

Abstract
Matricellular proteins are secreted, nonstructural proteins that regulate the extracellular matrix (ECM) and interactions between cells through modulation of growth factor signaling, cell adhesion, migration, and proliferation. Despite being well described in the context of nonneuronal tissues, recent studies have revealed that these molecules may also play instrumental roles in central nervous system (CNS) development and diseases. In this minireview, we discuss the matricellular protein families SPARC (secreted protein acidic and rich in cysteine), Hevin/SC1 (SPARC-like 1), TN-C (Tenascin C), TSP (Thrombospondin), and CCN (CYR61/CTGF/NOV), which are secreted by astrocytes during development. These proteins exhibit a reduced expression in adult CNS but are upregulated in reactive astrocytes following injury or disease, where they are well placed to modulate the repair processes such as tissue remodeling, axon regeneration, glial scar formation, angiogenesis, and rewiring of neural circuitry. Conversely, their reexpression in reactive astrocytes may also lead to detrimental effects and promote the progression of neurodegenerative diseases.
1. Introduction
Astrocytes secrete numerous factors and active molecules [1–4], which modulate synapse development, neuronal activity, and plasticity during development and in the mature brain [5–10]. In particular, immature astrocytes produce and secrete many types of proteins that allow them to remodel the extracellular matrix (ECM) surrounding neurons and synapses [11]. In addition, astrocytes have the ability to react to any kind of insult or change in their environment, physical, viral, or chronic disease, and work side by side with microglia, the immune cells of the brain, in order to contain and to repair the brain from injuries [12, 13]. Astrocyte reactivity is associated with striking changes in gene expression and morphology [14, 15]. It has been demonstrated that reactive astrocytes revert to a partially immature molecular profile [16], which allow them to reexpress a variety of factors/proteins required for tissue remodelling around injury sites [13]. Some of these proteins have direct roles on modulation of ECM and cell to cell interactions. One mechanism used by reactive astrocytes to reshape their microenvironment is through the secretion of matricellular proteins.
Matricellular proteins are a family of structurally unrelated proteins that are secreted into the extracellular space. They act as nonstructural regulators of the ECM and cell-matrix interactions through modulation of growth factor signaling, cytokines, hormones, and proteases [17]. In general, matricellular proteins are highly expressed during development and present at a lower level during adulthood. However, their expression is upregulated following injury or disease. Interestingly, mice with homozygous-null mutations for this family of molecules do not usually exhibit gross abnormalities and generally have subtle phenotypes until challenged [18]. In contrast with other extracellular matrix proteins, matricellular proteins have been shown to have deadhesive activity, which likely contributes to their role in tissue remodeling following injury or in disease states. The matricellular protein family comprises many members to date (recently reviewed in [19]). Although widely studied in nonneuronal tissues, their roles in the CNS are not well understood. This minireview will focus on the expression and function of matricellular proteins secreted from astrocytes of the CNS, both in development and in the context of glial reactivity and disease, namely, SPARC (secreted protein acidic and rich in cysteine, also known as osteonectin/BM-40), Hevin (also known as SPARC-like 1 or SC1), Tenascin C (TN-C), Thrombospondins (TSP), and the CCN (CYR61/CTGF/NOV) family.
2. SPARC and Hevin/SC1
SPARC/osteonectin/BM-40 (now referred to as SPARC) and SC1/Hevin, first discovered two decades ago, are part of a larger family of SPARC family proteins, which share a common E-F hand calcium binding (EC) domain [20–23].
Of the SPARC family proteins, Hevin has the greatest homology to SPARC and both proteins have three main domains; an N-terminal acidic calcium-binding domain, a central cysteine-rich follistatin domain, and the C-terminal EC domain [24] which confers collagen-binding [25–27]. SPARC has been shown to modulate several growth factor signaling cascades (i.e., VEGF (vascular endothelial growth factor), PDGF (platelet-derived growth factor), FGF2 (fibroblast growth factor-2), and TGFβ (transforming growth factor beta) and can regulate integrin-mediated adhesion (reviewed in [28]). Mice containing targeted deletions of either SPARC or Hevin are viable and fertile and generally exhibit relatively mild phenotypes until revealed by injury or challenge [20, 29, 30]. However, recent studies of SPARC and Hevin knock-out animals have revealed important roles for these molecules in CNS development [9, 31–33].
During embryonic development, SPARC and Hevin have somewhat overlapping expression profiles and are found in radial glia and the developing vasculature (recently described in detail in [34, 35]). Indeed, Hevin was shown to be important for appropriate termination of radial glia-guided neuronal migration and proper lamination of the cerebral cortex [32].
During the first few weeks of postnatal development, SPARC and Hevin are highly expressed in astrocytes [9, 31], with Hevin also being present in neurons [36]. Secretion of SPARC and Hevin from astrocytes has been shown to play an important role during synapse development (recently reviewed in [5, 11]). Kucukdereli and colleagues [31] demonstrated that astrocyte-secreted Hevin promotes the formation of excitatory synapses in the superior colliculus. They proposed that SPARC may compete with Hevin to regulate synapse development, since SPARC was found to antagonize the synaptogenic effects of Hevin on cultured RGC neurons. In contrast, morphological synapse development in hippocampal neurons cultured with SPARC null astrocytes versus wild type astrocytes was similar [9]. However, synapses in SPARC null cultures were found to have a greater number of AMPA receptors and an increase in synaptic strength, which was rescued through application of recombinant SPARC. Regulation of AMPA receptors by SPARC required β3-integrin signaling [9]. A recent study has shown that these synaptic changes can lead to alterations in hippocampal-related behaviours in the adult. Campolongo and colleagues demonstrated that SPARC knock-out mice exhibited an increase in anxiety and antidepressant-like behaviours [29].
Taken together, these studies suggest that SPARC may play an important role in excitatory synapse development by preventing premature maturation of synapses to facilitate proper formation of neural circuitry. How Hevin and SPARC might interact to exert their effects on synapses is not yet understood.
2.1. SPARC and Hevin Are Upregulated in Reactive Astrogliosis
In the mature CNS, the expression of SPARC and Hevin is reduced, and, in the case of SPARC, its expression is confined to radial-like glia cells such as Bergmann glia and Muller glia, resting microglia at low levels in astrocytes [35, 37]. In contrast, Hevin expression in the adult brain is restricted to mature astrocytes and neurons and is not present in microglia [34, 38]. However, upon injury or disease, the expression of both proteins is upregulated in reactive astrocytes [39, 40]. Hevin is found in the cell bodies of reactive, dividing astrocytes and in hypertrophied processes surrounding neurons and blood vessels following status epilepticus in rat [41]. Hevin is also highly expressed in reactive glia following lesion injury [42, 43] and ischemic stroke [44], where Hevin expression was shown to be upregulated in astrocytes surrounding the lesion at 1 day after stroke and was sustained for up to a week. Similarly, SPARC has been shown to be upregulated in reactive astrocytes following injury [39] and stroke [45]. Interestingly, SPARC upregulation occurred in a slow sustained manner in both of these studies, suggesting that SPARC produced from reactive astrocytes may play a role in later, rather than immediate, processes of recovery and repair.
In the human brain, SPARC is expressed in reactive human astrocytes proximal to brain tumours [46] and was shown to be increased in cases of human epilepsy [47]. SPARC is expressed in several types of brain tumours, and its expression in astrocytomas and gliomas is generally associated with increased invasion, angiogenesis, and a negative prognosis [48, 49]. The role of Hevin in cancer is likely complex: its expression has been shown to be correlated with invasive gliomas [50] yet it is highly downregulated in other malignancies and has been described as a tumour suppressor gene [24, 51].
2.2. Cleavage of SPARC and Hevin and the Generation of Functional Protein Fragments
The role of Hevin and SPARC in cancer, brain injury, and disease is likely dependent on the tissue type and other molecules in the ECM environment, such as the presence of proteases. Both SPARC and Hevin have been shown to be cleaved by members of the matrix metalloproteinases (MMP) family to release functional protein fragments. Hevin can be cleaved by MMP-3 and ADAMTS4 (a disintegrin and metalloproteinase with Thrombospondin motifs) to release a shorter, C-terminal fragment that is highly homologous to SPARC, known as “SPARC-like fragment” (SLF) [38, 52]. Interestingly, the SLF was shown to oppose the synaptogenic action of full length Hevin [31, 52] and was found associated with SPARC in neovasculature of gliomas [52]. Furthermore, it is possible that SPARC may play a role in the proteolysis of Hevin, since SPARC has been shown to induce the expression of MMPs [53, 54]. SPARC is regulated by MMP proteolysis and is cleaved by MMP-3 in vitro to produce protein fragments that have differential effects on cell proliferation and migration [55]. Peptides encompassing different domains of SPARC (such as would be released by MMP cleavage) have been shown to have potent effects. For example, “peptide 2.3,” which contains part of the follistatin-like domain of SPARC, regulates SPARC-β-integrin interactions in nonneuronal cells [56], can stimulate angiogenesis activity [57], and rescues β-integrin and AMPA receptor overaccumulation at synapses in SPARC null hippocampal cultures [9].
