

Abstract
Papillomaviruses (PVs) are widespread pathogens. However, the extent of PV infections in bats remains largely unknown. This work represents the first comprehensive study of PVs in Iberian bats. We identified four novel PVs in the mucosa of free-ranging Eptesicus serotinus (EserPV1, EserPV2, and EserPV3) and Rhinolophus ferrumequinum (RferPV1) individuals and analyzed their phylogenetic relationships within the viral family. We further assessed their prevalence in different populations of E. serotinus and its close relative E. isabellinus. Although it is frequent to read that PVs co-evolve with their host, that PVs are highly species-specific, and that PVs do not usually recombine, our results suggest otherwise. First, strict virus–host co-evolution is rejected by the existence of five, distantly related bat PV lineages and by the lack of congruence between bats and bat PVs phylogenies. Second, the ability of EserPV2 and EserPV3 to infect two different bat species (E. serotinus and E. isabellinus) argues against strict host specificity. Finally, the description of a second noncoding region in the RferPV1 genome reinforces the view of an increased susceptibility to recombination in the E2-L2 genomic region. These findings prompt the question of whether the prevailing paradigms regarding PVs evolution should be reconsidered.
Keywords: bats, papillomavirus, evolution, phylogeny, biodiversity, wildlife
Introduction
Members within the order Chiroptera are extremely successful in terms of ecological diversity, accounting for more than one-fifth (∼1,000) of all extant mammal species (Simmons 2005). Bats play a key role in terrestrial ecosystems as pollinators, insect controllers, seed dispersers, and reforesters (Kunz et al. 2011). They are also instrumental as vectors of zoonotic pathogens, being the reservoir for a number of infectious agents capable of crossing species barriers and of infecting human and nonhuman hosts (Calisher et al. 2006). Recent studies have contributed to enlarge the list of viruses infecting bats (Chu et al. 2008; Falcón et al. 2011; Drexler et al. 2012; Kurth et al. 2012; Wu et al. 2012), and so far, more than 80 different viral agents have been identified including the highly pathogenic rabies virus (Turner 1975) and related lyssaviruses (Samaratunga et al. 1998), Nipah and Hendra viruses (Field et al. 2000; Chua et al. 2002), Ebola and Marburg viruses (Monath 1999; Leroy et al. 2005), and SARS virus (Li et al. 2005). Moreover, beyond their role as potential reservoirs of infectious agents, there is growing concern about bat disease and mortality. Approximately 25% of the world’s bat species are threatened with extinction and yet, little is known about actual bat pathogens (Rector et al. 2006; Mühldorfer et al. 2011).
Papillomaviridae are a family of small, nonenveloped, epitheliotrophic dsDNA viruses. The approximately 8,000 bp genome includes an upstream regulatory region, and up to eight open reading frames (ORFs) named after their expression timing, with an early region encoding for the proteins involved in viral replication and immunomodulation and a late region encoding for the capsid proteins. Papillomaviruses (PVs) infect the skin and mucosa of mammals, but they have also been found in birds, turtles, and snakes and probably infect all amniotes (Bravo et al. 2010). Although most PVs cause asymptomatic infections, some PVs can provoke malignant cell transformations. Certain human PVs are responsible for over one-third of all infection-associated cancers in humans, including different types of anogenital cancers, head and neck cancers, and skin cancers in genetically susceptible individuals (Zur Hausen 2009). Further, neoplastic malignant lesions have also been linked to animal PVs in several different hosts: bats (RaPV1) (Rector et al. 2006), cats (FcaPV2 and FcaPV3) (Lange CE, Tobler K, Markau T, et al. 2009), dogs (CPV1, CPV3, and CPV7) (Lange CE, Tobler K, Ackermann M, et al. 2009), horses (BPV1, BPV2 and EcPV2) (Nasir and Campo 2008; Scase et al. 2010), rodents (McPV2) (Nafz et al. 2008), rabbits (SfPV1) (Giri et al. 1985), and sheep (OvPV3) (Alberti et al. 2010).
