

Abstract
Myotonic dystrophy (DM) is a multi-systemic disease that impacts cardiac and skeletal muscle as well as the central nervous system (CNS). DM is unusual because it is an RNA-mediated disorder due to the expression of toxic microsatellite expansion RNAs that alter the activities of RNA processing factors, including the muscleblind-like (MBNL) proteins. While these mutant RNAs inhibit MBNL1 splicing activity in heart and skeletal muscles, Mbnl1 knockout mice fail to recapitulate the full-range of DM symptoms in these tissues. Here, we generate mouse Mbnl compound knockouts to test the hypothesis that Mbnl2 functionally compensates for Mbnl1 loss. Although Mbnl1−/−; Mbnl2−/− double knockouts (DKOs) are embryonic lethal, Mbnl1−/−; Mbnl2+/− mice are viable but develop cardinal features of DM muscle disease including reduced lifespan, heart conduction block, severe myotonia and progressive skeletal muscle weakness. Mbnl2 protein levels are elevated in Mbnl1−/− knockouts where Mbnl2 targets Mbnl1-regulated exons. These findings support the hypothesis that compound loss of MBNL function is a critical event in DM pathogenesis and provide novel mouse models to investigate additional pathways disrupted in this RNA-mediated disease.
Keywords: Mbnl1, Mbnl2, muscleblind-like, myotonic dystrophy, RNA-mediated disease
INTRODUCTION
Myotonic dystrophy (DM) is an autosomal dominant neuromuscular disease characterized by a multi-systemic phenotype, including skeletal muscle myotonia and progressive weakness/wasting, cardiac arrhythmias, ocular ‘dust-like’ cataracts, insulin insensitivity, hypogammaglobulinemia, hypersomnia and cerebral atrophy (Ranum & Cooper, 2006; Udd & Krahe, 2012). DM is also one of the most variable human hereditary diseases with the age-of-onset ranging from neonates to older adults with nearly all organ systems affected to different degrees.
DM is caused by expansions of either CTG trinucleotide repeats in the 3′ untranslated region (3′ UTR) of the DMPK gene on chromosome 19 (DM type 1, DM1) or CCTG repeats in the first intron of the CNBP gene on chromosome 3 (DM type 2, DM2) (Ranum & Cooper, 2006; Udd & Krahe, 2012). These non-coding C(C)TG expansion, or C(C)TGexp, mutations generate pathogenic C(C)UGexp RNAs that are toxic because they perturb the normal cellular activities of several RNA binding factors, including the muscleblind-like (MBNL), CUGBP1 and ETR3-like (CELF), hnRNP H and STAU1 proteins (Ho et al, 2004; Kanadia et al, 2003; Miller et al, 2000; Paul et al, 2006; Philips et al, 1998; Ravel-Chapuis et al, 2012; Timchenko et al, 1996). The MBNL proteins appear to play a particularly prominent role in DM pathogenesis since each of the three MBNL genes (MBNL1, MBNL2, MBNL3) produce multiple isoforms with different, but relatively high, binding affinities for C(C)UGexp RNAs in vitro (Kino et al, 2004; Warf & Berglund, 2007; Yuan et al, 2007) and MBNL proteins are sequestered by mutant DM transcripts in nuclear RNA foci in vivo (Jiang et al, 2004; Mankodi et al, 2001; Miller et al, 2000). Mbnl1ΔE3/ΔE3, Mbnl2ΔE2/ΔE2 and Mbnl3ΔE2/ΔE2 isoform knockout (KO) mice recapitulate DM-relevant phenotypes with skeletal muscle myotonia, myopathy and particulate cataracts in Mbnl1ΔE3/ΔE3 KOs, increased REM sleep propensity and learning/memory deficits in Mbnl2ΔE2/ΔE2 KOs and an age-associated decline in skeletal muscle regeneration in Mbnl3ΔE2/ΔE2 KO mice (Charizanis et al, 2012; Kanadia et al, 2003; Poulos et al, 2013). Mbnl1ΔE3/ΔE3 and Mbnl2ΔE2/ΔE2 isoform knockout alleles do not express detectable Mbnl1 or Mbnl2 protein, respectively, so for simplicity they will be subsequently referred to as Mbnl1 and Mbnl2 KOs or Mbnl1−/− and Mbnl2−/−. Additionally, Mbnl1 overexpression reverses myotonia in transgenic HSALR mice, which express a CUGexp RNA in skeletal muscle, and transgenic MBNL1 overexpression mice are viable with normal muscle structure and function (Chamberlain & Ranum, 2012; Kanadia et al, 2006).
Sequestration and functional inhibition of all three MBNL paralogues depends on C(C)UGexp RNA repeat length and expression level, which are highly variable between tissues. Therefore, we propose that the full range of DM phenotypes cannot be adequately modelled in Mbnl single KO mice. Indeed, some prominent DM-relevant manifestations, including skeletal muscle weakness/wasting and cardiac arrhythmias, are not routinely observed in Mbnl1 and Mbnl2 single KOs. To test this proposal, we investigated the effects of combined Mbnl1 and Mbnl2 deficiency on cardiac and skeletal muscle structure and function. While Mbnl1; Mbnl2 homozygous double KOs (DKOs) were embryonic lethal, Mbnl1−/−; Mbnl2+/− mice survived until adulthood but developed skeletal and cardiac muscle defects, as well as alternative splicing changes, characteristic of DM. More severe manifestations of DM skeletal muscle disease were observed in Mbnl1−/−; Mbnl2c/c; Myo-Cre+/− KO mice, with complete absence of Mbnl2 protein expression restricted to muscle fibres. High-throughput sequencing combined with crosslinking/immunoprecipitation (HITS-CLIP), a technique that identifies binding sites for RNA-binding proteins in vivo, demonstrated that Mbnl2 upregulation following the loss of Mbnl1 expression resulted in an increase in Mbnl2 binding to Mbnl1 muscle RNA targets and partial correction of DM-relevant mis-splicing. Our results demonstrate that the major symptoms of cardiac and skeletal muscle disease in DM can be recapitulated in Mbnl compound knockout models and that sequestration of both Mbnl1 and Mbnl2 by C(C)UGexp RNAs is an important pathogenic feature of DM.
RESULTS
Reduced lifespan in Mbnl1; Mbnl2 compound KO mice
Mbnl1 KO adults develop skeletal muscle myotonia and DM-associated muscle pathology, including centralized myonuclei and split fibres, but do not display either the hypotonia or adult-onset muscle wasting features of congenital or adult-onset DM, respectively (Kanadia et al, 2003). In contrast, Mbnl2 KO mice display characteristic manifestations of DM central nervous system (CNS) disease, such as REM sleep propensity disturbance and memory deficits, but do not exhibit muscle abnormalities (Charizanis et al, 2012). Interestingly, we noted that Mbnl2 protein levels increased strikingly in Mbnl1 KOs, particularly in skeletal muscle (Fig 1A), in agreement with a recent study that reported Mbnl2 protein upregulation following shRNA-mediated knockdown of Mbnl1 in C2C12 myoblasts (Wang et al, 2012). Other factors implicated in DM pathogenesis (Mbnl3, Hnrnph1, Stau1) did not show similar increases in protein level (Supporting Information Fig S1A). The increase in Mbnl2 protein was likely due to a corresponding increase in Mbnl2 mRNA level (Fig 1B). To determine if combined loss of Mbnl1 and Mbnl2 would recapitulate neonatal hypotonia and/or progressive muscle wasting in adults, we attempted to generate Mbnl1; Mbnl2 DKO mice but loss of both proteins resulted in embryonic lethality (Supporting Information Fig S1B). In contrast, Mbnl1−/−; Mbnl2+/−, as well as Mbnl1+/−; Mbnl2−/−, KOs were viable although the latter line showed reduced embryonic viability so it was not characterized further. While Mbnl2 is low in WT adult muscle, the upregulated Mbnl2 protein in Mbnl1 KO muscle localized primarily to the nuclear compartment (Fig 1C). Elevated nuclear levels of Mbnl2 in the Mbnl1 KO correlated with increased inclusion of Mbnl2 exons 6 and 8 in skeletal muscle (Supporting Information Fig S2A, B) (Zhang et al, 2013). While the function of Mbnl2 exon 8 is unknown, Mbnl2 exon 6 (54 nt) is homologous to Mbnl1 exon 7 that may encode a nuclear localization sequence (Lin et al, 2006).
Figure 1.
Reduced viability of Mbnl1; Mbnl2 KO mice.
A. Immunoblot analysis of Mbnl1+/+ (WT), Mbnl1+/−, Mbnl1−/− and Mbnl1−/−; Mbnl2+/− KO quadriceps muscle (quad), heart and brain tissues.
B. Upregulation of Mbnl2 RNA in Mbnl1−/− KO quadriceps muscle (n = 3 per genotype, *p = 0.0008, Student's t-test).
C. Immunoblots showing Mbnl2 upregulation in myonuclei of Mbnl1−/− KO muscle. Nucleolin (Ncl) and lactate dehydrogenase A (Ldha) were included as nuclear/nucleolar and cytoplasmic markers, respectively.
D. Kaplan–Meier survival analysis of Mbnl1−/− KO and WT (red line) versus Mbnl1−/−; Mbnl2+/− (blue) KO mice (n = 10 per genotype).
E. Body weight analysis showed Mbnl1−/−; Mbnl2+/− KOs were ∼30% smaller than Mbnl1−/− single KOs between 8 and 12 weeks of age (n ≥ 7 except for Mbnl1−/−; Mbnl2+/− at 12 weeks of age n = 5, *p = 0.00132 (week 4), 0.00047 (week 5), 4.6E-05 (week 6), 0.00013 (week 7), 2.03E-06 (week 8), 2.89E-05 (week 9), 0.00014 (week 10), 0.0002 (week 11), 0.00013 (week 12), Student's t-test).
