

Abstract
Atherosclerosis is a complex inflammatory disease involving extensive vascular vessel remodelling and migration of vascular cells. As RCAN1 is implicated in cell migration, we investigated its contribution to atherosclerosis. We show RCAN1 induction in atherosclerotic human and mouse tissues. Rcan1 was expressed in lesional macrophages, endothelial cells and vascular smooth muscle cells and was induced by treatment of these cells with oxidized LDLs (oxLDLs). Rcan1 regulates CD36 expression and its genetic inactivation reduced atherosclerosis extension and severity in Apoe−/− mice. This effect was mechanistically linked to diminished oxLDL uptake, resistance to oxLDL-mediated inhibition of macrophage migration and increased lesional IL-10 and mannose receptor expression. Moreover, Apoe−/−Rcan1−/− macrophages expressed higher-than-Apoe−/− levels of anti-inflammatory markers. We previously showed that Rcan1 mediates aneurysm development and that its expression is not required in haematopoietic cells for this process. However, transplantation of Apoe−/−Rcan1−/− bone-marrow (BM) cells into Apoe−/− recipients confers atherosclerosis resistance. Our data define a major role for haematopoietic Rcan1 in atherosclerosis and suggest that therapies aimed at inhibiting RCAN1 expression or function might significantly reduce atherosclerosis burden.
Keywords: atherosclerosis, hypercholesterolemia, inflammation, macrophage, RCAN1
INTRODUCTION
Atherosclerosis, the underlying cause of myocardial infarction, stroke and peripheral vascular disease, is the major cause of morbidity and mortality in the developed world. The initial steps of atherosclerosis are characterized by the subendothelial accumulation of apolipoprotein B-containing low-density lipoproteins (LDLs) in the artery wall. The oxidative modification of these lipoproteins [oxidized LDL (oxLDL)] triggers the activation of the vascular endothelium and drives an influx of monocytes to the vascular intima, where they differentiate into macrophages and phagocytose oxLDL (Hansson & Hermansson, 2011). Although in other contexts macrophages egress from the inflammation site after engulfing unwanted material, in an atherosclerotic plaque the loading of oxLDL into macrophages shifts them to a more sessile, foam-cell phenotype, and these foam cells do not leave the lesion after clearing the lipids (Angeli et al, 2004; Randolph, 2008). The trapping of cholesterol-engorged foam cells causes the plaque to expand through the recruitment of additional leukocytes and vascular smooth muscle cells (VSMCs). As these lesions mature they continue to accumulate extracellular lipids, and the central core of the mature plaque becomes necrotic. Rupture of plaques produce acute coronary syndromes, unstable angina, myocardial infarction and sudden death (Libby, 2002).
Monocytes/macrophages are a relatively heterogeneous population, and the existence of at least two broad classes of macrophage phenotype has been proposed: proinflammatory macrophages (classically activated or M1) and those involved in resolution and repair (alternatively activated or M2) (Gordon & Taylor, 2005). M2 macrophages produce low levels of pro-inflammatory cytokines but high levels of arginase1 (Arg1), mannose receptor (Mrc1 or CD206) and IL10, and have a higher phagocytic capacity and a lower antigen presentation capacity than M1 macrophages (Gordon & Taylor, 2005). The existence of other macrophage phenotypes has been proposed that fit neither the classical nor the alternative activation pattern (Mosser & Edwards, 2008). The heterogeneity of atherosclerotic plaque macrophages has been recognized for many years and several types of macrophages have been found in atherosclerotic lesions (Bouhlel et al, 2007; Khallou-Laschet et al, 2010).
Regulator of calcineurin 1 (RCAN1) belongs to a family of endogenous regulators of calcineurin activity (RCAN; previously known as DSCR/MCIP/calcipressin/Adapt78 in mammals) (Davies et al, 2007). The RCAN1 protein is highly conserved (Davies et al, 2007), displaying 96% identity between human and mouse (Strippoli et al, 2000). The human and mouse RCAN1 genes are expressed as two isoforms, RCAN1-1 and RCAN1-4, that differ at their N terminus as a consequence of alternative promoter usage and first exon usage (Davies et al, 2007; Fuentes et al, 1997). RCAN1-1 and RCAN1-4 have different expression patterns and different regulation mechanisms control their expression. While RCAN1-1 seems to be constitutively expressed in most tissues, transcription of the RCAN1-4 variant is induced de novo by several stimuli that activate the calcineurin-NFAT pathway (Cano et al, 2005; Crawford et al, 1997; Ermak et al, 2002; Esteban et al, 2011; Minami et al, 2004; Wang et al, 2002; Yang et al, 2000). RCAN1 has been implicated in important physiological and pathological processes, including tumour growth and angiogenesis, sepsis, cardiac hypertrophy, mast-cell function, T-cell survival, and synaptic plasticity and memory (Baek et al, 2009; Harris et al, 2005; Hoeffer et al, 2007; Ryeom et al, 2008; Yang et al, 2009). Rcan1 additionally plays an essential role in the migration of VSMCs in response to angiotensin II stimulation; moreover, Rcan1 genetic ablation in the mouse confers resistance to abdominal aortic aneurysm and to neointima formation in a restenosis model (Esteban et al, 2011). Rcan1 in endothelial cells inhibits VEGF-induced migration and in vitro tube formation (Iizuka et al, 2004; Minami et al, 2004). In contrast, Rcan1 knockdown in cancer cell lines increases motility while its forced expression reduces their motility and CN activity (Espinosa et al, 2009). Rcan1 thus appears to have opposite roles in cell migration in different settings. Since most studies have involved the simultaneous inactivation of Rcan1-1 and Rcan1-4 or have examined the effect of over-expressing or knocking-down only one of these isoforms, it has not yet been possible to ascribe specific roles to one Rcan1 isoform and not the other.
Here, we investigated the contribution of Rcan1 to atherosclerosis development. We show that RCAN1 is induced in human and mouse atherosclerotic tissues and, using a mouse model of atherosclerosis and bone-marrow (BM) transplantation assays, we demonstrate that Rcan1 in haematopoietic cells promotes atherosclerosis. We also present evidence that the pro-atherogenic action of Rcan1 is mediated by oxLDL-uptake and that macrophage polarization and trapping are central to its pro-atherogenic role.
RESULTS
RCAN1-4 is upregulated in human and mouse atherosclerosis lesions
To assess RCAN1 expression in human atherosclerotic lesions, we compared human atherosclerotic coronary arteries with non-atherosclerotic coronary arteries and internal mammary arteries, a vessel that does not develop atherosclerosis. RCAN1-4 protein expression was markedly higher in atherosclerotic vessels than in non-atherosclerotic coronary arteries and internal mammary arteries (Fig 1A). Although RCAN1-1 expression is usually constitutive, its level also appeared to be higher in atherosclerotic arteries, but the difference was less marked than for RCAN1-4 (Fig 1A). The protein expression differences were accompanied by correspondingly higher expression of RCAN1-1 and RCAN1-4 mRNA in atherosclerotic arteries (Fig S1 of Supporting Information).
Figure 1.
RCAN1-4 is induced in atherosclerotic human and mouse arteries
Immunoblot of RCAN1 in atherosclerotic and non-atherosclerotic human coronary artery and in human internal mammary artery (hIMA). Protein bands corresponding to RCAN1-1 and RCAN1-4 are indicated. α-ACTIN was used as loading control.
The aortic sinus of Apoe−/− mice fed standard chow or a high-fat diet (HFD) was immunostained for Rcan1. Representative images are shown of Rcan1 immunohistochemistry (top panels) and fluorescence microscopy (bottom panels). Bar, 50 µm.
Representative confocal immunofluorescence images of Rcan1 (red), lectin (green), αSMA (green) and Mac3 (green) in aortic sinus lesions of HFD-fed Apoe−/− mice. Merged images are also shown. Bar, 5 µm.
Quantitative PCR analysis of Rcan1 mRNA expression in the aortic arch of C57BL/6 and Apoe−/− mice fed standard chow or an HFD. mRNA amounts were normalized to Hprt1 expression (means ± SEM; n = 3). Student's t-test, *p = 0.013, **p = 0.0019.
Representative Rcan1 immunoblot in the same tissues as in D.
