

Non-ketotic hyperglycinaemia usually presents in neonates or infants with seizures, hypotonia, and developmental delays. Baker et al. describe a variant form with atypical features including leukodystrophy in patients without AMT, GLDC or GCSH mutations, and report defects in lipoic acid synthesis and related iron-sulphur cluster biogenesis genes including LIAS, BOLA3 and GLRX5.
Keywords: nonketotic hyperglycinemia, leukodystrophy, iron-sulphur cluster, lipoic acid
Abstract
Patients with nonketotic hyperglycinemia and deficient glycine cleavage enzyme activity, but without mutations in AMT, GLDC or GCSH, the genes encoding its constituent proteins, constitute a clinical group which we call ‘variant nonketotic hyperglycinemia’. We hypothesize that in some patients the aetiology involves genetic mutations that result in a deficiency of the cofactor lipoate, and sequenced genes involved in lipoate synthesis and iron-sulphur cluster biogenesis. Of 11 individuals identified with variant nonketotic hyperglycinemia, we were able to determine the genetic aetiology in eight patients and delineate the clinical and biochemical phenotypes. Mutations were identified in the genes for lipoate synthase (LIAS), BolA type 3 (BOLA3), and a novel gene glutaredoxin 5 (GLRX5). Patients with GLRX5-associated variant nonketotic hyperglycinemia had normal development with childhood-onset spastic paraplegia, spinal lesion, and optic atrophy. Clinical features of BOLA3-associated variant nonketotic hyperglycinemia include severe neurodegeneration after a period of normal development. Additional features include leukodystrophy, cardiomyopathy and optic atrophy. Patients with lipoate synthase-deficient variant nonketotic hyperglycinemia varied in severity from mild static encephalopathy to Leigh disease and cortical involvement. All patients had high serum and borderline elevated cerebrospinal fluid glycine and cerebrospinal fluid:plasma glycine ratio, and deficient glycine cleavage enzyme activity. They had low pyruvate dehydrogenase enzyme activity but most did not have lactic acidosis. Patients were deficient in lipoylation of mitochondrial proteins. There were minimal and inconsistent changes in cellular iron handling, and respiratory chain activity was unaffected. Identified mutations were phylogenetically conserved, and transfection with native genes corrected the biochemical deficiency proving pathogenicity. Treatments of cells with lipoate and with mitochondrially-targeted lipoate were unsuccessful at correcting the deficiency. The recognition of variant nonketotic hyperglycinemia is important for physicians evaluating patients with abnormalities in glycine as this will affect the genetic causation and genetic counselling, and provide prognostic information on the expected phenotypic course.
Introduction
Nonketotic hyperglycinemia (NKH, MIM# 605899) is a disorder of glycine metabolism defined by deficient enzyme activity of the glycine cleavage enzyme system and biochemically characterized by elevated glycine in serum and CSF, with an increased CSF:plasma glycine ratio. Typically patients present neonatally with lethargy evolving into coma, apnoea requiring ventilator support, hypotonia, hiccupping, and myoclonic jerks (Hennermann et al., 2012). Patients regain ventilatory drive after 2 weeks, but develop profound intellectual disability, spasticity, and intractable seizures. Some patients have brain malformations such as agenesis of the corpus callosum or hydrocephalus. Patients with attenuated NKH often present in infancy, have developmental delays, seizures, hyperactivity, and chorea (Hennermann et al., 2012). Biochemically, the glycine cleavage enzyme system is composed of the P-protein (GLDC gene, MIM# 238300), which removes CO2 from glycine and transfers the amino-methyl group to lipoate on the H-protein (GCSH gene, MIM# 238330), and the T-protein (AMT gene, MIM# 238310) which releases ammonia and forms methylenetetrahydrofolate, after which the reduced lipoate is re-oxidized by the L-protein (Fig. 1) (Kikuchi et al., 2008). In typical NKH, 72% of patients have a causative mutation in GLDC, and 24% in AMT, with no mutations identified in GCSH (Kure et al., 2006; Hamosh et al., 2009). In 4% of patients, no mutations were identified in a gene encoding a constituent of the glycine cleavage enzyme, despite proven deficient glycine cleavage enzyme activity and elevated glycine levels. We will refer to this group as ‘variant NKH’. A previous report of a patient with a regressive form of NKH who had deficient H-protein activity and H-protein devoid of lipoate focused our attention on the cofactor lipoate in variant NKH (Hirage et al., 1981; Trauner et al., 1981). After normal development for 6 months, this patient lost all milestones, became lethargic, hypotonic, irritable, and developed seizures, dying at 15 months of age. The lipoate synthase enzyme uses an iron-sulphur cluster in a radical S-adenosylmethionine dependent reaction (Fig. 1) (Cicchillo et al., 2004; Schonauer et al., 2009). Recently, a patient with lactic acidosis was identified with mutations in the lipoate synthase gene (LIAS, MIM# 607031) (Mayr et al., 2011). This patient presented with convulsions, progressive hypotonia, somnolence and major lactic acidosis. He developed spastic tetraparesis, multicystic encephalopathy and mild hypertrophic cardiomyopathy. Rare patients presenting in infancy with severe fatal primary lactic acidosis had mutations in BOLA3 (MIM# 613183) and NFU1 (MIM# 608100) genes affecting iron-sulphur cluster metabolism, and causing impaired lipoylation of the E2 subunit of pyruvate dehydrogenase and 2-ketoglutarate dehydrogenase (αKGDH) (Cameron et al., 2011; Navarro-Sastre et al., 2011; Haack et al., 2013). They also had elevated plasma glycine levels. Patients with BOLA3 mutation presented in infancy with large lactic acidosis, progressive developmental delay, epileptic encephalopathy, white matter disease, cardiomyopathy, vomiting and distress, usually fatal in the first year of life (Seyda et al., 2001; Cameron et al., 2011; Haack et al., 2013). Patients with NFU1 mutations developed lactic acidosis, lethargy, feeding difficulties and failure to thrive, weakness and hypotonia, irritability, white matter disease, and spongiform cerebral degeneration with necrosis, or with pulmonary hypertension and neurological regression, usually fatal in infancy (Seyda et al., 2001; Cameron et al., 2011; Navarro-Sastre et al., 2011). All patients had elevated lactate and glycine levels. In this study, we identified 11 patients from 10 families with variant NKH. We hypothesized that defects in lipoate synthesis could be causative in some patients with variant NKH. For these patients, we review the clinical and biochemical spectrum and the genetic aetiology, including the first patients with neurological disease as a result of mutations in GLRX5.
Figure 1.

Metabolic pathway of the glycine cleavage enzyme system and the synthesis of its lipoate cofactor. Disorders of components of the glycine cleavage enzyme (GLDC and AMT) cause classic NKH. Disorders of the biosynthesis of lipoyl-H (LIAS, GLRX5, BOLA3) cause variant NKH.
Materials and methods
Clinical studies
Patients with a biochemical phenotype of NKH, including deficient glycine cleavage system activity, but without mutations in GLDC, AMT and GCSH on sequencing and without deletions or duplications in the GLDC gene, were consented on an Institutional Review Board-approved protocol (05-0790), and clinical data and radiological images were collected.