MMPs and their inhibitors, TIMPs (tissue inhibitors of metalloproteinases), are upregulated in reactive astrocytes in a range of disease contexts such as following ischemic stroke or in Alzheimer's disease (AD), where they are thought to have an important role (reviewed in [58, 59]). In ischemic stroke or injury, MMP expression has been shown to have a detrimental role in the initial stages [60], but it is protective in the later recovery phases where it is important for promoting angiogenesis of new blood vessels and restoration of the blood brain barrier [61]. The role of MMPs in angiogenesis is thought to involve VEGF signaling [58], which has also been shown to be regulated by SPARC (reviewed in [28]). Given that MMP can regulate SPARC and vice versa, it would be interesting to determine whether MMP and SPARC could coordinate regulation of tissue remodeling and angiogenesis following injury to the CNS.
Currently, the role of SPARC and Hevin in the CNS has been mostly studied in the context of development. One interesting question is whether these proteins will have similar functions in the adult CNS following injury or disease. For example, are SPARC and Hevin involved in de novo synapse formation and circuit rewiring following CNS injury? Does their function require proteolysis? Answers to these questions will help provide insight into the functions of SPARC and Hevin produced from reactive astrocytes and their role in modulating the ECM environment.
3. Tenascin C
Tenascin C (or TN-C with C for cytotactin) is a secreted extracellular matrix glycoprotein that is part of a family of three homologs along with Tenascin R and Tenascin X. TN-C was discovered in the early 1980s and has carried numerous names like myotendinous antigen, glioma mesenchymal extracellular matrix, hexabrachion, tenascin, Jl-200/220, and cytotactin [62]. TN-C is composed of several distinct domains containing epidermal growth factor-like repeats, fibronectin type III repeats, and a segment of great homology with the α and β chains of fibrinogen [62, 63]. It is transiently expressed by neural and nonneural cells during development and plays a role in the ECM remodelling during tissue repair. During brain development, the expression of TN-C by radial glia and subpopulations of astrocytes has been shown to affect multiple processes such as cell migration and proliferation, axonal guidance, and synaptic plasticity.
3.1. Astrocyte-Secreted TN-C Is a Key Molecule in the Establishment of Neuronal Circuitry
TN-C was first described as a protein produced and secreted in vitro by astrocytes in culture [64, 65]. It was demonstrated that its expression was limited to specific astrocyte cell lines or subpopulations during CNS development [66, 67]. Paradoxically, TN-C was considered both as a repulsive substrate for neuronal and astrocytic growth [68, 69] and as a permissive one, by providing axonal guidance cues [66, 67, 70, 71]. Depending on the context, such as the expression of other ECM molecules, TN-C can have the opposite differential effects on neuronal growth. TN-C is also known to influence astrocyte proliferation and induces process elongation through an autocrine/paracrine mechanism [63]. In addition, TN-C has been shown to act through β1 and α9 integrin-dependent cell adhesion [69, 72] and to interact physically with the neuronal GPI—membrane-anchored adhesion glycoprotein F3/contactin of the Ig superfamily [71].
TN-C is expressed in vivo in various brain regions and exhibits a precise temporal and spatial distribution. In the somatosensory cortex of rodents, TN-C was detected by immunohistochemistry in populations of astrocytes delineating the boundaries of whisker barrel fields in the early postnatal development (P1–P7) [73]. By P9, TN-C expression in barrels has almost disappeared. TN-C was also reported to be transiently expressed by cortical radial glia cells [74] and by astrocytes of the optic nerves [75]. In the cerebellar cortex, TN-C was found to be highly expressed in the first four postnatal weeks before being downregulated [75]. Using in situ hybridization and electron microscopy, the authors showed that TN-C was mainly produced and secreted by astrocytes and epithelial cells and was only detected at the cellular surface of neurons. However, in the developing hippocampus, TN-C not only is expressed by radial glia cells and immature astrocytes but also by a subset of neurons of the stratum oriens [76]. In the spinal cord, TN-C is synthesized by a subset of gliogenic precursors in the late phase of embryogenesis and has been shown to influence proliferation and migration of subpopulation of astrocytes [77].
Surprisingly, TN-C null mice do not show a major phenotype or alteration in the gross histoarchitecture of the CNS but do display subtle morphological alterations in some subtypes of neurons [78, 79]. In addition, TN-C null mice exhibited an enhanced proliferation but a delayed migration of immature astrocytes toward the ventral spinal cord white matter [77]. Furthermore, it has been shown that the number of cells is changed in the cerebral cortex of TN-C null mice when compared to the WT animals, with an abnormally high neuronal density and increased astrogliosis but low density of parvalbumin-positive interneurons and reduced ratio of oligodendrocytes [79]. Evers and colleagues observed plasticity impairments in the hippocampus of one-month-old TN-C null mice. Indeed, intertheta burst stimulation-(TBS-) induced LTP (long term potentiation) were shown to be significantly reduced and LTD (long term depression) abolished at Schaffer collateral—CA1 synapses whereas LTP in the dentate gyrus or CA3 were described as normal [78]. The authors demonstrated a causal effect between the deficiency in TN-C and an impairment of the L-type Ca2+ channel-dependent forms of synaptic plasticity in the CA1 that could be mediated by interactions with integrins or proteoglycans. In addition, TN-C −/− mice trained in the step-down avoidance test also showed clear deficits in contextual memory [80]. Interestingly, different domains of TN-C have distinct functional properties. Indeed, the intrahippocampal injections in wild type animals of the recombinant fragment of TN-C containing the fibronectin type III repeats 6–8, but not 3–5, blocked memory formation and suggests that TN-C can directly modulate synaptic plasticity [80]. Therefore, the reexpression of TN-C by reactive astrocytes during injuries or brain diseases could impact the processes of cell proliferation, migration, and synaptic plasticity.
3.2. TN-C in Astrogliosis and Brain Diseases
Tenascin C is poorly expressed in adult brain. However, it has been observed to be upregulated at injury sites in correlation with astrocyte reactivity and glial scar formation. Indeed, stab wound in cerebellar and cerebral cortices structures have been shown, by in situ hybridization and immunohistochemistry, to enhance the production of TN-C in a discrete population of GFAP-(glial fibrillary acidic protein) positive astrocytes proximal to the injury site [81]. TN-C is also intensely expressed by astrocytes in culture in scratch wound assays [82]. Interestingly, TN-C could be involved in the maintenance of astrogliosis surrounding site of severe injuries. Indeed, stab wound assays in cerebral cortex revealed that GFAP expression, which is usually correlated with severity of reactivity, was significantly weaker in TN-C null versus wild type mice, one week after the stab [83]. However, in the same study, IgG leakage persisted much longer in TN-C-deficient mice and RNA levels of proinflammatory cytokines TNFα (tumour necrosis factor alpha), IL-6 (Interleukin-6), and IL-1β levels were higher [83]. Thus, the production of TN-C may influence blood-brain barrier (BBB) repair and the regulation of inflammatory cytokine levels either directly or indirectly through modulation of BBB integrity. In addition, TN-C production in astrocytes can be modulated by inflammatory cytokines and growth factors. Indeed, astrocytes increased their production of TN-C when cultured in presence of activated macrophage- and microglial-conditioned media [84]. Smith and colleagues have also demonstrated that the synergic action of both TGF-β1 and basic fibroblast growth factor (bFGF), both factors known to be upregulated in injury, stimulates the expression of TN-C [84].
Interestingly, TN-C upregulation has been described in numerous neurodegenerative diseases. TN-C expression is induced in the hippocampi of both epileptic rat brains [85, 86] and human patients with temporal lobe epilepsy (TLE) [87]. In brains of TLE patients, the regions exhibiting a diffuse and elevated expression of TN-C were also characterized by an extended area of reactive gliosis and synaptic reorganization. In addition, TN-C was recently reported as a plasma biomarker for neurodegenerative diseases, as levels were found to be significantly increased in blood of Alzheimer's disease (AD), mild cognitive impairments patients [88, 89], and in the amniotic fluid of Down syndrome-affected pregnancies [90]. In addition, TN-C level was shown to be upregulated in the AD-like mouse model CRND8 when compared to the wild type littermates [91]. Furthermore, TN-C deficiency in transgenic CRND8 mice provoked a reduction of β and γ secretase activity, Aβ oligomerization, plaque load, and synaptic impairments. In addition, loss of TN-C in CRND8 Tg mice enhanced production of anti-inflammatory cytokines and reduced proinflammatory cytokines [91]. This contrasts with the effect of TN-C deficiency on inflammation levels in the context of physical injury [83]. Therefore, TN-C may act as a promoter or inhibitor of the inflammation depending on the type of insult. In support of this, TN-C has also been demonstrated to induce production of proinflammatory cytokines via activation of Toll-like receptor 4 (TLR4) in nonneuronal tissue [92]. Interestingly, TLR4 is known to be expressed at the surface of astrocytes [93, 94] and microglia [95] and is directly implicated in the induction of inflammation in various neurodegenerative diseases [95–97].
In conclusion, the upregulation of TN-C by reactive astrocytes in response to an insult to the brain is context specific and may be a key factor in multiple processes such as the maintenance of astrocyte reactivity, BBB repair, and the potentiation of inflammatory processes. It could also directly affect neuronal plasticity and lead to memory impairments.
4. Thrombospondins
There are five known thrombospondins (TSP, also known as THBS) in vertebrates, which can be classified into two groups: the trimeric TSP1 and -2 and the pentameric TSP3, -4, and 5. All of the TSPs have epidermal growth factor-(EGF-) like repeats followed by calcium-binding type 3 repeats, and all share a highly conserved C-terminal region [98]. Thrombospondins are secreted molecules and interact with structural components of the ECM as well as proteases, cytokines, and growth factors to modulate cell signaling, adhesion, migration, and other cellular processes [99]. TSPs have many known binding partners, including integrins, neuroligins, Reelin receptors ApoER and VLDLR (very low density lipoprotein receptor), the calcium channel subunit α2δ-1, and several growth factor ligands and their receptors (summarized in [100, 101]).