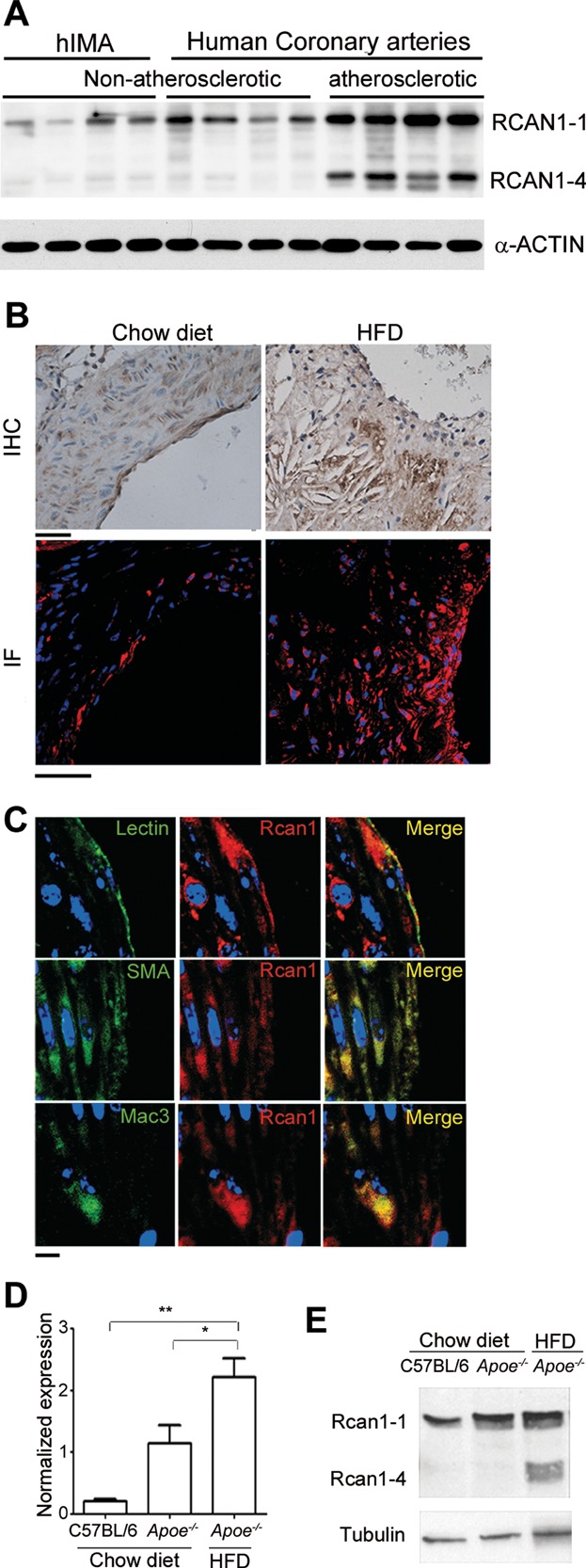
To investigate the role of RCAN1 in atherogenesis, we used the Apoe−/− mouse model (Plump et al, 1992; Zhang et al, 1992). These mice develop atherosclerosis spontaneously, and the appearance of lesions is accelerated by feeding them a cholesterol-rich diet. Like human familial hypercholesterolemia patients, these mice develop lesions in the aortic valves (Getz & Reardon, 2012). We fed 3-month old Apoe−/− mice a high-fat diet (HFD) for 6 weeks and compared Rcan1 expression in the aortic valves with that in wild-type and Apoe−/− mice fed a standard chow diet. Aortic valves of Apoe−/− mice fed an HFD showed marked Rcan1 staining in cells close to areas of lipid deposition, while staining was much weaker in the non-atherosclerotic valves of mice fed the control diet (Fig 1B, top panels). More intense Rcan1 expression in the valves of HFD-fed mice was also evident upon analysis by confocal immunofluorescence (Fig 1B, lower panels). Immunofluorescent staining of plaques for markers of endothelial cells, VSMCs and macrophages revealed elevated Rcan1 expression in all three cell types (Fig 1C). Quantitative PCR analysis of the aortic arch, which is also predisposed to lesion formation in mice, revealed higher Rcan1 expression in chow-fed Apoe−/− mice than in wt C57BL/6 mice, and expression was higher still in Apoe−/− mice fed an HFD (Fig 1D). As in human samples, western blot analysis of Rcan1 expression in the aortic arch of these animals indicated that Rcan1-4 was strongly induced in atherosclerotic mice (Fig 1E). In contrast, Rcan1-1 expression was barely affected in atherosclerosis (Fig 1E).
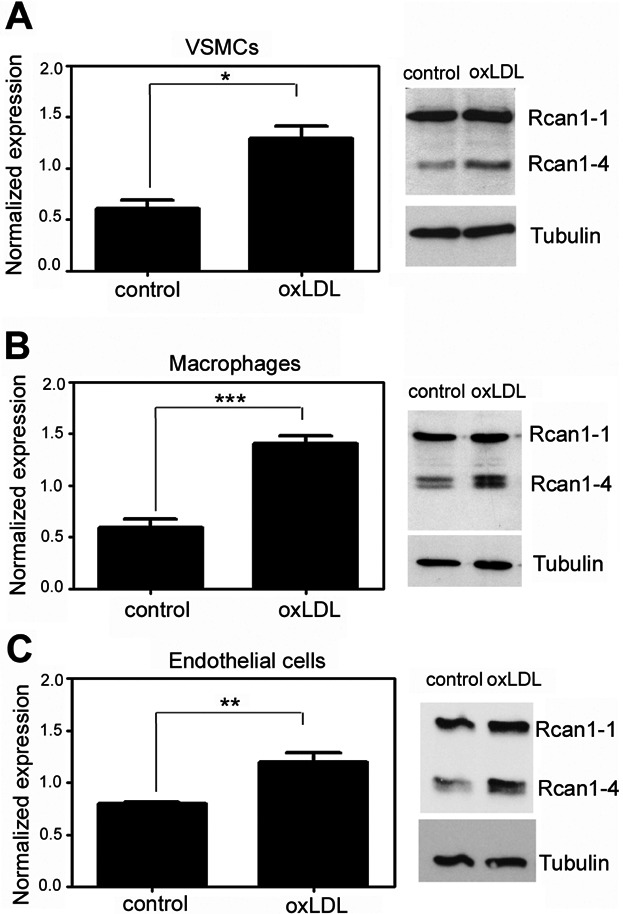
The subendothelial accumulation of oxLDLs is a crucial early event in plaque formation. We therefore determined whether oxLDL regulates Rcan1 expression in the major cell compartments present in the plaque. Treatment of mouse primary macrophages, VSMCs, and endothelial cells with oxLDL induced the expression of Rcan1 mRNA and Rcan1-4 protein in all three cells types, but did not induce Rcan1-1 protein (Fig 2).
Figure 2.
Induction of Rcan1-4 expression by oxLDL Quantitative PCR and representative immunoblot analysis of Rcan1 expression in cells isolated from Apoe−/− mice and treated with 50 µg/ml oxLDL:
VSMCs (n = 3),
Peritoneal macrophages (n = 3) and
Endothelial cells (n = 6). mRNA amounts were normalized to Hprt1 expression (means ± SEM). Student's t-test, *p = 0.007, **p = 0.001, ***p = 0.0003.
Rcan1 inactivation reduces atherosclerotic lesion burden
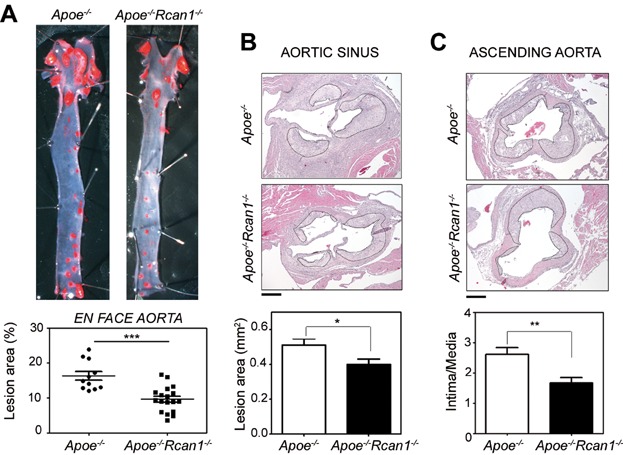
To investigate the contribution of Rcan1 to atheroma formation we compared Apoe−/− with Apoe−/−Rcan1−/− (double knockout) mice. Rcan1 targeting affected both isoforms and lack of expression was confirmed in aorta and heart (Fig S2 of Supporting Information). Apoe−/− and Apoe−/−Rcan1−/− mice showed similar body weight increases after 6 weeks on an HFD (Fig S3A of Supporting Information), and there were no significant inter-group differences in serum concentrations of triglyceride, total and free cholesterol, high-density lipoprotein cholesterol and LDL cholesterol (Fig S3B–C of Supporting Information). En face analysis of Oil Red O stained atherosclerotic plaques revealed that aortic lesion size in Apoe−/−Rcan1−/− mice was significantly smaller than in Apoe−/− mice (Fig 3A). Since the aortic sinus and ascending aorta are particularly prone to atherosclerosis, we also compared atheroma formation in these regions. Haematoxylin and eosin (H&E) staining of cross-sections revealed a significantly smaller lesion area in both regions in Apoe−/−Rcan1−/− mice (Fig 3B–C).
Figure 3.
Rcan1 deficiency decreases atherogenesis burden
Representative Oil red O staining and quantification of lesion area in the aortas of Apoe−/− (n = 11) and Apoe−/−Rcan1−/− mice (n = 18) fed an HFD for 6 weeks. Each data point denotes an individual mouse, and the horizontal bars denote the mean (long bar) and the SEM.
Representative H&E staining of the aortic sinus and quantification of the lesion area in this region (mean ± SEM).
Representative H&E staining of the ascending aorta and quantification of lesion measured as intima/media ratio in this region in the same cohort of animals (mean ± SEM). Bars, 200 µm. Student's t-test, *p = 0.02, **p = 0.0025, ***p = 0.0001.
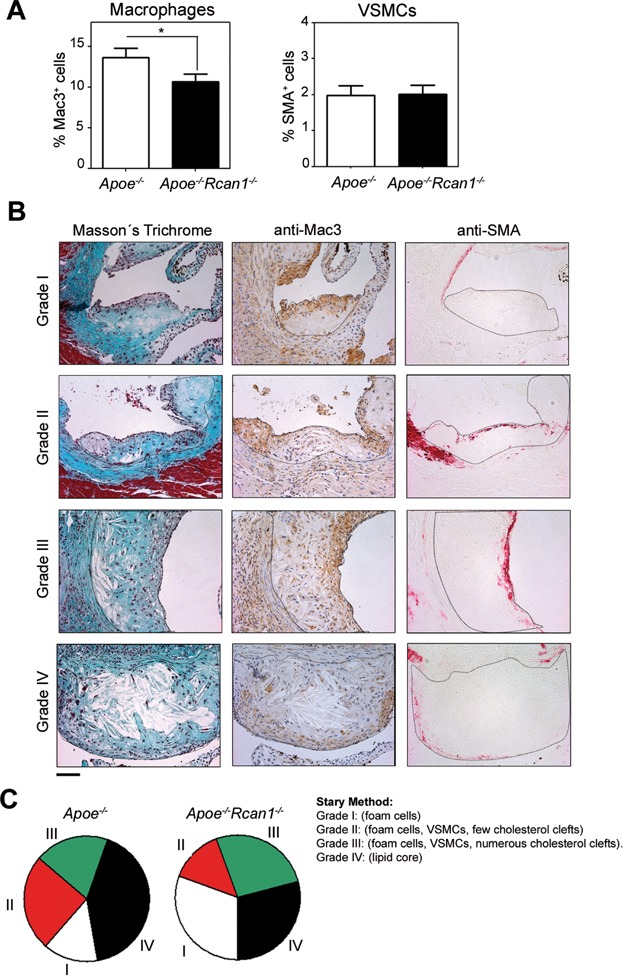
To assess the progression of atheromas in Apoe−/− and Apoe−/−Rcan1−/− mice, we determined the presence of macrophages and VSMCs and the lipid deposition pattern in the aortic cusps, the region that contains the most advanced lesions in the Apoe-deficient model (Nakashima et al, 1994). The relative content of macrophages was lower in Apoe−/−Rcan1−/− plaques, whereas numbers of VSMCs were similar in the two genotypes (Fig 4A). To confirm that Apoe−/−Rcan1−/− mice had less-advanced plaques, we classified lesions according to the Stary method (Stary et al, 1994) into early plaques (grade I) containing only macrophages; grade II lesions containing macrophages, VSMCs and a few scattered cholesterol clefts; grade III lesions containing macrophages, VSMCs and numerous cholesterol clefts and advanced plaques (grade IV) containing macrophages, VSMCs and a large lipid core (Fig 4B). After 6 weeks of HFD, ≈ 42% plaques in Apoe−/− mice were grade IV and only ≈ 14% were grade I (Fig 4C). In contrast, the proportion of grade IV plaques in Apoe−/−Rcan1−/− mice was ≈ 28% and that of grade I plaques was ≈ 31% (Fig 4C). These results therefore indicate that Rcan1 plays a key role in atherosclerosis progression.