Molecular studies
Genomic DNA was extracted using the QuickGene-810 Robot (Autogen). Patient samples were genotyped using Affymetrix (Santa Clara) SNP6.0 chips according to manufacturer’s protocol by the Genomics and Microarray Core at the University of Colorado. The .CEL files generated after array scanning were imported into Affymetrix Genotyping Console to generate a .CHP file using Birdseed v2 algorithm. Homozygosity mapping was carried out by first performing loss of heterozygosity analysis using Partek Genomic Suite. Regions >1 Mb (million base pairs) with very low rates of heterozygosity were selected for further analysis. Candidate genes were identified as mitochondrially targeted using the programs Mitoprot, TargetP, Predotar and Wolf-PSORT and retained if mitochondrial localization was identified by two programs, or if they were present in MitoCarta (Pagliarini et al., 2008; Ghezzi et al., 2010). Candidate genes were sequenced following PCR amplification with M13-tagged primers located at least 20 bp in the intronic regions (primers available upon request), and mutation analysis was performed using CodonCode Aligner (Dedham) and Mutation Surveyor (SoftGenetics). Candidate genes sequenced include the iron-sulphur cluster biogenesis genes: LYRM4, NFS1, NFU1, ISCU, BOLA3, GRPEL1, HSPA9, HSCB, ISCA1, ISCA2, IBA57, IND1 and GLRX5 (Cameron et al., 2011); the lipoate synthesis genes: LIAS, LIPT1, LIPT2 and ACSM1, and select genes of intramitochondrial fatty acid synthesis (Hiltunen et al., 2009, 2010). Mutations were analysed in silico for pathogenicity using Sift, Mutation Taster, and PolyPhen2 (Adzubei et al., 2010; Schwarz et al., 2010; Sim et al., 2012). A Sequenom multiplexed MALDI-TOF (Matrix-Assisted Laser Desorption/Ionization Time-Of-Flight) mass spectrometry assay was used for analysis of the GLRX5 mutation in 86 control subjects of Lebanese descent living in Australia, and sequencing for the mutation in 94 Chinese control samples obtained from Coriell (Camden). RNA from a GLRX5 patient was isolated using the SV Total RNA Isolation System from Promega, reverse transcribed using SuperScript® III First-Strand Synthesis System for RT-PCR by Invitrogen, and the complementary DNA sequenced as above.
Biochemical and proteomic studies
Modelling of the p.K51del mutation on the 3D structure of GLRX5 was carried out in silico using the known crystal structure (Johansson et al., 2011) and viewed and modified with Cn3D v4.3 software (Madej et al., 2012). Modelling of the mutations in the lipoate synthase was carried out using orthologous yeast Lip5 and E. coli biotin synthase (PDB ID: 1R30) structures (Sulo and Martin, 1993). Western blot analysis for GLRX5 was done in fibroblast lysate using an antibody from Abcam, and lipoylated proteins using an antibody from EMD Chemicals, Inc. This method showed a band of the lipoylated E2 subunits of pyruvate dehydrogenase (dihydrolipoamide S-acetyltransferase, DLAT, 70 kDa) and of αKGDH (dihydrolipoamide succinyltransferase, DLST, 48.7 kDa) (Cameron et al., 2011). Human skin fibroblasts were grown in α-minimal essential media (unless otherwise specified) with 10% HyClone FetalClone III (Thermo Scientific) at 37°C and 5% CO2. Respiratory chain enzyme complexes I, II, II + III, III, IV and citrate synthase were determined spectrophotometrically on a Cary 300 spectrophotometer as described (Rahman et al., 1996; Kendrick et al., 2011), with some modifications. Enzyme activities of pyruvate dehydrogenase and of the glycine cleavage enzyme were assayed by following the release of radiolabeled CO2 in fibroblasts and liver, respectively as described previously (Hyland and Leonard, 1983; Wicking et al., 1986; Rolland et al., 1993; Toone et al., 1994).
Pathophysiology and iron metabolism studies
Aconitase enzyme activity was measured after separating the cytosolic and mitochondrial enzyme in an acrylamide gel followed by in-gel activity stain using cis-aconitic acid, phenazone methosulphate and isocitrate dehydrogenase (Tong and Rouault, 2006). After culturing fibroblasts in iron-rich Richter’s modified minimal essential medium, they were stained using a Prussian Blue iron stain. Quantitative measurement of iron in patient fibroblast cells was performed using the colorimetric ferrozine-based assay as described (Riemer et al., 2004) with the following modification: lysis of fibroblast cells with 200 μl of 50 mM NaOH was followed by sonication to break denatured DNA, and absorbance at 550 nm was measured using a Shimadzu UV-2401 PC spectrophotometer. Real time-quantitative PCR of genes involved in iron regulation in patient fibroblasts was carried out using a 7500 Fast quantitative PCR instrument (Applied Biosystems) and TaqMan® Gene Expression Assays (HMOX1: Hs01110250_m1, IRP2: Hs00386293_m1, SLC40A1: Hs00205888_m1) with GOLPH3 as the endogenous control chosen for its stable expression between all cell lines.
Fibroblast transfection and treatment studies
The full length complementary DNA of GLRX5 and of LIAS obtained from Origene was cloned into the retroviral transfection vector Gryphon pCHAC-mWasabi (Allele Biotech) and transfected into the Gryphon amphotropic packaging cell line using Lipofectamine® LTX reagent (Invitrogen). After transfection of primary patient fibroblasts with virus-containing supernatants using GFP fluorescence of the vector as a control, the corrective effect was followed by pyruvate dehydrogenase enzyme activity and by western blot for lipoylated proteins. At 80% confluence, fibroblasts were incubated for 72 h in medium containing 50 µM lipoate or 4-(triphenylphosphonium)-butyllipoate, synthesized as previously described (Ripcke et al., 2009), before harvesting and analysis.
Results
Clinical history
Of the 11 patients from 10 families with a biochemical diagnosis of NKH but normal molecular analysis of AMT, GLDC and GCSH, a genetic cause was identified in eight patients from eight families. The varying clinical presentations of these patients are summarized below, the biochemical findings are provided in Table 1, and neuroimaging in Fig. 2. Except for glycine, alanine, and lactate as listed, other amino acids and organic acids were normal in all children. Because of the persistently elevated glycine levels and deficient glycine cleavage enzyme activity, all patients were initially diagnosed with NKH.
Table 1.
Biochemical phenotype of variant nonketotic hyperglycinemia patients
| Patient ethnicity | Gene mutation | Glycine plasma µM | Glycine CSF µM | Ratio | Lactate mM | Alanine µM | GCS assay* nmol/min/mg protein | PDH assay** nmol/min/mg protein | Respiratory chain enzymes |
|---|---|---|---|---|---|---|---|---|---|
| 1 LEB | GLRX5 p.K51del | 844 | 23 | 0.03 | 0.2–1.8 B | 354–526 B | Low | 0.07 (M) | Normal fibs |
| 1.4–1.5 S | 29–32 S | ||||||||
| 2 LEB | GLRX5 p.K51del | 804 | 15 | 0.02 | 0.6–2.6 B | 343–523 B | Low | 0.54 (D) | Normal fibs & muscle |
| 1.4–1.5 S | 26–27 S | 0.14 (M) | |||||||
| 3 CHI | GLRX5 p.K51del/ c.82ins[GCGTGCGG] | 829 | 25 | 0.03 | 1.2 B | 324 B | Low | NA | |
| 1.25 S | 25.5 S | ||||||||
| 4 AUS | BOLA3 p.R46X | 735 | 21 | 0.03 | 4.2 B | 403 B | 0.5 (11.4–14.2) | 0.07 (O) | Normal fibs |
| 3.8 S | 42 S | ||||||||
| 5 IND | BOLA3 p.R46X | 801 | 19 | 0.02 | 3.9 B | 493/663 B | Low | NA | Low SDH liver |
| 2.6 S | 43/63 S | ||||||||
| 6 AFR | BOLA3 p.R46X | 445 | 40 | 0.09 | 2.15 B | 342–782 B | Low | 0.94 | Normal fibs |
| 1.3 S | 40–46 S | ||||||||
| 7 SOM | LIAS p.E159K | 890/1035 | 180/94 | 0.20/0.09 | 10.2 B | 456 B | No activity | 0.51 (D) | Normal fibs |
| 1.49 S | |||||||||
| 8 TUR | LIAS p.D215E | 759 | 16 | 0.02 | NA | 350–511 B | No activity | 0.65 (D) | Normal fibs |
| 0.56 (C)† | |||||||||
| Control | <343 | <10/20 | <0.02 | <2.2 B | <520 B | 0.87–3.34 (D) | |||
| 0.18–0.42(M) | |||||||||
| 0.7–1.1 (O) | |||||||||
| <2.0 S | <46 S | ||||||||
| 0.56–1.23 (C) | |||||||||
| PUBLISHED CASES | |||||||||
| Cameron et al., 2011 | BOLA3 p.E42RfsX13 | 571 | 28 | 0.05 | 11–19 | NA | 0.36 (1.16 ± 0.16) | Low 1,2,3 | |
| Mayr et al., 2011 | LIAS p.R249H | 906 | NA | NA | 4.6–58 | NA | 0.5 (6–19.7) | Normal | |
| Haack et al., 2013 | BOLA3 p.I67N | 533 | NA | NA | 7.7–33 | NA | 1.5 (6–19.7) | Low 1,2 | |
| BOLA3 p.I67N | 541 | NA | NA | 2.4–23 | NA | 0.7 (1.5–3.9) | Low 1,2,4 |
Enzyme activity in nmol/min/mg protein.