Reconstructing the evolutionary history of pathogens such as PVs requires a taxonomic sampling with a balanced description of the pathogens’ diversity in all possible hosts. However, more than half of all known PVs correspond to human PVs (PAvE, Papillomavirus Episteme Database, http://pave.niaid.nih.gov/, last accessed December, 2013). Considering that there are over 23,000 amniote species serving as potential hosts (Bravo et al. 2010; International Union for Conservation of Nature 2013, http://www.iucnredlist.org/, last accessed December, 2013), we have little insight of nonhuman PV diversity. The development of a comprehensive and unbiased scenario for PVs evolution demands the consideration of poorly studied groups such as bats. To date, only five different bat PVs have been fully sequenced. MschPV1, MschPV2, and MrPV1 were isolated, respectively, from oropharyngeal and/or anal swabs from healthy free-ranging Miniopterus schreibersii and Myotis ricketti individuals (Tse et al. 2012; Wu et al. 2012), while EhelPV1 was retrieved from hair bulbs from a healthy captive Eidolon helvum individual (García-Pérez et al. 2013). RaPV1 was recovered from a basosquamous carcinoma on the wing of a Rousettus aegyptiacus individual (Rector et al. 2006). Partial sequences of the E1 and L1 genes have also been retrieved from hair bulbs from a healthy captive Pteropus giganteus (García-Pérez et al. 2013). Further, Baker et al. (2013) conducted a metagenomic study on African Ei. helvum bats, resulting in the identification of hundreds of short PV-like sequences, isolated from throat, lung, and urine samples.
In this study, we have systematically surveyed the presence of PVs in oropharyngeal swabs from 22 out of the 31 extant Iberian bat species (VV.AA. 2010). We have identified and completely sequenced the complete genomes of four novel PVs isolated from two different bat species, namely Eptesicus serotinus and Rhinolophus ferrumequinum. Moreover, we have assessed the prevalence of the novel PVs in Iberian colonies of E. serotinus and the morphologically very similar, closely related species E. isabellinus (Juste et al. 2013).
Material and Methods
Ethics Statement
All persons responsible for samples collection were qualified and experienced bat researchers who had bat capture and sampling permits issued by the competent environmental authority of their study regions as follows: Dirección General de Gestión del Medio Natural, Consejería de Medio Ambiente, Junta de Andalucía, Spain (code #201230E040); Dirección General de Montes y Espacios Naturales, Consejería de Agricultura, Junta de Comunidades de Castilla-La Mancha, Spain (code #DGMEN/SEN_avp_12083_aut); Dirección General del Medio Natural, Consejería de Fomento y Medio Ambiente, Junta de Castilla y León, Spain (code #EP/CYL/201/2012); Dirección General de Medio Ambiente, Consejería de Agricultura, Desarrollo Rural y Medio Ambiente y Energía, Gobierno de Extremadura, Spain (code #CN009/12/ACA). The sampling protocol was approved by the Bioethical and Animal Welfare Committee (CEBA-EBD) of the Estación Biológica de Doñana (EBD-CSIC), study code #CEBA-EBD_11_30, adhering to the guidelines in the RD1201/2005 on the protection of animals used for experimentation and other scientific purposes.
Sample Collection and Nucleic Acid Extraction
A total of 44 oropharyngeal swabs were taken from free-ranging bats belonging to 22 different species through Spain during 2002–2008. An exhaustive list of the samples is provided in supplementary table S1, Supplementary Material online. Bats identification was mainly based on morphological characters. For sibling or morphologically cryptic complexes, the identification was based on the sequencing of a diagnostic mitochondrial fragment (Cytb ∼500 bp) following the protocol established by Ibáñez et al. (2006). Sample collection and DNA extraction were performed as described previously (Echevarría et al. 2001). Several contention measurements were undertaken in order to prevent contamination: all pipetting steps were performed under safety hood cabinets; DNA extraction and subsequent DNA amplification were performed in different locations (i.e., Madrid and Barcelona); all pre- and post-PCR manipulations were performed in different facilities.
Viral Genome Amplification, Sequencing, and Cloning
Rolling circle amplification (RCA) was performed to generate full-length PV genomes following an optimized protocol (Schulz et al. 2009). PV presence was tested using the PV degenerated FAP (Forslund et al. 1999) and CP primers (Iftner et al. 2003). The FAP and CP primers amplify DNA fragments of approximately 450 bp of the L1 and E1 genes, respectively. Complete viral genome amplification, sequencing, and cloning were performed as described previously (García-Pérez et al. 2013). The complete genomes were sequenced by primer walking using forward and reverse primers, and each position was read at least twice in each direction. Overlapping fragments were generated and used to clone each of the four novel PVs using the CloneJet PCR Cloning Kit (Fermentas) and DH5α cells (Invitrogen). Two clones per overlapping fragment were selected and sequenced to further confirm the PVs genome sequence. Novel PVs were designated EserPV1, EserPV2, EserPV3, and RferPV1, following suggestions to avoid naming ambiguities (García-Pérez et al. 2013).