Figure 4.
Rcan1 deficiency results in less-advanced plaques
Quantification of neointimal macrophage and VSMC content in lesions of the aortic sinus of Apoe−/− (n = 11) and Apoe−/−Rcan1−/− mice (n = 18) fed an HFD for 6 weeks. Student's t-test, *p = 0.017.
Representative images of Masson's trichrome, anti-Mac3 and anti-SMA staining of lesions of the aortic sinus classified according to the Stary method: grade I (mostly foam cells), grade II (foam cells, VSMCs and few cholesterol clefts), grade III (foam cells, VSMCs and numerous cholesterol clefts) and grade IV (lipid core). Bars, 50 µm.
Classification of aortic sinus lesions according to the Stary method in the cohort of Apoe−/− and Apoe−/−Rcan1−/− animals shown in Fig 3.
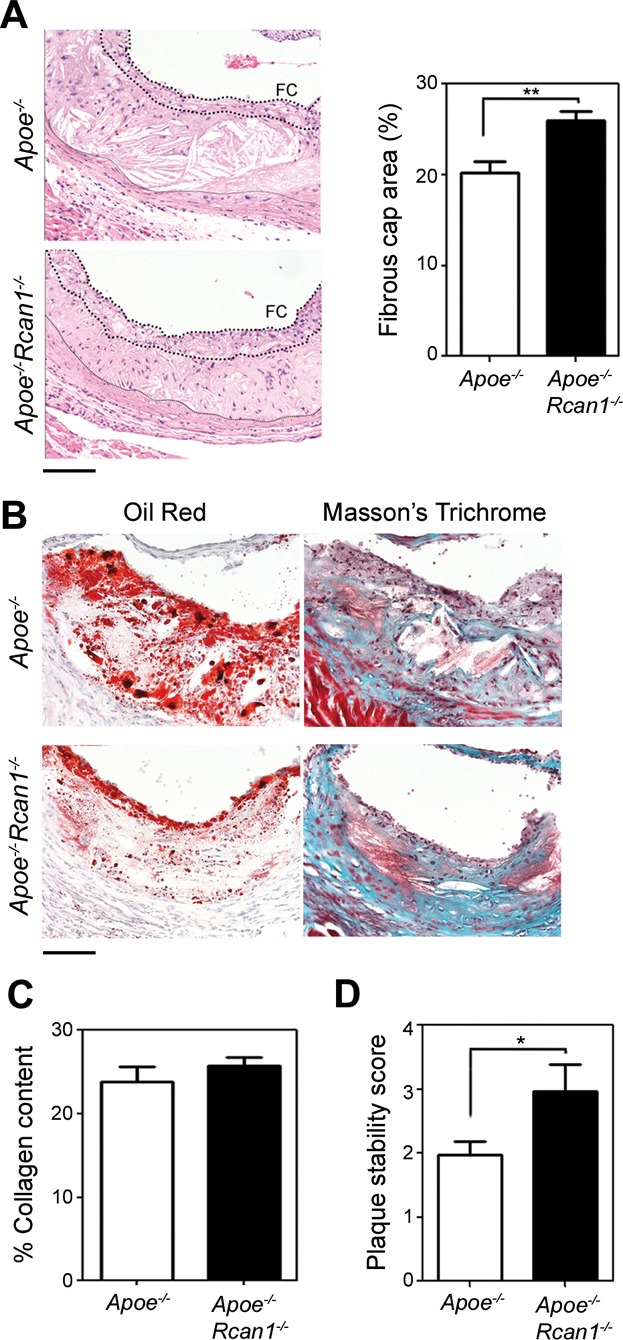
While Rcan1 targeting generates less advanced atherosclerotic plaques, it might as well potentially decrease their stability. Although the Apoe−/− mouse is not particularly prone to develop unstable plaques in the aorta (Getz & Reardon, 2012), it was formally possible that Rcan1 deficiency might increase plaque vulnerability. We therefore determined the effect of Rcan1 targeting on plaque stability. A key feature of unstable plaques is thinning of the fibrous cap, usually accompanied by a reduction in collagen content. The extent and thickness of the fibrous cap, relative to plaque size, were larger in Apoe−/−Rcan1−/− mice than in Apoe−/− mice (Fig 5A and Fig S4 of Supporting Information), whereas the collagen content of the fibrous cap was similar in both genotypes (Fig 5B–C). Moreover, lipid content, determined by Oil Red staining, was lower in plaques of Apoe−/−Rcan1−/− mice (Fig 5B) and these plaques had a higher stability score (Fig 5D). Expression of metalloproteases MMP2 and MMP9, another index of plaque instability, was almost identical in Apoe−/− and Apoe−/−Rcan1−/− plaques (Fig S5 of Supporting Information) and no plaques displayed evidence of haemorrhage (Fig S6 of Supporting Information). These data, together with the lower macrophage content of Rcan1-deficient plaques, thus indicate that Rcan1 targeting does not induce characteristics of unstable atherosclerotic plaques, and suggest instead that Rcan1 inactivation might increase plaque stability.
Figure 5.
Rcan1 deficiency does not decrease plaque stability
Representative H&E staining and quantification of fibrous cap area of lesions in the aortic sinus of Apoe−/− (n = 25 valves) and Apoe−/−Rcan1−/− mice (n = 45 valves) fed an HFD for 6 weeks. Data are shown relative to the lesion area (means ± SEM). Student's t-test, **p = 0.0012. Thick and thin dotted lines, respectively, delimit the fibrous cap (FC) and lesion.
Representative images of Oil Red and Masson's trichrome staining of cryocut cross-sections of the aortic arch lesions of Apoe−/− (n = 15) and Apoe−/−Rcan1−/− mice (n = 17) fed an HFD for 6 weeks. Bar, 50 µm.
Quantification of collagen content in the fibrous cap of these lesions (mean ± SEM).
Plaque stability score determined by collagen content divided by Oil Red staining area of the same lesions (mean ± SEM; Student t-test, *p = 0.048).
Rcan1 regulates CD36 expression and mediates foam-cell formation
Oil Red O staining of intracellular lipids indicated that while macrophages isolated from Apoe−/− and Apoe−/−Rcan1−/− mice engulfed unmodified LDL similarly (Fig 6A), fewer Apoe−/−Rcan1−/− macrophages engulfed oxLDL, and Apoe−/−Rcan1−/− macrophages also appeared to load less oxLDL per cell (Fig 6B). We measured intracellular oxLDL particles by Laurdan generalized polarization (Laurdan GP) (Sanchez et al, 2007; Sanchez et al, 2010), a confocal technique that measures water content inside lipid compartments. Intracellular oxLDLs are tightly packed and shift Laurdan GP towards red frequencies (Ferretti et al, 2002), whereas other lipid compartments, including intracellular membranes, contain more water and give a yellow or green signal (Sanchez et al, 2007; Sanchez et al, 2010). Macrophages exposed to oxLDL thus show its presence in the cytosol as red dots (Fig 6C). Quantification of the area occupied by oxLDL revealed that Apoe−/− macrophages took up ≈ 2.5 times more oxLDL than Apoe−/−Rcan1−/− cells (Fig 6C).
Figure 6.
Rcan1 mediates macrophage uptake of oxLDL
A,B. Apoe−/− and Apoe−/−Rcan1−/− peritoneal macrophages were exposed to (A) native LDL or (B) oxLDL for 24 h and then Oil Red O stained. Representative images and quantification are shown of stained cells in three independent experiments (mean ± SEM; Student's t-test, **p = 0.0018). Bars, 20 µm.
C. Apoe−/− and Apoe−/−Rcan1−/− peritoneal macrophages cultured in medium alone (control) or containing oxLDL were stained with Laurdan. Representative Laurdan GP images are shown with quantification of the area covered by oxLDL (red dots) (n = 20; mean ± SEM; Student's t-test, ***p = 0.0001). Black bar, 10 µm.
D. Quantitative PCR analysis of CD36 expression in the aortic arch of atherosclerotic Apoe−/− (n = 12) and Apoe−/−Rcan1−/− mice (n = 13) pooled from three independent experiments. mRNA amounts were normalized to m36B4 expression (means ± SEM). Student's t-test *p = 0.039.
E. Representative flow cytometry fluorescence histograms of CD36-stained macrophages of the indicated genotypes and quantification of the median fluorescence levels (n = 3; mean ± SEM; Student's t-test, *p = 0.036).