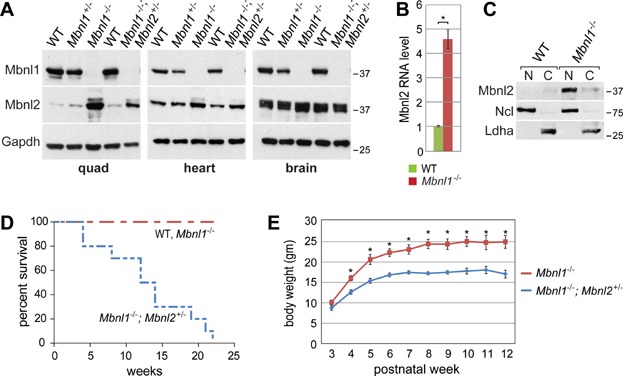
Despite the compensatory increase in Mbnl2 expression in skeletal muscle, lifespan was severely compromised in Mbnl1−/−; Mbnl2+/− KO animals with no mice surviving beyond 23 weeks of age (Fig 1D) and body weight was maintained at ∼30% less than Mbnl1−/− (Fig 1E). Since the MBNL loss-of-function model for DM proposes that both MBNL1 and MBNL2 are sequestered to varying levels depending on C(C)UGexp RNA length and expression level, Mbnl1−/−; Mbnl2+/− KO mice were further evaluated for DM-relevant skeletal muscle defects to test the hypothesis that loss of both Mbnl1 and Mbnl2 proteins would increase the severity of skeletal muscle deficits.
Progressive muscle weakness/wasting and NMJ defects in Mbnl1−/−; Mbnl2+/− KOs
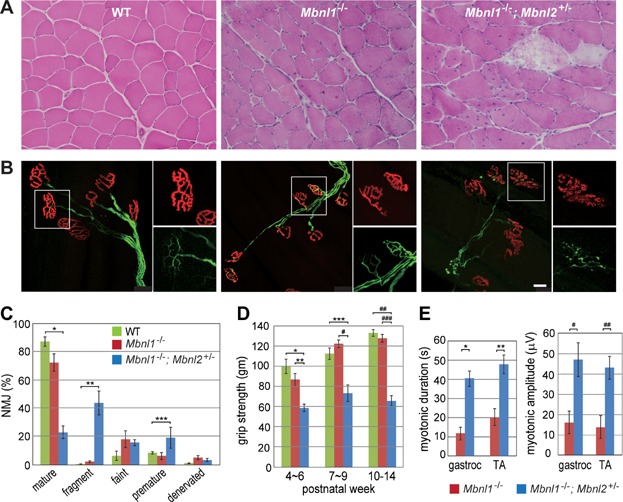
Histological hallmarks of DM patient muscle, which include centralized nuclei, fibre size heterogeneity and split fibres, have been previously noted in older (>6 months of age) Mbnl1 KO quadriceps muscle (Kanadia et al, 2003; Kanadia et al, 2006). While these muscle abnormalities are considerably less severe in younger Mbnl1 KO mice, more severe pathological changes (increased fibre size variation, atrophic and splitting fibres, increased numbers of central nuclei indicative of muscle degeneration/regeneration) were observed in Mbnl1−/−; Mbnl2+/− muscles at 10–16 weeks of age (Fig 2A). Additionally, young (8–10 weeks of age) Mbnl1−/−; Mbnl2+/− mice developed severe mobility problems and showed impaired rotarod performance (Supporting Information Fig S1C). Abnormalities in diaphragm neuromuscular junctions (NMJs), including changes in end-plate size and shape, have been documented in a poly(CUG) transgenic mouse model for DM1 (Panaite et al, 2008). Similar, but more profound, changes to NMJ structures were observed in Mbnl1−/−; Mbnl2+/− tibialis anterior (TA) muscles but not WT and Mbnl1−/− (Fig 2B). Significant loss of mature, and an increase in degenerated (fragmented endplates) and premature, NMJs was observed in Mbnl1−/−; Mbnl2+/− mice (Fig 2B, C). Moreover, grip strength analysis revealed muscle weakness as early as 4 weeks of age, which was half of the WT level by 10 weeks of age (Fig 2D). Myotonia, a characteristic feature of DM muscle, was also dramatically increased in Mbnl1−/−; Mbnl2+/− muscles compared to single Mbnl1 KO mice with both the amplitude (>4-fold) and duration of discharges increased 2.5 to ∼4-fold (Fig 2E and Supporting Information Fig S1D). Thus, Mbnl1−/−; Mbnl2+/− KOs recapitulate DM-relevant muscle weakness/wasting and myotonia.
Figure 2.
DM-associated muscle wasting/weakness in Mbnl1; Mbnl2 KOs.
A. H&E staining of WT, Mbnl1−/− and Mbnl1−/−; Mbnl2+/− TA muscles showing centralized myonuclei, split fibres and myofibre degeneration in young (10–16 weeks of age) Mbnl1−/−; Mbnl2+/− KOs.
B. Mbnl1 and Mbnl2 depletion leads to NMJ defects. WT, Mbnl1−/− and Mbnl1−/−; Mbnl2+/− TA muscles were dissected and labelled with α-bungarotoxin (red) and anti-neurofilament H (NFH-1, green). WT NMJs showed mature pretzel-shaped endplates, Mbnl1−/− KOs displayed similar features as WT although moderate changes in NMJ structure were noted whereas Mbnl1−/−; Mbnl2+/− KOs showed NMJ fragmentation and a decrease in mature NMJs (scale bar = 25 μm).
C. Percent of NMJs with various morphologies including mature, fragmented (fragment), faint, premature and denervated (n = 5 per genotype, *p = 1.9E-07, **p = 1.4E-06, ***p < 0.00827, Student's t-test).
D. Age-dependent increase in muscle weakness in Mbnl1−/−; Mbnl2+/− KOs. Grip strength test (forelimb) showed an increase in muscle weakness between 4 and 14 weeks of age (n ≥ 6 per genotype, *p = 0.0009, **p = 0.00198, ***p = 0.00161, #p = 2.7E-05, ##p = 4.6E-06, ###p = 1.2E-06, Student's t-test).
E. Myotonic duration and amplitude are significantly increased in Mbnl1−/−; Mbnl2+/− KO muscle (n = 4, *p = 0.00141, **p = 0.00554, #p = 0.02137, ##p = 0.0101, Student's t-test).
Combined depletion of Mbnl1 and Mbnl2 exacerbates DM-associated splicing defects
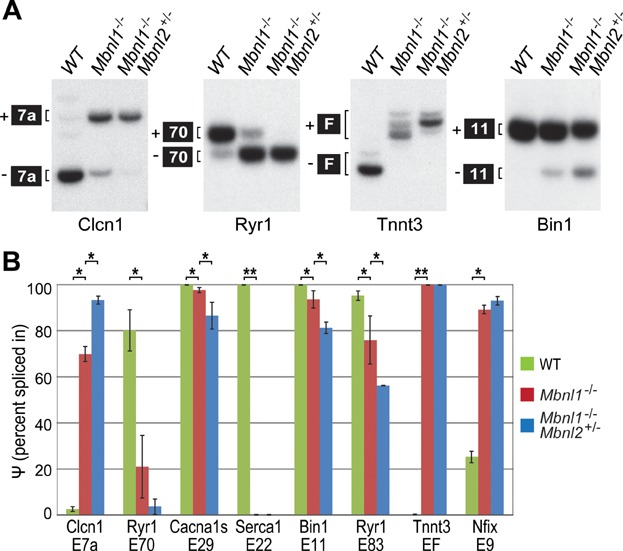
DM-associated myotonia is caused by mis-regulated splicing of Clcn1 pre-mRNA and depletion of full-length CLCN1 protein from the sarcolemma. Because myotonia was more severe following the depletion of Mbnl2 (Fig 2E), we first compared the Clcn1 splicing pattern in WT, Mbnl1 KO and Mbnl1−/−; Mbnl2+/− KO skeletal muscle. Depletion of Mbnl2 in the Mbnl1 KO background led to a nearly complete loss of the adult Clcn1 isoform (−exon 7a) (Fig 3A). Gene profiling studies have highlighted an important role for Mbnl proteins in calcium homeostasis and regulation (Osborne et al, 2009). Several gene transcripts important for calcium regulation, including Ryr1, Serca1 and Cacna1s, were more severely mis-spliced in Mbnl1−/−; Mbnl2+/− KO muscle compared to Mbnl1 KO alone and this degree of mis-splicing was similar to that observed previously in DM1 patient muscle (Kimura et al, 2005; Tang et al, 2012). In addition, Bin1, Tnnt3 and Nfix mis-splicing events, which occur in Mbnl1 KOs and human DM muscle (Du et al, 2010; Fugier et al, 2011; Kanadia et al, 2003), were also tested. Mbnl1−/−; Mbnl2+/− KO muscle showed either more pronounced splicing changes, or were equivalent to Mbnl1 KOs (e.g. Serca1, Tnnt3) (Fig 3B and Fig S1E). As reported previously, global effects on alternative splicing regulation are not observed in Mbnl1 KO mice (Du et al, 2010; Kanadia et al, 2003; Wang et al, 2012) and this was also observed for Mbnl1−/−; Mbnl2+/− KOs (Fig S1F). Collectively, these results demonstrated that combined depletion of Mbnl1 and Mbnl2 recapitulated muscle myotonia, weakness/wasting and the splicing patterns characteristic of severely affected DM1 skeletal muscle (Fugier et al, 2011; Wheeler & Thornton, 2007).