The different TSPs are expressed throughout the organism at various stages of mammalian development [98]. In the CNS, TSPs 1 and 2 are expressed in cultured and developing astrocytes, with expression peaking during the first postnatal week in mice [102, 103]. In addition, high levels of TSP1 can be found in cultured human astrocytes [104, 105]. Astrocyte-secreted TSP1/2 has been shown to be important for the formation of excitatory synapses in vitro and in vivo ([102, 106], recently reviewed extensively in [11, 101]). Briefly, TSP treatment of cultured neurons led to an increase in the number of synapses [102] and rate of synaptogenesis [107]. Synapses induced by TSPs are morphologically normal but postsynaptically silent, indicating that TSP is important for the initiation of synapse formation, but other secreted factors are required for completion of synapse maturation [5, 9], reviewed in [108].
4.1. Expression of TSPs in the Mature CNS and Following Injury
In mature astrocytes, the expression of TSP1/2 is decreased, although a low level is maintained throughout the brain and is particularly concentrated in areas of neurogenesis in the adult (i.e., subventricular zone (SVZ) and subgranular zone (SGZ)) [102, 109–111]. Indeed, loss of TSP1 was associated with a decrease in the number and distribution of SVZ neural precursors entering the olfactory bulb from the rostral migratory stream [112]. In contrast to TSP1/2, expression of TSP4 is low during development but present in adulthood, where it is expressed in spinal cord astrocytes [113] and mature forebrain astrocytes [114] and was found localized to synapses at neuromuscular junctions in the peripheral nervous system [115].
Similar to the other astrocytic matricellular proteins, TSPs are upregulated in reactive astrocytes following ischemic [116, 117] and mechanical [118] injury. It is likely that several signaling pathways can stimulate TSP production in reactive astrocytes. For example, in an in vivo trauma model, TSP1 expression was induced by extracellular ATP (Adenosine triphosphate) via P2Y purinergic receptors and ERK (extracellular signal-regulated kinase)/p38 MAPK (mitogen-activated protein kinase) signaling [119]. Additionally, TSP1 expression was activated by collagen-stimulated integrin signaling in reactive astrocytes following brain injury [120].
Following stroke, TSP1 and -2 display a differential temporal expression pattern; TSP1 was shown to be upregulated in the penumbra (the region surrounding the lesion core) within 3 days, whereas changes in TSP2 expression were delayed and levels were only increased at one week after stroke [116, 117]. Interestingly, the peak in expression of TSP1 and TSP2 was inversely correlated with angiogenic activity in the penumbra, consistent with a role for TSP1/2 in regulation of angiogenesis [121]. Conversely, Liauw and colleagues [116] reported that loss of TSP1/2 did not affect blood vessel density after stroke. Furthermore, they suggested that TSPs play a beneficial role in synaptic recovery, since TSP1/2 null mice exhibited a reduction in synapse number and axonal sprouting (and consequent motor function) when compared to wild type mice. This is particularly interesting considering that several studies have shown that TSPs can promote neurite outgrowth of cultured neurons in vitro [122–125] and is expressed around regenerating axons following injury in vivo [126, 127]. In addition, TSPs have been shown to induce dendritic spine formation [105] and are important for synaptogenesis ([102, 107] discussed in text above). In support of this, TSP expression is downregulated in diseases characterized by synaptic loss or abnormal synapse formation, such as in AD [128] and Down syndrome [105]. Taken together, upregulation of TSP1/2 expression in reactive astrocytes may contribute to tissue recovery following injury by promoting de novo synapse formation and rewiring of neural circuitry.
One key question in the field is where reactive astrocytes come from? Do they develop from local mature astrocytes or do they originate from neurogenic niches and migrate to the site of injury? It is possible that both of these processes occur depending on injury context [129]. In addition, it is thought that the molecular profile of reactive astrocytes overlaps with that of immature astrocytes or radial glial cells [16]. Recently, recruitment of newborn astrocytes and neurons from the SVZ was shown to play an important role following ischemic injury and stroke [130, 131]. Importantly, TSP4 was shown to be upregulated in SVZ-derived astrocytes surrounding lesions induced by photothrombotic injury and was required for injury-induced astrogenesis of neural stem cells, which was mediated through Notch signaling. Loss of TSP4 led to an increase in hemorrhaging and changes in glial scar formation [131]. In addition, expression of TSP4 was increased following peripheral nerve injury in spinal cord astrocytes, which morphologically resembled immature astrocytes [113]. Further study of TSPs may assist in the understanding of astrocyte heterogeneity and the process of astrogliosis.
5. CCN Family
The matricellular protein family CCN, which stands for CYR61/CTGF/NOV, are a family of six homolog members containing CYR61/CCN1 (cysteine-rich 61), CTGF/CCN2 (connective tissue growth factor), NOV/CCN3 (nephroblastoma overexpressed), and WISP-1/CCN4, WISP-2/CCN5, and WISP-3/CCN6 (Wnt-inducible-secreted proteins). All CCN proteins have a modular structure, which consists of four conserved cysteine rich-domains, with sequence homology to the insulin-like growth factor-binding proteins (IGFBP), the von Willebrand factor C (VWC) domain, Thrombospondin type 1 repeat (TSR), and a C-terminal domain with a cysteine-knot motif. CCN proteins can bind to integrin receptors and coreceptors such as heparan sulfate proteoglycans (HSPGs), low-density lipoprotein receptors-related proteins (LPRs), and TRKA [132]. They have been reported to participate in multiple functions in non-CNS organs, such as in migration, proliferation, and apoptosis (reviewed in [132]). They are upregulated in chronic inflammatory diseases, where they are suggested to be important players in the modulation of inflammatory cytokines and chemokines production (reviewed in [133]). Despite established roles in nonneuronal tissues, the expression and function of CCN family proteins in the CNS remain poorly understood.
5.1. CCN2/CTGF, Astrocytes, and Brain Insults
CCN2/CTGF is currently the only CCN family member in which expression has been demonstrated in vivo in CNS astrocytes. CCN2 expression has been detected not only in astrocyte somas and processes but also in a subpopulation of cortical neurons in adult rat brain, in tanycytes, and in the grey matter of the spinal cord [134]. CCN2 has been reported to be highly expressed in human astrocyte cell cultures [135] but was predominantly detected in neurons in healthy human brain and partially in subtypes of glial cells such as the glia limitans [136, 137].
Furthermore, CCN2 upregulation has been widely observed in reactive gliosis adjacent to the site of mechanical injuries caused by stab wound in rodents and also in the brain tissue of stroke patients [137] and following traumatic brain injury (TBI) [136]. Interestingly, in neurodegenerative diseases, CCN2 elevation has been detected both in neurons and reactive astrocytes. This dual expression was reported after excitotoxic damages provoked by kainic lesions in rat hippocampi [138] and in CNS tissue of multiple sclerosis (MS) [139], amyotrophic lateral sclerosis (ALS), [140] and AD [141] patients. In brains of AD patients, this increase was particularly significant around the amyloid plaques and neurofibrillary tangles [141]. CCN2 increase has also been highly correlated with glioblastoma (reviewed in [142]). Interestingly, CCN proteins are also proposed as potent modulators of cytokines and chemokines in different organs [133]. In primary cultured rat astrocytes, CCN2 mRNA levels were reported to be differentially modulated by distinct cytokines, showing an upregulation following treatment with TGFβ and downregulation by TNFα [133]. Since both TGFβ and TNFα are known to dramatically modify the glial and neuronal environment in lesioned tissue and neurodegenerative diseases [143–147], it is interesting to speculate that CCN2 production and release by reactive astrocytes may participate in the cascade of inflammatory events occurring in injuries and neurodegenerative processes. Finally, CCN2 has been demonstrated to bind to TrkA (neurotrophic tyrosine kinase receptor type 1) and p75NTR (p75 neurotrophin receptor), receptors which transduce neurotrophin signals [148]. How CCN2 elevation might influence neurotrophin signalling in brain diseases remains to be evaluated.
6. Perspectives: Astrocyte-Derived Matricellular Proteins as Tools to Reshape the ECM and Cell-Cell Interactions Following Injury and Disease
Reactive astrocytes are found in nearly all situations following central nervous system injury and disease. Astrocyte reactivity is contextual [13] and leads to numerous morphological changes, such as an increase in the number and volume of processes [149], extension and elongation [150], and polarisation of processes around sites of injury [151]. This process requires dynamic modifications in cell-cell and cell-ECM interactions and involves the recruitment of other cell types, including immune cells [13]. Furthermore, the microenvironment surrounding the insult may modulate reactive astrogliosis and formation of the glial scar [152]. In this review, we have discussed several nonstructurally related families of matricellular proteins which are upregulated in reactive astrocytes. A common feature among these proteins is that they exhibit high levels of expression during development which is reduced to a lower level in the adult. However, their expression is reactivated following injury or disease. This pattern of expression is consistent with the fact that reactive astrocytes upregulate expression of proteins (GFAP, Vimentin, BLBP (brain lipid-binding protein), and Nestin) that are also present at high levels in radial glia or immature astrocytes [153]. Despite this, recent studies have shown that reactive astrocytes exhibit significant heterogeneity and the type of astrocyte reactivity occurring is dependent on the type and extent of CNS injury and disease [154–156]. For example, there is an increase in proliferating reactive astrocytes following lesions such as stroke but not in diseases such as Alzheimer's disease [129, 157]. Matricellular proteins have been described as dynamic regulators of the ECM and cell-cell interactions and are well placed to regulate the process of astrocyte reactivity and tissue remodelling occurring in the CNS following injury and in neurodegenerative disease (see Figure 1).