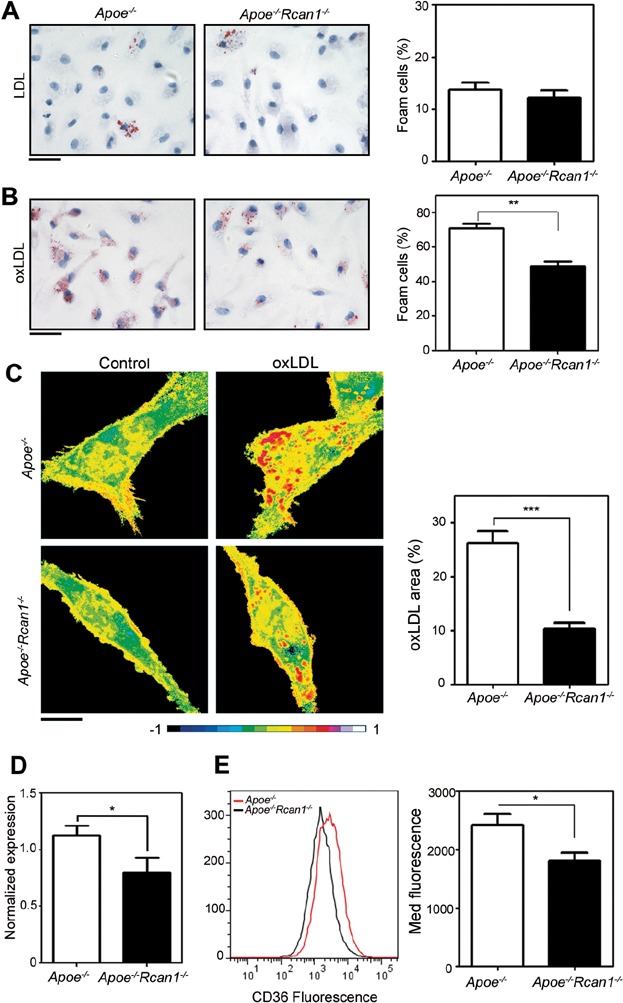
OxLDL uptake and foam-cell formation are mediated by the class A and B scavenger receptors SR-A and CD36 (Febbraio et al, 2000; Suzuki et al, 1997). Real-time PCR analysis detected significantly higher levels of CD36 in the aortic arches of Apoe−/− mice compared with Apoe−/−Rcan1−/− mice (Fig 6D), whereas expression of SR-A was similar in the two genotypes (Fig S7A of Supporting Information). Accordingly, flow cytometry analysis revealed that CD36 levels, but not those of SR-A, were significantly downregulated in Apoe−/−Rcan1−/− macrophages (Fig 6E and Fig S7B of Supporting Information). Lentiviral re-expression of Rcan1-1 and Rcan1-4 in Apoe−/−Rcan1−/− peritoneal macrophages (Fig 7A) increased cell surface expression of CD36 (Fig 7B) and concomitantly increased the numbers of oxLDL particles taken up by Apoe−/−Rcan1−/− peritoneal macrophages (Fig 7C–D). These results thus support that Rcan1 contributes to foam-cell formation by regulating CD36 expression.
Figure 7.
Rcan1 re-expression in Apoe−/−Rcan1−/− macrophages increases CD36 expression and the uptake of oxLDL Apoe−/−Rcan1−/− peritoneal macrophages were transduced with lentiviruses encoding GFP or Rcan1-1-IRES-GFP plus Rcan1-4-IRES-GFP.
A. Representative immunoblot analysis of Rcan1 expression in these cells.
B. Quantification of the normalized median fluorescence levels of CD36 determined by flow cytometry in the GFP+ population (n = 4; mean ± SEM; Student's t-test *p = 0.02).
C,D. (C) Representative Laurdan GP images and (D) quantification of the area covered by oxLDL (red dots) in GFP-F4/80 double-positive cells after culture in the presence of oxLDL (n = 3; mean ± SEM; Student's t-test, *p = 0.01). Scale bar, 10 µm.

Lipid accumulation by lesional macrophages can also reflect altered cholesterol efflux. To investigate the contribution of Rcan1 to cholesterol efflux, foam-cell formation was induced in Apoe−/− and Apoe−/−Rcan1−/− peritoneal macrophages by incubating them with particles of acetylated LDL (acLDL), a non-atherosclerotic modified form of LDL, in the presence of 3H-cholesterol. Apoe−/−Rcan1−/− macrophages accumulated less 3H-cholesterol (Fig S8A of Supporting Information). Addition of HDL to the foam-cell cultures promoted cholesterol efflux, and this effect was modestly stronger in Rcan1-deficient cells (Fig S8B of Supporting Information). Consistent with this effect, ABC transporter expression was slightly higher in Apoe−/−Rcan1−/− peritoneal macrophages and in atherosclerotic lesions in the aortic arch of these animals (Fig S8C–D of Supporting Information).
Rcan1 regulates oxLDL-mediated inhibition of macrophage migration
The presence of macrophages in atheroma plaques depends not only on their recruitment, but also on their capacity to exit the plaque, a process strongly impaired by oxLDL (Angeli et al, 2004; Randolph, 2008). Since Rcan1 can either promote or repress cell migration (Espinosa et al, 2009; Iizuka et al, 2004; Minami et al, 2004), we first determined whether Rcan1 was required for chemotactic macrophage migration. Apoe−/− and Apoe−/−Rcan1−/− macrophages migrated similarly in Boyden Transwell chambers in response to MCP-1 or a combination of MCP-1 and fetal bovine serum (FBS) (Fig S9 of Supporting Information). We next investigated whether Rcan1 participated in oxLDL-elicited inhibition of macrophage chemotaxis. While exposure of Apoe−/− macrophages to oxLDL sharply reduced their migration towards the chemotactic stimulus, Rcan1-deficient macrophages were not affected (Fig 8A–B). Accordingly, in wound-healing assays oxLDL inhibited random migration of MCP1-treated Apoe−/− macrophages, but not that of Apoe−/−Rcan1−/− cells, as revealed by larger numbers of Apoe−/−Rcan1−/− cells invading the denuded area (Fig S10 of Supporting Information). In the Transwell assays neither LDL nor acLDL significantly altered migration of macrophages of either genotype towards the chemoattractant (Fig 8A–B). These data thus suggest that Rcan1 does not directly regulate macrophage migration, but is a central mediator of oxLDL-elicited inhibition of their egress from atherosclerotic plaques.
Figure 8.
Macrophage migration is inhibited by oxLDL in an Rcan1-dependent manner
A. Representative images of Hoechst-stained Apoe−/− and Apoe−/−Rcan1−/− macrophages, treated as indicated, on the lower filter surface of chemotaxis chambers (Bar, 100 µm).
B. Quantification of migrated macrophages in 10 fields per condition. Data are shown relative to non-treated cells (mean ± SEM, n = 4; one-way ANOVA, **p = 0.002, ***p < 0.00001).
C,D. Apoe−/− and Apoe−/−Rcan1−/− macrophages were plated on serum-coated coverslips, incubated with oxLDL at 37°C and then photographed at the indicated times. (C) Representative photomicroscopy images are shown (Bar, 20 µm) and (D) quantification of the spreading of these cells (mean ± SEM, n = 3; two-way ANOVA, *p = 0.019, **p = 0.002, ##p = 0.003).
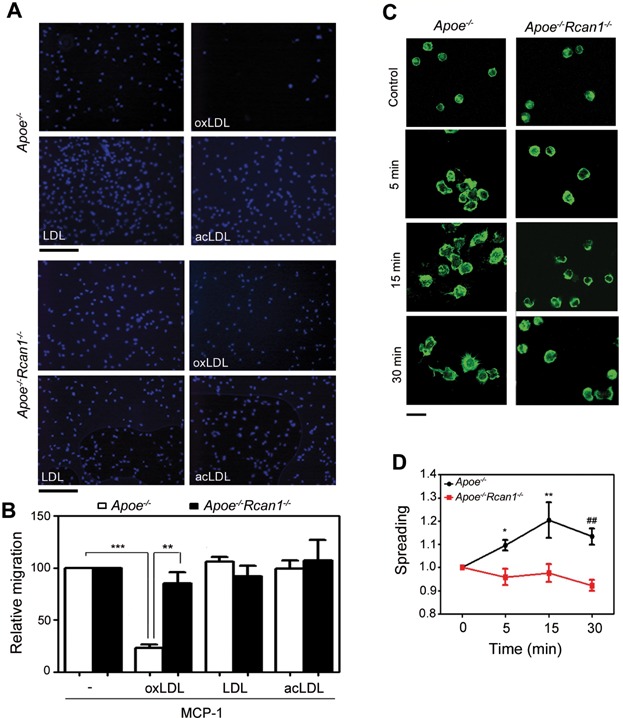
Cell migration is a complex process that involves cellular spreading and cycles of formation and disruption of focal adhesion contacts (Stossel, 1994). To characterize the mechanisms by which Rcan1 mediates oxLDL-induced inhibition of macrophage chemotaxis, we measured its role in oxLDL-induced cell spreading. Spreading of Apoe−/− macrophages, but not that of Apoe−/−Rcan1−/− cells, was readily induced within 30 min of exposure to oxLDL (Fig 8C). Automated quantification of macrophage spreading revealed significant differences between Apoe−/− and Apoe−/−Rcan1−/− macrophages from early time points (Fig 8D).
Rcan1−/− macrophages express anti-inflammatory phenotype markers
Although several types of macrophages have been detected in atherosclerotic lesions, macrophages with a classic M1-like pro-inflammatory phenotype appear to be the most abundant (Waldo et al, 2008). Given that oxLDL increases the expression of M1 markers (Chase et al, 2002) and Apoe−/−Rcan1−/− macrophages ingest oxLDL less efficiently than Apoe−/− macrophages, we postulated that atherosclerotic lesions of Apoe−/−Rcan1−/− mice might contain macrophages with a phenotype different from those of Apoe−/− mice. To test this, we analysed the expression of the anti-inflammatory phenotype markers Mrc1 and IL-10 in aortic cusps of Apoe−/− and Apoe−/−Rcan1−/− mice. Expression of both markers was higher in lesions from Apoe−/−Rcan1−/− mice (Fig 9A–B), and IL-10 levels were also higher in the aortic arch of atherosclerotic Apoe−/−Rcan1−/− mice (Fig S11 of Supporting Information). These data suggest that lesions in Apoe−/−Rcan1−/− mice are enriched in macrophages with a rather anti-inflammatory phenotype.