Figure 3.
DM-associated splicing deficits following Mbnl1 and Mbnl2 depletion.
A. RT-PCR analysis of WT, Mbnl1−/−, Mbnl1−/−; Mbnl2+/− splicing for several pre-mRNA targets that are mis-spliced in DM skeletal muscle. All targets showed enhancement of the fetal isoform splicing pattern in Mbnl1−/−; Mbnl2+/− KOs compared to WT and Mbnl1−/− single KOs. The apparent decrease in Clcn1 levels between WT and Mbnl1−/− is reproducible (Charizanis et al, 2012) and likely the result of differences in RNA stability. The multiple Tnnt3 +F and −F isoforms result from alternative splicing of exons 4–8 in the absence of BsrB1 digestion of the F exon (Kanadia et al, 2003).
B. Percent spliced-in (Ψ) values for eight target pre-mRNAs in WT, Mbnl1−/−, Mbnl1−/−; Mbnl2+/− quadriceps muscles (n = 3 per genotype, *p = 4.4E-06 [Clcn1, WT vs. Mbnl1−/−], 0.00039 [Clcn1, Mbnl1−/− vs. Mbnl1−/−; Mbnl2+/−], 0.00323 [Ryr1 E70, WT vs. Mbnl1−/−], 0.02506 [Cacna1s, WT vs. Mbnl1−/−], 0.0318 [Cacna1s, Mbnl1−/− vs. Mbnl1−/−; Mbnl2+/−], 0.04219 [Bin1, WT vs. Mbnl1−/−], 0.00891 [Bin1, Mbnl1−/− vs. Mbnl1−/−; Mbnl2+/−], 0.01377 [Ryr1 E83, WT vs. Mbnl1−/−], 0.03117 [Ryr1 E83, Mbnl1−/− vs. Mbnl1−/−; Mbnl2+/−], 3.7E-06 [Nfix, WT vs. Mbnl1−/−], Student's t-test; ** no Ψ variance was observed for Serca1 and Tnnt3 for WT, Mbnl1−/− and Mbnl1−/−; Mbnl2+/−).
Mbnl1−/−; Mbnl2+/− KOs model cardiac conduction defects in DM
Heart conduction abnormalities are common in DM1 patients and cardiac-related sudden death is a leading cause of mortality in this disease (Groh et al, 2008; McNally & Sparano, 2011). Endomyocardial biopsies as well as postmortem examination of DM1 hearts have noted interstitial fibrosis, fatty infiltration, cardiomyocyte hypertrophy and focal myocarditis (Pelargonio et al, 2002). Similarly, interstitial myocardial fibrosis was detected in all Mbnl1−/−; Mbnl2+/−, but not all Mbnl1−/−, KO mice (Supporting Information Fig S3A, B). Fibrosis was also observed in enlarged right atria.
DM1 patients with cardiac abnormalities generally show various extents of heart chamber dilation and hypertrophy (Hermans et al, 2012; McNally & Sparano, 2011; Pelargonio et al, 2002). MRI analysis of Mbnl1−/−; Mbnl2+/− KOs revealed atrial dilatation and left ventricular hypertrophy (Fig 4A and B). Although compromised myocardial contractility in Mbnl1−/−; Mbnl2+/− KOs was anticipated, the ejection fraction was not significantly different from WT (Supporting Information Fig S3C). Mbnl1−/−; Mbnl2+/− KO hearts were enlarged and heart/body weight (HW/BW) ratios increased ∼60% in Mbnl1−/−; Mbnl2+/− KOs versus WT (Fig 4B).
Figure 4.
Compound mouse knockout model for DM heart disease.
A. MRI cardiac images revealed right atrial (RA) dilatation and thickening of the posterior left ventricular wall in Mbnl1−/−; Mbnl2+/− KOs compared to WT mice while Mbnl1−/− mice displayed similar, but less severe, morphological changes.
B. End diastolic volume (EDV), right atrial (RA) area, the ratio of left ventricular wall volume (WV) to end diastolic volume (WV/EDV) and heart/body weight ratio are shown (n = 5 per genotype, *p = 0.0043, **p = 0.0139, ***p = 0.001, #p = 0.0006, ##p = 0.0075, ###p = 0.0042, ^p = 0.0054, ^^p = 0.0035, ^^^p = 3.9E-08, Student's t-test).
C. Electrocardiography (ECG) evidence for first-degree AV block. Increases in PR interval and QRS broadening were noted compared to WT mice.
D. Significant ECG changes in PR interval, QRS duration and QT interval in WT, Mbnl1−/− and Mbnl1−/−; Mbnl2+/− KO hearts are shown (n = 5 per genotype, *p = 0.0006, **p = 0.0064, ***p = 0.0001, #p = 0.0035, ##p = 0.0117, Student's t-test).
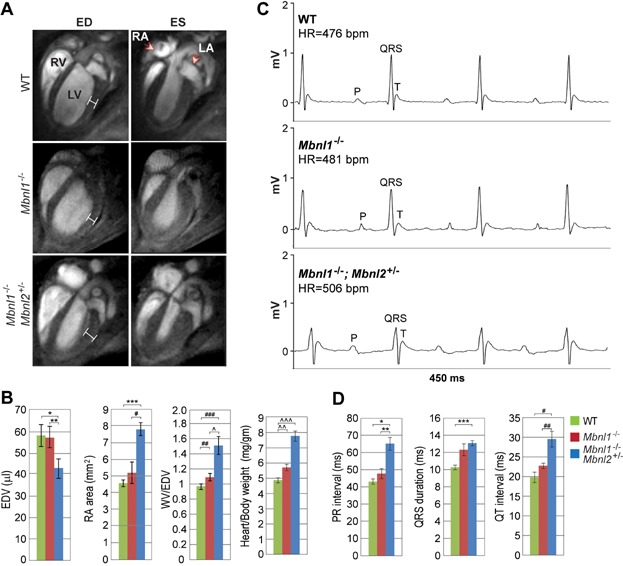
The majority of DM1 patients are abnormal as assessed by electrocardiography (ECG) with prolonged PR interval (20–40% of patients) and QRS widening (5–25%) (McNally & Sparano, 2011). Indeed, a large-scale clinical study revealed a direct correlation between CTG repeat length, arrhythmias, PR prolongation and QRS widening (Groh et al, 2002). ECG analysis of Mbnl1−/−; Mbnl2+/− KOs showed PR interval prolongation, or an ∼51% increase compared to WT, indicating a first degree AV block in these mutants (Fig 4C and D and Supporting Information Fig S3D). Both QRS duration and QT interval were also significantly increased.
Since depletion of Mbnl2 in the absence of Mbnl1 resulted in structural and functional alterations to the heart, DM-associated mis-splicing events for Mbnl1 heart splicing targets were evaluated by RT-PCR. For all targets examined, Mbnl1−/−; Mbnl2+/− KOs showed a dramatic shift towards fetal isoforms (Fig 5A and Supporting Information Fig S3E). For example, the fetal isoform of Tnnt2 includes exon 5 and splicing of this exon increased from ∼0% in WT to >70% in Mbnl1−/−; Mbnl2+/− KOs. Enhanced mis-splicing also occurred for Ryr2, which regulates calcium release from the sarcoplasmic reticulum and RYR2 mutations cause catecholaminergic polymorphic ventricular tachycardia (Venetucci et al, 2012), and Cacna1s, which was recently shown to be mis-spliced in cardiac and skeletal muscle leading to aberrant gating of this Ca(V)1.1 calcium channel (Tang et al, 2012). Both of these Mbnl1 splicing targets, as well as additional targets (Sorbs1, Tmem63b, Spag9, Mbnl1), showed augmentation of mis-splicing changes in Mbnl1−/−; Mbnl2+/− KOs compared to Mbnl1 KO alone with the single exception of Arhgef7 (Fig 5B).
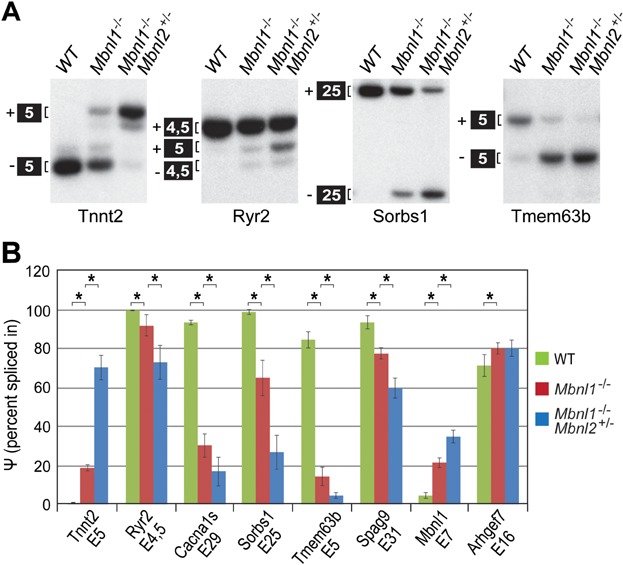
Figure 5.
Splicing alterations of cardiac RNA targets in Mbnl1−/−; Mbnl2+/− KOs.
A. Increased mis-splicing of the Mbnl1 pre-mRNA targets Tnnt2, Ryr2, Sorbs1 and Tmem63b occurs in Mbnl1−/−; Mbnl2+/− compound KOs compared to WT or Mbnl1 single KO mice.