Figure 1.
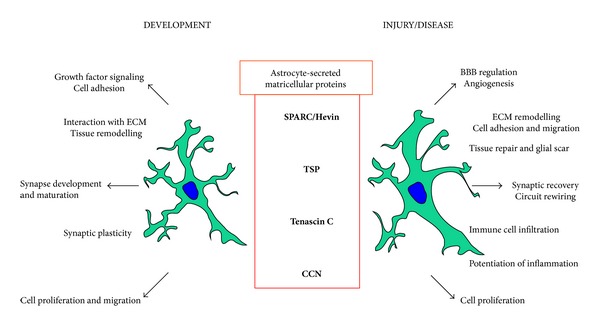
Astrocyte-secreted matricellular proteins: Developmental tools for reactivity purposes. Astrocyte-secreted matricellular proteins are highly expressed during the development of the CNS where they have multiple complex roles (left panel). In general, the expression of these molecules in the adult nervous system is reduced, but they are reexpressed at a high level in reactive astrocytes following injury or in disease states. Here, we summarise the possible roles of matricellular proteins secreted from reactive astrocytes (right panel).
Ultimately, reactive astrogliosis can progress to form a glial scar, which has been shown to have both positive and negative effects on tissue recovery. The glial scar, which contains proliferating reactive astrocytes, forms a dense physical barrier around an injury. This protects the healthy brain from damaged tissue and inflammation and has been shown to play a role in repair of the BBB [14]. It is conceivable that matricellular proteins play an important role in modulating the glial scar, perhaps through regulation of glial cell proliferation and the surrounding vasculature. For example, SPARC is antiproliferator and can regulate microglia proliferation in vitro and in vivo following photothrombotic stroke [21, 45]. Conversely, TN-C was found to induce astrocyte proliferation in vitro [63]. In addition, SPARC, CCN2, and TSP1/2 have been implicated in angiogenesis and leukocyte infiltration in nonneuronal tissues [101, 158, 159] and therefore may serve to repair the BBB and restrain an immune response [160–162].
It has been well documented that the glial scar can inhibit axon regeneration; reactive “scar” astrocytes produce proteoglycans and other ECM molecules to inhibit axonal outgrowth, which ultimately limits functional recovery [163]. Through modulation of structural ECM components and adhesion and growth factor signaling (e.g., integrin signaling), astrocyte-secreted matricellular proteins may control axonal growth. In support of this, TN-C has been shown to both promote and inhibit axonal outgrowth and TSPs are known to positively affect growth [164]. All four matricellular protein families modulate cell adhesion in vitro, where they have been reported to restructure actin-containing focal adhesions and stress fibers [165]. This may translate into an in vivo role of “opening up” the ECM to create a permissive microenvironment for facilitating cell migration, outgrowth, angiogenesis, synaptic remodelling, and morphological plasticity. At the same time, upregulation of matricellular proteins may have negative consequences for tissue recovery and progression of disease. For example, matricellular proteins could promote an inflammatory response [19] and chronic inflammation is thought to be detrimental in neurodegenerative diseases such as Alzheimer's disease [166–168].
Huge challenges are awaited in order to understand the secretion of matricellular proteins from reactive astrocytes and functions in the CNS. Given their potential to modulate the microenvironment surrounding regions of brain injury and disease, astrocyte-secreted matricellular proteins may represent important therapeutic targets for CNS repair.
Acknowledgments
This work was supported by funding from Fonds de recherche du Québec-Santé (FRQS, Grant no. 27507) and the Canadian Institutes of Health Research (CIHR, Grant no. MOP111152). The authors thank Dr. Keith Murai for helpful comments on the paper.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Bezzi P, Gundersen V, Galbete JL, et al. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nature Neuroscience. 2004;7(6):613–620. doi: 10.1038/nn1246. [DOI] [PubMed] [Google Scholar]
- 2.Hamilton NB, Attwell D. Do astrocytes really exocytose neurotransmitters? Nature Reviews Neuroscience. 2010;11(4):227–238. doi: 10.1038/nrn2803. [DOI] [PubMed] [Google Scholar]
- 3.Martineau M, Shi T, Puyal J, et al. Storage and uptake of D-serine into astrocytic synaptic-like vesicles specify gliotransmission. Journal of Neuroscience. 2013;33(8):3413–3423. doi: 10.1523/JNEUROSCI.3497-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schubert V, Bouvier D, Volterra A. SNARE protein expression in synaptic terminals and astrocytes in the adult hippocampus: a comparative analysis. Glia. 2011;59(10):1472–1488. doi: 10.1002/glia.21190. [DOI] [PubMed] [Google Scholar]
- 5.Allen NJ, Bennett ML, Foo LC, et al. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature. 2012;486(7403):410–414. doi: 10.1038/nature11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.di Castro MA, Chuquet J, Liaudet N, et al. Local Ca2+ detection and modulation of synaptic release by astrocytes. Nature Neuroscience. 2011;14(10):1276–1284. doi: 10.1038/nn.2929. [DOI] [PubMed] [Google Scholar]
- 7.Haber M, Murai KK. Reshaping neuron-glial communication at hippocampal synapses. Neuron Glia Biology. 2006;2(1):59–66. doi: 10.1017/S1740925X06000032. [DOI] [PubMed] [Google Scholar]
- 8.Henneberger C, Papouin T, Oliet SHR, Rusakov DA. Long-term potentiation depends on release of d-serine from astrocytes. Nature. 2010;463(7278):232–236. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones EV, Bernardinelli Y, Tse YC, Chierzi S, Wong TP, Murai KK. Astrocytes control glutamate receptor levels at developing synapses through SPARC-β-integrin interactions. Journal of Neuroscience. 2011;31(11):4154–4165. doi: 10.1523/JNEUROSCI.4757-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Panatier A, Vallée J, Haber M, Murai KK, Lacaille JC, Robitaille R. Astrocytes are endogenous regulators of basal transmission at central synapses. Cell. 2011;146(5):785–798. doi: 10.1016/j.cell.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 11.Clarke LE, Barres BA. Emerging roles of astrocytes in neural circuit development. Nature Reviews Neuroscience. 2013;14(5):311–321. doi: 10.1038/nrn3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends in Immunology. 2007;28(3):138–145. doi: 10.1016/j.it.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathologica. 2010;119(1):7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends in Neurosciences. 2009;32(12):638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11(5):400–407. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- 16.Ridet JL, Malhotra SK, Privat A, Gage FH. Reactive astrocytes: cellular and molecular cues to biological function. Trends in Neurosciences. 1997;20(12):570–577. doi: 10.1016/s0166-2236(97)01139-9. [DOI] [PubMed] [Google Scholar]
- 17.Bornstein P. Thrombospondins as matricellular modulators of cell function. Journal of Clinical Investigation. 2001;107(8):929–934. doi: 10.1172/JCI12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bornstein P, Sage EH. Matricellular proteins: extracellular modulators of cell function. Current Opinion in Cell Biology. 2002;14(5):608–616. doi: 10.1016/s0955-0674(02)00361-7. [DOI] [PubMed] [Google Scholar]
- 19.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiological Reviews. 2012;92(2):635–688. doi: 10.1152/physrev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bradshaw AD. Diverse biological functions of the SPARC family of proteins. International Journal of Biochemistry and Cell Biology. 2012;44(3):480–488. doi: 10.1016/j.biocel.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brekken RA, Sage EH. SPARC, a matricellular protein: at the crossroads of cell-matrix communication. Matrix Biology. 2001;19(8):816–827. doi: 10.1016/s0945-053x(00)00133-5. [DOI] [PubMed] [Google Scholar]
- 22.Johnston IG, Paladino T, Gurd JW, Brown IR. Molecular cloning of SC1: a putative brain extracellular matrix glycoprotein showing partial similarity to osteonectin/BM40/SPARC. Neuron. 1990;4(1):165–176. doi: 10.1016/0896-6273(90)90452-l. [DOI] [PubMed] [Google Scholar]
- 23.Mason IJ, Taylor A, Williams JG, Sage H, Hogan BL. Evidence from molecular cloning that SPARC, a major product of mouse embryo parietal endoderm, is related to an endothelial cell ’culture shock’ glycoprotein of Mr 43,000. The EMBO Journal. 1986;5(7):1465–1472. doi: 10.1002/j.1460-2075.1986.tb04383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sullivan MM, Sage EH. Hevin/SC1, a matricellular glycoprotein and potential tumor-suppressor of the SPARC/BM-40/Osteonectin family. International Journal of Biochemistry and Cell Biology. 2004;36(6):991–996. doi: 10.1016/j.biocel.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 25.Hambrock HO, Kaufmann B, Müller S, et al. Structural characterization of TSC-36/Flik: analysis of two charge isoforms. Journal of Biological Chemistry. 2004;279(12):11727–11735. doi: 10.1074/jbc.M309318200. [DOI] [PubMed] [Google Scholar]