Genomic and Protein Sequence Annotation
The ORFs encoded in EserPV1, EserPV2, EserPV3, and RferPV1 were identified with the ORF Finder tool on the NCBI server. They were confirmed by comparison with a nonredundant protein sequences database in GenBank through the BLASTP server. The MEME algorithm (Bailey et al. 2009) was used to identify putative E2 binding site (E2BS) sequence patterns occurring in Lambda + MuPVs and previously described bat PVs. Pairwise identities and similarity values for nucleotide and protein sequences were calculated using the EMBOSS Needle software.
Phylogenetic Analysis
The data set used to examine the phylogenetic relationships of the four novel viruses comprised 143 PVs that covered their currently known diversity in terms of hosts. A comprehensive description of the PVs employed in this study is provided in supplementary table S2, Supplementary Material online. For this selection, the E1, E2, L2, and L1 genes were analyzed.
Amino acid alignments were constructed individually for each protein (E1, E2, L2, and L1) with MUSCLE (Edgar 2004), filtered with GBLOCKS (Castresana 2000), and concatenated. Maximum likelihood phylogenetic inference for the E1–E2–L2–L1 combination was conducted with RAxML v7.2.8 (Stamatakis 2006), using the GTR + Γ4 substitution model for nucleotide alignments and the LG substitution model for amino acid alignments (Gottschling, Göker et al. 2011). Phylogenetic analyses were calculated considering the corresponding partitions (12 for nucleotide alignments, corresponding to one per codon position per gene, and 4 for amino acid alignments), with individual per partition branch length optimization and running 1,000 bootstrap replicates. Additional phylogenetic analyses were performed as described above separately for the E1–E2 and L2–L1 combinations to investigate possible topological incongruences.
Individual phylogenies for the E6, E7, E1, E2, L2, and L1 genes were constructed exclusively for the Lambda + MuPVs and used to create a supernetwork using SPLITSTREE v4 (Huson and Bryant 2006). Bayesian inference was performed on the same alignments with PHYLOBAYES v3.3 (Lartillot et al. 2009) applying the GTR + Γ4 substitution model for nucleotide alignments and the LG substitution model for amino acid alignments, removing constant sites, running two independent chains, and checking for convergence comparing discrepancies among partitions. The convergence criterion between chains was that the largest discrepancy observed across all bipartitions should be below 0.1. Trees were rooted a posteriori using the sequences of PVs found in birds and in turtles.
A phylogeny including the bat species from which complete PV genome sequences have been retrieved was constructed using a concatenation of partial sequences from three mitochondrial markers, namely Cytochrome Oxidase I (COI, 592 bp), Cytochrome b (Cytb, 754 bp), and NADH dehydrogenase (ND1, 518 bp), and the nuclear gene recombination activating gene (RAG2, 759 bp). Bayesian inference was performed with BEAST applying the GTR substitution model, using four partitions (one per gene), running two independent chains and empirically estimating the gamma distribution parameter. The first 100,000 trees were discarded as burn-in, and posterior probabilities were calculated by sampling 3 × 106 generations every 300 trees.
All viral contig sequences obtained by Baker et al. (2013) were screened for PV origin using TBlastX searches, further identifying for each contig the coding frames and sequences and eliminating premature stop codons. The final curated amino acid sequences are provided in supplementary table S3, Supplementary Material online. An evolutionary placement algorithm (EPA) (Berger and Stamatakis 2011) was applied to introduce the sequences into the previously constructed well-resolved phylogeny as described (Mengual-Chuliá et al. 2012). A total of 381 sequences (E1 n = 151, E2 n = 40, L2 n = 55, and L1 n = 135), together with the partial E1 and L1 sequences of PgigPV1, were assigned a phylogenetic position. The same strategy was used to infer the phylogenetic positions of the partial E1 (n = 5) and L1 (n = 9) sequences obtained in this study.
Prevalence of EserPV1, EserPV2, and EserPV3 in Iberian Bat Populations
Prevalence was investigated by PCR screening of 267 additional samples, including oropharyngeal swabs (n = 78), anogenital swabs (n = 85), and hair bulbs (n = 104) obtained from 106 E. serotinus (n = 33) and E. isabellinus (n = 73) individuals. Samples were collected from central Spain during 2012. A detailed summary of the samples is provided in supplementary table S4, Supplementary Material online. DNA extraction from swabs was performed as described above. DNA extraction from hair bulbs was performed following a protocol for the isolation of genomic DNA from tissues (Qiagen). Full-length PV genomes were amplified using RCA, and RCA products were used as template for the PCR reactions. Primer sequences, gene targets, amplicon sizes, and PCR conditions are specified in supplementary table S5, Supplementary Material online. Presence of other unknown PVs was further studied using the PV-specific FAP and the CP primers, as described (Mengual-Chuliá et al. 2012). Obtained amplicons were also sequenced in both strands using the same amplification primers and analyzed through a BlastX search to confirm their PV origin.
