

Abstract
To reach cancer cells in a tumor, a blood-borne therapeutic molecule or cell must make its way into the blood vessels of the tumor and across the vessel wall into the interstitium, and finally migrate through the interstitium. Unfortunately, tumors often develop in ways that hinder each of these steps. Our research goals are to analyze each of these steps experimentally and theoretically, and then integrate the resulting information in a unified theoretical framework. This paradigm of analysis and synthesis has allowed us to obtain a better understanding of physiological barriers in solid tumors, and to develop novel strategies to exploit and/or to overcome these barriers for improved cancer detection and treatment.
Keywords: Tumor microcirculation, Angiogenesis, Blood flow, Vascular permeability, Diffusion and convection, Receptor-ligand binding, Interstitial pressure, Lymphatics, Cell adhesion and deformation, Cancer detection and treatment
1. Introduction
Cancer is the second leading cause of death in the United States and in many industrialized countries [1]. After the primary tumor has been surgically removed and/or sterilized by radiation, the residual disease is usually managed with a variety of systemic therapies (Table 1). For these therapies to be successful, they must satisfy two requirements: (a) the relevant agent must be effective in the in vivo orthotopic microenvironment of tumors, and (b) this agent must reach the target cells in vivo in optimal quantities. The goal of our research is to examine the latter issue – the delivery of diagnostic and therapeutic agents to solid tumors and normal host tissues.
Table 1.
Systemic therapy of cancer
| Therapy | Agent
|
||
|---|---|---|---|
| Molecules | Particles | Cells | |
| Radiotherapy | × | × | |
| Chemotherapy | × | × | |
| Immunotherapy | × | × | × |
| Gene therapy | × | × | × |
| Hyperthermia | × | ||
| Phototherapy | × | × | |
Agents used in various conventional and novel therapies can be divided in three categories: molecules, particles, and cells
All conventional and novel therapeutic agents can be divided into three categories – molecules, particles and cells (Table 1). A blood-borne molecule or particle that enters the tumor vasculature reaches cancer cells via distribution through the vascular compartment, transport across the microvascular wall, and transport through the interstitial compartment. For a molecule of given size, charge, and configuration, each of these transport processes may involve diffusion and convection. In addition, during the journey the molecule may bind nonspecifically to proteins or other tissue components, bind specifically to the target(s), or be metabolized [2]. Although lymphokine-activated killer (LAK) cells (lymphocytes activated by the lymphokine interleukin-2) or tumor-infiltrating lymphocytes (TIL) are capable of deformation, adhesion, and migration, they encounter the same barriers that restrict their movement in tumors. Some of these physiological parameters are also important for heat transfer in normal and tumor tissues during hyperthermic treatment of cancer [3].
The overall aim of our research is to develop a quantitative understanding of each of the abovementioned steps involved in the delivery of various agents. More specifically, our goals are to understand: (1) how angiogenesis takes place and what determines blood flow heterogeneities in tumors; (2) how blood flow influences the metabolic microenvironment in tumors, and how microenvironment affects the biological properties of tumors (e.g., vascular permeability; cell adhesion); (3) how material moves across the microvascular wall; and (4) how it moves through the interstitial compartment and the lymphatics. In addition, we are examining the role of cell deformation and adhesion in the delivery of cells. Following analysis of these processes for molecules, particles and cells, we integrate this information in a unified framework for scale-up from mice to men (Fig. 1). In this article, I will briefly describe various experimental and theoretical approaches used in our lab, our recent findings in these six areas, and finally, how we have taken some of these concepts from bench to bedside for potential improvement in cancer detection and treatment.
Fig. 1.

Quantitative understanding of various steps involved in the delivery of therapeutic agents is studied by analyzing the underlying processes and then integrating the resulting information in a unified framework. More specifically, our goal is to develop a quantitative understanding of (a) angiogenesis and blood flow; (b) metabolic microenvironment; (c) transvascular transport; (d) interstitial and lymphatic transport; (e) cell transport, and (f) systemic distribution and interspecies scale-up.
2. Experimental and theoretical approaches
We have utilized five approaches to gain insight into the pathophysiology of solid tumors:
A tissue-isolated tumor which is connected to the host’s circulation by a single artery and a single vein [4,5]. This technique was originally developed by P.M. Gullino at the National Cancer Institute in 1961 for rats [6]; we have recently adapted it to mice [7,8] and humans [9].
A modified Sandison rabbit ear chamber [10,11], a modified Algire mouse dorsal chamber [12,13], and a cranial window in mice and rats [14]. The ear chamber has the advantage of superior optical quality and the mice of working with immunodeficient and genetically engineered animals [15,16]. Recently we have developed a quantitative angiogenesis assay using these windows to study the physiology of vessels induced by individual growth factors [17] (Fig. 2). We also perfuse single vessels of tumors in these windows [18]. We also utilize two acute preparations: liver and mesentery.
In vitro methods to assess the deformability, adhesion and permeability of normal and neoplastic cells [19–22], as well as measurements of adhesion molecules’ expression in intact monolayers [23] (Fig. 3).
Routine molecular biology techniques (e.g., in situ hybridization, Southern, Northern and Western blotting).
Mathematical models to describe and integrate the data obtained from the above three approaches, to scale up biodistribution data from mice to men, and to design future experiments [24–37].
Fig. 2.

Various microcirculatory preparations used to study delivery of therapeutic agents in solid tumors: (a) Sandison window in the rabbit ear [11]; (b) Algire window in the dorsal skin of rodents [13]; (c) cranial window in rodents [14]; and (d) collagen I gel, containing angiogenic factors, sandwiched between nylon mesh (3 × 3 mm) to permit the growth of blood vessels [17]. These preparations allow noninvasive, continuous measurement of angiogenesis and blood flow; metabolites, such as pH, pO2; transport of molecules and particles; and cell–cell interactions in vivo.
Fig. 3.

Targeted sampling fluorometry (TSF) allows the quantification of adhesion molecule expression over an intact cell monolayer on a cell-by-cell basis. At top are the two images acquired for analysis: the red nuclei are stained with propidium iodide and the adhesion molecule is labeled with fluorescein (green) using double immunostaining. The nuclei are first located in the propidium iodided channel, and regions of interest (ROIs) formed around each nucleus (bottom left); these ROIs are then applied to the immunostain image to find the fluorescence intensity in each region, corresponding to one cell. The procedure yields a histogram of intensities for the monolayer (adapted from Ref. [23]).
While each of these approaches has its limitations, it is their combination that has permitted us to develop the framework for tumor microcirculation and drug delivery described in this article.
3. Distribution through vascular space
The tumor vasculature consists of both vessels recruited from the pre-existing network of the host vasculature, and vessels resulting from the angiogenic response of host vessels to cancer cells [38,39]. Movement of molecules through the vasculature is governed by the vascular morphology (i.e., the number, length, diameter, and geometric arrangement of various blood vessels) and the blood flow rate [25,40–42].
Although the tumor vasculature originates from the host vasculature and the mechanisms of angiogenesis are similar [38,43], its organization may be completely different depending on the tumor type, its growth rate, and its location [42]. The fractal dimensions and minimum path lengths of tumor vasculature are different from those of the normal host vessels [40,41]. The architecture and blood flow are different not only among various tumor types but also between a spontaneous tumor and its transplants [39,44]. For example, unlike normal tissue, where RBC velocity is dependent on vessel diameter, there is no such dependence in tumors [13,14]. Further-more, the RBC velocity may be an order of magnitude lower in tumors compared to the host vessels (Fig. 4). The temporal and spatial heterogeneity in tumor blood flow may, in part, be a result of elevated geometric and viscous resistance in tumor vessels [5,45,46], coupling between high vascular permeability and elevated interstitial fluid pressure [34], and vascular remodeling by intussusception [43].
Fig. 4.

Blood velocity as a function of vessel diameter in (a) normal pial vessels, and (b) a human glioma (U87) xenograft on the pial surface. Note that in normal microcirculation, blood velocity is dependent on vessel diameter, whereas in tumors there is no such dependence. Furthermore, the blood velocity in tumor vessels is about an order of magnitude lower than in host vessels (adapted from Ref. [14]).
Based on perfusion rates, four regions can be recognized in a tumor: an avascular, necrotic region; a seminecrotic region; a stabilized microcirculation region; and an advancing front [47] (Fig. 5a). Intratumor blood flow distributions in spontaneous animal and human tumors are now being investigated using nuclear magnetic resonance, positron emission tomography, and functional computed tomography [30,48,49]. While limited, these results are in concert with the transplanted tumor studies: blood flow rates in necrotic and seminecrotic regions of tumors are low, while those in non-necrotic regions are variable and can be substantially higher than in surrounding (contralateral) host normal tissues [50]. Considering these spatial and temporal heterogeneities in blood supply coupled with variations in the vascular morphology at both microscopic and macroscopic levels, it is not surprising that the spatial distribution of therapeutic agents in tumors is heterogeneous and that the average uptake decreases, in general, with an increase in tumor weight. This perfusion heterogeneity also makes it difficult to heat the tumor periphery during hyperthermia [3].