- 26.Maurer P, Hohenadl C, Hohenester E, Gohring W, Timpl R, Engel J. The C-terminal portion of BM-40 (SPARC/osteonectin) is an autonomously folding and crystallisable domain that binds calcium and collagen IV. Journal of Molecular Biology. 1995;253(2):347–357. doi: 10.1006/jmbi.1995.0557. [DOI] [PubMed] [Google Scholar]
- 27.Sasaki T, Miosge N, Timpl R. Immunochemical and tissue analysis of protease generated neoepitopes of BM-40 (osteonectin, SPARC) which are correlated to a higher affinity binding to collagens. Matrix Biology. 1999;18(5):499–508. doi: 10.1016/s0945-053x(99)00041-4. [DOI] [PubMed] [Google Scholar]
- 28.Rivera LB, Bradshaw AD, Brekken RA. The regulatory function of SPARC in vascular biology. Cellular and Molecular Life Sciences. 2011;68(19):3165–3173. doi: 10.1007/s00018-011-0781-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Campolongo M, Benedetti L, Podhajcer OL, Pitossi F, Depino AM. Hippocampal SPARC regulates depression-related behavior. Genes, Brain and Behavior. 2012 doi: 10.1111/j.1601-183X.2012.00848.x. [DOI] [PubMed] [Google Scholar]
- 30.Framson PE, Sage EH. SPARC and tumor growth: where the seed meets the soil? Journal of Cellular Biochemistry. 2004;92(4):679–690. doi: 10.1002/jcb.20091. [DOI] [PubMed] [Google Scholar]
- 31.Kucukdereli H, Allen NJ, Lee AT, et al. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins hevin and SPARC. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(32):E440–E449. doi: 10.1073/pnas.1104977108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gongidi V, Ring C, Moody M, et al. SPARC-like 1 regulates the terminal phase of radial glia-guided migration in the cerebral cortex. Neuron. 2004;41(1):57–69. doi: 10.1016/s0896-6273(03)00818-3. [DOI] [PubMed] [Google Scholar]
- 33.Eroglu C. The role of astrocyte-secreted matricellular proteins in central nervous system development and function. Journal of Cell Communication and Signaling. 2009;3(3-4):167–176. doi: 10.1007/s12079-009-0078-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lloyd-Burton S, Roskams AJ. SPARC-like 1 (SC1) is a diversely expressed and developmentally regulated matricellular protein that does not compensate for the absence of SPARC in the CNS. Journal of Comparative Neurology. 2012;520(12):2575–2590. doi: 10.1002/cne.23029. [DOI] [PubMed] [Google Scholar]
- 35.Vincent AJ, Lau PW, Roskams AJ. SPARC is expressed by macroglia and microglia in the developing and mature nervous system. Developmental Dynamics. 2008;237(5):1449–1462. doi: 10.1002/dvdy.21495. [DOI] [PubMed] [Google Scholar]
- 36.Lively S, Brown IR. Localization of the extracellular matrix protein SC1 coincides with synaptogenesis during rat postnatal development. Neurochemical Research. 2008;33(9):1692–1700. doi: 10.1007/s11064-008-9606-z. [DOI] [PubMed] [Google Scholar]
- 37.Mendis DB, Malaval L, Brown IR. SPARC, an extracellular matrix glycoprotein containing the follistatin module, is expressed by astrocytes in synaptic enriched regions of the adult brain. Brain Research. 1995;676(1):69–79. doi: 10.1016/0006-8993(95)00101-u. [DOI] [PubMed] [Google Scholar]
- 38.Weaver MS, Workman G, Cardo-Vila M, Arap W, Pasqualini R, Sage EH. Processing of the matricellular protein hevin in mouse brain is dependent on ADAMTS4. Journal of Biological Chemistry. 2010;285(8):5868–5877. doi: 10.1074/jbc.M109.070318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu X, Ying G, Wang W, et al. Entorhinal deafferentation induces upregulation of SPARC in the mouse hippocampus. Molecular Brain Research. 2005;141(1):58–65. doi: 10.1016/j.molbrainres.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 40.McKinnon PJ, Margolskee RF. SC1: a marker for astrocytes in the adult rodent brain is upregulated during reactive astrocytosis. Brain Research. 1996;709(1):27–36. doi: 10.1016/0006-8993(95)01224-9. [DOI] [PubMed] [Google Scholar]
- 41.Lively S, Brown IR. Analysis of the extracellular matrix protein SC1 during reactive gliosis in the rat lithium-pilocarpine seizure model. Brain Research. 2007;1163(1):1–9. doi: 10.1016/j.brainres.2007.05.052. [DOI] [PubMed] [Google Scholar]
- 42.Mendis DB, Ivy GO, Brown IR. Induction of SC1 mRNA encoding a brain extracellular matrix glycoprotein related to SPARC following lesioning of the adult rat forebrain. Neurochemical Research. 2000;25(12):1637–1644. doi: 10.1023/a:1026626805612. [DOI] [PubMed] [Google Scholar]
- 43.Mendis DB, Ivy GO, Brown IR. SC1, a brain extracellular matrix glycoprotein related to SPARC and follistatin, is expressed by rat cerebellar astrocytes following injury and during development. Brain Research. 1996;730(1-2):95–106. doi: 10.1016/0006-8993(96)00440-4. [DOI] [PubMed] [Google Scholar]
- 44.Lively S, Schlichter LC. SC1/hevin identifies early white matter injury after ischemia and intracerebral hemorrhage in young and aged rats. Journal of Neuropathology & Experimental Neurology. 2012;71(6):480–493. doi: 10.1097/NEN.0b013e318256901c. [DOI] [PubMed] [Google Scholar]
- 45.Lloyd-Burton SM, York EM, Anwar MA, Vincent AJ, Roskams AJ. SPARC regulates microgliosis and functional recovery following cortical ischemia. Journal of Neuroscience. 2013;33(10):4468–4481. doi: 10.1523/JNEUROSCI.3585-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang H, Colella S, Kurrer M, Yonekawa Y, Kleihues P, Ohgaki H. Gene expression profiling of low-grade diffuse astrocytomas by cDNA arrays. Cancer Research. 2000;60(24):6868–6874. [PubMed] [Google Scholar]
- 47.Özbas-Gerçeker F, Redeker S, Boer K, et al. Serial analysis of gene expression in the hippocampus of patients with mesial temporal lobe epilepsy. Neuroscience. 2006;138(2):457–474. doi: 10.1016/j.neuroscience.2005.11.043. [DOI] [PubMed] [Google Scholar]
- 48.Rempel SA, Golembieski WA, Ge S, et al. SPARC: a signal of astrocytic neoplastic transformation and reactive response in human primary and xenograft gliomas. Journal of Neuropathology and Experimental Neurology. 1998;57(12):1112–1121. doi: 10.1097/00005072-199812000-00002. [DOI] [PubMed] [Google Scholar]
- 49.Rich JN, Hans C, Jones B, et al. Gene expression profiling and genetic markers in glioblastoma survival. Cancer Research. 2005;65(10):4051–4058. doi: 10.1158/0008-5472.CAN-04-3936. [DOI] [PubMed] [Google Scholar]
- 50.Turtoi A, Musmeci D, Naccarato AG, et al. Sparc-like protein 1 is a new marker of human glioma progression. Journal of Proteome Research. 2012;11(10):5011–5021. doi: 10.1021/pr3005698. [DOI] [PubMed] [Google Scholar]
- 51.Esposito I, Kayed H, Keleg S, et al. Tumor-suppressor function of SPARC-like protein 1/hevin in pancreatic cancer. Neoplasia. 2007;9(1):8–17. doi: 10.1593/neo.06646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weaver M, Workman G, Schultz CR, Lemke N, Rempel SA, Sage EH. Proteolysis of the matricellular protein hevin by matrix metalloproteinase-3 produces a SPARC-like fragment (SLF) associated with neovasculature in a murine glioma model. Journal of Cellular Biochemistry. 2011;112(11):3093–3102. doi: 10.1002/jcb.23235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rich JN, Shi Q, Hjelmeland M, et al. Bone-related genes expressed in advanced malignancies induce invasion and metastasis in a genetically defined human cancer model. Journal of Biological Chemistry. 2003;278(18):15951–15957. doi: 10.1074/jbc.M211498200. [DOI] [PubMed] [Google Scholar]
- 54.Tremble PM, Lane TF, Sage EH, Werb Z. SPARC, a secreted protein associated with morphogenesis and tissue remodeling, induces expression of metalloproteinases in fibroblasts through a novel extracellular matrix-dependent pathway. Journal of Cell Biology. 1993;121(6):1433–1444. doi: 10.1083/jcb.121.6.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sage EH, Reed M, Funk SE, et al. Cleavage of the matricellular protein SPARC by matrix metalloproteinase 3 produces polypeptides that influence angiogenesis. Journal of Biological Chemistry. 2003;278(39):37849–37857. doi: 10.1074/jbc.M302946200. [DOI] [PubMed] [Google Scholar]
- 56.Weaver MS, Workman G, Sage EH. The copper binding domain of SPARC mediates cell survival in vitro via interaction with integrin β1 and activation of integrin-linked kinase. Journal of Biological Chemistry. 2008;283(33):22826–22837. doi: 10.1074/jbc.M706563200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lane TF, Iruela-Arispe ML, Johnson RS, Sage EH. SPARC is a source of copper-binding peptides that stimulate angiogenesis. Journal of Cell Biology. 1994;125(4):929–943. doi: 10.1083/jcb.125.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cunningham LA, Wetzel M, Rosenberg GA. Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia. 2005;50(4):329–339. doi: 10.1002/glia.20169. [DOI] [PubMed] [Google Scholar]