*glycine cleavage enzyme activity is measured in liver biopsy.
**pyruvate dehydrogenase enzyme assay is measured in fibroblasts (fibs).
B = blood (plasma or serum); S = spinal fluid; D = Denver; M = Melbourne, O = Oxford, C = Copenhagen.
†PDH/Citrate synthase 4.8 (normal values 6.1–13.4).
LEB = Lebanese; CHI = Chinese; AUS = Australian; IND = Indian; AFR = African–American; SOM = Somali; TUR = Turkish.
Figure 2.

Neuroimaging of patients with variant nonketotic hyperglycinemia. (A) Brain MRI of Patient 1 at 10 years of age shows multifocal lesions of increased signal affecting the cerebrum on T2 sequence of the brain on axial images (left), and lesions of decreased signal intensity in the upper cervical spinal cord as shown on T1 sequence (right). (B) Spinal MRI images of Patient 2 show central lesions of the upper spinal cord of increased intensity on T2 sequence on axial and sagittal series. (C) Brain MRI of Patient 4 at 6 months of age shows lesions of increased signal on T2 sequence in the periventricular white matter (left) and central cervical spinal cord (right). (D) Brain MRI of Patient 5 at age 18 months shows extensive central lesions of increased signal on T2 sequence on axial and coronal images. (E) Brain MRI of Patient 6 at 9 years of age shows mild cerebral and cerebellar atrophy on T2 sequences. (F) Brain MRI of Patient 8 at 20 days of age, shows cerebral atrophy particularly of the white matter and increased signal in thalami and basal ganglia on T2 sequences. In the diffusion weighted images, there is diffusion restriction in the long tracts of the brainstem (yellow arrow) and white matter of the cerebellum, the posterior crux of the internal capsule (yellow arrow) and the basal ganglia (red arrow), and in cortical areas (blue arrow).
Patient 1
Patient 1 is the previously-described daughter of non-consanguineous parents of Lebanese descent living in Sydney, Australia who had a normal birth and developmental history until 2.5 years of age, when she developed progressive spasticity and constipation (Chiong et al., 2007). She had borderline cognitive abilities, slow processing speed and behavioural problems at school. She had upper and lower extremity spasticity, pyramidal signs and dysarthria. Brain MRI showed signal abnormalities involving the frontal and parietal white matter, which over time progressed to involve the deep white matter, periaqueductal grey matter, and around the central canal in the cervical spine (Fig. 2A). There was no lactate peak on magnetic resonance spectroscopy. Now at the age of 11 years, she has age-appropriate mathematics and reading skills, but mild learning difficulties and poor concentration. Her neurological signs are static with spastic diplegia, hypertonicity in extremities, and she is able to walk with crutches.
Patient 2
Patient 2, the daughter of first cousin parents of Lebanese descent living in Sydney, had a normal birth and developmental history until 7 years of age, when she developed spasticity and worsening clumsiness. She was cognitively gifted with no regression, hypotonia or seizures. She has lower extremity spasticity and ataxia, but after therapy with botulinum toxin, she walks and can run independently. MRI shows lesions in the cervical spinal cord (Fig. 2B). At the age of 11 years, she continues to excel cognitively, but with prominent spasticity. Treatment with α-lipoic acid (300 mg twice daily) did not provide added improvement over standard treatment.
Patient 3
Patient 3 is a previously-described Chinese boy living in Taiwan with normal birth history, growth and development, who presented with gait abnormalities at the age of 2 years and has a 6-month history of visual disturbance (Wei et al., 2011). He had leg spasticity and nystagmus. Brain MRI showed optic atrophy and signal abnormalities in the central and subcortical frontal white matter and genu of the corpus callosum, with atrophy in the body and splenium of the corpus callosum and frontal gyri. At 1.5 years after onset, he had lesions in the lower medulla oblongata extending to the mid-thoracic spinal cord. At the age of 7 years, he is cognitively intact, but has slow deterioration of vision and increasing spasticity.
Patient 4
This Caucasian boy living in Sydney had a normal birth and development until aged 6 months, when he had regression in gross motor skills, hypotonia, and poor head control. He had visual inattention as a result of optic atrophy, increased irritability, poor feeding, and episodes of spasticity and myoclonus without correlate on EEG, and without seizures. An echocardiogram revealed mild left ventricular hypertrophy. MRI revealed signal abnormalities involving the deep white matter, optic nerve and cervical spine (Fig. 2C). He deteriorated precipitously and died at the age of 7 months.
Patient 5
At the age of 8 months, Patient 5—the daughter of first cousin consanguineous Indian parents with normal birth and early developmental history—had regression of skills including loss of head control and inability to roll over. She developed seizures, severe hypotonia and extrapyramidal signs. She had poor feeding and diarrhoea, optic atrophy and vision loss. MRI revealed signal abnormalities involving the deep white matter and sparing the subcortical fibers (Fig. 2D). She had increased lactate on magnetic resonance spectroscopy. She died at 22 months after an acute febrile illness. Post-mortem examination revealed hypertrophic cardiomyopathy with mitochondrial proliferation, and loss of all cellular material in the white matter. A previous sibling had died of a similar condition.
Patient 6
This African–American daughter of unrelated parents developed normally until 6 months of age, when development slowed with hypotonia and poor head control. At the age of 12 months she had myoclonus and choreoathetoid movements. By 2 years of age she had 7 to 10 words and walked independently. She had tonic-clonic seizures controlled on a single anti-epileptic medication and at age 3 years was diagnosed with NKH. She had a developmental quotient of 43% at 3.5 years. From ages 3 to 8 years she slowly developed spasticity and ataxia, lost her ability to walk, and became non-verbal. At 9 years of age, she developed recurrent status epilepticus, and severe deterioration after an apnoeic event with hypoglycaemia. She fed poorly resulting in severe malnutrition with loss of >30% body weight over 1 year. Her worsening spasticity was accompanied by contractures. She spent her time curled in the foetal position. She died at the age of 11 years. MRI revealed mild cerebral and cerebellar atrophy, but no leukodystrophy (Fig. 2E).
Patient 7
Patient 7 was the daughter of consanguineous Somali parents. She presented neonatally with hypotonia, seizures from day 3, and apnoea requiring ventilation on day 7. She developed myoclonus, hiccupping, and a burst suppression pattern on EEG. In infancy, there was failure to thrive, severe psychomotor retardation, acquired microcephaly, myoclonus, hiccupping, intractable seizures, acquired hearing loss, and persistent sleepiness. In late infancy, she developed spasticity with pyramidal tract signs, nystagmus, and absent development. By the age of 2 years, she received a G-tube for unsafe swallowing and had intermittent stridor. She died at the age of 2 years 8 months with persistent seizures in a deteriorating condition. On brain MRI at age of 20 days, she had atrophy of the cerebrum in both cortex and white matter (Fig. 2F). There was diffusion restriction in the long tracts of the brainstem and internal capsule (yellow arrow in Fig. 2F), in the basal ganglia (red arrow Fig. 2F), cerebellar white matter and peduncles, and in the cortex (blue arrow Fig. 2F). There was abnormal T2 signal in the caudate, putamen, thalami and dorsal pons. A sibling born later was a carrier.