Fig. 5.

Physiological barriers that a blood-borne molecule encounters before it reaches a cancer cell in a solid tumor. (a) Schematic of a heterogeneously perfused tumor showing well-vascularized periphery; a seminecrotic, intermediate zone; and an avascular, necrotic central region. Note that, immediately after i.v. injection, the molecules are delivered to perfused regions only. (b) Low interstitial pressure in the periphery permits adequate extravasation of fluid and macromolecules. (c) These macromolecules move toward the center by the slow process of diffusion. In addition, interstitial fluid oozing from tumor carries macromolecules with it by convection into the normal tissue. Note that the interstitial movement may be further retarded by binding. Products of metabolism may be cleared rapidly by blood (reproduced from Ref. [102]).
4. Metabolic microenvironment
The temporal and spatial heterogeneities in blood flow are expected to lead to a compromised metabolic microenvironment in tumors. To quantify the spatial gradients of key metabolites, we have recently adapted two optical techniques: fluorescence ratio-imaging microscopy (FRIM) and phosphorescence quenching microscopy (PQM) [51–55]. As shown in Fig. 6, both pH and pO2 decrease as one moves away from tumor vessels leading to acidic and hypoxic regions in tumors. While low pO2 and pH are detrimental to some therapies (e.g., radiation), they might enhance the effect of certain drugs, if the drug could be delivered in adequate quantities in those regions [56–58].
Fig. 6.
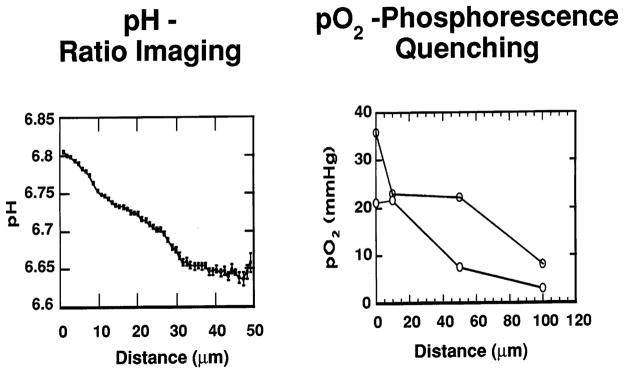
Spatial gradients of metabolites in tumors. (a) pH gradients measured using fluorescence ratio imaging microscopy (adapted from Ref. [54]). (b) pO2 gradients measured using phosphorescence quenching (adapted from Ref. [55]). Distance from the vessel wall, in microns, is shown on the x-axis, with zero being the vessel wall.
To gain further insight into tumor metabolism, we have combined two powerful approaches: magnetic resonance spectroscopy and tissue isolated tumors. The former allows us to measure the energy level in tumors while the latter allows us to control the supply of individual substrates (e.g., glucose, oxygen) to the tumor. Using this approach, we have recently shown that solid tumors depend more on glucose than oxygen to maintain their ATP level [59].
5. Transport across the microvascular wall
Once a blood-borne molecule has reached an exchange vessel, its extravasation, Js(g/s), occurs by diffusion and convection and, to some extent, presumably by transcytosis [60]. Diffusive flux is proportional to the exchange vessel’s surface area, S (cm2), and the difference between the plasma and interstitial concentrations, Cp−Ci(g/m). Convection is proportional to the rate of fluid leakage, Jf (m/s), from the vessel. Jf, in turn, is proportional to S and the difference between the vascular and interstitial hydrostatic pressures, pv−pi (mmHg), minus the osmotic reflection coefficient (s) times the difference between the vascular and interstitial osmotic pressures pv−pi (mmHg). The proportionality constant that relates transluminal diffusion flux to concentration gradients Cp−Ci is referred to as the vascular permeability coefficient, P (cm/s), and the constant that relates fluid leakage to pressure gradients is referred to as the hydraulic conductivity, Lp (cm/mmHg · s). The effectiveness of the transluminal osmotic pressure difference in producing fluid movement across a vessel wall is characterized by s, which is close to 1 for a macromolecule and close to zero for a small molecule. Thus, the transport of a molecule across normal or tumor vessels is governed by three transport parameters (P, Lp, and S), the surface area for exchange, and the transvascular concentration and pressure gradients.
Vascular permeability and hydraulic conductivity of tumors in general is significantly higher than that of various normal tissues [14,60–65] and, hence, these vessels may lack permselectivity [66] (Fig. 7a, b). Positively changed molecules have a higher permeability [67]. Despite increased overall permeability, not all blood vessels of a tumor are leaky (Fig. 7b). Even the leaky vessels have a finite pore size, which we have been able to measure in a variety of human and rodent tumors [68], including a human colon carcinoma (LS174T) xenografted in the dorsal window (Fig. 7c, d). Our hypothesis is that the large pore size in tumors represents wide inter-endothelial junctions [69]. Not only does the vascular permeability vary from one tumor to the next, but within the same tumor it varies both spatially and temporally [60]. The local microenvironment plays an important role in controlling vascular permeability. For example, a human glioma (HGL21) is fairly leaky when grown subcutaneously in immunodeficient mice, but it exhibits blood–brain barrier properties in the cranial window (Fig. 7e, f). We have not seen such site-dependent differences for other tumors. Our working hypothesis is that the host-tumor interactions control the production and secretion of cytokines associated with permeability changes (e.g., VPF/VEGF and its inhibitors). A better understanding of the molecular mechanisms of permeability-regulation in tumors is likely to yield strategies for improved drug delivery [70].
Fig. 7.

Transvascular transport in dorsal skin and tumors. (a) There is hardly any extravasation of 90-nm diameter liposomes from normal vessels; (b) heterogeneous extravasation of 90-nm diameter liposomes from LS174T tumor vessels, 48 h after injection. Note that some vessels are leaky as indicated by the yellow fluorescence for rhodamine, while others are not. Extravasated liposomes do not diffuse far from blood vessels (adapted from Ref. [14]). (c) Liposomes of 400 nm diameter (yellow fluorescent spots) extravasate adequately from LS174T tumor. (d) Liposomes of 600 nm diameter do not extravasate, suggesting that LS174T vessels have pore-size cut-off of about 500 nm (adapted from Ref. [66]). (e) The human glioma (HGL21) xenograft is permeable to Lissamin green (i.e., tumor tissue becomes green) when grown subcutaneously (Yuan and Jain, unpublished results). (f) The same glioma develops blood–brain barrier properties (i.e., impermeable to Lissamin green) when grown in the cranial window (adapted from Ref. [14]).
If tumor vessels are indeed ‘leaky’ to fluid and macromolecules, then what leads to the poor extravasation of these agents in various regions of tumors? As shown by us and others [71–84], experimental and human tumors exhibit high interstitial fluid pressure. Furthermore, the uniformly high pressure drops precipitously to normal values in the tumor’s periphery or in the peritumor region [24,31,72]. This may lower fluid extravasation in the high pressure regions, especially because the oncotic and hydrostatic pressures are also equal between the intravascular and extravascular space [73,85]. Because the transvascular transport of macromolecules in normal tissues occurs primarily by convection [60,86], convective transport of macromolecules in the center of tumors may be less than in the tumor periphery [18,24,31]. Additionally, the average vascular surface area per unit tissue weight decreases with tumor growth, hence reduced transvascular exchange would be expected in large tumors compared with small tumors [24,25].
6. Transport through interstitial space and lymphatics
Once a molecule has extravasated, its movement through the interstitial space occurs by diffusion and convection [79]. Diffusion is proportional to the concentration gradient in the interstitium, and convection is proportional to the interstitial fluid velocity, ui (cm/s). The latter, in turn, is proportional to the pressure gradient in the interstitium. Just as the interstitial diffusion coefficient, D (cm2/s), relates the diffusive flux to the concentration gradient, the interstitial hydraulic conductivity, K (cm2/mmHg · s), relates the interstitial velocity to the pressure gradient [79]. Values of these transport coefficients are determined by the structure and composition of the interstitial compartment as well as the physicochemical properties of the solute molecule [87–93].
Using fluorescence recovery after photobleaching (FRAP) we have found D of various molecules to be about 1/3 that in water [94] and similar to that in the host tissue [88]. Similarly, the value of K for a human colon carcinoma xenograft (LS174T), measured using two different methods [95,96], was found to be higher than that of a hepatoma [93], which in turn was higher than that of the liver. Given these relatively high values of D and K, why do exogenously injected macromolecules not distribute uniformly in tumors? As discussed next, there are two reasons for this apparent paradox.
