reported inconsistently on the link between these predictors and phenotype or age at onset (Holt et al., 1989; Moraes et al., 1989; Goto et al., 1990). However, we found that multiple regression identifies consistent correlation between disease phenotype or progression and both mitochondrial DNA heteroplasmy and deletion size. Furthermore, multiple regression of previously published data is revealing; for example, whereas López-Gallardo et al. (2009) found that mitochondrial DNA deletion size is not predictive of phenotype (P = 0.3953), if we perform logistic regression on their same data, but use multiple regression with deletion size and heteroplasmy, deletion size is indeed a significant predictor of phenotype (P = 0.0330); in this case, the negative correlation between heteroplasmy and deletion size leads to the masking of the effect of deletion size when simple linear regression is used.
The correlations between predictive factors are of interest in themselves. A negative correlation between muscle heteroplasmy levels and mitochondrial DNA deletion size was noted by López-Gallardo et al. (2009) who surmized from this that shorter mitochondrial DNA deletions do not have a replicative advantage. We would speculate that the observed correlation between muscle mitochondrial DNA heteroplasmy and mitochondrial DNA deletion size most likely reflects the spectrum of disease that presents with a clinically recognizable phenotype within the population, rather than any intrinsic biochemically, or otherwise, driven relationship. Thus, individuals with small mitochondrial DNA deletions present at low heteroplasmy levels would not present clinically with symptoms, which is consistent with the observation that potentially pathogenic mitochondrial DNA mutations are widespread in the non-diseased population but at sub-threshold levels (Elliott et al., 2008).
We have found that deletions including the MT-CYB gene are related to faster progression, which is consistent with the report by López-Gallardo et al. (2009) that deletion of the MT-CYB gene was linked to a more severe phenotype. However, the reason for this remains uncertain, but could potentially indicate that complex III deficiency has a particularly important role in the disease mechanism.
We also identified that MT-CO deletion, that is involvement of either MT-CO1, MT-CO2 or MT-CO3 in the deleted region, is predictive of COX-deficient fibre density. Though previous studies reported no such link (Goto et al., 1990; Oldfors et al., 1992), re-analysis of these studies using multiple regression reveals the same trend in all cases, and at statistical significance when the study is large enough (Goto et al., 1990). The impact of mitochondrial DNA deletion location on COX-deficient fibre density, and the consistent correlation between mitochondrial DNA deletion size and disease burden and progression, brings into question the hypothesis that mitochondrial–transfer RNA genes are the root of the pathogenicity of mitochondrial DNA deletions (Schon et al., 2012), which is predicated on the lack of correlation between the site of the mitochondrial DNA deletion and the pathogenicity. However, a more comprehensive data set would be required to elucidate the relative importance of mitochondrial–transfer RNA genes, specific oxidative phosphorylation protein genes, and other potential pathogenic mechanisms as regards biochemical defects and the resulting clinical phenotype.
An important implication of this study regards the threshold level for pathogenicity of a mitochondrial DNA deletion (Rossignol et al., 2003). The co-dependence of disease burden on mitochondrial DNA heteroplasmy and mitochondrial DNA deletion size and location, and in particular the interaction of these quantities, implies that the threshold for phenotypic expression is likely to be dependent on the size and location of the deletion.
Although our studies provide new insights into disease progression in patients with single, large-scale mitochondrial DNA deletions, further work is still required. First, it should be considered that both mitochondrial DNA deletion size and our location parameter are perhaps merely proxies for the underlying pathogenic nature of the mitochondrial DNA deletion; a more nuanced characterization of these mitochondrial DNA deletions may better predict pathogenesis and mitochondrial disease progression. However, such a model would potentially involve a large number of parameters, in which case a larger data set would be required to achieve reasonable statistical power. Second, it is notable that we do not have any predictor that encapsulates the sometimes multi-system nature of disease. In this regard, Auré et al. (2007) found that the presence of the mutation in blood was predictive of a neurological phenotype, and a similar trend in urine, a result confirmed by Blackwood et al. (2010) with regard to severe early onset disease as compared to milder phenotypic presentation. These reports suggest that blood or urine mitochondrial DNA heteroplasmy levels, together with skeletal muscle mitochondrial DNA heteroplasmy, may lead to an improved prediction of disease prognosis. Third, in this study we have only considered progression of the overall disease burden; modelling of progression in individually affected systems should provide useful understanding to clinicians with regard to patient care.