B. Percent spliced-in (Ψ) values for eight target pre-mRNAs in WT, Mbnl1−/−, Mbnl1−/−; Mbnl2+/− hearts (n = 3 per genotype, *p = 5.3E-05 [Tnnt2, WT vs. Mbnl1−/−], 0.00023 [Tnnt2, Mbnl1−/− vs. Mbnl1−/−; Mbnl2+/−], 0.03988 [Ryr2, WT vs. Mbnl1−/−], 0.028 [Ryr2, Mbnl1−/− vs. Mbnl1−/−; Mbnl2+/−], 5.5E-05 [Cacna1s, WT vs. Mbnl1−/−], 0.02365 [Cacna1s, Mbnl1−/− vs. Mbnl1−/−; Mbnl2+/−], 0.0035 [Sorbs1, WT vs. Mbnl1−/−], 0.0074 [Sorbs1, Mbnl1−/− vs. Mbnl1−/−; Mbnl2+/−], 4.8E-05 [Tmem63b, WT vs. Mbnl1−/−], 0.0296 [Tmem63b, Mbnl1−/− vs. Mbnl1−/−; Mbnl2+/−], 0.0035 [Spag9, WT vs. Mbnl1−/−], 0.0076 [Spag9, Mbnl1−/− vs. Mbnl1−/−; Mbnl2+/−], 0.00076 [Mbnl1, WT vs. Mbnl1−/−], 0.00692 [Mbnl1, Mbnl1−/− vs. Mbnl1−/−; Mbnl2+/−], 0.048 [Arhgef7, WT vs. Mbnl1−/−], Student's t-test). Targets are implicated in calcium homeostasis and contractility (Tnnt2, Ryr2, Cacna1s), insulin (Sorbs1), VEGF (Tmem63b) and JNK (Spag9) signalling pathways.
Mbnl conditional compound loss of function model for DM
Although Mbnl1−/−; Mbnl2+/− KOs displayed decreased lifespan and DM-associated changes in skeletal and cardiac muscle structure and function, Mbnl2 protein levels remained elevated over WT (Fig 1A). This result suggested that Mbnl2 levels increased as a compensatory response to Mbnl1 loss to maintain at least some expression of adult-specific isoforms. However, MBNL1 and MBNL2 have also been recently implicated as negative regulators of pluripotency since they repress ES cell-specific alternative splicing of key regulatory factors, including FOXP1 (Han et al, 2013). Since Mbnl2 protein expression normally declines during muscle development (Charizanis et al, 2012), elevated Mbnl2 levels in Mbnl1−/− and Mbnl1−/−; Mbnl2+/− KOs may not be due to compensatory upregulation but instead might reflect an early developmental deficiency and a more immature tissue phenotype in these mutant adults. This finding prompted us to examine the effect of muscle-specific elimination of Mbnl2 protein expression in the Mbnl1 KO background later in development using a Cre-mediated conditional knockout strategy. Similar to Mbnl1−/−; Mbnl2+/− KOs, Mbnl1−/−; Mbnl2c/c; Myo-Cre+/− KO mice were small at birth but developed kyphosis and severe motor deficits beginning at 12 weeks of age. In agreement with the hypothesis that complete loss of Mbnl1 and Mbnl2 protein expression would exacerbate muscle pathology and splicing defects, Mbnl1−/−; Mbnl2c/c; Myo-Cre+/− KO mice showed a profound deficiency of adult skeletal muscle (Fig 6A), severe muscle pathology (small myofibers, fibre size heterogeneity, centralized nuclei in nearly all myofibers) (Fig 6B, C) and reduced grip strength (Fig 6D). Moreover, the alternative splicing of specific pre-mRNAs, including Bin1 (Fugier et al, 2011) and Cacna1s (Tang et al, 2012), previously associated with defects in skeletal muscle excitation-contraction coupling and muscle weakness/wasting shifted to more CDM and DM1-like splicing patterns with only ∼25% of Bin1 (+exon 11) and ∼10% Cacna1s (+exon 29) adult isoforms expressed in Mbnl1−/−; Mbnl2c/c; Myo-Cre+/− KO muscle (Fig 6E, F). These results provided additional support for the MBNL compound loss-of-function model for DM and suggested that Mbnl conditional KOs will be valuable tools to study DM disease pathways.
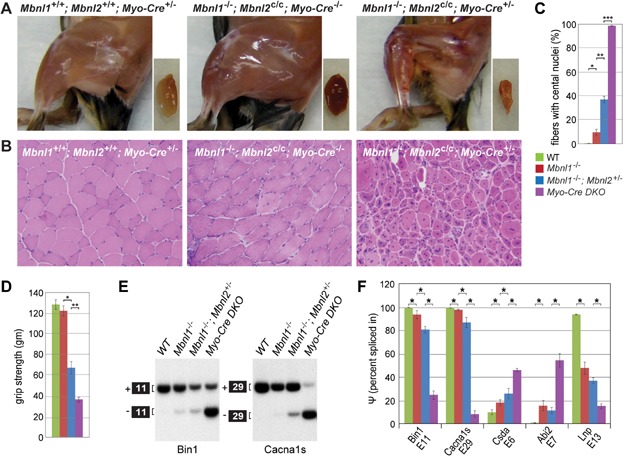
Figure 6.
Enhanced muscle pathology in Mbnl1; Mbnl2 muscle-specific KO mice.
A. Images of left hindlimb (left large panels), and dissected quadripceps muscles (small right panels), of WT (Mbnl1+/+; Mbnl2+/+; Myo-Cre+/−), Mbnl1 KO (Mbnl1−/−; Mbnl2c/c; Myo-Cre−/−) and Mbnl1−/−; Mbnl2c/c; Myo-Cre+/− DKOs.
B. H&E staining of transverse muscle sections in tibialis anterior (TA) muscles of WT (Mbnl1+/+; Mbnl2+/+; Myo-Cre+/−), Mbnl1 KO (Mbnl1−/−; Mbnl2c/c; Myo-Cre−/−) and Mbnl1−/−; Mbnl2c/c; Myo-Cre+/− DKOs.
C. Increased centralized myonuclei in Mbnl1−/−; Mbnl2c/c; Myo-Cre+/− DKOs (purple) versus WT (green, Mbnl1+/+; Mbnl2+/+; Myo-Cre+/−), Mbnl1−/− KO (red, Mbnl1−/−; Mbnl2c/c; Myo-Cre−/−) and Mbnl1−/−; Mbnl2+/− (blue) mice (n = 2 per genotype, *p = 0.00208, **p = 9.3E-06, ***p = 2.7E-10, Student's t-test).
D. Decreased grip strength in Mbnl1−/−; Mbnl2c/c; Myo-Cre+/− DKOs (purple) versus WT (green), Mbnl1 KO (red) and Mbnl1−/−; Mbnl2+/− (blue) mice (15 weeks of age, n = 11 for WT and Mbnl1−/−, 5 for Mbnl1−/−; Mbnl2+/− and 2 for Myo-Cre+/− DKO; *p = 7.7E-06, **p = 0.03337, Student's t-test).
E. RT-PCR analysis showing profound increases in Bin1 exon 11 and Cacna1s exon 29 skipping when the expression of both Mbnl1 and Mbnl2 is eliminated in Mbnl1−/−; Mbnl2c/c; Myo-Cre+/− DKO (Myo-cre DKO) skeletal muscle.
F. Percent spliced-in (Ψ) values for five target pre-mRNAs in WT (green), Mbnl1−/− (red), Mbnl1−/−; Mbnl2+/− (blue) and Mbnl1−/−; Mbnl2c/c; Myo-Cre+/− DKO (Myo-Cre DKO) quadriceps muscles (n ≥ 2, *p = 0.0069 [Bin1, WT vs. Mbnl1−/−], 0.00047 [Bin1, Mbnl1−/− vs. Mbnl1−/−; Mbnl2+/−], 8.7E-06 [Bin1, Mbnl1−/−; Mbnl2+/− vs. Myo-Cre+/− DKO], 0.00078 [Cacna1s, WT vs. Mbnl1−/−], 0.00158 [Cacna1s, Mbnl1−/− vs. Mbnl1−/−; Mbnl2+/−], 2.1E-06 [Cacna1s, Mbnl1−/−; Mbnl2+/− vs. Myo-Cre+/− DKO], 0.00055 [Csda, WT vs. Mbnl1−/−], 0.00809 [Csda, Mbnl1−/− vs. Mbnl1−/−; Mbnl2+/−], 0.00108 [Csda, Mbnl1−/−; Mbnl2+/− vs. Myo-Cre+/− DKO], 7.2E-05 [Abi2, WT vs. Mbnl1−/−], 0.00876 [Abi2, Mbnl1−/−; Mbnl2+/− vs. Myo-Cre+/− DKO], 0.006 [Lnp, WT vs. Mbnl1−/−], 0.00825 [Lnp, Mbnl1−/−; Mbnl2+/− vs. Myo-Cre+/− DKO], Student's t-test).
Splicing compensation between Mbnl proteins in vivo
Loss of Mbnl1 in Mbnl1 KO mice led to Mbnl2 protein upregulation in the nucleus while Mbnl1−/−; Mbnl2+/− KOs showed an enhancement of the skeletal and cardiac muscle phenotypes characteristic of DM. These results suggested that Mbnl2 functionally compensated for Mbnl1 loss by targeting Mbnl1-regulated splicing events. To test this possibility, Mbnl protein-RNA interactions were assessed in vivo using HITS-CLIP. Mbnl1+/+, Mbnl1−/− and Mbnl2−/− quadriceps muscles were dissected, immediately powdered in liquid nitrogen and irradiated with UV-light to form covalently crosslinked protein-RNA complexes. Following digestion with RNase A to generate short RNA tags and RNA end-labelling with 32P-ATP, Mbnl2-RNA complexes were immunopurified and resolved by SDS-PAGE (Fig 7A). At high RNase, Mbnl2 migrated at ∼40 kDa and this band was absent in Mbnl2 KO muscle but increased significantly in Mbnl1 KO quadriceps in agreement with the earlier immunoblot analysis (Fig 1A). Sequencing of RNA tags bound directly to Mbnl2 in muscle demonstrated a 2- 9-fold increase in Mbnl2 binding to previously documented Mbnl1 skeletal muscle RNA targets (Fig 7B) (Du et al, 2010). Of 667 significantly misspliced targets, 169 (∼25%) showed increased Mbnl2 binding in Mbnl1 KO mice as compared to WT. Gene ontology (GO) analysis of these targets revealed enrichment for phosphoproteins, alternative splicing factors, ion binding factors involved in calcium homeostasis, ion channel regulation and cytoskeletal organization. Mapping analysis of these targets showed elevated binding of Mbnl2 in the vicinity of alternatively spliced exons in Mbnl1 KO mice compared to WT (Fig 7C). Therefore, Mbnl2 compensates for the loss of Mbnl1 splicing activity by enhanced binding to Mbnl1-regulated splicing targets resulting in partial retention of the adult splicing pattern.