The time constant for a molecule with diffusion coefficient D to diffuse across distance L is approximately L2/4D. For diffusion of IgG in tumors, this time constant is on the order of 1 h for a 100-μm distance, days for a 1-mm distance, and months for a 1-cm distance. So, for a 1-mm tumor, diffusional transport would take days, and for a 1-cm tumor it would take months. If the central vessels have collapsed completely due to cellular proliferation and interstitial matrix rearrangement there would be no delivery of macromolecules by blood flow to this necrotic center. Binding may further retard the transport in tumors [26,27,94,97–101]. The role of binding is clearly illustrated in Fig. 8, which compares the rate of fluorescence recovery of a photobleached spot in tumor tissue injected with a non-specific vs. specific IgG. In addition to the heterogeneity in D in tumors, the most unexpected result of these photobleaching studies was the large extent (30–40%) of non-specific binding [94].
Fig. 8.

Role of binding in the interstitial transport in tumors, measured using fluorescence recovery after photobleaching. (a) Recovery of a photobleached spot is complete in about 100 s for a non-specific monoclonal antibody. (b) Recovery is incomplete for an antibody against carcino-embryonic antigen, present on the surface of many carcinoma cells (adapted from Ref. [94]).
As mentioned earlier, interstitial fluid pressure is high in the center of tumors and low in the periphery and surrounding tissue [24,31,72]. Therefore, one would expect interstitial fluid motion from the tumor’s periphery into the surrounding normal tissue (Fig. 5b, c). In various animal and human (xenograft) tumors studied to date, 6–14% of plasma entering the tumor has been found to leave from the tumor’s periphery [60,102]. This fluid leakage leads to a radially outward interstitial fluid velocity of 0.1–0.2 μm/s at the periphery of a 1-cm ‘tissue-isolated’ tumor [60]. (The radially outward velocity is likely to be an order of magnitude lower in a tumor grown in the subcutaneous tissue or muscle [24].) A macromolecule at the tumor periphery has to overcome this outward convection to diffuse into the tumor. The relative contribution of this mechanism of heterogeneous distribution of antibodies in tumors may be smaller than the contribution of heterogeneous extravasation due to elevated pressure and necrosis [24].
In most normal tissues, extravasated macromolecules are taken up by the lymphatics and brought back to the central circulation. Because of the lack of functional lymphatics within the tumor, the fluid and macromolecules oozing from the tumor surface must be picked by the peri-tumor host lymphatics [25]. To characterize the transport into and within the lymphatic capillaries, we have recently developed a mouse tail model [103]. We have measured uptake and transport in this model using a macroscopic approach (RTD analysis) and a microscopic approach (FRAP) [104,105]. Our current efforts are directed towards understanding changes in lymphatic transport in the presence of a tumor [106].
7. Transport of cells
So far we have discussed the parameters that govern the transport of molecules and particles (e.g., liposomes) in tumors. When a leukocyte enters a blood vessel, it may continue to move with flowing blood, collide with the vessel wall, adhere transiently or stably, and finally extravasate. These interactions are governed by both local hydrodynamic forces and adhesive forces. The former are determined by the vessel diameter and fluid velocity, and the latter by the expression, strength and kinetics of bond formation between adhesion molecules and by surface area of contact [107,108]. Deformability of cells affects both types of forces. Despite their importance in immunotherapy and gene therapy, the determinants of cell transport in tumors have not been examined.
Using intravital microscopy, we have recently shown that rolling of endogenous leukocytes is generally low in tumor vessels, whereas stable adhesion (> 30 s) is comparable between normal and tumor vessels (Fig. 9a, b) [109]. On the other hand, both rolling and stable adhesion are nearly zero in angiogenic vessels induced in collagen gels by bFGF or VEGF/VPF, two of the most potent angiogenic factors [17]. Whether the latter is due to a low flux of leukocytes into angiogenic vessels and/ or downregulation of adhesion molecules in these immature vessels is currently under investigation. The age of the animal also plays an important role in leukocyte-endothelial interactions [110].
Fig. 9.

Leukocyte-endothelial interactions in normal and tumor [109] and angiogenic [17] vessels in the dorsal skin window and the cranial window: (a) rolling, and (b) adhesion. Note that rolling is significantly reduced in tumor vessels compared to host vessels, while stable adhesion is similar in both vessel types. Both rolling and adhesion are negligible in angiogenic vessels.
To gain further insight into the type of cells that adhere to tumor vessels, we examined the localization of IL-2-activated natural killer (A-NK) cells in normal and tumor tissues in mice using positron emission tomography [19,111]. Following systemic injection, we found that these cells localized primarily in the lungs immediately after injection and a non-detectable number of cells arrived in the tumor [19]. These findings were consistent with our previous work on the deformability of these cells using micropipet aspiration technique, in which we showed that IL-2 activation makes these cells rigid, and predicted their mechanical entrapment in the lung microcirculation [21,112]. Constitutive expression of certain adhesion molecules in the lung vasculature also facilitates their localization in the lungs [113].
One approach to reduce lung entrapment is to reduce the rigidity of these cells [114]. Instead, to circumvent the lung, we decided to inject A-NK cells into the blood supply of tumors, and we found that A-NK cells, both xenogenic and syngeneic, adhered to blood vessels in three different tumor models [111,115,116]. These results also supported the hypothesis that the endogenous cells that adhere to tumor vessels after systemic IL-2 injection are mostly activated lymphocytes [117].
To find out the adhesion molecules involved in the A-NK cell adhesion to tumor vessels, we utilized two in vitro approaches. In the first approach, we simulated the tumor vasculature in vitro, by incubating the human umbilical vein endothelial cells (HUVECs) in the tumor interstitial fluid collected using a micropore chamber [6,33,56,118]. Using targeted sampling fluorometry (Fig. 3), we were able to quantify the expression of relevant adhesion molecules on the HUVEC monolayers [23]. To determine the relative contributions of these molecules in adhesion under physiological flow conditions, we utilized the flow chamber [20]. Using appropriate antibodies, we found that molecules upregulated on the HUVECs include ICAM-1 and VCAM-1, which bind to CD18 and VLA-4 on the A-NK cells. We also observed sporadic upregulation of E-selectin. We were able to confirm the role of these molecules in vivo by treating A-NK cells with antibodies against CD18 and VLA-4 prior to injecting them into the arterial supply of tumors. As in our in vitro studies, blocking these adhesion molecules nearly eliminated the adhesion of A-NK cells to tumor vessels [118].
What leads to the upregulation of these molecules in the tumor vasculature? We already knew that these molecules can be upregulated by TNFα and a protein of 90 kDa molecular weight (p90) secreted by some neoplastic cells [107,119,120], and downregulated by TGFβ [121–123]. We wanted to find out if there are other molecules present in the tumor milieu that are also inducing this upregulation. Since tumor growth and metastasis are angiogenesis dependent, we decided to focus on the two most potent angiogenic molecules — bFGF and VEGF/VPF [38,113,124]. We found that VEGF can mimic tumor interstitial fluid, and upregulate these molecules. bFGF, on the other hand, exhibited no effect when used alone, but abrogated the upregulation induced by VEGF or TNFα [118]. These findings were in concert with earlier reports that bFGF retards the transmigration of lymphocytes across endothelial monolayer [125] and reduces adhesion of endothelial cells to collagen [126]. They also offer a possible explanation for lower leukocyte-endothelial interactions in tumors; bFGF might have downregulated adhesion molecules in these tumors. Our current efforts are directed towards defining interactions between angiogenic and adhesion molecules using various in vitro and in vivo approaches, including genetically engineered mice [15,113].
8. Pharmacokinetic modeling: scale up from mouse to human
So far we have analyzed each of the steps in the delivery of molecules and cells to and within solid tumors. Can we take this information and integrate it in a unified framework? We have been successful to some extent in this endeavor, using physiologically-based pharmacokinetic modeling. This approach, pioneered by two chemical engineers, K. Bischoff and R.L. Dedrick, in the 1960s, has been applied successfully to describe and scale up the biodistribution of low-molecular weight agents (for a review, see Refs. [3,127,128]). We have extended this approach to macromolecules and cells [28,29,129–131].
In this approach, a mammalian body is represented by a number of physiological compartments interconnected anatomically (Fig. 10). The volume and blood flow rate to each of these compartments/ organs are known or can be measured. The parameters that characterize transport across the sub-compartments (i.e., vascular, interstitial and cellular) and the metabolism of various agents are not generally known and cannot be easily measured. Our philosophy has been to use as many measured parameters as possible and estimate the remaining parameters by fitting the model to the murine biodistribution data. By scaling-up the parameters using well-defined scale-up laws [127], we then predict the biodistribution in human patients and compare with clinical data. Discrepancies between predictions and actual data help us in identifying inter-species differences and force us to question our model assumptions. This is an evolutionary process — as our understanding of underlying physiology and biochemistry improves, the relevant parameters are modified and the model is refined further. The model is useful not only for designing murine experiments and/or clinical trials, but also in identifying the sensitive parameters that need careful measurement and analysis. If we need detailed spatial information about a tissue/organ, then we develop a distributed parameter model for that organ, e.g., tumor [24–29,32,132,133]. While simple in principle, this cyclic approach of analysis and synthesis has served as a useful paradigm for developing a deeper understanding of drug and cell distribution in normal and malignant tissues. The level of sophistication of these models is likely to improve with our understanding of underlying principles [40].