Results
Genomic Organization and Sequence Similarity to Other PVs
We have characterized the complete genomes of four novel PVs isolated from mucosal swabs of two free-ranging Iberian E. serotinus (EserPV1, EserPV2, and EserPV3) and one R. ferrumequinum (RferPV1) individuals.
The genomes of EserPV1, EserPV2, EserPV3, and RferPV1 consisted of 7,668, 7,574, 7,711, and 8,249 bp, respectively, and are available in GenBank under the following accession numbers: KC858263, KC858264, KC858265, and KC858266. The four genomes presented the typical PV ORFs coding for five early proteins (E6, E7, E1, E2, and E4) and two late proteins (L2 and L1) located on the same coding strand. Putative E4 ORFs were identified nested within E2, as they contained rich proline stretches that characterize E4 proteins. The four PVs presented a noncoding region (NCR1) spanning between the stop codon of L1 and the start codon of E6. Additionally, RferPV1 contained a second noncoding region between the early and late regions. The genomic arrangement of these four PVs and the location of some common amino acid motifs and regulatory elements are depicted in supplementary figure S1, Supplementary Material online. A detailed description of the genomes, including the precise location of ORFs, amino acid motifs, and regulatory elements, is provided in supplementary table S6, Supplementary Material online.
Two classical zinc-binding domains (CX2CX29CX2C) separated by 36 amino acids were located in all E6 oncoproteins. The same motif was found in the E7 oncoproteins of RferPV1 and EserPV2 and slightly modified (CX2CX30CX2C) in the E7 proteins of EserPV1 and EserPV3 and in those of EhelPV1, MschPV2, and RaPV1. The E7 oncoproteins of EserPV2 and RferPV1 contained a potential pRB-binding motif (LXCXE), absent in the E7 proteins of EserPV1, EserPV3, and MschPV2. The ATP-binding domain (GX4GKS) appeared in all E1 proteins. A leucine zipper domain (LX6LX6LX6L) was found in the E2 proteins of EserPV1 and EserPV3 but was absent in EserPV2, RferPV1 EhelPV1, MschPV1, and MschPV2 (supplementary table S6, Supplementary Material online).
Polyadenylation signals (AATAAA) for the early and late transcripts were found at the beginning of the L2 gene and in the NCR1, respectively. TATA boxes (TATAAA) of the E6 promoter were located close to the 3′ end of the NCR1. Canonical (ACCG-N4-CGGT) and noncanonical putative E2-binding sites (E2BS) were detected in the NCR1 and also in the L2 genes of the novel bat PVs. The canonical and noncanonical E2BS patterns occurring in the NCR1 and L2 genes of Lambda + MuPVs, and the E2BS pattern occurring in the NCR1 of bat PVs, are illustrated in supplementary figures S2 and S3, Supplementary Material online. The presence of putative E2BS had not been previously reported within the L2 genes of PVs, and their biological significance is unknown.
Sequence similarities among all bat PVs described to date were investigated by pairwise alignments of the E6, E7, E1, E2, L2, and L1 genes and of their respective proteins (supplementary table S7, Supplementary Material online). Sequence similarities for the L1 genes and proteins were also studied within the Lambda + MuPVs crown group (supplementary table S8, Supplementary Material online). EserPV1 and EserPV3 showed the highest similarity to each other while RferPV1 shared its highest similarity with both EserPV1 and EserPV3. EserPV2 was most similar to MschPV1. Low similarity percentages were found between the novel PVs and the bat PVs MrPV1 and RaPV1. Considering the ICTV guidelines for delineating PV taxonomy, based exclusively on nucleotide identity on the L1 gene (de Villiers et al. 2004), the novel PVs here described RferPV1, EserPV1, and EserPV3 could belong together into a single novel genus within the Lambda + MuPVs crown group, while EserPV2 and MschPV1 belong together into a different genus, branching close to the origin of the four PV crown groups (supplementary tables S7 and S8, Supplementary Material online).