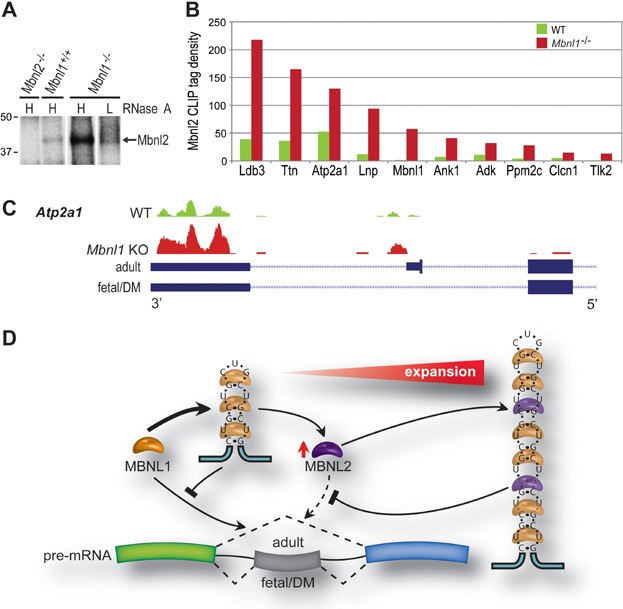
Figure 7.
HITS-CLIP analysis in Mbnl1 KO mice.
A. Autoradiography showing increased Mbnl2 protein crosslinking to P32-labelled RNA tags from Mbnl1 KO quadriceps muscle versus WT. RNA tags were generated using either high (H) or low (L) RNase A concentrations. Mbnl2 KO (Mbnl2−/−) muscle was included as a crosslinking control.
B. Increased Mbnl2 CLIP tag density following Mbnl1 loss in Mbnl1−/− KO muscle for 10 Mbnl1 splicing targets (n = 3 for each genotype).
C. UCSC Genome Browser snapshot of Atp2a1 with wiggle plots of Mbnl2 CLIP tags showing increased Mbnl2 binding on Mbnl1-regulated targets in Mbnl1−/− versus WT muscle.
D. MBNL protein titration model for DM. In muscle, MBNL1 is the primary MBNL protein that promotes adult splicing patterns (dotted lines depict adult skipping and fetal/DM inclusion of a fetal exon). Early in disease progression, relatively short C(C)UGexp RNAs sequester MBNL1 which, in turn, leads to MBNL2 upregulation (red arrow) and partial mitigation (dotted arrow) of the spliceopathy. However, as C(C)UGexp lengths increase, the splicing activities of both MBNL1 and MBNL2 are blocked (line with bar) leading to loss of adult splicing and more severe disease manifestations.
DISCUSSION
Defects in DNA replication, repair and recombination cause microsatellite expansions in DM1 and DM2 (Lopez Castel et al, 2010; Mirkin, 2007). Both the DMPK and CNBP genes are broadly expressed and microsatellite C(C)TG expansions are dynamic, tend to increase with age and show a high degree of somatic mosaicism (Higham et al, 2012; Morales et al, 2012). Because different tissues, and even individual cells within tissues, contain different repeat lengths and varying degrees of pathology, the development of animal models that faithfully recapitulate the consequences of these microsatellite expansions has been exceptionally challenging.
Transgenic poly(CUG) mouse models have provided key insights into the DM pathogenic mechanism (Sicot & Gomes-Pereira, 2013). For example, HSALR transgenic mice, which express an expanded (CUG)∼220 repeat only in skeletal muscles, develop multiple DM-like muscle abnormalities, including myotonia, fibre size variability, split fibres and centralized myonuclei although they fail to display DM-like severe muscle weakness and wasting perhaps due to the relatively short CUGexp length (Mankodi et al, 2000). Interestingly, a recent study implicated mis-splicing of exon 29 in CACNA1S, a calcium channel that regulates excitation-contraction coupling, in DM muscle weakness/wasting but poly(CUG) expression in HSALR muscle only moderately affects this splicing event (Tang et al, 2012). The multi-systemic features of DM are reproduced in the DM300 (CTG300-600) and DMSXL (CTG1000-1800) transgenic models although disease-relevant phenotypes and RNA mis-splicing are relatively mild (Sicot & Gomes-Pereira, 2013). In contrast, tissue-specific and Cre-inducible EpA960 transgenic mice, which express the DMPK 3′ UTR with an interrupted CTG960 repeat at relatively high levels, display severe cardiac arrhythmias, muscle wasting and splicing defects (Orengo et al, 2008; Wang et al, 2007).
Since transcription of mutant DMPK and CNBP genes results in the production of pathogenic C(C)UG repeat RNAs that dysregulate gene expression by altering the activities of several RNA-binding proteins, another approach has been to generate Celf1 gain-of-function (Koshelev et al, 2010; Ward et al, 2010) and Mbnl loss-of function models (Charizanis et al, 2012; Kanadia et al, 2003; Poulos et al, 2013). Because MBNL proteins are the only factors which have been shown to be sequestered by C(C)UG RNAs, the primary objective of this study was to determine if compound depletion of Mbnl function recapitulates disease-relevant skeletal and cardiac muscle manifestations. Previously, we demonstrated that Mbnl2 muscle expression decreases during postnatal development and Mbnl2 KO mice fail to exhibit the major muscle features of DM (Charizanis et al, 2012). Here, we show that loss of Mbnl1 gene expression leads to upregulation of Mbnl2, which subsequently targets those splicing events that are normally regulated primarily by Mbnl1. This is reminiscent of other alternative splicing factors, including the Rbfox family in which Rbfox2 expression is upregulated in the absence of Rbfox1 and vice versa (Gehman et al, 2012; Gehman et al, 2011). This compensatory Mbnl2 upregulation may be essential for Mbnl1 KO viability since Mbnl1; Mbnl2 DKOs were embryonic lethal. Additionally, Mbnl1−/−; Mbnl2+/− KOs displayed muscle weakness and progressive atrophy, two of the common symptoms observed in DM1 patients.
DM1 patients are prone to sudden death due to cardiac arrhythmias (Groh et al, 2008; Motta et al, 1979). Mbnl1−/−; Mbnl2+/− KOs also developed these cardiac symptoms and DM-associated mis-splicing events increased for a number of key target RNAs, including Tnnt2, Ryr2 and Cacna1s in the heart and Clcn1, Ryr1 and Tnnt3 in skeletal muscle. Interestingly, Mbnl1−/− KO mice showed a more modest increase of Mbnl2 in the brain (Fig 1A) and previous studies have reported that mis-splicing of several brain RNA targets in this knockout model was significantly less than observed in human DM1 brain (Suenaga et al, 2012) or in Mbnl2−/− KO mice (Charizanis et al, 2012). Although this study focused on the role of MBNL proteins in the regulation of alternative splicing in skeletal muscle and heart, the development of additional Mbnl1; Mbnl2 conditional KO models should allow assessment of the relative contributions of each Mbnl gene to CNS function. It is also interesting to note that similar to DMSXL homozygotes, which express large CUGexp RNAs, Mbnl2−/−, Mbnl1−/−; Mbnl2+/− and Mbnl1−/−; Mbnl2c/c; Myo-Cre+/− KO mice are small at birth whereas Mbnl1−/− KOs are not. Therefore, Mbnl2 expression in the embryonic muscle compartment appears to be a critical factor in the normal development of body size. Finally, MBNL1 has been implicated in altered miRNA biogenesis (Rau et al, 2011) and RNA localization (Wang et al, 2012) in DM so it will be important to examine the effects of compound Mbnl loss on these regulatory pathways.
MBNL1 is the most abundant MBNL protein in adult skeletal muscle and plays the predominant role in alternative splicing regulation in muscle while MBNL2 muscle levels decrease during postnatal development (Charizanis et al, 2012; Holt et al, 2009; Wang et al, 2012). These observations suggest a model in which sequestration of MBNL1 by relatively short C(C)UGexp RNAs in early-stage muscle disease results in MBNL2 upregulation and partial restoration of the normal adult splicing program (Fig 7D). However, progressive increases in C(C)UGexp lengths overwhelms this compensatory mechanism leading to impairment of MBNL-driven adult splicing patterns and more severe pathological consequences. In summary, our results provide important new experimental support for the MBNL loss-of-function model for DM and introduce new mouse models to examine the tissue-specific consequences of MBNL loss.