Fig. 10.

Schematic of physiologically-based kinetic model to describe the biodistribution of molecules and cells in a mammalian system. Such an approach permits interspecies scale-up of biodistribution (adapted from Ref. [28]).
9. Bench to bedside
The physiological factors that contribute to the poor delivery of therapeutic agents to tumors include heterogeneous blood supply, interstitial hypertension, relatively long transport distances in the interstitium, and cellular heterogeneities (Fig. 5). How can these physiological barriers be exploited or overcome? Can we take our findings about these barriers from the bench to the bedside? Two recently developed strategies that have the potential to improve the detection and treatment of solid tumors in patients are described here.
As stated earlier, all solid tumors in patients exhibit interstitial hypertension (Table 2), provided the patient has not received any anti-edema treatment [76]. We have also shown theoretically and confirmed experimentally that IFP rises quite steeply in the tumor boundary [31,72]. We have used this knowledge in improving the design of the needle used by radiologists to localize the tumor for surgical excision [134]. We can facilitate the needle placement in a tumor by placing a pressure-sensor in the needle. Since tumors begin to exhibit interstitial hypertension almost from the onset of angiogenesis [135], this needle may be able to help in localizing early disease. The same concept may be useful in optimizing location and infusion pressure of needles employed in intratumor infusion of therapeutic agents [95], and for monitoring response to therapy [83].
Table 2.
Interstitial fluid pressure (mmHg) in normal and neoplastic tissues in patients
| Tissue type | n | Mean | Range |
|---|---|---|---|
| Normal skin | 5 | 0.4 | −1.0–3.0 |
| Normal breast | 8 | 0.0 | − 0.5–3.0 |
| Head/neck carcinomas | 27 | 19.0 | 1.5–79.0 |
| Cervical carcinomas | 26 | 23.0 | 6.0–94.0 |
| Lung carcinomas | 26 | 10.0 | 1.0–27.0 |
| Metastatic melanomas | 14 | 21.0 | 0.0–60.0 |
| Metastatic melanomas | 12 | 14.5 | 2.0–41.0 |
| Breast carcinomas | 13 | 29.0 | 5.0–53.0 |
| Breast carcinomas | 8 | 15.0 | 4.0–33.0 |
| Brain tumorsa | 17 | 7.0 | 2.0–15.0 |
| Brain tumorsa | 11 | 1.0 | − 0.5–8.0 |
| Colorectal liver metastases | 8 | 21.0 | 6.0–45.0 |
| Lymphomas | 7 | 4.5 | 1.0–12.5 |
| Renal cell carcinoma | 1 | 38.0 | — |
Patients were treated with anti-edema therapy.
Several physical (e.g., radiation, heat) and chemical (e.g., vasoactive drugs) agents may lead to an increase in tumor blood flow or vascular permeability [44,60,136–140], or lower pH [56,58]. Another approach may be based on increasing the interstitial transport rate of molecules by increasing K or D enzymatically [93,95,102] or using multi-step approaches [29,37,141,142]. We have used several physical and chemical agents to lower IFP in tumors [13,96,143–148]. Since microvascular and interstitial pressures in tumors are approximately equal, any change in one is followed rapidly by a similar change in the other, and thus the convective enhancement disappears rapidly [35,73,149,150]. By adapting a poroelastic model to solid tumors, we have calculated theoretically and confirmed experimentally that the time constant of pressure transmission across the tumor vasculature is on the order of 10 s [35]. During such a short time, the convective enhancement is calculated to be very small (~ 1%). However, if the vascular pressure is increased repeatedly and if the transvascular transport is unidirectional, or if the molecule binds avidly in the extravascular region, then we can, in principle, increase drug delivery to solid tumors significantly (Fig. 11).
Fig. 11.

A novel approach to increase convective transport of molecules across tumor vessels based on the finding that there is a ~ 10-s delay in the transmission of intravascular pressure to the interstitial compartment. For this approach to work, the transvascular transport has to be uni-directional or the extravasated molecule must bind avidly so that it does not intravasate when the intravascular pressure is lower than interstitial pressure (adapted from Ref. [35]).
In contrast, the physiological barriers discussed here may be less of a problem for: (a) radioimmunodetection; (b) treating leukemias, lymphomas, and small tumors (e.g., micrometastases) in which the physiological barriers are not yet fully established; (c) treatment of adequately perfused, low-pressure regions of large tumors; and (d) treatment with antibodies or other agents directed against the host cells (e.g., tumor endothelial cells, fibroblasts) or the sub-endothelial matrix. These physiological barriers also may pose less problems for treatment with a molecule or cell that has nearly 100% specificity for cancer cells. Until such selective molecules or cells are developed, methods are urgently needed to overcome or exploit these physiological barriers in tumors. It is hoped that an improved understanding of transport in tumors will help in developing these strategies [151].
Acknowledgments
I thank Carol Lyons, Larry Baxter and Stuart Friedrich for their help with the references, Lance Munn for his help with Figures 1–3, Fan Yuan with Figures 4 and 7, Gabriel Helmlinger with Figure 6, David Berk with Figure 8, Marc Dellian with Figure 9, Larry Baxter with Figure 10, Paolo Netti with Figure 11, and Yves Boucher with Table 2. Research described here was primarily supported by grants from the National Cancer Institute, the National Science Foundation and the American Cancer Society.
An earlier version of this article was published as “1995 Whitaker Lecture: Delivery of Molecules, Particles and Cells to Solid Tumors,” in the Annals of Biomedical Engineering 24 (1996) 457–473. The author thanks the Biomedical Engineering Society for allowing him to reproduce this article.
Footnotes
PII of original article: S0169-409X(97)00027-6. The article was originally published in Advanced Drug Delivery Reviews 26 (1997) 71–90.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beardsley T. Trends in cancer epidemiology: a war not won. Sci Am. 1994;270:118–126. [Google Scholar]
- 2.Jain RK. Barriers to drug delivery in solid tumors. Sci Am. 1994;271:58–65. doi: 10.1038/scientificamerican0794-58. [DOI] [PubMed] [Google Scholar]
- 3.Jain RK. Transport phenomena in tumors. Adv Chem Eng. 1994;20:129–200. [Google Scholar]
- 4.Sevick EM, Jain RK. Blood flow and venous pH of tissue-isolated Walker 256 carcinoma during hyperglycemia. Cancer Res. 1988;48:1201–1207. [PubMed] [Google Scholar]
- 5.Sevick EM, Jain RK. Geometric resistance to blood flow in solid tumors perfused ex vivo: effects of tumor size and perfusion pressure. Cancer Res. 1989;49:3506–3512. [PubMed] [Google Scholar]
- 6.Gullino PM. Techniques in Tumor Pathophysiology. In: Busch H, editor. Methods in Cancer Research. Academic Press; New York: 1970. [Google Scholar]
- 7.Kristjansen PE, Roberge S, Lee I, Jain RK. Tissue- isolated human tumor xenografts in athymic nude mice. Microvasc Res. 1994;48:389–402. doi: 10.1006/mvre.1994.1063. [DOI] [PubMed] [Google Scholar]
- 8.Kristjansen PEG, Brown TJ, Shipley LA, Jain RK. Intratumor pharmacokinetics, flow resistance, and metabolism during gemcitabine infusion in ex vivo perfused human small cell lung cancer. Clin Cancer Res. 1996;2:359–367. [PubMed] [Google Scholar]
- 9.Less JR, Posner MC, Wolmark N, Jain RK. Geometric resistance to blood flow and vascular network architecture in human colorectal carcinoma. Microcirculation. 1997;4:25–33. doi: 10.3109/10739689709148315. [DOI] [PubMed] [Google Scholar]
- 10.Dudar TE, Jain RK. Microcirculatory flow changes during tissue growth. Microvasc Res. 1983;25:1–21. doi: 10.1016/0026-2862(83)90040-7. [DOI] [PubMed] [Google Scholar]