Phylogenetic Analysis
Phylogenetic trees were calculated using maximum likelihood and Bayesian approaches, both at the nucleotide and at the amino acid levels. Details of the different alignments and partitions considered are provided in supplementary table S9, Supplementary Material online. Topogical incongruences between early (E1–E2) and late (L2–L1) phylogenies were not observed for the novel bat PVs. The E1–E2–L2–L1 concatenation was therefore used for subsequent analyses. All reconstructed phylogenetic trees are available in the supplementary material, Supplementary Material online, as suggested (Drew et al. 2013). The E1–E2–L2–L1 gene combination generated well-supported phylogenies (fig. 1) in which PVs segregated into four major groups, as previously described: Alpha + OmikronPVs (infecting Artiodactyla, Carnivora, Cetacea, Chiroptera, and Primates), Beta + XiPVs (infecting Artiodactyla, Carnivora, Eulipotyphla, Primates, and Rodentia), Delta + ZetaPVs (infecting Artiodactyla and Perissodactyla), and Lamba + MuPVs (infecting Carnivora, Chiroptera, Lagomorpha, Primates, and Rodentia). Bat PVs did not constitute a monophyletic group. Instead, phylogenetic analyses revealed the existence of at least five different bat PV lineages: 1) RaPV1 showed an uncertain position basal to the tree; 2) EserPV2 and MschPV1 appeared as sister taxa, close to the basal branching events of the four crown groups; 3) MrPV1 was the sister taxon of the UmPV1, confidently nested within the Alpha + OmikronPVs crown group; 4) EserPV1, EserPV3, and RferPV1 constituted a monophyletic clade within the Lambda + MuPVs crown group; 5) EhelPV1 and MschPV2 showed an uncertain phylogenetic position within the Lambda + MuPVs crown group (fig. 1 and supplementary fig. S4, Supplementary Material online). The phylogenetic network reconstructed for Lambda + MuPVs confirmed the closer evolutionary relationships among EserPV1, EserPV3, and RferPV1 but could not resolve the ambiguity regarding EhelPV1 and MschPV2 (fig. 2). Bat PV-like sequences obtained by Baker et al. (2013) in their metagenomic study of the Ei. helvum virome appeared scattered throughout the PV phylogeny and belonged to all different crown groups (supplementary fig. S5, Supplementary Material online).
Fig. 1.—

Bayesian amino acid phylogenetic reconstruction for the E1–E2–L2–L1 concatenation. Branch lengths are drawn to scale, with the scale bar indicating the evolutionary distance in substitutions per site. Numbers above the branches indicate Bayesian posterior probabilities and ML bootstrap support values. Maximum support values are indicated with an asterisk (*), while values below 0.50 and 50 are indicated with a dash (-). Color code highlights the four PV crown groups: red, Alpha + OmikronPVs; green, Beta + XiPVs; blue, Delta + ZetaPVs; ochre, Lambda + MuPVs. Viruses whose detailed phylogenetic relationships could not be disentangled are labeled in black. Silhouettes represent the infected hosts. Taxonomic classification of both hosts (host order) and viruses (PV genera) are included. Gray dots highlight the five lineages encompassing bat PVs. Branches corresponding to clades or PVs that contain an E2–L2 region and may thus reflect individual recombination events are highlighted with a black star. The novel bat PVs described here are highlighted with black arrows.
Fig. 2.—

Lambda + MuPVs supernetwork. The network was constructed using the best-known maximum-likelihood trees of each individual nucleotide PV gene (E6, E7, E1, E2, L2, and L1). Color code represents the different orders of the hosts. Specific PVs tropisms and outcome of the corresponding infections are indicated in the inset. PV genera are specified in gray.
Prevalence of EserPV1, EserPV2, and EserPV3 in Iberian Bat Populations
Anogenital, oropharyngeal, and hair bulb samples from 33 E. serotinus and 73 E. isabellinus individuals were taken from seven Iberian bat colonies. All samples were tested for the presence of EserPV1, EserPV2, and EserPV3 DNA using specific primers. The amplicons and regions targeted for each virus are detailed in supplementary table S5, Supplementary Material online. In total, 10% (8/78) of oropharyngeal samples, 6% (5/85) of anogenital samples, and none of the hair bulbs tested positive for DNA of any of these viruses. Our results highlight the essentially mucosal tropism of these three PVs, also initially retrieved from oropharyngeal swabs. Regarding species specificity, 30% (10/33) among the E. serotinus individuals and 5% (3/62) among the E. isabellinus individuals tested positive for DNA of any of these three viruses (fig. 3 and supplementary table S10, Supplementary Material online). All amplified sequences corresponded exactly to the one isolated originally, independent of the host species. Additionally, presence of other unknown PVs on the same samples was studied using the PV-specific FAP and CP primers. In total, 21% (16/78) of oropharyngeal samples and 0% of anogenital samples tested positive for the presence of DNA of other PVs (fig. 3 and supplementary table S10, Supplementary Material online). The accession numbers for the partial L1 and E1 sequences amplified, respectively, by the FAP and CP primers are provided in supplementary table S11, Supplementary Material online, and pairwise nucleotide identities between these partial sequences and their closest relatives are provided in supplementary table S12, Supplementary Material online. The alignment of the novel fragments amplified with the FAP and CP primers is provided in supplementary table S13, Supplementary Material online. Phylogenetic analysis of these short PV sequences revealed that they were most closely related to the novel bat PVs reported here and to the Sigma-PV EdPV1 and the Nu-PV HPV41 (supplementary fig. S6, Supplementary Material online).