There are additional limitations to be acknowledged. First, though most of the findings from our cohort are corroborated by the meta-analysis, there are differences in the two data sets; in particular the correlations between predictors are generally stronger in our cohort than the meta-analysis. These differences may arise in part from the fact that our data are more homogeneous, but also perhaps because the make-up of our cohort is different; we have relatively few patients presenting with a Kearns-Sayre syndrome phenotype compared with the meta-analysis. Secondly, our studies, and those selected for the meta-analysis have concentrated on muscle heteroplasmy levels. Finally, though we do not employ Bonferroni adjustment in most of our analyses for reasons well documented in the literature and particularly as we are testing a priori hypotheses (Thomas, 1998), the large number of statistical tests used do open up the potential for more frequent type I statistical errors. However, consistent findings in both our cohort and the meta-analysis provide support for firm conclusions to be drawn.
Our finding that deletion size, heteroplasmy, and deletion location are predictors of disease progression is important for all clinicians looking after patients with single, large-scale mitochondrial DNA deletions. We have therefore developed a web-based tool (http://research.ncl.ac.uk/mitoresearch) to support clinicians in their management of these patients.
In conclusion, we have demonstrated that skeletal muscle mitochondrial DNA heteroplasmy, mitochondrial DNA deletion size and deletion location are predictive of disease severity and progression, and that applied with NMDAS, a quantitative measure of total disease burden, we are able to longitudinally model disease progression both at a population level and for individual patients. This means that advice and care plans for patients can be given on an individual basis. In addition, understanding the natural history of disease is crucial if we are to assess the benefits of therapeutic treatments. Thus we strongly recommend that all patients with single, large scale mitochondrial DNA deletion disease have both a detailed analysis of the muscle biopsy and clinical evaluation using the NMDAS rating scale.
Supplementary Material
Acknowledgements
Grateful thanks go to Dr Claudia Racca for helpful advice and guidance throughout the study. We would also like to acknowledge MRC Centre for Translational Research in Neuromuscular Disease: Mitochondrial Disease Cohort (Research Ethics Committee Reference 08/H045/72) for providing the NMDAS data.
Glossary
Abbreviations
- COX
cytochrome c oxidase
- CPEO
chronic progressive external ophthalmoplegia
- NMDAS
Newcastle Mitochondrial Disease Adult Scale
Funding
This work was funded by The Wellcome Trust Centre for Mitochondrial Research [G906919], Newcastle University Centre for Brain Ageing and Vitality (supported by the Biotechnology and Biological Sciences Research Council, Engineering and Physical Sciences Research Council, Economic and Social Research Council and Medical Research Council [G0700718]), MRC Centre for Neuromuscular Disease [G000608-1], The MRC Centre for Translational Research in Neuromuscular Disease Mitochondrial Disease Patient Cohort (UK) [G0800674], Lily Foundation, the UK NIHR Biomedical Research Centre in Age and Age Related Diseases award to the Newcastle upon Tyne Hospitals NHS Foundation Trust and UK NHS Specialist Commissioners ‘Rare Mitochondrial Disorders of Adults and Children’ Service. JPG was funded by a Wellcome Trust Studentship [090194/Z/09/Z].
Supplementary material
Supplementary material is available at Brain online.
References
- Akaike H. A new look at the statistical model identification. IEEE Trans Automat Contr. 1974;AC-19:716–23. [Google Scholar]
- Apabhai S, Gorman GS, Sutton L, Elson JL, Plötz T, Turnbull DM, et al. Habitual physical activity in mitochondrial disease. PLoS One. 2011;6:e22294. doi: 10.1371/journal.pone.0022294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auré K, Ogier de Baulny H, Laforêt P, Jardel C, Eymard B, Lombès A. Chronic progressive ophthalmoplegia with large-scale mtDNA rearrangement: can we predict progression? Brain. 2007;130:1516–24. doi: 10.1093/brain/awm067. [DOI] [PubMed] [Google Scholar]