MATERIALS AND METHODS
Mouse Mbnl KO models
Detailed information on the generation of Mbnl1 and Mbnl2 KO mice has been published (Charizanis et al, 2012; Kanadia et al, 2003). Compound KOs were generated by crossing Mbnl1+/ΔE3 and Mbnl2+/ΔE2 heterozygotes (C57BL/129 background) due to fertility issues with homozygous Mbnl1 and Mbnl2 KOs. Muscle-specific Mbnl2 KO mice were produced using Myo-Cre transgenic mice (gift of E. Olson, University of Texas Southwestern Medical Center) (Li et al, 2005). Mbnl1ΔE3/ΔE3; Mbnl2c/c; Myo-Cre+/− mice were generated by crossing Mbnl1+/ΔE3; Mbnl2c/c; Myo-Cre+/− males with Mbnl1+/ΔE3; Mbnl2c/c; Myo-Cre−/− females. Controls included Mbnl1+/+; Mbnl2+/+; Myo-Cre+/− (WT control) and Mbnl1ΔE3/ΔE3; Mbnl2c/c; Myo-Cre−/− (Mbnl1 KO control) mice. All animal procedures were reviewed and approved by the University of Florida IACUC.
Immunoblotting and subcellular fractionation
Dissected tissues from WT (Mbnl1+/+), Mbnl1+/ΔE3 (Mbnl1+/−), Mbnl1ΔE3/ΔE3; Mbnl2+/ΔE2 (Mbnl1−/−; Mbnl2+/−) (4–5 months of age) skeletal muscles (quadriceps) and hearts were homogenized (Polytron, Kinematica) in lysis buffer [20 mM HEPES-KOH, pH 8.0, 100 mM KCl, 0.1% Igepal CA-630 (Sigma), 0.5 mM phenylmethysulphonyl fluoride, 5 μg/ml pepstain A, 1 μg/ml chymostatin, 1 mM ɛ-aminocaproic acid, 1 mM p-aminobenzamidine, 1 μg/ml leupeptin, 2 μg/ml aprotinin] on ice, followed by sonication and centrifugation (16,100g, 15 min, 4°C) as described previously (Charizanis et al, 2012). Proteins were detected by immunoblotting (50 μg lysate/lane) using rabbit polyclonal antibody (rpAb) anti-Mbnl1 A2764 (gift of C. Thornton, University of Rochester), mouse monoclonal (mAb) anti-Mbnl2 3B4 (Santa Cruz Biotechnology), anti-Gapdh mAb 6C5 (Abcam), rpAb anti-nucleolin (Abcam), rpAb anti-Ldha (Cell Signaling Technology), rpAb anti-Hnrnph1 (Novus), rpAb anti-Stau1 (Abcam), rpAb anti-Mbnl3 (Poulos et al, 2013) and HRP-conjugated anti-mouse, or anti-rabbit, secondary antibody followed by ECL (GE Healthcare). Subcellular fractionations were performed using NE-PER Nuclear and Cytoplasmic Extraction Reagents according to the manufacturer's protocol (Thermo Scientific) using Mbnl1+/+ and Mbnl1−/− quadriceps muscle.
Grip strength and electromyography (EMG)
Mice (n = 6 for each genotype and time point) were assessed for forelimb grip strength using a grip strength meter (Columbia Instruments). EMG was performed on Mbnl1−/−, Mbnl1−/−; Mbnl2+/− (2–3 months of age, n = 4 per genotype) skeletal muscles (gastrocnemius, TA) as described previously (Chamberlain & Ranum, 2012; Kanadia et al, 2006). Mice were placed on a temperature-controlled heating pad and 30G concentric needle electrodes (CareFusion Teca Elite, n = 4–5 insertions/muscle) were used with the TECASynergy EMG system (VIASYS Healthcare). Myotonic amplitudes were measured 8 s post-insertion.
Muscle histology, NMJ analysis and immunohistochemistry
Frozen skeletal muscle transverse sections (10 μm) of TA muscle were stained with hematoxylin and eosin (H&E) to determine the extent of DM-relevant muscle pathology. For cardiac analysis, hearts were fixed with 4% paraformaldehyde and longitudinal sections (5 μm) were stained by H&E or Masson's trichrome.
For NMJ structural analysis, Mbnl1+/+, Mbnl1−/− and Mbnl1−/−; Mbnl2+/− (4–5 months of age) TA muscles were dissected and fixed in 2% paraformaldehyde at 4°C overnight and then the myofibers were teased off into 5–10 thinner muscle bundles. For histological analysis, acetylcholine receptors were stained with 1 μg/ml α-bungarotoxin conjugated with Alexa Fluor 594 (Invitrogen). To detect axons and synapses, muscle fibres were incubated with chicken polyclonal antibodies against neurofilament H (NF-H, EnCor Biotechnology, 1:2000 dilution) and followed by secondary antibody conjugated with Alexa Fluor 488 (1:500 dilution) (Invitrogen). For NMJ quantification, >100 NMJs per genotype were counted and categorized into four groups previously described for NMJ morphological development including mature (pretzel-shaped endplates with branched innervating axons), fragment (endplate fragmentation), faint (endplate with low α-bungarotoxin staining), premature (pre-maturation stages including plaque, perforated plaque, c-shape, branched) and denervated (no endplate synapses) (Kummer et al, 2004; Sahashi et al, 2012; Valdez et al, 2010).
RNA splicing and qRT-PCR
Mouse WT, Mbnl1−/−, Mbnl1−/−; Mbnl2+/− (4–5 months of age) skeletal muscle (quadriceps) and heart RNAs, as well as Mbnl1+/+; Mbnl2+/+; Myo-Cre+/− and Mbnl1−/−; Mbnl2c/c; Myo-Cre+/− skeletal muscle RNAs, were isolated using TRI Reagent (Sigma–Aldrich) and cDNAs were prepared using 5 µg of RNA and SuperScript III RT (Invitrogen) according to manufacturer's protocol. For analysis of alternative splicing, specific PCR primers were used (Supporting Information Table S1) and the products were labelled with [α32P]-dCTP (2.5 μCi/reaction, PerkinElmer Life Sciences) followed by autoradiography and phosphorimaging to calculate the Ψ value (percent spliced in) as described previously (Suenaga et al, 2012).
Quantitative RT-PCR to determine Mbnl2 RNA levels was performed using RNAs isolated from Mbnl1+/+ and Mbnl1−/− quadriceps muscle and the MyiQ real-time PCR detection system (Biorad). First-strand cDNA was prepared as described above and Mbnl2 transcripts containing exon 2 were amplified using 2XiQ SYBR Green Supermix (Biorad) and 250 nM of the MSS4595 and MSS4596 primers (Supporting Information Table S1). Gapdh amplicons, generated using MSS4178 and MSS4179, were used for normalization. Real-time PCR reaction conditions were 95.0°C for 10 min, 45 cycles of 95.0°C for 15 s, 55.0°C for 30 s and 72.0°C for 30 s followed by 7 min at 72°C.
ECG and MRI heart analysis
Detail procedures for ECG acquisition have been described previously (Gehrmann & Berul, 2000; Kasahara et al, 2001). Briefly, mice were anesthetized with 1.5% isoflurane and the ECG was recorded using six surface leads. ECG recordings were acquired using a multichannel amplifier followed by conversion to digital signals for analysis. (MAClab system, AD Instruments). Corrected QT interval was calculated using Mitchell's formula (Mitchell et al, 1998).
ECG-gated Cine Cardiac MRI has also been described previously (Slawson et al, 1998; Warren et al, 2012). Mice were anesthetized with 1.5% isoflurane and a warm air fan (SA Instruments) was used to maintain stable body temperature. MRI images were acquired using a 4.7 Tesla (bore size 33 cm) horizontal scanner (Agilent). This system includes a 12 cm inner diameter active-shield gradient coil and has a 40 G/cm gradient strength and rise-time of 135 µs. A home-made quadrature saddle-shaped transceiver surface coil of 20 × 30 mm2 in size was used. Mice were positioned with the heart in the magnet iso-centre and the long- and short-axis orientation of the heart was scouted using a gradient-echo sequence. Shimming and pulse calibration were performed automatically prior to each experiment. Cine-FLASH was used to acquire temporally resolved dynamic short-axis and long-axis images of the heart with the following parameters: TReff = RR-interval, TE = 1.8 ms, flip angle = 30°, field of view, 25 × 25 mm2; acquisition matrix, 128 × 128 pixels; slice thickness, 1 mm. The number of frames per cardiac cycle was adapted to the heart rate of each animal to encompass the entire cardiac cycle.
To calculate the left ventricle ejection fraction (LVEF), short axis images from apex to bottom were acquired and endocardial contours were manually traced in end-diastole and in end-systole using ImageJ. The left ventricle cavity volume was calculated as the sum of the cavity areas multiplied by the section thickness (Lalande et al, 2004; Malm et al, 2004). Papillary muscles and trabeculations were, according to the American Society of Echocardiography (ASE) criteria, included in the LV cavity (Malm et al, 2004). LVEF was calculated based on LVEF% = (EDVLV − ESVLV)/EDVLV × 100, where EDVLV is the end-diastolic volume of the left ventricle, ESVLV is the end-systolic volume of the left ventricle. Right atrial (RA) area was traced and calculated using the method described by Prakken (Prakken et al, 2011). The extent of LV hypertrophy was indicated by the ratio of left ventricular wall volume (WV) to EDVLV.
Rotarod analysis
Mice (8–10 weeks of age, n = 5 per group) were allowed to acclimate in a behavioural facility for 30 min and then placed on an accelerating rotarod (Accuscan Instruments) rotating at 4 RPM which was gradually increased to 40 RPM over 5 min and then continued at 40 RPM for an additional 5 min. Latency to fall (in s) from the rotating bar was recorded. Mice were rested for 10 min after each trial, which was repeated three additional times per day for four consecutive days. A mouse's latency to fall for each day was recorded as the mean latency of the four consecutive trials (***p < 0.001 for all groups, one-way ANOVA).