Fig. 3.—
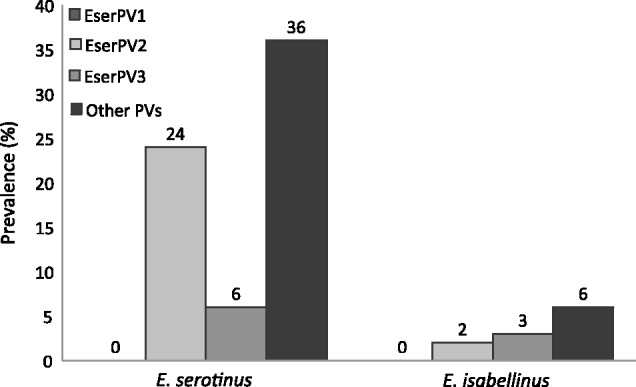
Prevalence of EserPV1, EserPV2, EserPV3, and other PVs DNA in the screened samples recovered from seven different Iberian E. serotinus and E. isabellinus colonies.
Discussion
There is an intrinsic value in the explicit introduction of the study of pathogen evolutionary history into molecular medicine research. Understanding the phylogenetic relationships and mechanisms driving PVs diversification will provide better insight into viral evolution and virus–host interactions. Three assumptions have traditionally dominated PV research: that PVs have co-evolved with their hosts (Van Ranst et al. 1995); that PVs are highly species-specific (Van Ranst et al. 1995; Halpern 2000; Bernard et al. 2006); and that there are minimal virus–virus interactions and therefore events such as recombination are rare (Plummer et al. 2011). Although all three dogmas have been recurrently challenged by recent results (Gottschling et al. 2007; Woolford et al. 2007; Gottschling, Göker et al. 2011; Sakakibara et al. 2013), it is still a commonplace to assume their validity and to address research on human PVs as if they were a monophyletic, distinct entity from animal PVs (Brody 2012; Bernard 2013; de Villiers 2013).
Virus–Host Co-evolution Is Not a Major Determinant of Mammalian PV Evolution
The PV–host co-evolution hypothesis has relied on the gross phylogenetic correspondence for certain PVs and for their mammalian hosts: Alpha- and BetaPVs infecting primates, DeltaPVs infecting ruminants, LambdaPVs infecting carnivores, and PiPVs infecting rodents. However, a systematic topology analysis reveals the absence of congruence between these PVs and their host phylogenies, without a single example of identical tree topologies for both PVs and hosts (Chan et al. 1997; Antonsson and Hansson 2002; Gottschling, Bravo et al. 2011; Gottschling, Göker et al. 2011). The contribution of the different mechanisms to the evolution of PVs can be quantified, and virus–host co-evolution explains only around 30% of all events needed to invoke reconciliation of PVs and hosts phylogenetic trees (Gottschling, Göker et al. 2011). This value should nevertheless be taken with caution, given the limited taxon of animal PVs and the focus on human PVs. The lack of global congruence between PVs and host trees is also reflected on the existence of several polyphyletic lineages for PVs infecting the same host. This is true for PVs isolated from humans, chimpanzees, gorillas, macaques, rodents, dogs, cats, cattle, sheep, horses, dolphins, and porpoises (fig. 1 and supplementary fig. S4, Supplementary Material online), which belong to different PV genera and appear scattered throughout the PV phylogeny in a highly polyphyletic pattern (Bravo et al. 2010; Gottschling, Göker et al. 2011).