Patient 8
This boy of Turkish non-consanguineous parents presented at 2 days of age with hypotonia and seizures evolving into tonic-clonic seizures and absences, which were effectively treated with topiramate and lamotrigine. His last seizure was at age 12 years. He had developmental delays with walking at age 18 months and talking at age 3.5 years. At the age of 14 years, he is functioning at an age equivalent level of 7 years. He was hyperactive until age 9 years, but at present is calm and inactive. His motor function is clumsy and immature, without specific movement disturbances. Brain MRI at age of 7 days was normal. His sister presented at birth with hypotonia and tonic-clonic seizures, and apnoea requiring ventilator support for 3 days. She had elevated plasma glycine but a normal CSF:plasma glycine ratio. She died at the age of 7 months after 1 month of fluctuating fever and vomiting.
Molecular results
GLRX5
Recognizing the same clinical phenotype and shared Lebanese origin in Patients 1 and 2, homozygosity for a rare recessive disease was assumed. Single-nucleotide polymorphism analysis identified 10 autosomal regions >1 Mb of shared homozygosity in both children for which their parents are heterozygous on chromosomes 3, 4, 8 and 14, containing 218 coding genes, of which 39 genes encode mitochondrially localized proteins. Two candidate genes, OXSM (MIM# 610324) and GLRX5 (MIM# 609588), were identified from intramitochondrial fatty acid synthesis, lipoate metabolism, and iron-sulphur cluster biogenesis. Sequencing did not identify a mutation in OXSM, but identified in GLRX5 in exon 1 a deletion c.151_153delAAG; p.K51del, homozygous in both probands, heterozygous in their parents, and absent in 172 alleles of Lebanese migrants living in Australia, hence unlikely to be a polymorphism in this matched population. Sequencing GLRX5 in Patient 3, with a similar phenotype but of Chinese descent, identified the same mutation c.151_153delAAG on the maternal allele, and on the paternal allele an 8 bp insertion c.82_83insGCGTGCGG; p.G28Gfs*25, resulting in a premature stop codon at amino acid 52, 25 amino acid residues downstream. Both parents were carriers, and neither abnormality was identified in 188 control Chinese alleles. The complementary DNA sequence developed by reverse transcriptase PCR from RNA extracted from skin fibroblasts of Patient 1 was identical to the exonic genomic DNA, making a splice alteration induced by the 3-bp deletion unlikely. Western blot analysis using antibodies to GLRX5 revealed normal protein levels versus control (Fig. 3B). The deleted lysine 51 lies in the highly conserved glutaredoxin domain, is conserved across species from human to C. elegans (Fig. 3A), and is predicted to be deleterious in silico. Modelling positions the deleted amino acid between the first α-helix and first β-sheet, eight residues from K59, which interacts directly with the iron-sulphur cluster at the enzyme active site (Fig. 3D). In the tertiary and quaternary structures, the deleted amino acid localizes on the outside of the molecule, opposite the active site, and is in close proximity to the side chains of D52 and Y88, and is predicted to disrupt the first α-helix and β-sheet interaction due to the loss of electrostatic or hydrogen bonding interactions with D52 and Y88 (Fig. 3E) (Madej et al., 2012). Located on the surface, K51 might also be important for intermolecular recognition. Western blotting analysis using an antibody specific for lipoylated proteins revealed reduced to absent levels of lipoate on the E2 subunits of pyruvate dehydrogenase and αKGDH versus control fibroblasts (Fig. 3C), which normalized after transfection with a retrovirus containing a full length GLRX5 complementary DNA construct, proving that the dysfunction in GLRX5 was causative (Fig. 3C).
Figure 3.

Mutations in GLRX5. (A) On phylogenetic alignment, the deleted amino acid K51 is completely conserved from human to Drosophila. (B) The amount of GLRX5 protein on western blot is not reduced. (C) Western blot of lipoylated proteins in fibroblast lysate shows normal signal for the lipoylated E2 components of pyruvate dehydrogenase (PDH) and αKGDH in controls. In fibroblasts from Patients (Pt) 1 and 2 this band is almost completely absent, but it is clearly present after transfection of the fibroblasts of Patient 2 with a full-length construct of GLRX5 (Pt2-T). Images of citrate synthase shown as a loading control. (D) Three-dimensional modelling of the GLRX5 protein (PDB: 2WUL) shows the deleted lysine K51 to be located at the interface of the first α-helix and the first β-sheet. The active site K59 and the iron-sulphur cluster are indicated. (E) K51 (deleted in Patients 1–3) is in close proximity to residues D52 and Y88 (ball and stick). Average distances between residues in all four chains of the tetrameric crystal structure as well as standard deviations are shown in the table (insert). Distances were measured between the NZ of K51, closest O of D52 and OH of Y88. The relative positions of C67, which ligates the iron-sulphur cluster (orange and yellow spheres), and the bound glutathione (stick) are also shown.
BOLA3
The remaining patients were analysed for all candidate genes involved in lipoate synthesis. Three patients, each of different ethnicity (Patients 4–6), were homozygous for the same truncating mutation c.136C>T; p.R46X in exon 2 of BOLA3 isoform 1, and their parents were heterozygous carriers. The stop site created by this mutation is eight amino acids upstream of the previously described truncating duplication and frame shift (Fig. 4A) (Cameron et al., 2011). Patients 4 and 6 had reduced lipoylated E2 subunits of pyruvate dehydrogenase and αKGDH, but to varying degrees (Fig. 4B).
Figure 4.
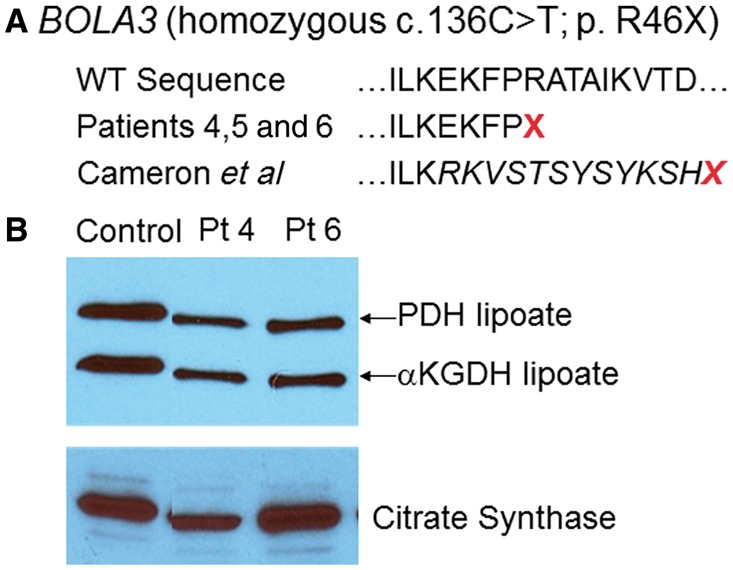
Mutations in BOLA3. (A) The location of the missense mutation in Patients 4, 5 and 6 at R46 is indicated in relation to the wild-type sequence and the mutation reported in Cameron et al. (2011). (B) Western blot of lipoylated proteins in fibroblast lysates shows normal signal for the lipoylated E2 components of pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase (αKGDH) in controls. In Patients 4 and 6 this band is substantially reduced in intensity. Images of citrate synthase shown as a loading control.