HITS-CLIP
HITS-CLIP cDNA libraries were generated as described previously with the following modifications (Charizanis et al, 2012). Quadriceps muscles were dissected from Mbnl1+/+, Mbnl1−/− and Mbnl2−/− mice (15–16 weeks of age), snap frozen and finely powdered in liquid N2 using a mortar and a pestle and crosslinked with UV-light using a Stratalinker 1800 (Stratagene). For cDNA library generation, WT and Mbnl1−/− (n = 3 each) were analysed and the required RNA size distributions were generated using RNase A concentrations of 38.6 and 0.0386 U/ml for high and low RNase, respectively. Immunoprecipitation was performed using mAb 3B4 (3.25 μg/mg lysate) and cDNA libraries were generated and sequenced as described previously (Charizanis et al, 2012). Raw reads were filtered to remove low quality reads and filtered reads were aligned to the mouse reference genome (mm9) using the Burrows-Wheeler Aligner (BWA [m1]). Sequences <20 nt were discarded and unique CLIP tags were identified after computationally removing potential PCR duplicates as described (Charizanis et al, 2012) (Supporting Information Table S2).
Accession number
HITS-CLIP data have been deposited in GEO under accession number GSE47794.
The paper explained
PROBLEM:
Myotonic dystrophy (DM), the most common adult-onset muscular dystrophy, is characterized by multi-systemic symptoms and reduced life expectancy. A current disease model proposes that DM is caused by the expression of toxic microsatellite expansion RNAs that inhibit the RNA splicing function of the MBNL proteins resulting in the mis-expression of fetal isoforms in adult tissues. However, the loss of individual Mbnl proteins in mouse knockout models only partially recapitulates DM symptoms in skeletal muscle and the heart suggesting that the expression of other factors may be critical for DM pathogenesis.
RESULTS:
While loss of Mbnl1 expression leads to compensatory Mbnl2 upregulation in skeletal muscle and heart, combined loss of Mbnl1 and Mbnl2 leads to reduced lifespan, severe myotonia together with muscle weakness and wasting, cardiac conduction block and significant enhancement of DM-associated RNA splicing errors. These findings demonstrate that the major clinical manifestations of DM in specific tissues can be modelled by compound loss of Mbnl protein activity.
IMPACT:
RNA splicing errors in DM have been proposed as biomarkers for disease progression and therapeutic trials, and Mbnl compound knockout mice provide a model system to identify additional RNA mis-splicing events. This study also serves as model for ongoing investigations focused on other RNA-mediated diseases, including chromosome 9-linked frontotemporal dementia and amyotrophic lateral sclerosis (C9ORF72 FTD/ALS), and investigations on the roles of MBNL proteins in ES cell-specific alternative splicing regulation.
Author contributions
Mouse lines were generated by KYL, KC, DF and HH. Experiments were designed and performed by KYL, ML, MM, RB and AM. Cardiac studies were planned and performed by ML, SAW, HA and HK and muscle analysis was carried out by KYL, ML, CMC and LPWR. The manuscript was prepared by KYL and MSS.
Acknowledgments
This work was supported by grants from the National Institutes of Health (AR046799 to MSS and NS058901 to MSS and LPWR). The authors thank E. Olson for Myo-Cre transgenic mice, C. Thornton for anti-MBNL1 antibody A2764, T. Hathorn for assistance with rotarod testing, H. Zeng for cardiac imaging and D. Falk for NMJ analysis. MRI data analysis was supported by the National High Magnetic Field Laboratory and obtained at the Advanced Magnetic Resonance Imaging and Spectroscopy facility in the University of Florida McKnight Brain Institute.
Supporting Information is available at EMBO Molecular Medicine Online.
The authors declare that they have no conflict of interest.
For more information
OMIM for Myotonic Dystrophy:
Muscular Dystrophy Association:
Myotonic Dystropy Foundation:
Myotonic Dystrophy Support Group:
Association Française contre les Myopathies:
Treat-NMD Neuromuscular Network:
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
References
- Chamberlain CM, Ranum LP. Mouse model of muscleblind-like 1 overexpression: Skeletal muscle effects and therapeutic promise. Hum Mol Genet. 2012;21:4645–4654. doi: 10.1093/hmg/dds306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G, et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron. 2012;75:437–450. doi: 10.1016/j.neuron.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Cline MS, Osborne RJ, Tuttle DL, Clark TA, Donohue JP, Hall MP, Shiue L, Swanson MS, Thornton CA, et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol. 2010;17:187–193. doi: 10.1038/nsmb.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N, et al. Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med. 2011;17:720–725. doi: 10.1038/nm.2374. [DOI] [PubMed] [Google Scholar]
- Gehman LT, Meera P, Stoilov P, Shiue L, O'Brien JE, Meisler MH, Ares M, Jr, Otis TS, Black DL. The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev. 2012;26:445–460. doi: 10.1101/gad.182477.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehman LT, Stoilov P, Maguire J, Damianov A, Lin CH, Shiue L, Ares M, Jr, Mody I, Black DL. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat Genet. 2011;43:706–711. doi: 10.1038/ng.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrmann J, Berul CI. Cardiac electrophysiology in genetically engineered mice. J Cardiovasc Electrophysiol. 2000;11:354–368. doi: 10.1111/j.1540-8167.2000.tb01806.x. [DOI] [PubMed] [Google Scholar]
- Groh WJ, Groh MR, Saha C, Kincaid JC, Simmons Z, Ciafaloni E, Pourmand R, Otten RF, Bhakta D, Nair GV, et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. New Engl J Med. 2008;358:2688–2697. doi: 10.1056/NEJMoa062800. [DOI] [PubMed] [Google Scholar]
- Groh WJ, Lowe MR, Zipes DP. Severity of cardiac conduction involvement and arrhythmias in myotonic dystrophy type 1 correlates with age and CTG repeat length. J Cardiovasc Electrophysiol. 2002;13:444–448. doi: 10.1046/j.1540-8167.2002.00444.x. [DOI] [PubMed] [Google Scholar]
- Han H, Irimia M, Ross PJ, Sung H-K, Alipanahi B, David L, Golipour A, Gabut M, Michael IP, Nachman EN, et al. MBNL proteins repress ES-cell-specific alternative splicing and reprogramming. Nature. 2013;498:241–245. doi: 10.1038/nature12270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans MC, Faber CG, Bekkers SC, de Die-Smulders CE, Gerrits MM, Merkies IS, Snoep G, Pinto YM, Schalla S. Structural and functional cardiac changes in myotonic dystrophy type 1: A cardiovascular magnetic resonance study. J Cardiov Magn Reson. 2012;14:48. doi: 10.1186/1532-429X-14-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higham CF, Morales F, Cobbold CA, Haydon DT, Monckton DG. High levels of somatic DNA diversity at the myotonic dystrophy type 1 locus are driven by ultra-frequent expansion and contraction mutations. Hum Mol Genet. 2012;21:2450–2463. doi: 10.1093/hmg/dds059. [DOI] [PubMed] [Google Scholar]
- Ho TH, Charlet BN, Poulos MG, Singh G, Swanson MS, Cooper TA. Muscleblind proteins regulate alternative splicing. EMBO J. 2004;23:3103–3112. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt I, Jacquemin V, Fardaei M, Sewry CA, Butler-Browne GS, Furling D, Brook JD, Morris GE. Muscleblind-like proteins: Similarities and differences in normal and myotonic dystrophy muscle. Am J Pathol. 2009;174:216–227. doi: 10.2353/ajpath.2009.080520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet. 2004;13:3079–3088. doi: 10.1093/hmg/ddh327. [DOI] [PubMed] [Google Scholar]
- Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, Swanson MS. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc Natl Acad Sci USA. 2006;103:11748–11753. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara H, Wakimoto H, Liu M, Maguire CT, Converso KL, Shioi T, Huang WY, Manning WJ, Paul D, Lawitts J, et al. Progressive atrioventricular conduction defects and heart failure in mice expressing a mutant Csx/Nkx2.5 homeoprotein. J Clin Invest. 2001;108:189–201. doi: 10.1172/JCI12694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Nakamori M, Lueck JD, Pouliquin P, Aoike F, Fujimura H, Dirksen RT, Takahashi MP, Dulhunty AF, Sakoda S. Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2 + -ATPase in myotonic dystrophy type 1. Hum Mol Genet. 2005;14:2189–2200. doi: 10.1093/hmg/ddi223. [DOI] [PubMed] [Google Scholar]
- Kino Y, Mori D, Oma Y, Takeshita Y, Sasagawa N, Ishiura S. Muscleblind protein, MBNL1/EXP, binds specifically to CHHG repeats. Hum Mol Genet. 2004;13:495–507. doi: 10.1093/hmg/ddh056. [DOI] [PubMed] [Google Scholar]
- Koshelev M, Sarma S, Price RE, Wehrens XH, Cooper TA. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum Mol Genet. 2010;19:1066–1075. doi: 10.1093/hmg/ddp570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer TT, Misgeld T, Lichtman JW, Sanes JR. Nerve-independent formation of a topologically complex postsynaptic apparatus. J Cell Biol. 2004;164:1077–1087. doi: 10.1083/jcb.200401115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalande A, Salve N, Comte A, Jaulent MC, Legrand L, Walker PM, Cottin Y, Wolf JE, Brunotte F. Left ventricular ejection fraction calculation from automatically selected and processed diastolic and systolic frames in short-axis cine-MRI. J Cardiov Magn Reson. 2004;6:817–827. doi: 10.1081/jcmr-200036143. [DOI] [PubMed] [Google Scholar]