- 59.Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S. Abeta-degrading enzymes in Alzheimer's disease. Brain Pathology. 2008;18(2):240–252. doi: 10.1111/j.1750-3639.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dejonckheere E, Vandenbroucke RE, Libert C. Matrix metalloproteinases as drug targets in ischemia/reperfusion injury. Drug Discovery Today. 2011;16(17-18):762–778. doi: 10.1016/j.drudis.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 61.Zhao BQ, Wang S, Kim HY, et al. Role of matrix metalloproteinases in delayed cortical responses after stroke. Nature Medicine. 2006;12(4):441–445. doi: 10.1038/nm1387. [DOI] [PubMed] [Google Scholar]
- 62.Erickson HP. Tenascin-C, tenascin-R and tenascin-X: a family of talented proteins in search of functions. Current Opinion in Cell Biology. 1993;5(5):869–876. doi: 10.1016/0955-0674(93)90037-q. [DOI] [PubMed] [Google Scholar]
- 63.Nishio T, Kawaguchi S, Iseda T, Kawasaki T, Hase T. Secretion of tenascin-C by cultured astrocytes: regulation of cell proliferation and process elongation. Brain Research. 2003;990(1-2):129–140. doi: 10.1016/s0006-8993(03)03448-6. [DOI] [PubMed] [Google Scholar]
- 64.Keilhauer G, Faissner A, Schachner M. Differential inhibition of neuron-neurone, neurone-astrocyte and astrocyte-astrocyte adhesion by L1, L2, and N-CAM antibodies. Nature. 1985;316(6030):728–730. doi: 10.1038/316728a0. [DOI] [PubMed] [Google Scholar]
- 65.Kruse J, Keilhauer G, Faissner A. The J1 glycoprotein—a novel nervous system cell adhesion molecule of the L2/HNK-1 family. Nature. 1985;316(6024):146–148. doi: 10.1038/316146a0. [DOI] [PubMed] [Google Scholar]
- 66.Meiners S, Powell EM, Geller HM. A distinct subset of tenascin/CS-6-PG-rich astrocytes restricts neuronal growth in vitro. Journal of Neuroscience. 1995;15(12):8096–8108. doi: 10.1523/JNEUROSCI.15-12-08096.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Powell EM, Geller HM. Dissection of astrocyte-mediated cues in neuronal guidance and process extension. Glia. 1999;26(1):73–83. doi: 10.1002/(sici)1098-1136(199903)26:1<73::aid-glia8>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 68.Faissner A, Kruse J. J1/tenascin is a repulsive substrate for central nervous system neurons. Neuron. 1990;5(5):627–637. doi: 10.1016/0896-6273(90)90217-4. [DOI] [PubMed] [Google Scholar]
- 69.Andrews MR, Czvitkovich S, Dassie E, et al. α9 integrin promotes neurite outgrowth on tenascin-C and enhances sensory axon regeneration. Journal of Neuroscience. 2009;29(17):5546–5557. doi: 10.1523/JNEUROSCI.0759-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meiners S, Powell EM, Geller HM. Neurite outgrowth promotion by the alternatively spliced region of tenascin-C is influenced by cell-type specific binding. Matrix Biology. 1999;18(1):75–87. doi: 10.1016/s0945-053x(98)00008-0. [DOI] [PubMed] [Google Scholar]
- 71.Rigato F, Garwood J, Calco V, Heck N, Faivre-Sarrailh C, Faissner A. Tenascin-C promotes neurite outgrowth of embryonic hippocampal neurons through the alternatively spliced fibronectin type III BD domains via activation of the cell adhesion molecule F3/contactin. Journal of Neuroscience. 2002;22(15):6596–6609. doi: 10.1523/JNEUROSCI.22-15-06596.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Probstmeier R, Pesheva P. Tenascin-C inhibits β1 integrin-dependent cell adhesion and neurite outgrowth on fibronectin by a disialoganglioside-mediated signaling mechanism. Glycobiology. 1999;9(2):101–114. doi: 10.1093/glycob/9.2.101. [DOI] [PubMed] [Google Scholar]
- 73.Steindler DA, Cooper NGF, Faissner A, Schachner M. Boundaries defined by adhesion molecules during development of the cerebral cortex: the J1/tenascin glycoprotein in the mouse somatosensory cortical barrel field. Developmental Biology. 1989;131(1):243–260. doi: 10.1016/s0012-1606(89)80056-9. [DOI] [PubMed] [Google Scholar]
- 74.Götz M, Stoykova A, Gruss P. Pax6 controls radial glia differentiation in the cerebral cortex. Neuron. 1998;21(5):1031–1044. doi: 10.1016/s0896-6273(00)80621-2. [DOI] [PubMed] [Google Scholar]
- 75.Bartsch S, Bartsch U, Dorries U, et al. Expression of tenascin in the developing and adult cerebellar cortex. Journal of Neuroscience. 1992;12(3):736–749. doi: 10.1523/JNEUROSCI.12-03-00736.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ferhat L, Chevassus Au Louis N, Jorquera I, et al. Transient increase of tenascin-C in immature hippocampus: astroglial and neuronal expression. Journal of Neurocytology. 1996;25(1):53–66. doi: 10.1007/BF02284785. [DOI] [PubMed] [Google Scholar]
- 77.Karus M, Denecke B, Ffrench-Constant C, Wiese S, Faissner A. The extracellular matrix molecule tenascin C modulates expression levels and territories of key patterning genes during spinal cord astrocyte specification. Development. 2011;138(24):5321–5331. doi: 10.1242/dev.067413. [DOI] [PubMed] [Google Scholar]
- 78.Evers MR, Salmen B, Bukalo O, et al. Impairment of L-type Ca2+ channel-dependent forms of hippocampal synaptic plasticity in mice deficient in the extracellular matrix glycoprotein tenascin-C. Journal of Neuroscience. 2002;22(16):7177–7194. doi: 10.1523/JNEUROSCI.22-16-07177.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Irintchev A, Rollenhagen A, Troncoso E, Kiss JZ, Schachner M. Structural and functional aberrations in the cerebral cortex of tenascin-C deficient mice. Cerebral Cortex. 2005;15(7):950–962. doi: 10.1093/cercor/bhh195. [DOI] [PubMed] [Google Scholar]
- 80.Strekalova T, Sun M, Sibbe M, et al. Fibronectin domains of extracellular matrix molecule tenascin-C modulate hippocampal learning and synaptic plasticity. Molecular and Cellular Neuroscience. 2002;21(1):173–187. doi: 10.1006/mcne.2002.1172. [DOI] [PubMed] [Google Scholar]
- 81.Laywell ED, Dorries U, Bartsch U, Faissner A, Schachner M, Steindler DA. Enhanced expression of the developmentally regulated extracellular matrix molecule tenascin following adult brain injury. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(7):2634–2638. doi: 10.1073/pnas.89.7.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nishio T, Kawaguchi S, Yamamoto M, Iseda T, Kawasaki T, Hase T. Tenascin-C regulates proliferation and migration of cultured astrocytes in a scratch wound assay. Neuroscience. 2005;132(1):87–102. doi: 10.1016/j.neuroscience.2004.12.028. [DOI] [PubMed] [Google Scholar]
- 83.Ikeshima-Kataoka H, Shen JS, Eto Y, Saito S, Yuasa S. Alteration of inflammatory cytokine production in the injured central nervous system of tenascin-deficient mice. In Vivo. 2008;22(4):409–413. [PubMed] [Google Scholar]
- 84.Smith GM, Hale JH. Macrophage/microglia regulation of astrocytic tenascin: synergistic action of transforming growth factor-β and basic fibroblast growth factor. Journal of Neuroscience. 1997;17(24):9624–9633. doi: 10.1523/JNEUROSCI.17-24-09624.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Niquet J, Jorquera I, Faissner A, Ben-Ari Y, Represa A. Gliosis and axonal sprouting in the hippocampus of epileptic rats are associated with an increase of tenascin-C immunoreactivity. Journal of Neurocytology. 1995;24(8):611–624. doi: 10.1007/BF01257376. [DOI] [PubMed] [Google Scholar]
- 86.Becker AJ, Chen J, Zien A, et al. Correlated stage- and subfield-associated hippocampal gene expression patterns in experimental and human temporal lobe epilepsy. European Journal of Neuroscience. 2003;18(10):2792–2802. doi: 10.1111/j.1460-9568.2003.02993.x. [DOI] [PubMed] [Google Scholar]
- 87.Scheffler B, Faissner A, Beck H, et al. Hippocampal loss of tenascin boundaries in Ammon's horn sclerosis. Glia. 1997;19(1):35–46. [PubMed] [Google Scholar]
- 88.Soares HD, Potter WZ, Pickering E, et al. Plasma biomarkers associated with the apolipoprotein E genotype and Alzheimer disease. Archives of Neurology. 2012;69(10):1310–1317. doi: 10.1001/archneurol.2012.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hall JR, Johnson LA, Barber RC, Vo HT, Winter AS, 'Bryant SE O. Biomarkers of basic activities of daily living in Alzheimer's disease. Journal of Alzheimer's Disease. 2012;31(2):429–437. doi: 10.3233/JAD-2012-111481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cho CKJ, Smith CR, Diamandis EP. Amniotic fluid proteome analysis from down syndrome pregnancies for biomarker discovery. Journal of Proteome Research. 2010;9(7):3574–3582. doi: 10.1021/pr100088k. [DOI] [PubMed] [Google Scholar]
- 91.Xie K, Liu Y, Hao W, et al. Tenascin-C deficiency ameliorates Alzheimer's disease-related pathology in mice. Neurobiology of Aging. 2013;34(10):2389–2398. doi: 10.1016/j.neurobiolaging.2013.04.013. [DOI] [PubMed] [Google Scholar]
- 92.Midwood K, Sacre S, Piccinini AM, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nature Medicine. 2009;15(7):774–780. doi: 10.1038/nm.1987. [DOI] [PubMed] [Google Scholar]
- 93.Gorina R, Font-Nieves M, Márquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFκB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59(2):242–255. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- 94.Holm TH, Draeby D, Owens T. Microglia are required for astroglial toll-like receptor 4 response and for optimal TLR2 and TLR3 response. Glia. 2012;60(4):630–638. doi: 10.1002/glia.22296. [DOI] [PubMed] [Google Scholar]