The description of the novel bat PVs communicated here provides further evidence to reject the hypothesis of exclusively PV–host co-evolution. Bats monophyly is widely accepted and supported both by morphological (Simmons 1994) and molecular studies (Murphy et al. 2001). The existence of several polyphyletic bat PVs lineages rejects the hypothesis of co-evolution between PVs and bats at a global scale. Likewise, co-evolution as a more recent event seems also unlikely. Under a strictly co-evolution pattern, one would expect pteropid and rhinolophid PVs (RaPV1, EhelPV1, and RferPV1) to be monophyletic, while PVs infecting the remaining bats should form a different clade. One would also expect serotine and myotid PVs to be more closely related and both to be sister taxa to the miniopterid PVs (fig. 4). Instead, PVs infecting pteropids, rhilonophids, and vespertilionids are found intermingled in several, only distantly related, PV lineages (fig. 1). In addition to the four novel genomes here communicated, we have identified fragments of ten possible novel PVs from E. serotinus and four from E. isabellinus (supplementary tables S4 and S11, Supplementary Material online). Furthermore, the phylogenetic positions assigned here to other short PV-like sequences isolated from bats (Baker et al. 2013) suggest that the number of different bat PV lineages might be even larger (supplementary fig. S5, Supplementary Material online). The performed analysis placed the metagenomic sequences in close proximity to the novel bat PVs described, but strikingly, some taxa appeared nested within all PVs crown groups. The appearance at the same position of sequences retrieved from different PV genes supports these different locations. Non-assembled sequences must however be regarded preliminary, and cloned, complete genome sequences would be required to fully confirm the relationships of the putative novel viruses. Yet, it seems likely that bats can be the hosts to a plethora of different PVs belonging to all different crown groups.
Fig. 4.—
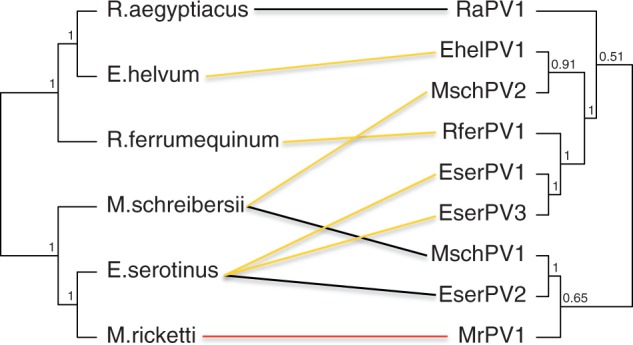
Tanglegram linking the phylograms of bat PVs and their hosts. Congruence between the phylogenetic relationships among host bat species after Bayesian inference (left) and bat PVs after maximum likelihood inference (right). Please note that the PV taxa depicted here are representatives of highly polyphyletic PV crown groups. Color code for the lines linking both phylograms corresponds to colors used in figure 2 for the different PV crown groups.
Remarkably, all isolates containing EserPV2 DNA corresponded to coinfections between this virus and a second PV (supplementary fig. S6 and table S4, Supplementary Material online). Interestingly, serotine bats from one of the sampled colonies (Casatejada) showed abundant ulcer-like wounds in their wings (data not shown). This was also the colony with the highest prevalence of PV infection, with 50% of the individuals testing positive for any PV (supplementary tables S4 and S10, Supplementary Material online). Further research is being carried out to establish the etiology of these lesions, unlikely to be associated to PV infections. The PV-specific FAP and CP primers failed to detect EserPV1, EserPV2, and EserPV3 in the screened samples. This fact suggests, in agreement with previous studies, that the efficiency of the universal consensus FAP and CP primers is limited and that they miss a considerable number of infections. It is therefore very likely that the prevalence of PV infection is larger than estimated here.
No Strict Species Specificity in PVs
PVs are usually considered as highly species-specific (Bernard et al. 2010; Bernard 2013). PVs are named after the host in which they were first isolated, following the ICTV code (Bernard et al. 2010). All subsequent analyses considering host–virus specificity commonly follow this naming convention as a hypothesis to assume host specificity or at least host preference. However, the results communicated here show that similar viruses can infect different hosts, which sums up to the several examples of heterologous PV infections between distantly related hosts (Munday et al. 2007; van Dyk et al. 2009; Munday and Knight 2010; Gottschling, Bravo et al. 2011). We initially isolated EserPV1, EserPV2, and EserPV3 from oropharyngeal swabs from two different E. serotinus individuals. Notably, the results of the screening performed in bat samples collected from seven different colonies revealed the presence of DNA from EserPV2 and EserPV3 not only in E. serotinus but also in E. isabellinus individuals. These are two morphologically cryptic but molecularly highly differentiated species (above 16% divergence in the cytochrome b gene) within the serotinus species group with different geographic distributions. E. serotinus is widely distributed throughout Europe, while E. isabellinus shows a patchy distribution restricted to the southern half of the Iberian Peninsula and North Africa (Ibáñez et al. 2006; Juste et al. 2013). The growing number of PV cross-infections descriptions suggests that certain PVs might exhibit a broader host range and/or that host switches between different species might occur more frequently than initially suspected. This potential to infect different hosts could help explain the highly polyphyletic and/or paraphyletic pattern that many PVs display.