- 11.Zawicki DF, Jain RK, Schmid-Schoenbein GW, Chien S. Dynamics of neovascularization in normal tissue. Microvasc Res. 1981;21:27–47. doi: 10.1016/0026-2862(81)90003-0. [DOI] [PubMed] [Google Scholar]
- 12.Leunig M, Yuan F, Berk DA, Gerweck LE, Jain RK. Angiogenesis and growth of isografted bone: quantitative in vivo assay in nude mice. Lab Invest. 1994;71:300–307. [PubMed] [Google Scholar]
- 13.Leunig M, Yuan F, Menger MD, Boucher Y, Goetz AE, Messmer K, Jain RK. Angiogenesis, microvascular architecture, microhemodynamics, and interstitial fluid pressure during early growth of human adenocarcinoma LS174T in SCID mice. Cancer Res. 1992;52:6553–6560. [PubMed] [Google Scholar]
- 14.Yuan F, Salehi HA, Boucher Y, Vasthare US, Tuma RF, Jain RK. Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res. 1994;54:4564–4568. [PubMed] [Google Scholar]
- 15.Yamada S, Mayadas T, Yuan F, Wagner D, Hynes R, Melder R, Jain RK. Rolling in P-selectin deficient mice is reduced but not eliminated in the dorsal skin. Blood. 1995;86:3487–3492. [PubMed] [Google Scholar]
- 16.Milstone DS, Fukumura D, Padget RC, O’Donnell PE, Davis VM, Benavidez OJ, Melder RJ, Jain RK, Gimbrone MA. Mice lacking E-selectin show normal rolling but reduced arrest of leukocytes on cytokine-activated microvascular endothelium. 1997. submitted. [PubMed] [Google Scholar]
- 17.Dellian M, Witwer BP, Salehi HA, Yuan F, Jain RK. Quantitation and physiological characterization of bFGF and VEGF/VPF induced vessels in mice: effect of microenvironment on angiogenesis. Am J Pathol. 1996;149:59–72. [PMC free article] [PubMed] [Google Scholar]
- 18.Lichtenbeld HC, Yuan F, Michel CC, Jain RK. Perfusion of single tumor microvessels: application to vascular permeability measurement. Microcirculation. 1996;3:349–357. doi: 10.3109/10739689609148307. [DOI] [PubMed] [Google Scholar]
- 19.Melder RJ, Brownell AL, Shoup TM, Brownell GL, Jain RK. Imaging of activated natural killer cells in mice by positron emission tomography: preferential uptake in tumors. Cancer Res. 1993;53:5867–5871. [PubMed] [Google Scholar]
- 20.Munn LL, Melder RJ, Jain RK. Analysis of cell flux in the parallel plate flow chamber: implications for cell capture studies. Biophys J. 1994;67:889–895. doi: 10.1016/S0006-3495(94)80550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sasaki A, Jain RK, Maghazachi AA, Goldfarb RH, Herberman RB. Low deformability of lymphokine-activated killer cells as a possible determinant of in vivo distribution. Cancer Res. 1989;49:3742–3746. [PubMed] [Google Scholar]
- 22.Traykov TT, Jain RK. Effect of glucose and galactose on red blood cell membrane deformability. Int J Microcirc Clin Exp. 1987;6:35–44. [PubMed] [Google Scholar]
- 23.Munn L, Koenig GC, Jain RK, Melder R. Kinetics of adhesion molecule expression and spatial organization using targeted sampling fluorometry. BioTechniques. 1995;19:622–631. [PubMed] [Google Scholar]
- 24.Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. I. Role of interstitial pressure and convection. Microvasc Res. 1989;37:77–104. doi: 10.1016/0026-2862(89)90074-5. [DOI] [PubMed] [Google Scholar]
- 25.Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. II. Role of heterogeneous perfusion and lymphatics. Microvasc Res. 1990;40:246–263. doi: 10.1016/0026-2862(90)90023-k. [DOI] [PubMed] [Google Scholar]
- 26.Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. III. Role of binding and metabolism. Microvasc Res. 1991;41:5–23. doi: 10.1016/0026-2862(91)90003-t. [DOI] [PubMed] [Google Scholar]
- 27.Baxter LT, Jain RK. Transport of fluid and macromolecules in tumors. IV. A microscopic model of the perivascular distribution. Microvasc Res. 1991;41:252–272. doi: 10.1016/0026-2862(91)90026-8. [DOI] [PubMed] [Google Scholar]
- 28.Baxter LT, Zhu H, Mackensen DG, Butler WF, Jain RK. Biodistribution of monoclonal antibodies: scale-up from mouse to man using a physiologically based pharmacokinetic model. Cancer Res. 1995;55:4611–4622. [PubMed] [Google Scholar]
- 29.Baxter LT, Zhu H, Mackensen DG, Jain RK. Physiologically based pharmacokinetic model for specific and nonspecific monoclonal antibodies and fragments in normal tissues and human tumor xenografts in nude mice. Cancer Res. 1994;54:1517–1528. [PubMed] [Google Scholar]
- 30.Eskey CJ, Wolmark N, McDowell CL, Domach MM, Jain RK. Residence time distributions of various tracers in tumors: implications for drug delivery and blood flow measurement. J Natl Cancer Inst. 1994;86:293–299. doi: 10.1093/jnci/86.4.293. [DOI] [PubMed] [Google Scholar]
- 31.Jain RK, Baxter LT. Mechanisms of heterogeneous distribution of monoclonal antibodies and other macromolecules in tumors: significance of elevated interstitial pressure. Cancer Res. 1988;48:7022–7032. [PubMed] [Google Scholar]
- 32.Jain RK, Wei J. Dynamics of drug transport in solid tumors: distributed parameter model. J Bioeng. 1977;1:313–329. [Google Scholar]
- 33.Jain RK, Wei J, Gullino PM. Pharmacokinetics of methotrexate in solid tumors. J Pharmacokinet Biopharm. 1979;7:181–194. doi: 10.1007/BF01059737. [DOI] [PubMed] [Google Scholar]
- 34.Netti PA, Roberge S, Boucher Y, Baxter LT, Jain RK. Effect of transvascular fluid exchange on arteriovenous pressure relationship: implication for temporal and spatial heterogeneities in tumor blood flow. Microvasc Res. 1996;52:27–46. doi: 10.1006/mvre.1996.0041. [DOI] [PubMed] [Google Scholar]
- 35.Netti PA, Baxter LT, Boucher Y, Skalak R, Jain RK. Time dependent behavior of interstitial pressure in solid tumors: implications for drug delivery. Cancer Res. 1995;55:5451–5458. [PubMed] [Google Scholar]
- 36.Pierson RN, Price DC, Wang J, Jain RK. Extracellular water measurements: organ tracer kinetics of bromide and sucrose in rats and man. Am J Physiol. 1978;235:254–264. doi: 10.1152/ajprenal.1978.235.3.F254. [DOI] [PubMed] [Google Scholar]
- 37.Yuan F, Baxter LT, Jain RK. Pharmacokinetic analysis of two-step approaches using bifunctional and enzyme-conjugated antibodies. Cancer Res. 1991;51:3119–3130. [PubMed] [Google Scholar]
- 38.Folkman J. Tumor angiogenesis. In: Mendelsohn PM, Howley MAP, Liotta LA, editors. The Molecular Basis of Cancer. W.B. Saunders; Philadelphia: 1995. [Google Scholar]
- 39.Jain RK. Determinants of tumor blood flow: a review. Cancer Res. 1988;48:2641–2658. [PubMed] [Google Scholar]
- 40.Baish JW, Gazit Y, Berk DA, Nozue M, Baxter LT, Jain RK. A novel approach to examine the role of vascular heterogeneity in nutrient and drug delivery for tumors: an invasion percolation model. Microvasc Res. 1996;51:327–346. doi: 10.1006/mvre.1996.0031. [DOI] [PubMed] [Google Scholar]
- 41.Gazit Y, Berk DA, Leunig M, Baxter LT, Jain RK. Scale-invariant behavior and vascular network formation in normal and tumor tissue. Phys Rev Lett. 1995;75:2428–2431. doi: 10.1103/PhysRevLett.75.2428. [DOI] [PubMed] [Google Scholar]
- 42.Less JR, Skalak TC, Sevick EM, Jain RK. Microvascular architecture in a mammary carcinoma: branching patterns and vessel dimensions. Cancer Res. 1991;51:265–273. [PubMed] [Google Scholar]
- 43.Patan S, Munn L, Jain RK. Intussusceptive microvascular growth in solid tumors: a novel mechanism of tumor angiogenesis. Microvasc Res. 1996;51:260–272. doi: 10.1006/mvre.1996.0025. [DOI] [PubMed] [Google Scholar]
- 44.Jain RK, Ward-Hartley KA. Tumor blood flow: characterization, modifications and role in hyperthermia. IEEE Trans Sonics Ultrasonics. 1984;31:504–526. [Google Scholar]
- 45.Sevick EM, Jain RK. Viscous resistance to blood flow in solid tumors: effect of hematocrit on intratumor blood viscosity. Cancer Res. 1989;49:3513–3519. [PubMed] [Google Scholar]