LIAS
Patient 7 was homozygous for the missense mutation c.475_477GAG>AAA; p.E159K in the lipoate synthase gene LIAS, Patient 8 was homozygous for the missense mutation c.645T>A; p.D214E. The parents of both patients were carriers each for the respective mutations. Both amino acids are completely conserved (Fig. 5A) and the mutations are predicted deleterious in silico. Modelling localizes E159 on the surface of LIAS away from the binding sites for S-adenosylmethionine and the iron-sulphur cluster and not at the dimerization site (Fig. 5B), whereas D215 is located deeper in the molecule closer to the active site. Patient 7 had no visible lipoylated E2 proteins (Fig. 5C), and Patient 8 had reduced bands of lipoylated proteins (Fig. 5D). Transfection with a full-length complementary DNA LIAS construct of fibroblasts from Patient 8 normalized pyruvate dehydrogenase enzyme activity (untransfected 0.75, after transfection 3.26, controls 0.87–3.34 nmol/min/mg protein).
Figure 5.

Mutations in lipoate synthase. (A) On phylogenetic alignment, the mutated amino acids E159 and D215 are completely conserved from human to Drosophila. (B) Three-dimensional modelling of the lipoate synthase protein shows the mutated amino acids in relation to the iron-sulphur cluster. (C) Western blot of lipoylated proteins in fibroblast lysate shows normal signal for the lipoylated E2 components of pyruvate dehydrogenase (PDH) and αKGDH in controls. In Patient 7 (Pt7), this band is absent (C), whereas in Patient 8 it is reduced (D). A ninth patient with variant non-ketotic hyperglycinaemia but with normal lipoylation is shown (P). Images of citrate synthase shown as a loading control.
Biochemical results
Liver glycine cleavage enzyme activity was deficient where measured. Cultured skin fibroblasts from available probands all had deficient pyruvate dehydrogenase enzyme activity and normal respiratory chain enzyme activities (Table 1). Disturbed cellular iron handling has previously been described in some patients with iron-sulphur cluster genetic abnormalities (Mochel et al., 2008; Ye et al., 2010). In contrast, both the mitochondrial and the cytosolic aconitase activities in our patients (n = 6) were normal (Supplementary Fig. 1). No intramitochondrial iron accumulation was visualized in the fibroblasts of the patients with mutant GLRX5 or BOLA3 (Supplementary Fig. 2). Quantification of iron per mg cellular protein in fibroblast lysates showed small differences: BOLA3-deficient Patient 6 had 133 ± 37% of normal control subjects (P = 0.003, n = 6), GLRX5-deficient Patient 1 had 119 ± 16% (P = 0.03, n = 6) and Patient 2 had 99 ± 13% (P = 0.9, n = 6) of normal control subject’s cellular iron (controls n = 30, 100 ± 20%). The previously reported GLRX5 patient had substantial transcriptional changes in iron metabolism-related genes with 15-fold elevation of heme oxygenase 1 (HMOX1), 5-fold elevation of ferroportin 1 (SLC40A1), and IRP2 was increased 2.4-fold. In our patients, IRP2 was not changed, and only in GLRX5-deficient Patient 1 were HMOX1 and SLC40A1 increased 2.9-fold compared with control fibroblasts; all other patients were inside the range seen in control subjects. Treatment of fibroblasts from Patient 7 for 3 days with either lipoic acid or with a mitochondrially targeted and cleavable form of lipoate did not correct lipoylation of mitochondrial proteins or increase pyruvate dehydrogenase enzyme activity (data not shown).
Discussion
The pathways for the biosynthesis of iron-sulphur clusters and for lipoate have recently been elucidated (Hiltunen et al., 2009, 2010; Lill et al., 2012; Rouault, 2012). Intramitochondrial fatty acid synthesis produces octanoyl-mt-acyl carrier protein (Hiltunen et al., 2009). The octanoyl group is then transferred to the H-protein of the glycine cleavage enzyme (Schonauer et al., 2009; Hiltunen et al., 2010), and sulphur is incorporated by the lipoate synthase enzyme (Nesbitt et al., 2008; Lill et al., 2012; Rouault, 2012). Lipoate is a critical cofactor for the glycine cleavage system, pyruvate dehydrogenase, αKGDH, and branched chain ketoacid dehydrogenase (Fig. 1). All patients presented here had reduced to absent lipoylation of proteins, and reduced activities of the lipoylated enzymes glycine cleavage enzyme system and pyruvate dehydrogenase. We identified mutations in three genes responsible for this deficiency in our patients: lipoic acid synthetase (LIAS), BolA family member 3 (BOLA3), and glutaredoxin 5 (GLRX5).
Pathogenic mutations in LIAS will result in reduced lipoylation of target proteins. The LIAS mutations changed highly conserved amino acids and were deleterious by in silico analyses. Reintroduction of the wild-type gene restored pyruvate dehydrogenase enzyme activity proving its causative role. Molecular modelling did not allow us to derive conclusive insights of the mechanism for this decreased enzymatic activity. It is not yet clear which genes contribute to the origin of the iron-sulphur cluster of LIAS, but BOLA3 has been implicated in this process (Cameron et al., 2011; Haack et al., 2013). In our patients, the nonsense nature of the stop mutation in BOLA3 and its recognition in multiple families suggest a causative role in this condition. Pathogenicity of a similar mutation, a frame-shift to stop codon eight residues upstream, has also been demonstrated (Cameron et al., 2011).
Mutations in GLRX5 are a newly discovered cause for reduced lipoylation. The proof of the pathogenicity rests on multiple lines of evidence. The deleted amino acid is located in the functionally important and conserved glutaredoxin domain. It resides in a domain of linkage in two geographically-related individuals, and is also found in an ethnically unrelated patient with the same phenotype, whereas it is absent in ethnically matched controls. Patient 3 also has an 8 bp insertion that frame shifts and truncates the message. Reintroduction of the normal gene into a patient’s fibroblasts corrects the lipoylation phenotype, providing compelling proof of a causal role. The molecular mechanism by which the deleted K51 impairs functionality is not yet clear. The protein is present in normal amounts and missplicing is unlikely given the normal RNA structure on reverse transcriptase PCR.
GLXR5 is a monothiol, mitochondrially located glutaredoxin that in yeast reduces disulphides using glutathione, including deglutathionylation of mitochondrial proteins (Lillig et al., 2008). This function is likely less relevant in humans as purified GLRX5 has 500-fold less deglutathionylation activity than GLRX2 (Johansson et al., 2011). Rather, GLRX5 is important for iron-sulphur cluster biogenesis, where in yeast it interacts with Isa1 in the transfer pathway to target enzymes after a basic [2Fe-2S] iron-sulphur cluster is generated on ISCU (Nesbitt et al., 2008; Lill et al., 2012; Rouault, 2012). Given the deficient lipoylation, GLRX5 likely functions in the pathway that provides an iron-sulphur cluster to lipoate synthase. A direct interaction with lipoate synthase has not been shown. The deleted K51 potentially disrupts the iron-sulphur cluster interacting lysine residue (K59) through a change in tertiary structure, rather than any direct interactions. Alternatively, given its location on the external surface, the deletion could affect interactions with partner proteins. Some studies have suggested a homotetrameric structure with two [2Fe-2S] clusters and four glutathione molecules bound (Johansson et al., 2011), whereas others propose a heterodimeric structure of Glrx5–Bola3 with two shared iron-sulphur clusters, similar to Glrx5–Bola3 (Li and Outten, 2012a; Li et al., 2012,b). but no conclusive data have been published (Lill et al., 2012). This will require further functional investigations.