- Bates MG, Hollingsworth KG, Newman JH, Jakovljevic DG, Blamire AM, MacGowan GA, et al. Concentric hypertrophic remodelling and subendocardial dysfunction in mitochondrial DNA point mutation carriers. Eur Heart J Cardiovasc Imaging. 2013;14:650–8. doi: 10.1093/ehjci/jes226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwood JK, Whittaker RG, Blakely EL, Alston CL, Turnbull DM, Taylor RW. The investigation and diagnosis of pathogenic mitochondrial DNA mutations in human urothelial cells. Biochem Biophys Res Commun. 2010;393:740–5. doi: 10.1016/j.bbrc.2010.02.072. [DOI] [PubMed] [Google Scholar]
- Box GEP, Cox DR. An analysis of transformations. J R Stat Soc B. 1964;26:211–52. [Google Scholar]
- de Laat P, Koene S, van den Heuvel LP, Rodenburg RJ, Janssen MC, Smeitink JA. Clinical features and heteroplasmy in blood, urine and saliva in 34 Dutch families carrying the m.3243A > G mutation. J Inherit Metab Dis. 2012;35:1059–69. doi: 10.1007/s10545-012-9465-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- den Heijer T, van der Lijn F, Koudstaal PJ, Hofman A, van der Lugt A, Krestin GP, et al. A 10-year follow-up of hippocampal volume on magnetic resonance imaging in early dementia and cognitive decline. Brain. 2010;133:1163–72. doi: 10.1093/brain/awq048. [DOI] [PubMed] [Google Scholar]
- Diggle PJ, Heagerty P, Liang KY, Zeger SL. Analysis of longitudinal data. Second edn. Oxford: Oxford University Press; 2002. [Google Scholar]
- Dobkin RD, Menza M, Allen LA, Gara MA, Mark MH, Tiu J, et al. Cognitive-behavioral therapy for depression in Parkinson's disease: a randomized, controlled trial. Am J Psychiatry. 2011;168:1066–74. doi: 10.1176/appi.ajp.2011.10111669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott HR, Samuels DC, Eden JA, Relton CL, Chinnery PF. Pathogenic mitochondrial DNA mutations are common in the general population. Am J Hum Genet. 2008;83:254–60. doi: 10.1016/j.ajhg.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enns GM, Kinsman SL, Perlman SL, Spicer KM, Abdenur JE, Cohen BH, et al. Initial experience in the treatment of inherited mitochondrial disease with EPI-743. Mol Genet Metab. 2012;105:91–102. doi: 10.1016/j.ymgme.2011.10.009. [DOI] [PubMed] [Google Scholar]
- Galvin JE, Powlishta KK, Wilkins K, McKeel DW, Jr, Xiong C, Grant E, et al. Predictors of preclinical alzheimer disease and dementia: A clinicopathologic study. Arch Neurol. 2005;62:758–65. doi: 10.1001/archneur.62.5.758. [DOI] [PubMed] [Google Scholar]
- Gellerich FN, Deschauer M, Chen Y, Müller T, Neudecker S, Zierz S. Mitochondrial respiratory rates and activities of respiratory chain complexes correlate linearly with heteroplasmy of deleted mtDNA without threshold and independently of deletion size. Biochim Biophys Acta. 2002;1556:41–52. doi: 10.1016/s0005-2728(02)00305-5. [DOI] [PubMed] [Google Scholar]
- Goto Y, Koga Y, Horai S, Nonaka I. Chronic progressive external ophthalmoplegia: a correlative study of mitochondrial DNA deletions and their phenotypic expression in muscle biopsies. J Neurol Sci. 1990;100:63–9. doi: 10.1016/0022-510x(90)90014-e. [DOI] [PubMed] [Google Scholar]
- Hassing LB, Grant MD, Hofer SM, Pedersen NL, Nilsson SE, Berg S, et al. Type 2 diabetes mellitus contributes to cognitive decline in old age: a longitudinal population-based study. J Int Neuropsychol Soc. 2004;10:599–607. doi: 10.1017/S1355617704104165. [DOI] [PubMed] [Google Scholar]
- He L, Chinnery PF, Durham SE, Blakely EL, Wardell TM, Borthwick GM, et al. Detection and quantification of mitochondrial DNA deletions in individual cells by real-time PCR. Nucleic Acids Res. 2002;30:e68. doi: 10.1093/nar/gnf067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–19. doi: 10.1038/331717a0. [DOI] [PubMed] [Google Scholar]
- Holt IJ, Harding AE, Cooper JM, Schapira AH, Toscano A, Clark JB, et al. Mitochondrial myopathies: clinical and biochemical features of 30 patients with major deletions of muscle mitochondrial DNA. Ann Neurol. 1989;26:699–708. doi: 10.1002/ana.410260603. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y, Goto YI, Minami R. Progression in a case of Kearns-Sayre syndrome. J Child Neurol. 2000;15:750–5. doi: 10.1177/088307380001501107. [DOI] [PubMed] [Google Scholar]