- 46.Sevick EM, Jain RK. Effect of red blood cell rigidity on tumor blood flow: increase in viscous resistance during hyperglycemia. Cancer Res. 1991;51:2727–2730. [PubMed] [Google Scholar]
- 47.Endrich B, Reinhold HS, Gross JF, Intaglietta M. Tissue perfusion inhomogeneity during early tumor growth in rats. J Natl Cancer Inst. 1979;62:387–395. [PubMed] [Google Scholar]
- 48.Eskey CJ, Koretsky AP, Domach MM, Jain RK. 2H-Nuclear magnetic resonance imaging of tumor blood flow: spatial and temporal heterogeneity in a tissue-isolated mammary adenocarcinoma. Cancer Res. 1992;52:6010–6019. [PubMed] [Google Scholar]
- 49.Hamberg LM, Kristjansen PE, Hunter GJ, Wolf GL, Jain RK. Spatial heterogeneity in tumor perfusion measured with functional computed tomography at 0.05 microliter resolution. Cancer Res. 1994;54:6032–6036. [PubMed] [Google Scholar]
- 50.Vaupel P, Jain RK, editors. Tumor Blood Supply and Metabolic Microenvironment: Characterization and Therapeutic Implications. Gustav Fischer Publications; Stuttgart: 1991. [Google Scholar]
- 51.Dellian M, Helmlinger G, Yuan F, Jain RK. Fluorescence ratio imaging and optical sectioning: effect of glucose on spatial and temporal gradients. Br J Cancer. 1996;74:1206–1215. doi: 10.1038/bjc.1996.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: simultaneous high-resolution measurements reveal a lack of correlation. Nat Med. 1996 doi: 10.1038/nm0297-177. in press. [DOI] [PubMed] [Google Scholar]
- 53.Martin GR, Jain RK. Fluorescence ratio imaging measurement of pH gradients: calibration and application in normal and tumor tissues. Microvasc Res. 1993;46:216–230. doi: 10.1006/mvre.1993.1048. [DOI] [PubMed] [Google Scholar]
- 54.Martin GR, Jain RK. Noninvasive measurement of interstitial pH profiles in normal and neoplastic tissue using fluorescence ratio imaging microscopy. Cancer Res. 1994;54:5670–5674. [PubMed] [Google Scholar]
- 55.Torres-Filho IP, Leunig M, Yuan F, Intaglietta M, Jain RK. Noninvasive measurement of microvascular and interstitial oxygen profiles in a human tumor in SCID mice. Proc Natl Acad Sci USA. 1994;91:2081–2085. doi: 10.1073/pnas.91.6.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jain RK, Shah SA, Finney PL. Continuous noninvasive monitoring of pH and temperature in rat Walker 256 carcinoma during normoglycemia and hyperglycemia. J Natl Cancer Inst. 1984;73:429–436. doi: 10.1093/jnci/73.2.429. [DOI] [PubMed] [Google Scholar]
- 57.Nozue M, Lee I, Manning JM, Manning LR, Jain RK. Oxygenation in tumors by modified hemoglobins. J Surg Oncol. 1996;62:109–114. doi: 10.1002/(SICI)1096-9098(199606)62:2<109::AID-JSO6>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 58.Ward KA, Jain RK. Response of tumours to hyperglycaemia: characterization, significance and role in hyperthermia. Int J Hyperthermia. 1988;4:223–250. doi: 10.3109/02656738809051100. [DOI] [PubMed] [Google Scholar]
- 59.Eskey CJ, Koretsky AP, Domach MM, Jain RK. Role of oxygen vs. glucose in energy metabolism in a mammary carcinoma perfused ex vivo: direct measurement by 31P NMR. Proc Natl Acad Sci USA. 1993;90:2646–2650. doi: 10.1073/pnas.90.7.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jain RK. Transport of molecules across tumor vasculature. Cancer Metastasis Rev. 1987;6:559–593. doi: 10.1007/BF00047468. [DOI] [PubMed] [Google Scholar]
- 61.Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol. 1995;146:1029–1039. [PMC free article] [PubMed] [Google Scholar]
- 62.Gerlowski LE, Jain RK. Microvascular permeability of normal and neoplastic tissues. Microvasc Res. 1986;31:288–305. doi: 10.1016/0026-2862(86)90018-x. [DOI] [PubMed] [Google Scholar]
- 63.Sevick EM, Jain RK. Measurement of capillary filtration coefficient in a solid tumor. Cancer Res. 1991;51:1352–1355. [PubMed] [Google Scholar]
- 64.Yuan F, Leunig M, Berk DA, Jain RK. Microvascular permeability of albumin, vascular surface area, and vascular volume measured in human adenocarcinoma LS174T using dorsal chamber in SCID mice. Microvasc Res. 1993;45:269–289. doi: 10.1006/mvre.1993.1024. [DOI] [PubMed] [Google Scholar]
- 65.Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK. Microvascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994;54:3352–3356. [PubMed] [Google Scholar]
- 66.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchiliin VP, Jain RK. Vascular permeability in a human tumor xenograft: molecular size-dependence and cut off size. Cancer Res. 1995;55:3752–3756. [PubMed] [Google Scholar]
- 67.Dellian M, Yuan F, Trubetskoy V, Torchilin VP, Jain RK. Dependence of microvascular permeability on molecular charge in a human tumor xenograft. Int J Microcirc Clin Exp. 1996;16(Suppl 1):152. [Google Scholar]
- 68.Hobbs SK, Yuan F, Cima LG, Jain RK. Tumor microvascular pore cutoff size: implications for macromolecules and particle drug delivery. Int J Microcirc Clin Exp. 1996;16(Suppl 1):218. [Google Scholar]
- 69.Roberts WG, Palade G. Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res. 1997;57:1207–1211. [PubMed] [Google Scholar]
- 70.Yuan F, Chen Y, Dellian M, Safabakhsh N, Ferrara N, Jain RK. Time-dependent changes in vascular permeability and morphology in established human tumor xenografts induced by an anti-VEGF/VPF antibody. Proc Natl Acad Sci USA. 1996;93:14765–14770. doi: 10.1073/pnas.93.25.14765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Arbit E, Lee J, DiResta G. Interstitial hypertension in human brain tumors: possible role in peritumoral edema formulation. In: Nagai H, Kamiya K, Ishi S, editors. Intracranial Pressure. Springer-Verlag; Tokyo: 1994. pp. 609–614. [Google Scholar]
- 72.Boucher Y, Baxter LT, Jain RK. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy. Cancer Res. 1990;50:4478–4484. [PubMed] [Google Scholar]
- 73.Boucher Y, Jain RK. Microvascular pressure is the principal driving force for interstitial hypertension in solid tumors: implications for vascular collapse. Cancer Res. 1992;52:5110–5114. [PubMed] [Google Scholar]
- 74.Boucher Y, Lee I, Jain RK. Lack of general correlation between interstitial fluid pressure and pO2 in tumors. Microvasc Res. 1995;50:175–182. doi: 10.1006/mvre.1995.1051. [DOI] [PubMed] [Google Scholar]
- 75.Boucher Y, Kirkwood JM, Opacic D, Desantis M, Jain RK. Interstitial hypertension in superficial metastatic melanomas in humans. Cancer Res. 1991;51:6691–6694. [PubMed] [Google Scholar]
- 76.Boucher Y, Salehi H, Witwer B, Harsh GR, Jain RK. Interstitial fluid pressure in intracranial tumors in patients and in rodents: effect of anti-edema therapy. Br J Cancer. 1997;75:829–836. doi: 10.1038/bjc.1997.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Curti BD, Urba WJ, Alvord WG, Janik JE, Smith JW, Madara K, Longo DL. Interstitial pressure of subcutaneous nodules in melanoma and lymphoma patients: changes during treatment. Cancer Res. 1993;53:2204–2207s. [PubMed] [Google Scholar]
- 78.Gutmann R, Leunig M, Feyh J, Goetz AE, Messmer K, Kastenbauer E, Jain RK. Interstitial hypertension in head and neck tumors in patients: correlation with tumor size. Cancer Res. 1992;52:1993–1995. [PubMed] [Google Scholar]
- 79.Jain RK. Transport of molecules in the tumor interstitium: a review. Cancer Res. 1987;47:3039–3051. [PubMed] [Google Scholar]
- 80.Less JR, Posner MC, Boucher Y, Borochovitz D, Wolmark N, Jain RK. Interstitial hypertension in human breast and colorectal tumors. Cancer Res. 1992;52:6371–6374. [PubMed] [Google Scholar]
- 81.Nathanson SD, Nelson L. Interstitial fluid pressure in breast cancer, benign breast conditions, and breast parenchyma. Ann Surg Oncol. 1994;1:333–338. doi: 10.1007/BF03187139. [DOI] [PubMed] [Google Scholar]
- 82.Roh HD, Boucher Y, Kalnicki S, Buchsbaum R, Bloomer WD, Jain RK. Interstitial hypertension in carcinoma of uterine cervix in patients: possible correlation with tumor oxygenation and radiation response. Cancer Res. 1991;51:6695–6698. [PubMed] [Google Scholar]