- 95.Lehnardt S, Massillon L, Follett P, et al. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(14):8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi KI. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. Journal of Neuroinflammation. 2008;5(article 23) doi: 10.1186/1742-2094-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stefanova N, Fellner L, Reindl M, Masliah E, Poewe W, Wenning GK. Toll-like receptor 4 promotes α-synuclein clearance and survival of nigral dopaminergic neurons. American Journal of Pathology. 2011;179(2):954–963. doi: 10.1016/j.ajpath.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Adams JC, Lawler J. The thrombospondins. Cold Spring Harbor Perspectives in Biology. 2011;3(10) doi: 10.1101/cshperspect.a009712.a009712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Adams JC. Functions of the conserved thrombospondin carboxy-terminal cassette in cell-extracellular matrix interactions and signaling. International Journal of Biochemistry and Cell Biology. 2004;36(6):1102–1114. doi: 10.1016/j.biocel.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 100.Murphy-Ullrich JE, Iozzo RV. Thrombospondins in physiology and disease: new tricks for old dogs. Matrix Biology. 2012;31(3):152–154. doi: 10.1016/j.matbio.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Risher WC, Eroglu C. Thrombospondins as key regulators of synaptogenesis in the central nervous system. Matrix Biology. 2012;31(3):170–177. doi: 10.1016/j.matbio.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Christopherson KS, Ullian EM, Stokes CCA, et al. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120(3):421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- 103.Scott-Drew S, ffrench-Constant C. Expression and function of thrombospondin-1 in myelinating glial cells of the central nervous system. Journal of Neuroscience Research. 1997;50(2):202–214. doi: 10.1002/(SICI)1097-4547(19971015)50:2<202::AID-JNR9>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 104.Asch AS, Leung LLK, Shapiro J, Nachman RL. Human brain glial cells synthesize thrombospondin. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(9):2904–2908. doi: 10.1073/pnas.83.9.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Garcia O, Torres M, Helguera P, Coskun P, Busciglio J. A role for thrombospondin-1 deficits in astrocyte-mediated spine and synaptic pathology in down’s syndrome. PLoS ONE. 2010;5(12) doi: 10.1371/journal.pone.0014200.e14200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Eroglu Ç, Allen NJ, Susman MW, et al. Gabapentin receptor α2δ-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139(2):380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xu J, Xiao N, Xia J. Thrombospondin 1 accelerates synaptogenesis in hippocampal neurons through neuroligin 1. Nature Neuroscience. 2010;13(1):22–24. doi: 10.1038/nn.2459. [DOI] [PubMed] [Google Scholar]
- 108.Allen NJ. Role of glia in developmental synapse formation. Current Opinion in Neurobiology. 2013;23(6):1027–1033. doi: 10.1016/j.conb.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 109.Hoffman JR, Dixit VM, O’Shea KS. Expression of thrombospondin in the adult nervous system. Journal of Comparative Neurology. 1994;340(1):126–139. doi: 10.1002/cne.903400109. [DOI] [PubMed] [Google Scholar]
- 110.Iruela-Arispe ML, Liska DJ, Sage EH, Bornstein P. Differential expression of thrombospondin 1, 2, and 3 during murine development. Developmental Dynamics. 1993;197(1):40–56. doi: 10.1002/aja.1001970105. [DOI] [PubMed] [Google Scholar]
- 111.Lu Z, Kipnis J. Thrombospondin 1—a key astrocyte-derived neurogenic factor. FASEB Journal. 2010;24(6):1925–1934. doi: 10.1096/fj.09-150573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Blake SM, Strasser V, Andrade N, et al. Thrombospondin-1 binds to ApoER2 and VLDL receptor and functions in postnatal neuronal migration. EMBO Journal. 2008;27(22):3069–3080. doi: 10.1038/emboj.2008.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kim DS, Li KW, Boroujerdi A, et al. Thrombospondin-4 contributes to spinal sensitization and neuropathic pain states. Journal of Neuroscience. 2012;32(26):8977–8987. doi: 10.1523/JNEUROSCI.6494-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cahoy JD, Emery B, Kaushal A, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. Journal of Neuroscience. 2008;28(1):264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Arber S, Caroni P. Thrombospondin-4, an extracellular matrix protein expressed in the developing and adult nervous system promotes neurite outgrowth. Journal of Cell Biology. 1995;131(4):1083–1094. doi: 10.1083/jcb.131.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Liauw J, Hoang S, Choi M, et al. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. Journal of Cerebral Blood Flow and Metabolism. 2008;28(10):1722–1732. doi: 10.1038/jcbfm.2008.65. [DOI] [PubMed] [Google Scholar]
- 117.Lin TN, Kim GM, Chen JJ, Cheung WM, He YY, Hsu CY. Differential regulation of thrombospondin-1 and thrombospondin-2 after focal cerebral ischemia/reperfusion. Stroke. 2003;34(1):177–186. doi: 10.1161/01.str.0000047100.84604.ba. [DOI] [PubMed] [Google Scholar]
- 118.Tran MD, Neary JT. Purinergic signaling induces thrombospondin-1 expression in astrocytes. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(24):9321–9326. doi: 10.1073/pnas.0603146103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tran MD, Furones-Alonso O, Sanchez-Molano J, Bramlett HM. Trauma-induced expression of astrocytic thrombospondin-1 is regulated by P2 receptors coupled to protein kinase cascades. Neuroreport. 2012;23(12):721–726. doi: 10.1097/WNR.0b013e32835688fe. [DOI] [PubMed] [Google Scholar]
- 120.Yonezawa T, Hattori S, Inagaki J, et al. Type IV collagen induces expression of thrombospondin-1 that is mediated by integrin α1β1 in astrocytes. Glia. 2010;58(7):755–767. doi: 10.1002/glia.20959. [DOI] [PubMed] [Google Scholar]
- 121.Lawler PR, Lawler J. Molecular basis for the regulation of angiogenesis by thrombospondin-1 and -2. Cold Spring Harbor Perspectives in Medicine. 2012;2(5) doi: 10.1101/cshperspect.a006627.a006627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Neugebauer KM, Emmett CJ, Venstrom KA, Reichardt LF. Vitronectin and thrombospondin promote retinal neurite outgrowth: developmental regulation and role of integrins. Neuron. 1991;6(3):345–358. doi: 10.1016/0896-6273(91)90244-t. [DOI] [PubMed] [Google Scholar]
- 123.DeFreitas MF, Yoshida CK, Frazier WA, Mendrick DL, Kypta RM, Reichardt LF. Identification of integrin α3β1 as a neuronal thrombospondin receptor mediating neurite outgrowth. Neuron. 1995;15(2):333–343. doi: 10.1016/0896-6273(95)90038-1. [DOI] [PubMed] [Google Scholar]
- 124.O’Shea KS, Liu LHJ, Dixit VM. Thrombospondin and a 140 kd fragment promote adhesion and neurite outgrowth from embryonic central and peripheral neurons and from PC12 cells. Neuron. 1991;7(2):231–237. doi: 10.1016/0896-6273(91)90261-w. [DOI] [PubMed] [Google Scholar]
- 125.Osterhout DJ, Frazier WA, Higgins D. Thrombospondin promotes process outgrowth in neurons from the peripheral and central nervous systems. Developmental Biology. 1992;150(2):256–265. doi: 10.1016/0012-1606(92)90240-h. [DOI] [PubMed] [Google Scholar]
- 126.Moller JC, Klein MA, Haas S, Jones LL, Kreutzberg GW, Raivich G. Regulation of thrombospondin in the regenerating mouse facial motor nucleus. Glia. 1996;17(2):121–132. doi: 10.1002/(SICI)1098-1136(199606)17:2<121::AID-GLIA4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 127.Hoffman JR, O’Shea KS. Thrombospondin expression in nerve regeneration II. Comparison of optic nerve crush in the mouse and goldfish. Brain Research Bulletin. 1999;48(4):421–427. doi: 10.1016/s0361-9230(99)00022-2. [DOI] [PubMed] [Google Scholar]
- 128.Buee L, Hof PR, Roberts DD, Delacourte A, Morrison JH, Fillit HM. Immunohistochemical identification of thrombospondin in normal human brain and in Alzheimer’s disease. American Journal of Pathology. 1992;141(4):783–788. [PMC free article] [PubMed] [Google Scholar]
- 129.Sirko S, Behrendt G, Johansson PA, et al. Reactive glia in the injured brain acquire stem cell properties in response to sonic hedgehog. Cell Stem Cell. 2013;12(4):426–439. doi: 10.1016/j.stem.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 130.Thored P, Arvidsson A, Cacci E, et al. Persistent production of neurons from adult brain stem cells during recovery after stroke. Stem Cells. 2006;24(3):739–747. doi: 10.1634/stemcells.2005-0281. [DOI] [PubMed] [Google Scholar]