- Jacobs LJ, Jongbloed RJ, Wijburg FA, de Klerk JB, Geraedts JP, Nijland JG, et al. Pearson syndrome and the role of deletion dimers and duplications in the mtDNA. J Inherit Metab Dis. 2004;27:47–55. doi: 10.1023/B:BOLI.0000016601.49372.18. [DOI] [PubMed] [Google Scholar]
- Johns DR, Rutledge SL, Stine OC, Hurko O. Directly repeated sequences associated with pathogenic mitochondrial DNA deletions. Proc Natl Acad Sci USA. 1989;86:8059–62. doi: 10.1073/pnas.86.20.8059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DK, Storandt M, Morris JC, Galvin JE. Longitudinal study of the transition from healthy aging to Alzheimer disease. Arch Neurol. 2009;66:1254–9. doi: 10.1001/archneurol.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DK, Galvin JE. Longitudinal changes in cognition in Parkinson's disease with and without dementia. Dement Geriatr Cogn Disord. 2011;31:98–108. doi: 10.1159/000323570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns TP, Sayre GP. Retinitis pigmentosa, external ophthalmoplegia and complete heart block. Arch Ophthalmol. 1958;60:280–289. [PubMed] [Google Scholar]
- Khrapko K, Bodyak N, Thilly WG, van Orsouw NJ, Zhang X, Coller HA, et al. Cell-by-cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 1999;27:2434–41. doi: 10.1093/nar/27.11.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS, Mosley TH, Catellier DJ, Coker LH Atherosclerosis Risk in Communities Study Brain MRI Study. Fourteen-year longitudinal study of vascular risk factors, APOE genotype, and cognition: The ARIC MRI Study. Alzheimer's Dement. 2009;5:207–14. doi: 10.1016/j.jalz.2009.01.027. [DOI] [PubMed] [Google Scholar]
- Kornblum C, Nicholls TJ, Haack TB, Schöler S, Peeva V, Danhauser K, et al. Loss-of-function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nat Genet. 2013;45:214–19. doi: 10.1038/ng.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan KJ, Bender A, Taylor RW, Turnbull DM. A multiplex real-time PCR method to detect and quantify mitochondrial DNA deletions in individual cells. Anal Biochem. 2007;370:127–9. doi: 10.1016/j.ab.2007.06.024. [DOI] [PubMed] [Google Scholar]
- Kuha J. AIC and BIC. Sociol Methods Res. 2004;33:188–229. [Google Scholar]
- Larsson NG, Holme E. Multiple short direct repeats associated with single mtDNA deletions. Biochim Biophys Acta. 1992;1139:311–4. doi: 10.1016/0925-4439(92)90106-w. [DOI] [PubMed] [Google Scholar]
- Lax NZ, Hepplewhite PD, Reeve AK, Nesbitt V, McFarland R, Jaros E, et al. Cerebellar ataxia in patients with mitochondrial DNA disease: a molecular clinicopathological study. J Neuropathol Exp Neurol. 2012;71:148–61. doi: 10.1097/NEN.0b013e318244477d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Gallardo E, López-Pérez MJ, Montoya J, Ruiz-Pesini E. CPEO and KSS differ in the percentage and location of the mtDNA deletion. Mitochondrion. 2009;9:314–17. doi: 10.1016/j.mito.2009.04.005. [DOI] [PubMed] [Google Scholar]
- Mancuso M, Orsucci D, Ienco EC, Pini E, Choub A, Siciliano G. Psychiatric involvement in adult patients with mitochondrial disease. Neurol Sci. 2013;34:71–4. doi: 10.1007/s10072-011-0901-0. [DOI] [PubMed] [Google Scholar]
- McFarland R, Taylor RW, Turnbull DM. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010;9:829–40. doi: 10.1016/S1474-4422(10)70116-2. [DOI] [PubMed] [Google Scholar]
- Meier DS, Weiner HL, Guttmann CR. Time-series modeling of multiple sclerosis disease activity: a promising window on disease progression and repair potential? Neurotherapeutics. 2007;4:485–98. doi: 10.1016/j.nurt.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mita S, Schmidt B, Schon EA, DiMauro S, Bonilla E. Detection of ‘deleted' mitochondrial genomes in cytochrome-c oxidase-deficient muscle fibers of a patient with Kearns-Sayre syndrome. Proc Natl Acad Sci USA. 1989;86:9509–13. doi: 10.1073/pnas.86.23.9509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraes CT, DiMauro S, Zeviani M, Lombes A, Shanske S, Miranda AF, et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearne-Sayre syndrome. N Engl J Med. 1989;320:1293–9. doi: 10.1056/NEJM198905183202001. [DOI] [PubMed] [Google Scholar]
- Moser EB. Repeated measures modeling with PROC MIXED. Proceedings of the 29th SAS Users Group International Conference, 2004. [Google Scholar]