Figure 9.
Increased expression of alternative macrophage polarization markers in Apoe−/−Rcan1−/− lesions and isolated macrophages
A,B. Representative immunofluorescence staining of (A) IL-10 and (B) Mrc1 in aortic sinus lesions of Apoe−/− and Apoe−/−Rcan1−/− mice fed an HFD. Bars, 20 µm.
C. Quantitative PCR analysis of IL-10, Mrc1, Mcp-1 and iNOS mRNA expression in Apoe−/− and Apoe−/−Rcan1−/− peritoneal macrophages. mRNA amounts were normalized to m36B4 expression (mean ± SEM; n = 3). Student's t-test, **p = 0.0078, ##p = 0.0058, ***p = 0.0007, ###p = 0.0009.

We next investigated the possible participation of Rcan1 in macrophage polarization. Expression of IL-10 and Mrc1 was significantly higher in Apoe−/−Rcan1−/− macrophages compared with Apoe−/− cells (Fig 9C), whereas Apoe−/−Rcan1−/− macrophages expressed lower levels of Mcp-1 levels (Fig 9C). Further consistent with an anti-inflammatory phenotype, Apoe−/−Rcan1−/− macrophages showed higher expression of Arg1 (Fig S12A of Supporting Information), modestly lower antigen presentation (Fig S12B of Supporting Information) and higher phagocytic activity (Fig S12C of Supporting Information). Apoe−/−Rcan1−/− cells also expressed higher levels of iNos, a marker of pro-inflammatory M1 macrophages (Fig 9C), but iNos expression in atherosclerotic lesions of the aortic arch was almost identical in Apoe−/− and Apoe−/−Rcan1−/− mice (Fig S13 of Supporting Information). It thus seems that Rcan1 ablation might contribute to iNos induction by thioglycolate ex vivo, but not to its induction by proatherogenic stimuli in vivo.
Transplantation of Rcan1−/− BM cells confers resistance to atherosclerosis
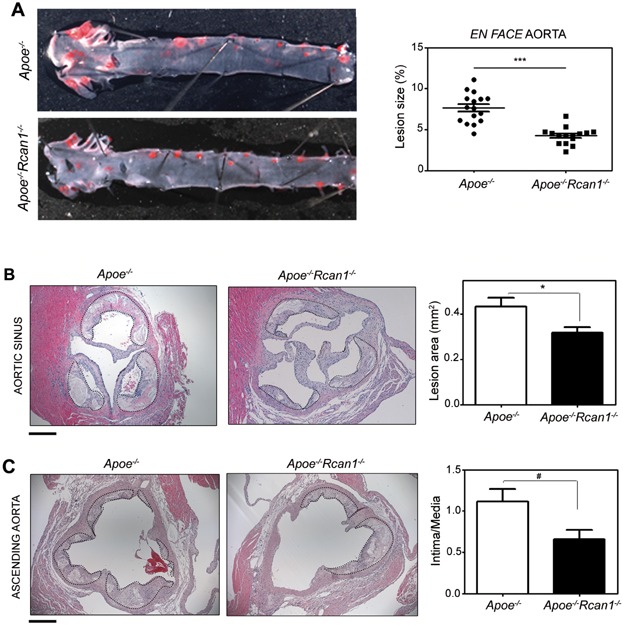
To test the possible promotion of atherosclerosis by macrophage-expressed Rcan1, we reconstituted the BM of 2-month old lethally irradiated Apoe−/− mice with BM-derived cells from Apoe−/−Rcan1−/− or Apoe−/− mice, thereby generating Apoe−/− mice with either Apoe−/−Rcan1−/− macrophages (Rcan1−/− → Apoe−/−) or Apoe−/−Rcan1+/+ macrophages (Rcan1+/+ → Apoe−/−). Since RCAN1 has been reported to induce GSK3β expression (Ermak et al, 2006) and this protein modulates haematopoietic progenitor cell function (Lapid et al, 2013), it is formally possible that Rcan1 ablation influences the nature of BM reconstitution. However, chimeric Rcan1−/− → Apoe−/− mice showed no significant reduction in Gsk3β expression (Fig S14A of Supporting Information), and blood cell populations of reconstituted Rcan1+/+ → Apoe−/− and Rcan1−/− → Apoe−/− mice were indistinguishable (Fig S14B–D of Supporting Information).
After reconstitution for 4 week, mice were fed an HFD for 6 weeks. Chimeric Rcan1−/− → Apoe−/− and Rcan1+/+ → Apoe−/− mice showed no significant differences in the body weight gain or in the serum concentrations of triglyceride, total cholesterol, HDL or LDL (Fig S15 of Supporting Information). En face analysis of Oil Red O aorta staining revealed markedly smaller lesion area in mice lacking expression of Rcan1 in BM-derived cells (Fig 10A). Cross-sectional analysis by H&E staining indicated that lesion size in the aortic sinus and the intima/media ratio in the ascending aorta were significantly smaller in Rcan1−/− → Apoe−/− mice than in Rcan1+/+ → Apoe−/− mice (Fig 10B–C). These data demonstrate that Rcan1 expression in the haematopoietic cell compartment plays a major role in atherosclerosis progression.
Figure 10.
Atherosclerosis requires Rcan1 expression in BM-derived cells
Representative Oil red O staining and quantification of the lesion area in the aortas of Apoe−/− mice transplanted with BM from Apoe−/− (n = 16) or Apoe−/−Rcan1−/− (n = 15) mice after 6 weeks on an HFD. Each data point denotes an individual mouse, and the horizontal bars denote the mean (long bar) and the SEM.
Representative H&E images and lesion area quantification in aortic sinus sections of these mice. Quantification is shown as mean ± SEM (n = 13). Bar, 200 µm.
Representative H&E images and quantification of lesion measured as intima/media ratio. Data are means ± SEM (n = 12). Bar, 200 µm. Student's t-test, *p = 0.02, #p = 0.024, ***p = 0.0001.
DISCUSSION
Atherosclerosis is a complex disease involving lipid accumulation and the central participation of endothelial cells, VSMCs and monocyte-derived macrophages. Our results demonstrate that RCAN1 is induced in human and mouse atherosclerosis and strongly suggest that Rcan1 promotes disease progression. In addition, we have identified several mechanisms underlying Rcan1-dependent atherosclerosis development. In particular, we have shown that Rcan1 mediates CD36 expression, foam-cell formation and oxLDL-elicited inhibition of macrophage migration. Moreover, Rcan1 ablation promotes expression of IL-10 and other anti-inflammatory markers, including Mrc1, in macrophages and lesions.
Rcan1 plays a critical role in vascular wall remodelling associated with aneurysm and neointima formation after angioplasty; and notably, BM transplantation experiments showed that haematopoietic-cell expression of Rcan1 is not required for aneurysm (Esteban et al, 2011). In contrast, our current findings show that Rcan1 expression in BM-cells is critical for atherogenesis. Our results do not however exclude a proatherogenic role of Rcan1 in VSMCs or endothelial cells. The eventual production of tissue-specific Rcan1-targeted mice will help to resolve this question. The different requirements for BM-cell expression of Rcan1 in atherosclerosis and aneurysm might be attributable to the pivotal role of Rcan1 in foam-cell formation, a central feature of atherosclerosis but not of aneurysm.
Although human and mouse atherosclerosis differ in several aspects, critical features are shared, and our results indicate that one such feature is RCAN1 induction. Moreover, we demonstrate that oxLDL, one of main stimuli of atherosclerosis, strongly induces Rcan1-4 not only in macrophages and VSMCs, but also in endothelial cells. Although most leukocytes in human and mouse atherosclerotic lesions are macrophages, plaques also contain lymphocytes, neutrophils and mast cells (Woollard & Geissmann, 2010). Whether Rcan1 is also induced in these cells remains to be determined.
Whereas RCAN1-1 is usually constitutive, RCAN1-4 expression is induced by several stimuli. Our data suggest that both RCAN1 isoforms might be upregulated in atherosclerotic lesions. However, only Rcan1-4 appears to be upregulated by atherogenic oxLDL in macrophages, endothelial cells and VSMCs. The presence of RCAN1-1 in lesions might indicate that in vivo this isoform is induced together with RCAN1-4 in macrophages, endothelial cells or VSMCs. Alternatively, the high RCAN1-1 levels in lesions might reflect recruitment of other cell populations with constitutively high RCAN1-1 expression.
Our findings provide in vivo and in vitro evidence that Rcan1 is pro-atherogenic and that its genetic ablation deactivates several important pathological mechanisms. Rcan1 deficiency inhibits macrophage engulfment of oxLDL and hence foam-cell formation, and Rcan1−/− macrophages are resistent to oxLDL-mediated inhibition of migration towards chemotactic stimuli, a feature related to foam-cell egress from the plaque in vivo. Finally, in the absence of Rcan1, macrophages appear to acquire features of a predominantly anti-inflammatory phenotype. Consistent with this view, Rcan1 inactivation in the Apoe-deficient mouse atherosclerosis model attenuates atherosclerotic lesion burden in terms both of lesion area and severity. While most lesions of Apoe−/− mice were advanced (grade IV), Apoe−/−Rcan1−/− mice exhibited a greater number of early lesions (grade I) without showing any evidence of increased plaque instability.