- 131.Benner EJ, Luciano D, Jo R, et al. Protective astrogenesis from the SVZ niche after injury is controlled by Notch modulator Thbs4. Nature. 2013;497(7449):369–373. doi: 10.1038/nature12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jun JI, Lau LF. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nature Reviews Drug Discovery. 2011;10(12):945–963. doi: 10.1038/nrd3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kular L, Pakradouni J, Kitabgi P, Laurent M, Martinerie C. The CCN family: a new class of inflammation modulators? Biochimie. 2011;93(3):377–388. doi: 10.1016/j.biochi.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 134.Kondo Y, Nakanishi T, Takigawa M, Ogawa N. Immunohistochemical localization of connective tissue growth factor in the rat central nervous system. Brain Research. 1999;834(1-2):146–151. doi: 10.1016/s0006-8993(99)01517-6. [DOI] [PubMed] [Google Scholar]
- 135.Meeuwsen S, Persoon-Deen C, Bsibsi M, Ravid R, Van Noort JM. Cytokine, chemokine and growth factor gene profiling of cultured human astrocytes after exposure to proinflammatory stimuli. Glia. 2003;43(3):243–253. doi: 10.1002/glia.10259. [DOI] [PubMed] [Google Scholar]
- 136.Schwab JM, Beschorner R, Nguyen TD, Meyermann R, Schluesener HJ. Differential cellular accumulation of connective tissue growth factor defines a subset of reactive astrocytes, invading fibroblasts, and endothelial cells following central nervous system injury in rats and humans. Journal of Neurotrauma. 2001;18(4):377–388. doi: 10.1089/089771501750170930. [DOI] [PubMed] [Google Scholar]
- 137.Schwab JM, Postler E, Nguyen TD, Mittelbronn M, Meyermann R, Schluesener HJ. Connective tissue growth factor is expressed by a subset of reactive astrocytes in human cerebral infarction. Neuropathology and Applied Neurobiology. 2000;26(5):434–440. doi: 10.1046/j.1365-2990.2000.00271.x. [DOI] [PubMed] [Google Scholar]
- 138.Hertel M, Tretter Y, Alzheimer C, Werner S. Connective tissue growth factor: a novel player in tissue reorganization after brain injury? European Journal of Neuroscience. 2000;12(1):376–380. doi: 10.1046/j.1460-9568.2000.00930.x. [DOI] [PubMed] [Google Scholar]
- 139.Holley JE, Gveric D, Newcombe J, Cuzner ML, Gutowski NJ. Astrocyte characterization in the multiple sclerosis glial scar. Neuropathology and Applied Neurobiology. 2003;29(5):434–444. doi: 10.1046/j.1365-2990.2003.00491.x. [DOI] [PubMed] [Google Scholar]
- 140.Spliet WGM, Aronica E, Ramkema M, Aten J, Troost D. Increased expression of connective tissue growth factor in amyotrophic lateral sclerosis human spinal cord. Acta Neuropathologica. 2003;106(5):449–457. doi: 10.1007/s00401-003-0741-y. [DOI] [PubMed] [Google Scholar]
- 141.Ueberham U, Ueberham E, Gruschka H, Arendt T. Connective tissue growth factor in Alzheimer’s disease. Neuroscience. 2003;116(1):1–6. doi: 10.1016/s0306-4522(02)00670-x. [DOI] [PubMed] [Google Scholar]
- 142.Halliday JJ, Holland EC. Connective tissue growth factor and the parallels between brain injury and brain tumors. Journal of the National Cancer Institute. 2011;103(15):1141–1143. doi: 10.1093/jnci/djr261. [DOI] [PubMed] [Google Scholar]
- 143.Buss A, Pech K, Kakulas BA, et al. TGF-β1 and TGF-β2 expression after traumatic human spinal cord injury. Spinal Cord. 2008;46(5):364–371. doi: 10.1038/sj.sc.3102148. [DOI] [PubMed] [Google Scholar]
- 144.Lindholm D, Castren E, Kiefer R, Zafra F, Thoenen H. Transforming growth factor-β1 in the rat brain: increase after injury and inhibition of astrocyte proliferation. Journal of Cell Biology. 1992;117(2):395–400. doi: 10.1083/jcb.117.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Logan A, Berry M, Gonzalez AM, Frautschy SA, Sporn MB, Baird A. Effects of transforming growth factor β1 on scar production in the injured central nervous system of the rat. European Journal of Neuroscience. 1994;6(3):355–363. doi: 10.1111/j.1460-9568.1994.tb00278.x. [DOI] [PubMed] [Google Scholar]
- 146.Montgomery SL, Bowers WJ. Tumor necrosis factor-alpha and the roles it plays in homeostatic and degenerative processes within the central nervous system. Journal of Neuroimmune Pharmacology. 2012;7(1):42–59. doi: 10.1007/s11481-011-9287-2. [DOI] [PubMed] [Google Scholar]
- 147.Santello M, Volterra A. TNFalpha in synaptic function: switching gears. Trends in Neurosciences. 2012;35(10):638–647. doi: 10.1016/j.tins.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 148.Wahab NA, Weston BS, Mason RM. Connective tissue growth factor CCN2 interacts with and activates the tyrosine kinase receptor TrkA. Journal of the American Society of Nephrology. 2005;16(2):340–351. doi: 10.1681/ASN.2003100905. [DOI] [PubMed] [Google Scholar]
- 149.Wilhelmsson U, Bushong EA, Price DL, et al. Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(46):17513–17518. doi: 10.1073/pnas.0602841103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wanner IB, Anderson MA, Song B, et al. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. Journal of Neuroscience. 2013;33(31):12870–12886. doi: 10.1523/JNEUROSCI.2121-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bardehle S, Krüger M, Buggenthin F, et al. Live imaging of astrocyte responses to acute injury reveals selective juxtavascular proliferation. Nature Neuroscience. 2013;16(5):580–586. doi: 10.1038/nn.3371. [DOI] [PubMed] [Google Scholar]
- 152.Wiese S, Karus M, Faissner A. Astrocytes as a source for extracellular matrix molecules and cytokines. Frontiers in Pharmacology. 2012;3(article 120) doi: 10.3389/fphar.2012.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Robel S, Berninger B, Götz M. The stem cell potential of glia: lessons from reactive gliosis. Nature Reviews Neuroscience. 2011;12(2):88–104. doi: 10.1038/nrn2978. [DOI] [PubMed] [Google Scholar]
- 154.Chaboub LS, Deneen B. Developmental origins of astrocyte heterogeneity: the final frontier of CNS development. Developmental Neuroscience. 2012;34(5):379–388. doi: 10.1159/000343723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rusnakova V, Honsa P, Dzamba D, Ståhlberg A, Kubista M, Anderova M. Heterogeneity of astrocytes: from development to injury—single cell gene expression. PLoS ONE. 2013;8(8) doi: 10.1371/journal.pone.0069734.e69734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.García-Marín V, García-López P, Freire M. Cajal’s contributions to glia research. Trends in Neurosciences. 2007;30(9):479–487. doi: 10.1016/j.tins.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 157.Kamphuis W, Orre M, Kooijman L, Dahmen M, Hol EM. Differential cell proliferation in the cortex of the APPswePS1dE9 Alzheimer’s disease mouse model. Glia. 2012;60(4):615–629. doi: 10.1002/glia.22295. [DOI] [PubMed] [Google Scholar]
- 158.Savani RC, Zhou Z, Arguiri E, et al. Bleomycin-induced pulmonary injury in mice deficient in SPARC. American Journal of Physiology—Lung Cellular and Molecular Physiology. 2000;279(4):L743–L750. doi: 10.1152/ajplung.2000.279.4.L743. [DOI] [PubMed] [Google Scholar]
- 159.Kubota S, Takigawa M. CCN family proteins and angiogenesis: from embryo to adulthood. Angiogenesis. 2007;10(1):1–11. doi: 10.1007/s10456-006-9058-5. [DOI] [PubMed] [Google Scholar]
- 160.Bush TG, Puvanachandra N, Horner CH, et al. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23(2):297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- 161.Herrmann JE, Imura T, Song B, et al. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. Journal of Neuroscience. 2008;28(28):7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Voskuhl RR, Peterson RS, Song B, et al. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. Journal of Neuroscience. 2009;29(37):11511–11522. doi: 10.1523/JNEUROSCI.1514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Silver J, Miller JH. Regeneration beyond the glial scar. Nature Reviews Neuroscience. 2004;5(2):146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 164.Barros CS, Franco SJ, Müller U. Extracellular matrix: functions in the nervous system. Cold Spring Harbor Perspectives in Biology. 2011;3(1) doi: 10.1101/cshperspect.a005108.a005108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Murphy-Ullrich JE. The de-adhesive activity of matricellular proteins: is intermediate cell adhesion an adaptive state? Journal of Clinical Investigation. 2001;107(7):785–790. doi: 10.1172/JCI12609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Zipp F, Aktas O. The brain as a target of inflammation: common pathways link inflammatory and neurodegenerative diseases. Trends in Neurosciences. 2006;29(9):518–527. doi: 10.1016/j.tins.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 167.Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease—a double-edged sword. Neuron. 2002;35(3):419–432. doi: 10.1016/s0896-6273(02)00794-8. [DOI] [PubMed] [Google Scholar]
- 168.Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harbor Perspectives in Medicine. 2012;2(1) doi: 10.1101/cshperspect.a006346.006346 [DOI] [PMC free article] [PubMed] [Google Scholar]