In this study, we delineate the clinical and biochemical phenotype of variant NKH. Biochemically, all patients had elevated plasma glycine and had been diagnosed as NKH. Most, but not all, had elevated CSF glycine but the increase was less than that seen in classic NKH, or even in the normal range, which complicates the diagnostic approach. Despite deficient pyruvate dehydrogenase activity in all, only some patients had elevated alanine and few had elevated lactate, which is strikingly different from the primary lactic acidosis presentation of multiple mitochondrial dysfunction syndrome. Deficient lipoylation of proteins was present in 8 of 10 families, making this a possible primary test for this heterogenous disorder. Although it is important to note that not all patients with variant NKH have defects in lipoylation (see Patient P in Fig. 5D).
Clinically, homogenous phenotypes in variant NKH have emerged. The patients with mutations in GLRX5 have relatively preserved cognition, but develop progressive spasticity and all remained alive. They developed signal abnormalities in the white matter hyperintense on T2 and hypointense on T1, progressive in time course and genetic in origin consistent with a leukodystrophy. Characteristically they also developed lesions affecting the periaqueductal zone in the upper spinal cord. Previous case reports of patients with biochemically diagnosed NKH with a phenotype of isolated progressive spastic paraparesis, sometimes accompanied by anterior horn cell disease or with ataxia, occurred in patients of similar Lebanese descent (Bank and Morrow, 1972; Steinman et al., 1979). This presentation differs from that of classic NKH, and differs dramatically from the single previously reported patient with a GLRX5 mutation, who presented at the age of 60 years with pyridoxine-refractory sideroblastic anaemia, but without neurological symptoms (MIM# 205950) (Camaschella et al., 2007). This patient also had insulin-dependent diabetes, cirrhosis, hypogonadism, and increased ferritin, transferrin saturation, and ringed sideroblasts and ferritin-positive erythroblasts.
The patients with BOLA3 mutations were characterized by neurodegeneration after an initially normal early development, followed by early demise with or without a period of stabilization. Two patients developed MRI signal abnormalities in the white matter of increased intensity on T2, decreased intensity on T1, progressive in time course, and on post-mortem pathology revealing loss of cellular material in the white matter, likely reflective of a leukodystrophic process. Additional symptoms include optic atrophy, hypotonia and spasticity, and cardiomyopathy, and sometimes lactic acidosis, although not to the dramatic degree noted in previous patients (Cameron et al., 2011; Haack et al., 2013). Two previously described NKH patients had similar findings: neuroregression and mildly elevated alanine in a patient with deficient liver H-protein lipoylation, and cardiomyopathy in a patient with NKH (Hirage et al., 1981; Trauner et al., 1981; Al-Shareef et al., 2011).
The phenotype induced by mutations in LIAS seems related to the severity of the functional impact on protein lipoylation. Patient 8, with some residual lipoylated enzymes present, had a mild and stable phenotype of developmental delay and mild seizure disorder, whereas Patient 7, with virtually absent lipoylation, presented with features of severe NKH such as diffusion restriction in the long tracts of the brainstem and internal capsule (yellow arrow in Fig. 2F), with Leigh-like lesions such as diffusion restriction in the basal ganglia (red arrow Fig. 2F), and cortical lesions as seen with αKGDH deficiency (blue arrow Fig. 2F). In mice, complete absence of lipoate synthase is embryonically lethal (Yi and Maeda, 2005).
The pathophysiology of variant NKH is complex. Iron-sulphur clusters are incorporated in many enzymes including respiratory chain complexes I, II and III and the aconitases. Some previously reported BOLA3 patients had reduced activities of some respiratory chain enzymes (Cameron et al., 2011; Haack et al., 2013). None of the patients in this study had a deficiency of a respiratory chain enzyme, other than reduced liver succinate dehydrogenase activity of Patient 5. Cytosolic aconitase lacking an iron-sulphur cluster loses its aconitase activity and becomes the iron response protein IRP1, binding iron response elements on messenger RNAs involved in cellular iron regulation (Pantopoulos, 2004). It stabilizes the transferrin receptor messenger RNA increasing iron import, and prevents translation of the transferrin and mitochondrial aconitase messenger RNAs (Pantopoulos, 2004). Dysfunctional cellular iron regulation was noted in patients with iron-sulphur cluster defects such as deficient aconitase activities in patients with ISCU mutations (Mochel et al., 2008). A previously reported patient with near absent GLRX5 protein, as a result of the splicing mutation c.294A>G and fibroblasts with RNA interference-mediated knock down of GLRX5, had deficient mitochondrial aconitase activity, IRP1 activation, decreased ferritin, increased expression of IRP2, HMOX1 and SLC40A1, and iron accumulation in mitochondria (Camaschella et al., 2007; Ye et al., 2010). In contrast, the currently studied GLRX5- and BOLA3-deficient fibroblasts had normal mitochondrial and cytosolic aconitase activities, and no mitochondrial iron accumulation in a high iron environment. Two cell lines showed a mild increase in cellular iron and one cell line showed a mild increase in HOMX1 and SLC40A1 gene expression, much less than previously described, whereas other cell lines did not show any effect. This discrepancy may be because of the different mutations in GLRX5, but the effect in BOLA3 was equally minimal. Thus, our study supports the conclusion that intracellular iron mishandling, although valid in some patients, cannot be generalized to the entirety of iron-sulphur cluster biogenesis defects, in contrast with the previous hypothesis (Rouault, 2012).
Instead of dysfunctional cellular iron metabolism, the key pathophysiological component in variant NKH appears to be deficient intramitochondrial lipoylation. Therefore restoration of intramitochondrial lipoate may be a therapeutic target. Treatment with lipoate has only a limited effect and, where it occurs, will likely act through activation of pathways of AMPK and PGC1-α, upregulating the transcription of some mitochondrial enzymes including pyruvate dehydrogenase, and could stabilize certain mutations with some residual activity (Koh et al., 2008; Packer and Cadenas, 2011). Lipoic acid is not transported into the mitochondria but synthesized de novo, therefore a mitochondrially targeted and releasable form of lipoate (Ripcke et al., 2009) was tried in our study, but also found to be ineffective. Consequently, a different therapeutic approach will be needed to correct the observed defects.
In conclusion, variant NKH involves in most, but not all patients studied to date, defects of lipoate biosynthesis and related iron-sulphur cluster biogenesis genes, including LIAS, BOLA3 and GLRX5. These patients present with a biochemistry of elevated glycine in plasma and CSF, less than expected for classic NKH. Symptoms such as leukodystrophy, secondary neurodegeneration after a period of normal development, Leigh disease, optic atrophy, cardiomyopathy and deafness are not part of classic NKH, and suggest the presence of variant NKH (Table 2). The presence of elevated alanine or lactate can also be indicative, but is not required. The absence of lipoylated proteins on western blot in fibroblasts and possibly leucocytes may become a valuable screening tool, but will need further clinical validation. The pathophysiology reflects the deficiency of lipoate on intra-mitochondrial enzymes rather than perturbation of cellular iron handling. This information expands our understanding of the disorder non-ketotic hyperglycinaemia, which can now be classified as ‘classic NKH’ with mutations in AMT, GLDC or GCSH, or ‘variant NKH’ with mutations in LIAS, BOLA3 and GLRX5, each with a specific phenotype. This information is important for physicians evaluating patients with abnormalities in glycine as it will affect the genetic causation and genetic counselling, and provide prognostic information on the expected phenotypic course.
Table 2.