The lower Oil Red O surface staining and the lower content of extracellular lipids in atherosclerotic lesions of Apoe−/−Rcan1−/− mice are consistent with the relatively low uptake of oxLDL by Apoe−/−Rcan1−/− macrophages. Interestingly, these macrophages have a similar capacity to engulf native LDL. Given that endocytic uptake of native LDL and oxLDL occurs via different receptors (Greaves & Gordon, 2009), our data suggest that Rcan1 promotes the expression or activity of receptors specific for chemically modified forms of LDL. This conclusion is supported by our findings that CD36, a scavenger receptor involved in the uptake of modified forms of LDL and foam-cell formation (Febbraio et al, 2000), is downregulated in Rcan1 deficient macrophages, while re-expression of Rcan1-1 and Rcan1-4 in these cells increased CD36 expression and foam-cell formation. Cholesterol efflux was weakly increased in Apoe−/−Rcan1−/− macrophages and the levels of the cellular transporters involved in this process were not significantly increased in these cells or in atherosclerotic lesions. Thus, the contribution of cholesterol efflux to Rcan1-mediated regulation of foam-cell formation remains uncertain.
The inflammatory nature of atherosclerotic disease is widely accepted. During the resolution of inflammation, macrophages egress from the inflamed site after engulfing pathogens, toxins or apoptotic cells. However, cholesterol-engorged macrophages fail to egress after clearing lipids and hence fail to resolve the inflammatory process (Angeli et al, 2004). Egress of macrophages from plaques is actively inhibited during hypercholesterolemia and this inhibition has been attributed, at least in part, to oxLDL loading into macrophages (Park et al, 2009). Our data suggest that Rcan1 might facilitate the trapping of lipid-laden macrophages in atherosclerotic lesions by mediating oxLDL uptake, macrophage spreading and inhibition of their egress. Our data thus suggest that Rcan1 might participate in a positive feedback loop in which Rcan1-mediated accumulation of oxLDL particles inhibits foam-cell egress from the plaque, resulting in increased exposure of the trapped foam cells to oxLDL and increased engulfment of these particles.
Most lesional macrophages in advanced plaques display a proinflammatory M1 phenotype, whereas alternatively activated M2 macrophages are more abundant in early lesions (Khallou-Laschet et al, 2010; Waldo et al, 2008). An additional macrophage subtype, whose gene expression program differs from that of M1 and M2 cells and which shows enhanced phagocytotic and chemotactic capacities, has also been reported in advanced plaques (Kadl et al, 2010), and analysis of CD163 and CD206 (MRC1) distribution in human carotid plaque macrophages also suggested that there may be at least three macrophage phenotypes present in human plaques (Bouhlel et al, 2007). Our results show elevated expression of IL-10 and Mrc1 in lesions of Rcan1 deficient mice and no increase of iNos. Since IL-10 is a powerful anti-inflammatory cytokine and its genetic ablation in mice promotes atherosclerotic lesion formation (Mallat et al, 1999), these results suggest that Rcan1 deficiency promotes the presence of macrophages with anti-inflammatory properties in the atheroma plaque, a conclusion consistent with the retarded progression of lesions in Apoe−/−Rcan1−/− mice. Whether these IL-10-producing cells are M2-like macrophages or an additional macrophage type remains to be determined. Supporting an M2-like identity, Apoe−/−Rcan1−/− macrophages expressed high levels of Mrc1 and arginase-1 and low levels of Mcp1, and showed elevated phagocytic activity and a low antigen presentation capacity. However, M2 macrophages appear to take up oxLDL more efficiently than M1 cells (van Tits et al, 2011), whereas Apoe−/−Rcan1−/− macrophages engulfed less oxLDL than Apoe−/− cells. Together, our data suggest that Rcan1 deficiency promotes a macrophage polarization distinct from M1 and M2.
The precise molecular mechanism involved in Rcan1 regulation of oxLDL uptake, foam-cell trapping and macrophage polarization has yet to be identified. Rcan1 was first identified as a negative regulator of CN activity (Rothermel et al, 2000). However, additional studies indicated that Rcan1 can also activate CN (Kingsbury & Cunningham, 2000; Vega et al, 2003) and our previous studies showed that Rcan1 neither activates nor inhibits CN in aortic tissues or primary VSMC cultures (Esteban et al, 2011). Our present results show that the absence of Rcan1 has no significant effect on CN activity in unstimulated cells or lipopolysaccharide-stimulated macrophages (Fig S16 of Supporting Information). Rcan1 thus appears to promote atherosclerosis without interfering with CN activity, perhaps through its interaction with other Rcan1-interacting proteins implicated in gene activation, such as Raf-1 (Cho et al, 2005), 14-3-3 (Abbasi et al, 2006) and NF-kB-inducing kinase (Lee et al, 2008).
Our results show that RCAN1 is induced in human and mouse atherosclerotic vessels, and suggest that Rcan1 regulates CD36 expression and thus contributes to the uptake of pathogenic forms of LDL cholesterol by macrophages in the plaque (Fig 11). The lower lipid uptake by Rcan1-deficient cells would attenuate their differentiation into pro-inflammatory macrophages and their accumulation in the plaque (Fig 11). Instead, expression by Rcan1-deficient macrophages of the anti-inflammatory cytokine IL-10 might inhibit further expansion of the plaque (Fig 11). These findings identify RCAN1 as an important regulator of atherosclerosis and strongly suggest that therapies aimed at inhibiting RCAN1 expression or function might significantly reduce atherosclerosis burden.
Figure 11.
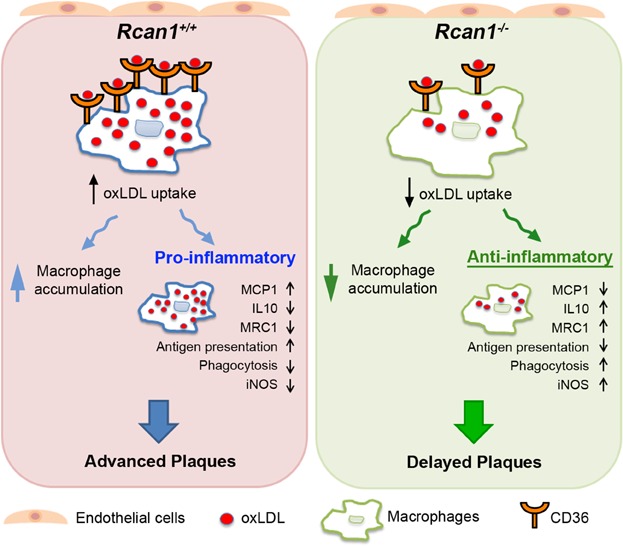
Model depicting the contribution of Rcan1 to plaque formation Rcan1 mediates expression of the oxLDL receptor CD36 in macrophages, thus regulating oxLDL uptake. Low uptake of oxLDL by Rcan1-deficient macrophages results in macrophage polarization to a phenotype distinct from M1 or M2. These cells display numerous anti-inflammatory features, including high levels of IL10 and MRC1, low levels of MCP1, pronounced phagocytosis activity and weak antigen presentation (Ag pres). However, these macrophages also show increased iNOS expression. Rcan1 ablation also decreases macrophage accumulation, likely by preventing their entrapment and/or the MCP1-mediated recruitment of additional macrophages. The lower accumulation of macrophages in the plaque and their conversion into predominantly anti-inflammatory cells might account for the delayed plaque progression.
MATERIALS AND METHODS
An expanded Materials and Methods Section is available in the Supporting Information.
Animal procedures
Animal studies were in accordance with the guidelines of the EU on animal care and approved by the institutional ethics committee. Double-knockout Apoe−/−Rcan1−/− mice were previously described (Esteban et al, 2011). Rcan1 targeting constitutively ablates the expression of both Rcan1-1 and Rcan1-4 in every cell (Porta et al, 2007). To accelerate atherosclerosis, 3-month-old mice were fed an HFD (10.8% total fat, 0.75% cholesterol) for 6 weeks. BM transplantation was performed as previously described (Esteban et al, 2011). After 4 weeks on chow diet, transplanted mice were placed on the HFD for 6 weeks. Plasma concentrations of free cholesterol, total cholesterol, LDL-cholesterol, HDL-cholesterol and triglycerides were measured enzymatically.
Human samples
Human coronary arteries and internal mammary arteries were collected from patients undergoing heart transplant and coronary artery bypass-graft surgery, respectively, at the Hospital de la Santa Creu i Sant Pau (Barcelona, Spain). The studies were approved by the Ethics Committee and were conducted in accordance with the Helsinki Declaration.
Histological analysis
Cross-sections (5-µm) of paraffin-embedded or cryopreserved samples of mouse hearts and aortae were immunostained or evaluated by conventional Masson's Trichrome, H&E or Oil Red staining. Cross-sections were stained with antibodies and reagents specific for Rcan1, SMA, Mac3, Mrc1, IL-10, Ter-119 or endothelial cells and processed for either immunohistochemistry or immunofluorescence using standard procedures.
Cell procedures
VSMCs were extracted from abdominal and thoracic aortas. Mouse lung endothelial cells were obtained from lung by selection with magnetic beads. Mouse peritoneal macrophages were collected by peritoneal lavage. Before stimulation, cells were rendered quiescent by culture in DMEM without FBS.
Western blot analysis
Human specimens and mouse aorta samples for western blotting were snap-frozen in liquid nitrogen and stored at −80°C. Protein extracts were obtained by tissue lysis in ice-cold lysis buffer, separated under reducing conditions on SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Proteins were detected with anti-Rcan1, anti-Gsk3-β, anti-α-actin, anti-alpha-tubulin, anti-PSF primary antibodies and HRP-conjugated secondary antibodies. Immunocomplexes were detected by chemiluminescence.
Atherosclerotic lesion analysis
Hearts from euthanized mice were perfused through the left ventricle with PBS. After fixing in 4% paraformaldehyde overnight at 4°, the aortas were thoroughly cleaned to remove all adventitial fat and connective tissue. Aortas were whole-mount stained with 0.2% Oil Red O in 80% methanol, opened longitudinally and pinned to black wax to expose the entire luminal surface. Images were acquired and the area of atherosclerotic plaques was measured using ImageJ software.