- 83.Znati CA, Karasek K, Faul C, Roh H-D, Boucher Y, Rosenstein M, Kalnicki S, Buchsbaum R, Chen A, Bloomer WD, Jain RK. Interstitial fluid pressure changes in cervical carcinomas in patients undergoing radiation therapy: a potential prognostic factor. 1996. submitted. [Google Scholar]
- 84.Znati CA, Rosenstein M, Boucher Y, Epperly MW, Bloomer WD, Jain RK. Effect of radiation on interstitial fluid pressure and oxygenation in a human colon carcinoma xenograft. Cancer Res. 1996;56:964–968. [PubMed] [Google Scholar]
- 85.Stohrer M, Boucher Y, Stangassinger M, Jain RK. Oncotic pressure in human tumor xenografts. Proc Am Assoc Cancer Res. 1995;36:311. [PubMed] [Google Scholar]
- 86.Rippe B, Haraldsson B. Fluid and protein fluxes across small and large pores in the microvasculature. Application of two-pore equations. Acta Physiol Scand. 1987;131:411–428. doi: 10.1111/j.1748-1716.1987.tb08257.x. [DOI] [PubMed] [Google Scholar]
- 87.Berk DA, Yuan F, Leunig M, Jain RK. Fluorescence photobleaching with spatial Fourier analysis: measurement of diffusion in light-scattering media. Biophys J. 1993;65:2428–2436. doi: 10.1016/S0006-3495(93)81326-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chary SR, Jain RK. Direct measurement of interstitial convection and diffusion of albumin in normal and neoplastic tissues by fluorescence photobleaching. Proc Natl Acad Sci USA. 1989;86:5385–5389. doi: 10.1073/pnas.86.14.5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Johnson EM, Berk DA, Jain RK, Deen WM. Diffusion and partitioning of proteins in charged agarose gels. Bio-phys J. 1995;68:1561–1568. doi: 10.1016/S0006-3495(95)80328-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Johnson EM, Berk DA, Jain RK, Deen WM. Hindered diffusion in agarose gels: test of effective medium model. Biophys J. 1996;70:1017–1026. doi: 10.1016/S0006-3495(96)79645-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Johnson M, Berk DA, Blankschtein D, Golan DE, Jain RK, Langer R. Lateral diffusion of small compounds in human stratum corneum and model lipid bilayer systems. Biophys J. 1996;71:2656–2668. doi: 10.1016/S0006-3495(96)79457-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nugent LJ, Jain RK. Extravascular diffusion in normal and neoplastic tissues. Cancer Res. 1984;44:238–244. [PubMed] [Google Scholar]
- 93.Swabb EA, Wei J, Gullino PM. Diffusion and convection in normal and neoplastic tissues. Cancer Res. 1974;34:2814. [PubMed] [Google Scholar]
- 94.Berk DA, Yuan F, Leunig M, Jain RK. Direct in vivo measurement of targeted binding in a human tumor xenograft. Proc Natl Acad Sci USA. 1997;94:1785. doi: 10.1073/pnas.94.5.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boucher Y, Brekken C, Netti PA, Baxter LT, Jain RK. Hydraulic conductivity of solid tumors: a novel in vivo measurement technique and implications for drug delivery. 1997. submitted. [Google Scholar]
- 96.Znati CA, Boucher Y, Rosenstein M, Turner D, Watkins S, Jain RK. Effect of radiation on the interstitial matrix and hydraulic conductivity of tumors. Cancer Res. 1996;56:964–968. [PubMed] [Google Scholar]
- 97.Juweid M, Neumann R, Paik C, Perez-Bacete MJ, Sato J, Van Osdol W, Weinstein JN. Micropharmacology of monoclonal antibodies in solid tumor: direct experimental evidence for a binding site barrier. Cancer Res. 1992;52:5144. [PubMed] [Google Scholar]
- 98.Kaufman EN, Jain RK. Quantification of transport and binding parameters using fluorescence recovery after photobleaching. Potential for in vivo applications. Biophys J. 1990;58:873–885. doi: 10.1016/S0006-3495(90)82432-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kaufman EN, Jain RK. Measurement of mass transport and reaction parameters in bulk solution using photobleaching. Reaction limited binding regime. Biophys J. 1991;60:596–610. doi: 10.1016/S0006-3495(91)82089-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kaufman EN, Jain RK. Effect of bivalent interaction upon apparent antibody affinity: experimental confirmation of theory using fluorescence photobleaching and implications for antibody binding assays. Cancer Res. 1992;52:4157–4167. [PubMed] [Google Scholar]
- 101.Kaufman EN, Jain RK. In vitro measurement and screening of monoclonal antibody affinity using fluorescence photobleaching. J Immunol Methods. 1992;155:1–17. doi: 10.1016/0022-1759(92)90265-u. [DOI] [PubMed] [Google Scholar]
- 102.Jain RK. Delivery of novel therapeutic agents in tumors: physiological barriers and strategies. J Natl Cancer Inst. 1989;81:570–576. doi: 10.1093/jnci/81.8.570. [DOI] [PubMed] [Google Scholar]
- 103.Leu AJ, Berk DA, Yuan F, Jain RK. Flow velocity in the superficial lymphatic network of the mouse tail. Am J Physiol. 1994;267:H1507–1513. doi: 10.1152/ajpheart.1994.267.4.H1507. [DOI] [PubMed] [Google Scholar]
- 104.Berk DA, Swartz MA, Leu AJ, Jain RK. Transport in lymphatic capillaries: II. Microscopic velocity measurement with fluorescence recovery after photobleaching. Am J Physiol. 1996;270:H330–H337. doi: 10.1152/ajpheart.1996.270.1.H330. [DOI] [PubMed] [Google Scholar]
- 105.Swartz MA, Berk DA, Jain RK. Transport in lymphatic capillaries: I. Macroscopic measurements using residence time distribution theory. Am J Physiol. 1996;270:H324–H329. doi: 10.1152/ajpheart.1996.270.1.H324. [DOI] [PubMed] [Google Scholar]
- 106.Leu A, Berk D, Jain RK. Search for initial lymphatics in solid tumors. 1997. submitted. [Google Scholar]
- 107.Melder RJ, Munn LL, Yamada S, Ohkubo C, Jain RK. Selectin and integrin mediated T lymphocyte rolling and arrest on TNFa-activated endothelium is augmented by erythrocytes. Biophys J. 1995;69:2131–2138. doi: 10.1016/S0006-3495(95)80087-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Munn LL, Melder RJ, Jain RK. Role of erythrocytes in leukocyte-endothelial interactions: mathematical model and experimental validation. Biophys J. 1996;71:466–478. doi: 10.1016/S0006-3495(96)79248-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Fukumura D, Salehi H, Witwer B, Tuma RF, Melder RJ, Jain RK. TNFa-induced leukocyte-adhesion in normal and tumor vessels: effect of tumor type. Transplantation site and host. Cancer Res. 1995;55:4824–4829. [PubMed] [Google Scholar]
- 110.Yamada S, Melder RJ, Leunig M, Ohkubo C, Jain RK. Leukocyte-rolling increases with age. Blood. 1995;86:4707–4708. [PubMed] [Google Scholar]
- 111.Melder RJ, Elmaleh D, Brownell AL, Brownell GL, Jain RK. A method for labeling cells for positron emission tomography (PET) studies. J Immunol Methods. 1994;175:79–87. doi: 10.1016/0022-1759(94)90333-6. [DOI] [PubMed] [Google Scholar]
- 112.Melder RJ, Jain RK. Kinetics of interleukin-2 induced changes in rigidity of human natural killer cells. Cell Biophys. 1992;20:161–176. doi: 10.1007/BF02823656. [DOI] [PubMed] [Google Scholar]
- 113.Jain RK, Koenig G, Dellian M, Fukumura D, Munn LL, Melder RJ. Leukocyte-endothelial adhesion and angiogenesis in tumors. Cancer Metastasis Rev. 1996;15:195–204. doi: 10.1007/BF00437472. [DOI] [PubMed] [Google Scholar]
- 114.Melder RJ, Jain RK. Reduction of rigidity in human activated natural killer cells by thioglycollate treatment. J Immunol Methods. 1994;175:69–77. doi: 10.1016/0022-1759(94)90332-8. [DOI] [PubMed] [Google Scholar]
- 115.Melder RJ, Salehi HA, Jain RK. Localization of activated natural killer cells in MCaIV mammary carcinoma grown in cranial windows in C3H mice. Microvasc Res. 1995;50:35–44. doi: 10.1006/mvre.1995.1036. [DOI] [PubMed] [Google Scholar]
- 116.Sasaki A, Melder RJ, Whiteside TL, Herberman RB, Jain RK. Preferential localization of human adherent lymphokine-activated killer cells in tumor microcirculation. J Natl Cancer Inst. 1991;83:433–437. doi: 10.1093/jnci/83.6.433. [DOI] [PubMed] [Google Scholar]