- Li S, Czubryt MP, McAnally J, Bassel-Duby R, Richardson JA, Wiebel FF, Nordheim A, Olson EN. Requirement for serum response factor for skeletal muscle growth and maturation revealed by tissue-specific gene deletion in mice. Proc Natl Acad Sci USA. 2005;102:1082–1087. doi: 10.1073/pnas.0409103102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA. Failure of MBNL1-dependent post-natal transitions in myotonic dystrophy. Hum Mol Genet. 2006;15:2087–2097. doi: 10.1093/hmg/ddl132. [DOI] [PubMed] [Google Scholar]
- Lopez Castel A, Cleary JD, Pearson CE. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat Rev Mol Cell Biol. 2010;11:165–170. doi: 10.1038/nrm2854. [DOI] [PubMed] [Google Scholar]
- Malm S, Frigstad S, Sagberg E, Larsson H, Skjaerpe T. Accurate and reproducible measurement of left ventricular volume and ejection fraction by contrast echocardiography: A comparison with magnetic resonance imaging. J Am Coll Cardiol. 2004;44:1030–1035. doi: 10.1016/j.jacc.2004.05.068. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Logigian E, Callahan L, McClain C, White R, Henderson D, Krym M, Thornton CA. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. 2000;289:1769–1773. doi: 10.1126/science.289.5485.1769. [DOI] [PubMed] [Google Scholar]
- Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS, Thornton CA. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet. 2001;10:2165–2170. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- McNally EM, Sparano D. Mechanisms and management of the heart in myotonic dystrophy. Heart. 2011;97:1094–1100. doi: 10.1136/hrt.2010.214197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirkin SM. Expandable DNA repeats and human disease. Nature. 2007;447:932–940. doi: 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol. 1998;274:H747–751. doi: 10.1152/ajpheart.1998.274.3.H747. [DOI] [PubMed] [Google Scholar]
- Morales F, Couto JM, Higham CF, Hogg G, Cuenca P, Braida C, Wilson RH, Adam B, del Valle G, Brian R, et al. Somatic instability of the expanded CTG triplet repeat in myotonic dystrophy type 1 is a heritable quantitative trait and modifier of disease severity. Hum Mol Genet. 2012;21:3558–3567. doi: 10.1093/hmg/dds185. [DOI] [PubMed] [Google Scholar]
- Motta J, Guilleminault C, Billingham M, Barry W, Mason J. Cardiac abnormalities in myotonic dystrophy. Electrophysiologic and histopathologic studies. Am J Med. 1979;67:467–473. doi: 10.1016/0002-9343(79)90795-2. [DOI] [PubMed] [Google Scholar]
- Orengo JP, Chambon P, Metzger D, Mosier DR, Snipes GJ, Cooper TA. Expanded CTG repeats within the DMPK 3' UTR causes severe skeletal muscle wasting in an inducible mouse model for myotonic dystrophy. Proc Natl Acad Sci USA. 2008;105:2646–2651. doi: 10.1073/pnas.0708519105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne RJ, Lin X, Welle S, Sobczak K, O'Rourke JR, Swanson MS, Thornton CA. Transcriptional and post-transcriptional impact of toxic RNA in myotonic dystrophy. Hum Mol Genet. 2009;18:1471–1481. doi: 10.1093/hmg/ddp058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panaite PA, Gantelet E, Kraftsik R, Gourdon G, Kuntzer T, Barakat-Walter I. Myotonic dystrophy transgenic mice exhibit pathologic abnormalities in diaphragm neuromuscular junctions and phrenic nerves. J Neuropath Exp Neurol. 2008;67:763–772. doi: 10.1097/NEN.0b013e318180ec64. [DOI] [PubMed] [Google Scholar]
- Paul S, Dansithong W, Kim D, Rossi J, Webster NJ, Comai L, Reddy S. Interaction of muscleblind, CUG-BP1 and hnRNP H proteins in DM1-associated aberrant IR splicing. EMBO J. 2006;25:4271–4283. doi: 10.1038/sj.emboj.7601296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelargonio G, Dello Russo A, Sanna T, De Martino G, Bellocci F. Myotonic dystrophy and the heart. Heart. 2002;88:665–670. doi: 10.1136/heart.88.6.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–741. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- Poulos MG, Batra R, Li M, Yuan Y, Zhang C, Darnell RB, Swanson MS. Progressive Impairment of muscle regeneration in muscleblind-like 3 isoform knockout mice. Hum Mol Genet. 2013;22:3547–3558. doi: 10.1093/hmg/ddt209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakken NH, Velthuis BK, Bosker AC, Mosterd A, Teske AJ, Mali WP, Cramer MJ. Relationship of ventricular and atrial dilatation to valvular function in endurance athletes. Br J Sports Med. 2011;45:178–184. doi: 10.1136/bjsm.2009.059188. [DOI] [PubMed] [Google Scholar]
- Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. Ann Rev Neurosci. 2006;29:259–277. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- Rau F, Freyermuth F, Fugier C, Villemin JP, Fischer MC, Jost B, Dembele D, Gourdon G, Nicole A, Duboc D, et al. Misregulation of miR-1 processing is associated with heart defects in myotonic dystrophy. Nat Struct Mol Biol. 2011;18:840–845. doi: 10.1038/nsmb.2067. [DOI] [PubMed] [Google Scholar]
- Ravel-Chapuis A, Belanger G, Yadava RS, Mahadevan MS, DesGroseillers L, Cote J, Jasmin BJ. The RNA-binding protein Staufen1 is increased in DM1 skeletal muscle and promotes alternative pre-mRNA splicing. J Cell Biol. 2012;196:699–712. doi: 10.1083/jcb.201108113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahashi K, Hua Y, Ling KK, Hung G, Rigo F, Horev G, Katsuno M, Sobue G, Ko CP, Bennett CF, et al. TSUNAMI: an antisense method to phenocopy splicing-associated diseases in animals. Genes Dev. 2012;26:1874–1884. doi: 10.1101/gad.197418.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicot G, Gomes-Pereira M. RNA toxicity in human disease and animal models: From the uncovering of a new mechanism to the development of promising therapies. Biochim Biophys Acta. 2013;1832:1390–1409. doi: 10.1016/j.bbadis.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Slawson SE, Roman BB, Williams DS, Koretsky AP. Cardiac MRI of the normal and hypertrophied mouse heart. Magnet Reson Med. 1998;39:980–987. doi: 10.1002/mrm.1910390616. [DOI] [PubMed] [Google Scholar]
- Suenaga K, Lee KY, Nakamori M, Tatsumi Y, Takahashi MP, Fujimura H, Jinnai K, Yoshikawa H, Du H, Ares M, Jr, et al. Muscleblind-like 1 knockout mice reveal novel splicing defects in the myotonic dystrophy brain. PloS One. 2012;7:e33218. doi: 10.1371/journal.pone.0033218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ZZ, Yarotskyy V, Wei L, Sobczak K, Nakamori M, Eichinger K, Moxley RT, Dirksen RT, Thornton CA. Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca(V)1.1 calcium channel. Hum Mol Genet. 2012;21:1312–11324. doi: 10.1093/hmg/ddr568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko LT, Miller JW, Timchenko NA, DeVore DR, Datar KV, Lin L, Roberts R, Caskey CT, Swanson MS. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucl Acids Res. 1996;24:4407–4414. doi: 10.1093/nar/24.22.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udd B, Krahe R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11:891–905. doi: 10.1016/S1474-4422(12)70204-1. [DOI] [PubMed] [Google Scholar]
- Valdez G, Tapia JC, Kang H, Clemenson GD, Jr, Gage FH, Lichtman JW, Sanes JR. Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc Natl Acad Sci USA. 2010;107:14863–14868. doi: 10.1073/pnas.1002220107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venetucci L, Denegri M, Napolitano C, Priori SG. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat Rev Cardiol. 2012;9:561–575. doi: 10.1038/nrcardio.2012.93. [DOI] [PubMed] [Google Scholar]
- Wang ET, Cody NA, Jog S, Biancolella M, Wang TT, Treacy DJ, Luo S, Schroth GP, Housman DE, Reddy S, et al. Transcriptome-wide regulation of pre-mRNA splicing and mRNA localization by muscleblind proteins. Cell. 2012;150:710–724. doi: 10.1016/j.cell.2012.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GS, Kearney DL, De Biasi M, Taffet G, Cooper TA. Elevation of RNA-binding protein CUGBP1 is an early event in an inducible heart-specific mouse model of myotonic dystrophy. J Clin Invest. 2007;117:2802–2811. doi: 10.1172/JCI32308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward AJ, Rimer M, Killian JM, Dowling JJ, Cooper TA. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum Mol Genet. 2010;19:3614–3622. doi: 10.1093/hmg/ddq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf MB, Berglund JA. MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. RNA. 2007;13:2238–2251. doi: 10.1261/rna.610607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren SA, Briggs LE, Zeng H, Chuang J, Chang EI, Terada R, Li M, Swanson MS, Lecker SH, Willis MS, et al. Myosin light chain phosphorylation is critical for adaptation to cardiac stress. Circulation. 2012;126:2575–2588. doi: 10.1161/CIRCULATIONAHA.112.116202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler TM, Thornton CA. Myotonic dystrophy: RNA-mediated muscle disease. Curr Opin Neurol. 2007;20:572–576. doi: 10.1097/WCO.0b013e3282ef6064. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Compton SA, Sobczak K, Stenberg MG, Thornton CA, Griffith JD, Swanson MS. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucl Acids Res. 2007;35:5474–5486. doi: 10.1093/nar/gkm601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Lee KY, Swanson MS, Darnell RB. Prediction of clustered RNA-binding protein motif sites in the mammalian genome. Nucleic Acids Res. 2013;41:6793–6807. doi: 10.1093/nar/gkt421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.