Migration assays
Migration of peritoneal macrophages was measured in a modified Boyden chamber using Transwell inserts with a 5 µm-pore membrane. Cells (1–2 × 105 per well) in AlphaMEM supplemented with 0.1% BSA were loaded into the migration chamber with 50 µg/ml of lipoprotein (LDL, oxLDL or acLDL). The number of migrated cells was counted on fluorescence microscopy photographs of 10 randomly selected fields.
For wound healing assays, a single scrape wound was made on peritoneal macrophage monolayers. After washing with PBS, cells were incubated with 2% FBS plus 100 ng/ml MCP-1 with or without 50 µg/ml oxLDL. Macrophage motility was monitored by time-lapse videomicroscopy. The number of cells that migrated into the denuded area was counted using ImageJ.
Spreading assays
Peritoneal macrophages were placed on serum-coated slides and allowed to attach. After stimulation with 50 µg/ml oxLDL, macrophages were fixed in 4% paraformaldheyde and stained with fluorescein-conjugated phalloidin. Cell perimeter and surface area were measured to determine cell spreading according to the formula
Foam-cell formation
Peritoneal macrophages were plated on coverslips, incubated with 50 µg/ml LDL or oxLDL for 24 h, fixed in 4% paraformaldehyde and stained with Oil red O and counterstained with haematoxylin.
Laurdan GP microscopy
Laurdan GP microscopy has been described previously (Bagatolli et al, 2003; Sanchez et al, 2007). Peritoneal macrophages were cultured in the presence of 1 µM Laurdan in serum-free medium. Laurdan fluorescence was excited with a mode-locked titanium-sapphire laser set at 780 nm and its emission collected at 445–465 nm and 474–514 nm. GP images were obtained with an ALBA microscope equipped with a 63× water objective and analysed using Image-J software.
Quantitative PCR
Real-time quantitative RT-PCR was performed using a Prime Time qPCR assay specific for human GAPDH and TaqMan Gene Expression assays specific for human RCAN1-1, RCAN1-4 and mouse Rcan1 and Hprt1. SYBR Green was used for RT-PCR detection of mouse CD-36, SR-A, ABCA, ABCG, IL-10, Mrc1, Arg1, Mcp-1, iNos and m36B4. Calculations were made from measurements of 3 replicates of each sample. The amount of target mRNA in samples was estimated by the 2CT relative quantification method using GAPDH, m36B4 or Hprt1 for normalization.
Statistical analysis
All values are expressed as means ± SEM. Differences were evaluated using one-way or two-way analysis of variance (ANOVA) and Bonferroni's post hoc test (experiments with ≥3 groups) or the Student's t-test, as appropriate for the data. Statistical significance was assigned at p < 0.05.
The paper explained
PROBLEM:
Atherosclerosis, the underlying cause of myocardial infarction, stroke and peripheral vascular disease, is the major cause of morbidity and mortality in the developed world. It is a complex inflammatory disease characterized by accumulation of oxidized LDL (oxLDL) that triggers activation of the vascular endothelium and migration of monocytes into the lesion. These monocytes take up oxLDL and become lipid-laden foam cells that recruit smooth muscle cells and additional leukocytes. As RCAN1 is a signalling intermediate implicated in cell migration, we hypothesized that RCAN1 might contribute to atherosclerosis development.
RESULTS:
We show that RCAN1 is induced in atherosclerotic human vessels and in the atherosclerotic arteries of a mouse model of atherosclerosis. Rcan1 is expressed in vivo in lesional macrophages, endothelial cells and vascular smooth muscle cells and was induced in vitro by treatment of these cells with oxLDLs. Rcan1 genetic deletion reduced the extent and severity of atherosclerosis in mice, and this effect was mechanistically linked to diminished expression of the oxLDL receptor in macrophages (CD36), decreased oxLDL uptake, resistance to oxLDL-mediated inhibition of macrophage migration and a shift of macrophage polarization towards an anti-inflammatory phenotype. Importantly, transplantation of Rcan1-deficient bone-marrow-derived cells greatly inhibited atherosclerosis.
IMPACT:Our data define a major role for haematopoietic Rcan1 in atherosclerosis and suggest that Rcan1 might facilitate the trapping of lipid-laden macrophages in atherosclerotic lesions by upregulating CD36-mediated oxLDL uptake and thereby trapping proinflammatory macrophages in atherosclerotic lesions. Our findings strongly suggest that future therapies aimed at inhibiting RCAN1 expression or function might significantly reduce atherosclerosis burden.
Author contributions
The study was conceived by JMR and MRC. NMB, VE, JMR and MRC designed the study and analysed the data. NMB and VE performed most of the experiments with contributions from SV, AE, KU and AA. CR and JMG assessed RCAN1 expression in human samples. SAS performed Laurdan GP microscopy and analysed the data obtained. VA, TO and TM provided experimental support and ideas for the project. JMR and MRC wrote the manuscript with contributions of NMB, SAS and JMG. All authors read and approved the manuscript.
Acknowledgments
We thank Dr. S. de la Luna for kindly providing pHA-CALP1L and pHA-CALP1S plasmids, Dr. S. Bartlett for English editing and R. Alberca for technical support. The Spanish Ministry of Economy and Competitiveness (Ministerio de Economía y Competitividad) supports MRC, JMR and JM-G with grants SAF2010-15126, SAF2009-10708 and SAF2012-40127, respectively; MRC is also supported by the Spanish Council for Scientific Research (CSIC); JMR is also supported by Fundación La Marató TV3 (080731), and Fundación Genoma España (GENOMA). The Red de Investigacion Cardiovascular (RIC) of the Spanish Ministry of Health (Ministerio de Sanidad) supports the research of JMR, VA and JM-G with grants RD12/0042/0022, RD12/0042/0028 and RD12/0042/0053, respectively. The Centro Nacional de Investigaciones Cardiovasculares (CNIC) is supported by the Spanish Ministry of Economy and Competitiveness and the Pro-CNIC Foundation. TM was supported in part by the Leading-edge Research Promotion Fund (LS038, TM). VE was an investigator of the Sara Borrell Program (CD06/00232), and NM-B holds an FPU fellowship (FPU2008-1500).
Supporting Information is available at EMBO Molecular Medicine Online.
The authors declare that they have no conflict of interest.
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
References
- Abbasi S, Lee JD, Su B, Chen X, Alcon JL, Yang J, Kellems RE, Xia Y. Protein kinase-mediated regulation of calcineurin through the phosphorylation of modulatory calcineurin-interacting protein 1. J Biol Chem. 2006;281:7717–7726. doi: 10.1074/jbc.M510775200. [DOI] [PubMed] [Google Scholar]
- Angeli V, Llodra J, Rong JX, Satoh K, Ishii S, Shimizu T, Fisher EA, Randolph GJ. Dyslipidemia associated with atherosclerotic disease systemically alters dendritic cell mobilization. Immunity. 2004;21:561–574. doi: 10.1016/j.immuni.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Baek KH, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, Lensch MW, Park IH, Yoon SS, Minami T, et al. Down's syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature. 2009;459:1126–1130. doi: 10.1038/nature08062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagatolli LA, Sanchez SA, Hazlett T, Gratton E. Giant vesicles, Laurdan, and two-photon fluorescence microscopy: Evidence of lipid lateral separation in bilayers. Methods Enzymol. 2003;360:481–500. doi: 10.1016/s0076-6879(03)60124-2. [DOI] [PubMed] [Google Scholar]
- Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S, Zawadzki C, Jude B, Torpier G, Marx N, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Cano E, Canellada A, Minami T, Iglesias T, Redondo JM. Depolarization of neural cells induces transcription of the Down syndrome critical region 1 isoform 4 via a calcineurin/nuclear factor of activated T cells-dependent pathway. J Biol Chem. 2005;280:29435–29443. doi: 10.1074/jbc.M506205200. [DOI] [PubMed] [Google Scholar]
- Chase AJ, Bond M, Crook MF, Newby AC. Role of nuclear factor-kappa B activation in metalloproteinase-1, -3, and -9 secretion by human macrophages in vitro and rabbit foam cells produced in vivo. Arterioscler Thromb Vasc Biol. 2002;22:765–771. doi: 10.1161/01.atv.0000015078.09208.92. [DOI] [PubMed] [Google Scholar]
- Cho YJ, Abe M, Kim SY, Sato Y. Raf-1 is a binding partner of DSCR1. Arch Biochem Biophys. 2005;439:121–128. doi: 10.1016/j.abb.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Crawford DR, Leahy KP, Abramova N, Lan L, Wang Y, Davies KJ. Hamster adapt78 mRNA is a Down syndrome critical region homologue that is inducible by oxidative stress. Arch Biochem Biophys. 1997;342:6–12. doi: 10.1006/abbi.1997.0109. [DOI] [PubMed] [Google Scholar]
- Davies KJ, Ermak G, Rothermel BA, Pritchard M, Heitman J, Ahnn J, Henrique-Silva F, Crawford D, Canaider S, Strippoli P, et al. Renaming the DSCR1/Adapt78 gene family as RCAN: Regulators of calcineurin. FASEB J. 2007;21:3023–3028. doi: 10.1096/fj.06-7246com. [DOI] [PubMed] [Google Scholar]
- Ermak G, Harris CD, Battocchio D, Davies KJ. RCAN1 (DSCR1 or Adapt78) stimulates expression of GSK-3beta. FEBS J. 2006;273:2100–2109. doi: 10.1111/j.1742-4658.2006.05217.x. [DOI] [PubMed] [Google Scholar]
- Ermak G, Harris CD, Davies KJ. The DSCR1 (Adapt78) isoform 1 protein calcipressin 1 inhibits calcineurin and protects against acute calcium-mediated stress damage, including transient oxidative stress. FASEB J. 2002;16:814–824. doi: 10.1096/fj.01-0846com. [DOI] [PubMed] [Google Scholar]