Clinical indications for variant non-ketotic hyperglycinaemia
| Gene | Comments | |
|---|---|---|
| Primary indications | ||
| Elevated plasma glycine level | GLRX5, BOLA3, LIAS | Most plasma glycine levels are >700 µM |
| Mild elevation of CSF glycine level | GLRX5, BOLA3, LIAS | Most are elevated |
| No mutations in GLDC, AMT, GCSH | GLRX5, BOLA3, LIAS | |
| Secondary indications: clinical | ||
| Leukodystrophy | GLRX5, BOLA3, LIAS | Common |
| Central lesions in cervical spinal cord | GLRX5 | High specificity |
| Neurodegeneration after normal development | BOLA3 | Not present in classic NKH |
| Optic atrophy | GLRX5, BOLA3, LIAS | |
| Leigh disease | LIAS | severely affected patients |
| Cardiomyopathy | BOLA3 | Both dilated and hypertrophic reported |
| Deafness | BOLA3, LIAS | Infrequent |
| Elevated alanine | GLRX5, BOLA3, LIAS | |
| Elevated lactate | BOLA3, LIAS | Infrequently present |
| Confirmatory testing: biochemical | ||
| Deficient pyruvate dehydrogenase enzyme activity | GLRX5, BOLA3, LIAS | Fibroblasts, usually partial deficiency |
| Deficient glycine cleavage enzyme activity | GLRX5, BOLA3, LIAS | Liver tissue required |
| Deficient lipoylated proteins on western blot | GLRX5, BOLA3, LIAS | Can be variable in BOLA3 |
| Sequencing variant NKH genes | GLRX5, BOLA3, LIAS | |
Supplementary Material
Acknowledgements
The authors acknowledge and thank the following individuals on this project: Flemming Wibrand and Ernst Christensen, Copenhagen University Hospital, Copenhagen, Denmark; Tracy Rouault, National Institutes of Health, Washington DC, USA; David Thorburn, Murdoch Childrens Research Institute, Melbourne, Victoria 3052, Australia; Kristina Williams, Molecular Discovery Core, University of Colorado, Aurora, CO, USA, and Sara Williams of the Children’s Hospital Colorado Clinical Pathology Laboratory, Aurora, CO, USA.
Glossary
Abbreviations
- αKGDH
2-ketoglutarate dehydrogenase
- NKH
nonketotic hyperglycinemia
Funding
This work was supported by the Hope for NKH Fund, the NKH Crusaders Fund, and by a pilot grant of the Colorado Clinical and Translational Science Institute, via the National Institute of Health [UL1RR025780 to P.R.B.] and the National Centre of Research Resources. Its contents are the author’s sole responsibility and do not necessarily represent official National Institute of Health views.
Supplementary material
Supplementary material is available at Brain online.
Web Resources
OMIM: http://www.ncbi.nlm.nih.gov/omim
Mitoprot: http://ihg.gsf.de/ihg/mitoprot.html
TargetP: http://www.cbs.dtu.dk/services/TargetP/
Predotar: http://urgi.versailles.inra.fr/predotar/predotar.html
Wolf-PSORT: http://wolfpsort.org/
MitoCarta: http://www.broadinstitute.org/pubs/MitoCarta/human.mitocarta.html
Sift: http://sift.jcvi.org/
Mutation Taster: http://www.mutationtaster.org/
PolyPhen2: http://genetics.bwh.harvard.edu/pph2/
Ensembl: http://uswest.ensembl.org/index.html
References
- Adzubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-shareef I, Arabi M, Dabbagh O. Cardiac involvement in nonketotic hyperglycinemia. J Child Neurol. 2011;26:970–3. doi: 10.1177/0883073811399150. [DOI] [PubMed] [Google Scholar]
- Bank WJ, Morrow G. A familial spinal cord disorder with hyperglycinemia. Arch Neurol. 1972;27:136–44. doi: 10.1001/archneur.1972.00490140040007. [DOI] [PubMed] [Google Scholar]
- Camaschella C, Campanella A, De Falco L, Boschetto L, Merlini R, Silvestri L, et al. The human counterpart of zebrafish shiraz shows sideroblastic-like microcytic anemia and iron overload. Blood. 2007;110:1353–8. doi: 10.1182/blood-2007-02-072520. [DOI] [PubMed] [Google Scholar]
- Cameron JM, Janer A, Levandovskiy V, MacKay N, Rouault TA, Tong W-H, et al. Mutations in iron-sulfur cluster scaffold genes NFU1 and BOLA3 cause a fatal deficiency of multiple respiratory chain and 2-oxoacid dehydrogenase enzymes. Am J Hum Genet. 2011;89:486–95. doi: 10.1016/j.ajhg.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiong MA, Procopis P, Carpenter K, Wilcken B. Late-onset nonketotic hyperglycinemia with leukodystrophy and an unusual clinical course. Pediatr Neurol. 2007;37:283–6. doi: 10.1016/j.pediatrneurol.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Cicchillo RM, Iwig DF, Nesbitt NM, Baleanu-Gogonea C, Souder MG, Tu L, et al. Lipoyl synthase requires two equivalents of S-adenosyl-L-methionine to synthesize one equivalent of lipoic acid. Biochemistry. 2004;43:6378–86. doi: 10.1021/bi049528x. [DOI] [PubMed] [Google Scholar]
- Ghezzi D, Sevrioukova I, Invernizzi F, Lamperti C, Mora M, D’Adamo P, et al. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2010;86:639–49. doi: 10.1016/j.ajhg.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack TB, Rolinski B, Haberberger B, Zimmermann F, Graf E, Athing U, et al. Homozygous missense mutation in BOLA3 causes multiple mitochondrial dysfunctions syndrome in two siblings. J Inherit Metab Dis. 2013;36:55–62. doi: 10.1007/s10545-012-9489-7. [DOI] [PubMed] [Google Scholar]
- Hamosh A, Scharer G, Van Hove J. Glycine encephalopathy. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP, editors. GeneReviews. Seattle: Updated 2009, Nov 24, Available online at http://www.ncbi.nlm.nih.gov/books/NBK1357/ (15 December 2012 date last accessed) [Google Scholar]
- Hennermann JB, Berger JM, Grieben U, Scharer G, Van Hove JLK. Prediction of long-term outcome in glycine encephalopathy: a clinical survey. J Inherit Metab Dis. 2012;35:253–61. doi: 10.1007/s10545-011-9398-1. [DOI] [PubMed] [Google Scholar]
- Hiltunen JK, Schonauer MS, Autio KJ, Mittelmeier TM, Kastaniotis AJ, Dieckmann CL. Mitochondrial fatty acid synthesis type II: more than just fatty acids. J Biol Chem. 2009;284:9011–5. doi: 10.1074/jbc.R800068200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiltunen JK, Autio KJ, Schonauer MS, Kursu VAS, Dieckmann CL, Kastaniotis AJ. Mitochondrial fatty acid synthesis and respiration. Biochim Biophys Acta. 2010;1797:1195–202. doi: 10.1016/j.bbabio.2010.03.006. [DOI] [PubMed] [Google Scholar]
- Hirage K, Kochi H, Hayasaka K, Kikuchi G. Defective glycine cleavage system in nonketotic hyperglycinemia. Occurrence of a less active glycine decarboxylase and an abnormal aminomethyl carrier protein. J Clin Invest. 1981;68:525–34. doi: 10.1172/JCI110284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyland K, Leonard JV. Revised assays for the investigation of congenital lactic acidosis using 14C keto acids, eliminating problems associated with spontaneous decarboxylation. Clin Chim Acta. 1983;133:177–87. doi: 10.1016/0009-8981(83)90403-5. [DOI] [PubMed] [Google Scholar]
- Johansson C, Roos AK, Montano SJ, Sengupta R, Filippakopoulos P, Guo K, et al. The crystal structure of human GLRX5: iron-sulfur cluster co-ordination, tetrameric assembly and monomer activity. Biochem J. 2011;433:303–11. doi: 10.1042/BJ20101286. [DOI] [PubMed] [Google Scholar]
- Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JL, et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J. 2011;433:505–14. doi: 10.1042/BJ20100791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi G, Motokawa Y, Yoshida T, Hiraga K. Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proc Jpn Acad Ser B Physiol Biol Sci. 2008;84:246–63. doi: 10.2183/pjab/84.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh EH, Cho EH, Kim M-S, Park J-Y, Lee K-U. Effects of alpha-lipoic acid on AMP-activated protein kinase in different tissues: therapeutic implications for the metabolic syndrome. In: Patel MS, Packer L, editors. Lipoic acid. Energy production, antioxidant activity and health effects. Boca Raton, FL: CRC Press; 2008. pp. 495–519. [Google Scholar]
- Kure S, Kato K, Dinopoulos A, Gail C, DeGrauw TJ, Christodoulou J, et al. Comprehensive mutation analysis of GLDC, AMT, and GCSH in nonketotic hyperglycinemia. Hum Mutat. 2006;27:343–52. doi: 10.1002/humu.20293. [DOI] [PubMed] [Google Scholar]
- Li H, Outten CE. Monothiol CGFS Glutaredoxins and BolA-like Proteins: [2Fe-2S] Binding Partners in Iron Homeostasis. Biochemistry. 2012a;51:4377–89. doi: 10.1021/bi300393z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Mapolelo DT, Randeniya S, Johnson MK, Outten CE. Human glutaredoxin 3 forms [2Fe-2S]-bridged complexes with human BolA2. Biochemistry. 2012b;51:1687–96. doi: 10.1021/bi2019089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill R, Hoffmann B, Molik S, Pierik AJ, Rietzschel N, Stehling O, et al. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim Biophys Acta. 2012;1823:1491–508. doi: 10.1016/j.bbamcr.2012.05.009. [DOI] [PubMed] [Google Scholar]
- Lillig CH, Berndt C, Holmgren A. Glutaredoxin systems. Biochim Biophys Acta. 2008;1780:1304–17. doi: 10.1016/j.bbagen.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Madej T, Addess KJ, Fong JH, Geer LY, Geer RC, Lanczycki CJ, et al. MMDB: 3D structures and macromolecular interactions. Nucleic Acids Res. 2012;40:D461–4. doi: 10.1093/nar/gkr1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr JA, Zimmermann FA, Fauth C, Bergheim C, Meierhofer D, Radmayr D, et al. Lipoic acid synthetase deficiency causes neonatal-onset epilepsy, defective mitochondrial energy metabolism, and glycine elevation. Am J Hum Genet. 2011;89:792–7. doi: 10.1016/j.ajhg.2011.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochel F, Knight MA, Tong W-H, Hernandez D, Ayyad K, Taivassalo T, et al. Splice mutation in the iron-sulfur cluster scaffold protein ISCU causes myopathy with exercise intolerance. Am J Hum Genet. 2008;82:652–60. doi: 10.1016/j.ajhg.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Sastre A, Tort F, Stehling O, Uzarska MA, Arranz JA, del Toro M, et al. A fatal mitochondrial disease is associated with defective NFU1 function in the maturation of a subset of mitochondrial Fe-S proteins. Am J Hum Genet. 2011;89:656–67. doi: 10.1016/j.ajhg.2011.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesbitt NM, Cicchillo RM, Lee K-H, Grove TL, Booker SJ. Lipoic acid biosynthesis. In: Patel MS, Packer L, editors. Lipoic acid. Energy production, antioxidant activity and health effects. Boca Raton, FL: CRC Press; 2008. pp. 11–56. [Google Scholar]
- Packer L, Cadenas E. Lipoic acid: energy metabolism and redox regulation of transcription and cell signaling. J Clin Biochem Nutr. 2011;48:26–32. doi: 10.3164/jcbn.11-005FR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;143:112–23. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantopoulos K. Iron metabolism and the IRE/IRP regulatory system. An update. Ann NY Acad Sci. 2004;1012:1–13. doi: 10.1196/annals.1306.001. [DOI] [PubMed] [Google Scholar]
- Rahman S, Blok RB, Dahl HH, Danks DM, Kirby DM, Chow CW, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol. 1996;39:343–51. doi: 10.1002/ana.410390311. [DOI] [PubMed] [Google Scholar]
- Riemer J, Hoepken HH, Czerwinska H, Robinson S, Dringen R. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal Biochem. 2004;331:370–5. doi: 10.1016/j.ab.2004.03.049. [DOI] [PubMed] [Google Scholar]
- Ripcke J, Zarse K, Ristow M, Birringer M. Small-molecular targeting of the mitochondrial compartment with an endogenously cleaved reversible tag. Chembiochem. 2009;10:1689–96. doi: 10.1002/cbic.200900159. [DOI] [PubMed] [Google Scholar]
- Rolland MO, Mandon G, Mathieu M. First trimester prenatal diagnosis of non-ketotic hyperglycinaemia by a micro assay of glycine cleavage enzyme. Prenat Diagn. 1993;13:771–2. doi: 10.1002/pd.1970130814. [DOI] [PubMed] [Google Scholar]
- Rouault TA. Biogenesis of iron-sulfur clusters in mammalian cells: new insights and relevance to human disease. Dis Model Mech. 2012;5:155–64. doi: 10.1242/dmm.009019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonauer MS, Kastaniotis AJ, Kursu VAS, Hiltunen JK, Dieckmann CL. Lipoic acid synthesis and attachment in yeast mitochondria. J Biol Chem. 2009;284:23234–42. doi: 10.1074/jbc.M109.015594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. Mutation Taster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–6. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- Seyda A, Newbold RF, Hudson TJ, Verner A, MacKay N, Winter S, et al. A novel syndrome affecting multiple mitochondrial functions, located by microcell-mediated transfer to chromosome 2p14-2p13. Am J Hum Genet. 2001;68:386–96. doi: 10.1086/318196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:W452–7. doi: 10.1093/nar/gks539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman GS, Yudkoff M, Berman PH, Blazer-Yost B, Segal S. Late-onset nonketotic hyperglycinemia and spinocerebellar degeneration. J Pediatr. 1979;94:907–11. doi: 10.1016/s0022-3476(79)80211-5. [DOI] [PubMed] [Google Scholar]
- Sulo P, Martin NC. Isolation and characterization of LIP5. A lipoate biosynthetic locus of Saccharomyces cerevisiae. J Biol Chem. 1993;268:17634–9. [PubMed] [Google Scholar]
- Tong WH, Rouault TA. Functions of mitochondrial ISCU and cytosolic ISCU in mammalian iron-sulfur cluster biogenesis and iron homeostasis. Cell Metab. 2006;3:199–210. doi: 10.1016/j.cmet.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Toone JR, Applegarth DA, Levy HL. Prenatal diagnosis of non-ketotic hyperglycinaemia: experience in 50 at-risk pregnancies. J Inherit Metab Dis. 1994;17:342–4. doi: 10.1007/BF00711825. [DOI] [PubMed] [Google Scholar]
- Trauner DA, Page T, Greco C, Sweetman L, Kulovich S, Nyhan WL. Progressive neurodegenerative disorder in a patient with nonketotic hyperglycinemia. J Pediatr. 1981;98:272–5. doi: 10.1016/s0022-3476(81)80659-2. [DOI] [PubMed] [Google Scholar]
- Wei SH, Weng W-C, Lee N-C, Hwu W-L, Lee W-T. Unusual spinal cord lesions in late-onset non-ketotic hyperglycinemia. J Child Neurol. 2011;26:900–3. doi: 10.1177/0883073810393965. [DOI] [PubMed] [Google Scholar]
- Wicking CA, Scholem RD, Hunt SM, Brown GK. Immunochemical analysis of normal and mutant forms of human pyruvate dehydrogenase. Biochem J. 1986;239:89–96. doi: 10.1042/bj2390089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye H, Jeong SY, Gosh MC, Kovtunovych G, Silvestri L, Ortillo D, et al. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human fibroblasts. J Clin Invest. 2010;120:1749–61. doi: 10.1172/JCI40372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi X, Maeda N. Endogenous production of lipoic acid is essential for mouse development. Mol Cell Biol. 2005;25:8387–92. doi: 10.1128/MCB.25.18.8387-8392.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.