- 117.Ohkubo C, Bigos D, Jain RK. Interleukin 2 induced leukocyte adhesion to the normal and tumor microvascular endothelium in vivo and its inhibition by dextran sulfate: implications for vascular leak syndrome. Cancer Res. 1991;51:1561–1563. [PubMed] [Google Scholar]
- 118.Melder RJ, Koenig G, Witwer BP, Safabakhsh N, Munn LL, Jain RK. During angiogenesis, vascular endothelial growth factor and basic fibroblast growth factor regulate natural killer cell adhesion to tumor endothelium. Nat Med. 1996;2:992–997. doi: 10.1038/nm0996-992. [DOI] [PubMed] [Google Scholar]
- 119.Jallal B, Powell F, Zachwieja J, Brakebusch C, Germain L, Jacobs J, Iacobelli S, Ullrich A. Suppression of tumor growth in vivo by local and systemic 90K level increase. Cancer Res. 1995;55:3223–3227. [PubMed] [Google Scholar]
- 120.Melder RJ, Koenig G, Munn LL, Jain RK. Adhesion of activated natural killer cells to TNF-alpha treated endothelium under physiological flow conditions. Natural Immunity. 1997;15:154. [PubMed] [Google Scholar]
- 121.Gamble JR, Vadas MA. Endothelial adhesiveness for blood neutrophils is inhibited by transforming growth factor-beta. Science. 1988;242:97–99. doi: 10.1126/science.3175638. [DOI] [PubMed] [Google Scholar]
- 122.Gamble JR, Vadas MA. Endothelial cell adhesiveness for human T lymphocytes is inhibited by transforming growth factor-beta. J Immunol. 1991;146:1149–1154. [PubMed] [Google Scholar]
- 123.Gamble JR, Khew-Goodall Y. Transforming growth factor-beta inhibits E-selectin expression on human endothelial cells. J Immunol. 1993;150:4494–4503. [PubMed] [Google Scholar]
- 124.Fidler IJ. Modulation of the organ microenvironment for treatment of cancer metastasis. J Natl Cancer Inst. 1995;87:1588–1592. doi: 10.1093/jnci/87.21.1588. [DOI] [PubMed] [Google Scholar]
- 125.Kitayama J, Nagawa J, Yasuhara H, Tsuno N, Kimura W, Shibata Y, Muto T. Suppressive effect of basic fibroblast growth factor on transendothelial emigration of CD4(+) T-lymphocyte. Cancer Res. 1994;54:4729–4733. [PubMed] [Google Scholar]
- 126.Haying JB, Williams SK. Reduced adhesion of human microvascular endothelial cells to collagen in response to basic FGF is mediated by b1 integrin. FASEB J. 1994;8:263. (Abstr.) [Google Scholar]
- 127.Dedrick RL. Animal scale-up. J Pharmacokinet Bio-pharm. 1973;1:435–461. doi: 10.1007/BF01059667. [DOI] [PubMed] [Google Scholar]
- 128.Gerlowski LE, Jain RK. Physiologically based pharmacokinetic modeling: principles and applications. J Pharm Sci. 1983;72:1103–1127. doi: 10.1002/jps.2600721003. [DOI] [PubMed] [Google Scholar]
- 129.Zhu H, Melder R, Baxter L, Jain RK. Physiologically based kinetic model of effector cell biodistribution in mammals: implications for adoptive immunotherapy. Cancer Res. 1996;56:3771–3781. [PubMed] [Google Scholar]
- 130.Zhu H, Baxter L, Jain RK. Potential and limitations of radioimmunodetection and radioimmunotherapy with monoclonal antibodies: evaluation using a physiologically-based pharmacokinetic model. J Nucl Med. 1997;96:256. [PubMed] [Google Scholar]
- 131.Zhu H, Jain RK, Baxter LT. Tumor pretargeting for radioimmunodetection and radioimmunotherapy: evaluation using a physiologically-based pharmacokinetic model. J Nucl Med. 1997 in press. [PubMed] [Google Scholar]
- 132.Jain RK. Effect of inhomogeneities and finite boundaries on temperature distribution in a perfused medium with application to tumors. Trans ASME J Biomech Eng. 1978;100:235–241. [Google Scholar]
- 133.Jain RK. Transient temperature distributions in an infinite perfused medium due to a time-dependent, spherical heat source. Trans ASME J Biomech Eng. 1979;101:82–86. [Google Scholar]
- 134.Jain RK, Boucher Y, Stacey-Clear A, Moore R, Kopans D. US Patent Number 5,396,897. Method for locating tumors prior to needle biopsy. 1995 Mar 14;
- 135.Boucher Y, Leunig M, Jain RK. Tumor angiogenesis and interstitial hypertension. Cancer Res. 1996;56:4264–4266. [PubMed] [Google Scholar]
- 136.Dudar TE, Jain RK. Differential response of normal and tumor microcirculation to hyperthermia. Cancer Res. 1984;44:605–612. [PubMed] [Google Scholar]
- 137.Fukumura D, Yuan F, Endo M, Jain RK. Role of nitric oxide in tumor microcirculation: blood flow, vascular permeability, and leukocyte-endothelial interactions. Am J Pathol. 1997;150:713–725. [PMC free article] [PubMed] [Google Scholar]
- 138.Gerlowski LE, Jain RK. Effect of hyperthermia on microvascular permeability to macromolecules in normal and tumor tissues. Int J Microcir Clin Exp. 1985;4:363–372. [PubMed] [Google Scholar]
- 139.Kristensen CA, Nozue M, Boucher Y, Jain RK. Reduction of interstitial fluid pressure after TNF-a treatment of human melanoma xenografts. Br J Cancer. 1996;74:533–536. doi: 10.1038/bjc.1996.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kristensen CA, Roberge S, Jain RK. Effect of tumor necrosis factor-alpha on vascular resistance, nitric oxide production, glucose and oxygen consumption in perfused, tissue-isolated human melanoma xenografts. Clin Cancer Res. 1997;3:319–324. [PubMed] [Google Scholar]
- 141.Baxter LT, Jain RK. Pharmacokinetic analysis of the microscopic distribution of enzyme-conjugated antibodies and prodrugs: comparison with experimental data. Br J Cancer. 1996;73:447–456. doi: 10.1038/bjc.1996.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Baxter LT, Yuan F, Jain RK. Pharmacokinetic analysis of the perivascular distribution of bifunctional antibodies and haptens: comparison with experimental data. Cancer Res. 1992;52:5838–5844. [PubMed] [Google Scholar]
- 143.Kristjansen PE, Boucher Y, Jain RK. Dexamethasone reduces the interstitial fluid pressure in a human colon adenocarcinoma xenograft. Cancer Res. 1993;53:4764–4766. [PubMed] [Google Scholar]
- 144.Lee I, Boucher Y, Jain RK. Nicotinamide can lower tumor interstitial fluid pressure: mechanistic and therapeutic implications. Cancer Res. 1992;52:3237–3240. [PubMed] [Google Scholar]
- 145.Lee I, Boucher Y, Demhartner TJ, Jain RK. Changes in tumour blood flow, oxygenation and interstitial fluid pressure induced by pentoxifylline. Br J Cancer. 1994;69:492–496. doi: 10.1038/bjc.1994.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lee I, Demhartner TJ, Boucher Y, Jain RK, Intaglietta M. Effect of hemodilution and resuscitation on tumor interstitial fluid pressure, blood flow, and oxygenation. Microvasc Res. 1994;48:1–12. doi: 10.1006/mvre.1994.1034. [DOI] [PubMed] [Google Scholar]
- 147.Leunig M, Goetz AE, Dellian M, Zetterer G, Gamarra F, Jain RK, Messmer K. Interstitial fluid pressure in solid tumors following hyperthermia: possible correlation with therapeutic response. Cancer Res. 1992;52:487–490. [PubMed] [Google Scholar]
- 148.Leunig M, Goetz AE, Gamarra F, Zetterer G, Messmer K, Jain RK. Photodynamic therapy-induced alterations in interstitial fluid pressure, volume and water content of an amelanotic melanoma in the hamster. Br J Cancer. 1994;69:101–103. doi: 10.1038/bjc.1994.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zlotecki RA, Baxter LT, Boucher Y, Jain RK. Pharmacologic modification of tumor blood flow and interstitial fluid pressure in a human tumor xenograft: network analysis and mechanistic interpretation. Microvasc Res. 1995;50:429–443. doi: 10.1006/mvre.1995.1069. [DOI] [PubMed] [Google Scholar]
- 150.Zlotecki RA, Boucher Y, Lee I, Baxter LT, Jain RK. Effect of angiotensin II induced hypertension on tumor blood flow and interstitial fluid pressure. Cancer Res. 1993;53:2466–2468. [PubMed] [Google Scholar]
- 151.Jain RK. Delivery of molecular medicine to solid tumors. Science. 1996;271:1079–1080. doi: 10.1126/science.271.5252.1079. [DOI] [PubMed] [Google Scholar]