- Espinosa AV, Shinohara M, Porchia LM, Chung YJ, McCarty S, Saji M, Ringel MD. Regulator of calcineurin 1 modulates cancer cell migration in vitro. Clin Exp Metastasis. 2009;26:517–526. doi: 10.1007/s10585-009-9251-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban V, Mendez-Barbero N, Jimenez-Borreguero LJ, Roque M, Novensa L, Garcia-Redondo AB, Salaices M, Vila L, Arbones ML, Campanero MR, et al. Regulator of calcineurin 1 mediates pathological vascular wall remodeling. J Exp Med. 2011;208:2125–2139. doi: 10.1084/jem.20110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, Sharma K, Silverstein RL. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105:1049–1056. doi: 10.1172/JCI9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti G, Rabini RA, Bacchetti T, Vignini A, Salvolini E, Ravaglia F, Curatola G, Mazzanti L. Glycated low density lipoproteins modify platelet properties: A compositional and functional study. J Clin Endocrinol Metab. 2002;87:2180–2184. doi: 10.1210/jcem.87.5.8466. [DOI] [PubMed] [Google Scholar]
- Fuentes JJ, Pritchard MA, Estivill X. Genomic organization, alternative splicing, and expression patterns of the DSCR1 (Down syndrome candidate region 1) gene. Genomics. 1997;44:358–361. doi: 10.1006/geno.1997.4866. [DOI] [PubMed] [Google Scholar]
- Getz GS, Reardon CA. Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:1104–1115. doi: 10.1161/ATVBAHA.111.237693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- Greaves DR, Gordon S. The macrophage scavenger receptor at 30 years of age: Current knowledge and future challenges. J Lipid Res. 2009;50:S282–286. doi: 10.1194/jlr.R800066-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12:204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- Harris CD, Ermak G, Davies KJ. Multiple roles of the DSCR1 (Adapt78 or RCAN1) gene and its protein product calcipressin 1 (or RCAN1) in disease. Cell Mol Life Sci. 2005;62:2477–2486. doi: 10.1007/s00018-005-5085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, Dey A, Sachan N, Wong H, Patterson RJ, Shelton JM, Richardson JA, Klann E, Rothermel BA. The Down syndrome critical region protein RCAN1 regulates long-term potentiation and memory via inhibition of phosphatase signaling. J Neurosci. 2007;27:13161–13172. doi: 10.1523/JNEUROSCI.3974-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka M, Abe M, Shiiba K, Sasaki I, Sato Y. Down syndrome candidate region 1,a downstream target of VEGF, participates in endothelial cell migration and angiogenesis. J Vasc Res. 2004;41:334–344. doi: 10.1159/000079832. [DOI] [PubMed] [Google Scholar]
- Kadl A, Meher AK, Sharma PR, Lee MY, Doran AC, Johnstone SR, Elliott MR, Gruber F, Han J, Chen W, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107:737–746. doi: 10.1161/CIRCRESAHA.109.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, Gaston AT, Clement M, Dussiot M, Levillain O, Graff-Dubois S, Nicoletti A, et al. Macrophage plasticity in experimental atherosclerosis. PLoS One. 2010;5:e8852. doi: 10.1371/journal.pone.0008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsbury TJ, Cunningham KW. A conserved family of calcineurin regulators. Genes Dev. 2000;14:1595–1604. [PMC free article] [PubMed] [Google Scholar]
- Lapid K, Itkin T, D'Uva G, Ovadya Y, Ludin A, Caglio G, Kalinkovich A, Golan K, Porat Z, Zollo M, et al. GSK3beta regulates physiological migration of stem/progenitor cells via cytoskeletal rearrangement. J Clin Invest. 2013;123:1705–1717. doi: 10.1172/JCI64149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EJ, Seo SR, Um JW, Park J, Oh Y, Chung KC. NF-kappaB-inducing kinase phosphorylates and blocks the degradation of Down syndrome candidate region 1. J Biol Chem. 2008;283:3392–3400. doi: 10.1074/jbc.M706707200. [DOI] [PubMed] [Google Scholar]
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- Mallat Z, Besnard S, Duriez M, Deleuze V, Emmanuel F, Bureau MF, Soubrier F, Esposito B, Duez H, Fievet C, et al. Protective role of interleukin-10 in atherosclerosis. Circ Res. 1999;85:e17–24. doi: 10.1161/01.res.85.8.e17. [DOI] [PubMed] [Google Scholar]
- Minami T, Horiuchi K, Miura M, Abid MR, Takabe W, Noguchi N, Kohro T, Ge X, Aburatani H, Hamakubo T, et al. Vascular endothelial growth factor- and thrombin-induced termination factor, Down syndrome critical region-1, attenuates endothelial cell proliferation and angiogenesis. J Biol Chem. 2004;279:50537–50554. doi: 10.1074/jbc.M406454200. [DOI] [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- Park YM, Febbraio M, Silverstein RL. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J Clin Invest. 2009;119:136–145. doi: 10.1172/JCI35535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plump AS, Smith JD, Hayek T, Aalto-Setala K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- Porta S, Serra SA, Huch M, Valverde MA, Llorens F, Estivill X, Arbones ML, Marti E. RCAN1 (DSCR1) increases neuronal susceptibility to oxidative stress: A potential pathogenic process in neurodegeneration. Hum Mol Genet. 2007;16:1039–1050. doi: 10.1093/hmg/ddm049. [DOI] [PubMed] [Google Scholar]
- Randolph GJ. Emigration of monocyte-derived cells to lymph nodes during resolution of inflammation and its failure in atherosclerosis. Curr Opin Lipidol. 2008;19:462–468. doi: 10.1097/MOL.0b013e32830d5f09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothermel B, Vega RB, Yang J, Wu H, Bassel-Duby R, Williams RS. A protein encoded within the Down syndrome critical region is enriched in striated muscles and inhibits calcineurin signaling. J Biol Chem. 2000;275:8719–8725. doi: 10.1074/jbc.275.12.8719. [DOI] [PubMed] [Google Scholar]
- Ryeom S, Baek KH, Rioth MJ, Lynch RC, Zaslavsky A, Birsner A, Yoon SS, McKeon F. Targeted deletion of the calcineurin inhibitor DSCR1 suppresses tumor growth. Cancer Cell. 2008;13:420–431. doi: 10.1016/j.ccr.2008.02.018. [DOI] [PubMed] [Google Scholar]
- Sanchez SA, Tricerri MA, Gunther G, Gratton E. Laurdan generalized polarization: From cuvette to microscope. In: Mendez-Vilas A, Diaz J, editors. Modern Research and Educational Topics in Microscopy: Applications in Physical/Chemical Sciences. Badajoz, Spain: Formatex; 2007. pp. 1007–1014. [Google Scholar]
- Sanchez SA, Tricerri MA, Ossato G, Gratton E. Lipid packing determines protein-membrane interactions: Challenges for apolipoprotein A-I and high density lipoproteins. Biochim Biophys Acta. 2010;1798:1399–1408. doi: 10.1016/j.bbamem.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W, Jr, Rosenfeld ME, Schaffer SA, Schwartz CJ, Wagner WD, Wissler RW. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb. 1994;14:840–856. doi: 10.1161/01.atv.14.5.840. [DOI] [PubMed] [Google Scholar]
- Stossel TP. The machinery of cell crawling. Sci Am. 1994;271:54–55. doi: 10.1038/scientificamerican0994-54. , 58-63. [DOI] [PubMed] [Google Scholar]
- Strippoli P, Petrini M, Lenzi L, Carinci P, Zannotti M. The murine DSCR1-like (Down syndrome candidate region 1) gene family: Conserved synteny with the human orthologous genes. Gene. 2000;257:223–232. doi: 10.1016/s0378-1119(00)00407-8. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Kurihara Y, Takeya M, Kamada N, Kataoka M, Jishage K, Ueda O, Sakaguchi H, Higashi T, Suzuki T, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386:292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- van Tits LJ, Stienstra R, van Lent PL, Netea MG, Joosten LA, Stalenhoef AF. Oxidized LDL enhances pro-inflammatory responses of alternatively activated M2 macrophages: A crucial role for Kruppel-like factor 2. Atherosclerosis. 2011;214:345–349. doi: 10.1016/j.atherosclerosis.2010.11.018. [DOI] [PubMed] [Google Scholar]
- Vega RB, Rothermel BA, Weinheimer CJ, Kovacs A, Naseem RH, Bassel-Duby R, Williams RS, Olson EN. Dual roles of modulatory calcineurin-interacting protein 1 in cardiac hypertrophy. Proc Natl Acad Sci USA. 2003;100:669–674. doi: 10.1073/pnas.0237225100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldo SW, Li Y, Buono C, Zhao B, Billings EM, Chang J, Kruth HS. Heterogeneity of human macrophages in culture and in atherosclerotic plaques. Am J Pathol. 2008;172:1112–1126. doi: 10.2353/ajpath.2008.070513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, De Keulenaer GW, Weinberg EO, Muangman S, Gualberto A, Landschulz KT, Turi TG, Thompson JF, Lee RT. Direct biomechanical induction of endogenous calcineurin inhibitor Down Syndrome Critical Region-1 in cardiac myocytes. Am J Physiol Heart Circ Physiol. 2002;283:H533–539. doi: 10.1152/ajpheart.00002.2002. [DOI] [PubMed] [Google Scholar]
- Woollard KJ, Geissmann F. Monocytes in atherosclerosis: Subsets and functions. Nat Rev Cardiol. 2010;7:77–86. doi: 10.1038/nrcardio.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Rothermel B, Vega RB, Frey N, McKinsey TA, Olson EN, Bassel-Duby R, Williams RS. Independent signals control expression of the calcineurin inhibitory proteins MCIP1 and MCIP2 in striated muscles. Circ Res. 2000;87:E61–68. doi: 10.1161/01.res.87.12.e61. [DOI] [PubMed] [Google Scholar]
- Yang YJ, Chen W, Edgar A, Li B, Molkentin JD, Berman JN, Lin TJ. Rcan1 negatively regulates Fc epsilonRI-mediated signaling and mast cell function. J Exp Med. 2009;206:195–207. doi: 10.1084/jem.20081140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.