- Nandhagopal R, Kuramoto L, Schulzer M, Mak E, Cragg J, Lee CS, et al. Longitudinal progression of sporadic Parkinson's disease: a multi-tracer positron emission tomography study. Brain. 2009;132:2970–9. doi: 10.1093/brain/awp209. [DOI] [PubMed] [Google Scholar]
- Old SL, Johnson MA. Methods of microphotometric assay of succinate dehydrogenase and cytochrome c oxidase activities for use on human skeletal muscle. Histochem J. 1989;21:545–55. doi: 10.1007/BF01753355. [DOI] [PubMed] [Google Scholar]
- Oldfors A, Larsson NG, Holme E, Tulinius M, Kadenbach B, Droste M. Mitochondrial DNA deletions and cytochrome c oxidase deficiency in muscle fibers. Journal of the Neurological Sciences. 1992;110:169–177. doi: 10.1016/0022-510x(92)90025-g. [DOI] [PubMed] [Google Scholar]
- Orsucci D, Calsolaro V, Siciliano G, Mancuso M. Quality of life in adult patients with mitochondrial myopathy. Neuroepidemiology. 2012;38:194–5. doi: 10.1159/000337161. [DOI] [PubMed] [Google Scholar]
- Pearson HA, Lobel JS, Kocoshis SA, Naiman JL, Windmiller J, Lammi AT, Hoffman R, Marsh JC. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr. 1979;95:976–984. doi: 10.1016/s0022-3476(79)80286-3. [DOI] [PubMed] [Google Scholar]
- Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, Letellier T. Mitochondrial threshold effects. Biochem J. 2003;370:751–62. doi: 10.1042/BJ20021594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotig A, Bourgeron T, Chretien D, Rustin P, Munnich A. Spectrum of mitochondrial DNA rearrangements in the Pearson marrow-pancreas syndrome. Hum Mol Genet. 1995;4:1327–30. doi: 10.1093/hmg/4.8.1327. [DOI] [PubMed] [Google Scholar]
- Sadikovic B, Wang J, El-Hattab A, Landsverk M, Douglas G, Brundage EK. Sequence homology at the breakpoint and clinical phenotype of mitochondrial DNA deletion syndromes. PLoS One. 2010;5:e15687. doi: 10.1371/journal.pone.0015687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer AM, Phoenix C, Elson JL, McFarland R, Chinnery PF, Turnbull DM. Mitochondrial disease in adults: a scale to monitor progression and treatment. Neurology. 2006;66:1932–4. doi: 10.1212/01.wnl.0000219759.72195.41. [DOI] [PubMed] [Google Scholar]
- Schon EA, Rizzuto R, Moraes CT, Nakase H, Zeviani M, DiMauro S. A direct repeat is a hotspot for large-scale deletion of human mitochondrial DNA. Science. 1989;244:346–9. doi: 10.1126/science.2711184. [DOI] [PubMed] [Google Scholar]
- Schon EA, Dimauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nature Reviews Genetics. 2012;13:878–90. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer JD. Using SAS PROC MIXED to fit multilevel models, hierarchical models, and individual growth models. Journal of Educational and Behavioral Statistics. 1998;23:323–55. [Google Scholar]
- Thomas VP. What's wrong with Bonferroni adjustments. BMJ. 1998;316:1236–8. doi: 10.1136/bmj.316.7139.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornatore JB, Grant LA. Burden among family caregivers of persons with Alzheimer's disease in nursing homes. Gerontologist. 2002;42:497–506. doi: 10.1093/geront/42.4.497. [DOI] [PubMed] [Google Scholar]
- Vu TC, Nutt JG, Holford HG. Progression of motor and nonmotor features of Parkinson's disease and their response to treatment. British Journal of Clinical Pharmacology. 2012;74:267–83. doi: 10.1111/j.1365-2125.2012.04192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittaker RG, Blackwood JK, Alston CL, Blakely EL, Elson JL, McFarland R, et al. Urine heteroplasmy is the best predictor of clinical outcome in the m.3243A>G mtDNA mutation. Neurology. 2009;72:568–9. doi: 10.1212/01.wnl.0000342121.91336.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita S, Nishino I, Nonaka I, Goto Y. Genotype and phenotype analyses in 136 patients with single large-scale mitochondrial DNA deletions. Journal of Human Genetics. 2008;53:598–606. doi: 10.1007/s10038-008-0289-8. [DOI] [PubMed] [Google Scholar]
- Yatsuga S, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T, et al. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012;1820:619–24. doi: 10.1016/j.bbagen.2011.03.015. [DOI] [PubMed] [Google Scholar]
- Zeviani M, Moraes CT, DiMauro S, Nakase H, Bonilla E, Schon EA, et al. Deletions of mitochondrial DNA in Kearns-Sayre syndrome. Neurology. 1988;38:1339–46. doi: 10.1212/wnl.38.9.1339. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.