Presence of a Second NCR in RferPV1
During evolution, and independently for different PV clades, the E2–L2 intergenic region has often behaved as a target for recombination: during viral integration in cancers (Doorbar et al. 2012); early in the evolution of AlphaPVs (Bravo and Alonso 2004; Varsani et al. 2006; Angulo and Carvajal-Rodríguez 2007); between PVs belonging to different genera (Gottschling, Bravo et al. 2011; Gottschling, Göker et al. 2011; Robles-Sikisaka et al. 2012); and even between members of two viral families, Papillomaviridae and Polyomaviridae (Woolford et al. 2007). The E2–L2 region can also accommodate both coding and noncoding genomic segments, which may have gained access to the PV genomes through recombination events with hitherto nonidentified donors. On the one hand, Alpha- and DeltaPVs encode in their E2-L2 region for different nonevolutionarily related E5 proteins (Bravo and Alonso 2004). On the other hand, LambdaPVs, EePV1, isolated from an European hedgehog, and the RferPV1 reported here, all incorporate a large noncoding region (NCR2) in the intergenic E2–L2 region. No regulatory, promoter, or coding elements can be identified in these NCR2, and the presence and conservation of such a long segment of around 1 kb, i.e., 15% of the viral genome, is puzzling. All these putative recombination events in the intergenic E2–L2 region of the PVs have likely occurred as individual, independent events. The alternative hypothesis of all clades containing elements between E2 and L2 to be monophyletic is rejected (Shimodaira-Hasegawa test, P value <0.01) when compared with nonconstrained trees. Our interpretation implies thus that at least five independent recombination events (identified in fig. 1) may have occurred in between the E2 and L2 genes throughout the evolutionary history of PVs.
Conclusion
The description here reported of four novel PVs and the assessment of their prevalence in Iberian bat populations reinforce the notion that the evolutionary dynamics of PVs are complex. Our results strongly point toward multiple forces as drivers of PVs evolution, including co-evolution, adaptive radiation, broad host range, host switch, and recombination. However, as long as our knowledge of PVs, diversity is biased toward a specific host (humans) and thus unbalanced, sound conclusions about PVs evolution cannot be reached. The number of nonhuman PVs has increased in recent years (currently 114 known nonhuman PVs isolated from 55 different species). Yet, taxonomic sampling is insufficient and clades such as Afrotheria or Xenarthra remain largely unexplored. A systematic sampling would eventually allow the development of a comprehensive evolutionary framework reconciling the biology, epidemiology, and genomic structure of PVs. Only with this scenario we will be able to provide an answer to many of the still unsolved questions such as the presence of signatures in different PVs that account for their oncogenic potential, their cell tropism, or their host range.
Supplementary Material
Supplementary figures S1–S6 and tables S1–S13 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Acknowledgments
I.G.B. conceived the study; R.G.P. designed and performed experiments; J.J., J.E.E., C.I., N.A., and I.G. retrieved and identified samples; J.M.G. performed experiments; R.G.P., I.G.B., and J.J. analyzed the data; R.G.P. and I.G.B. drafted the manuscript. All authors revised and approved the last version of the manuscript. The authors thank Dr Susana Martínez-Alós for her work during animal retrieval and identification and Ana Esteban for her technical assistance in PVs detection. R.G.P. was the recipient of grants from the Red Temática de Investigación Cooperativa en Cáncer (RTICC) and “La Caixa” Foundation. This work was supported by the disappeared Spanish Ministry for Science and Innovation (MICINN) grants CGL2010-16713, SAF2006-12784-CO2, and SAF2009-09172.
Literature Cited
- Alberti A, et al. Ovis aries Papillomavirus 3: a prototype of a novel genus in the family Papillomaviridae associated with ovine squamous cell carcinoma. Virology. 2010;407:352–359. doi: 10.1016/j.virol.2010.08.034. [DOI] [PubMed] [Google Scholar]
- Angulo M, Carvajal-Rodríguez A. Evidence of recombination within human alpha-papillomavirus. Virol J. 2007;4:33. doi: 10.1186/1743-422X-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonsson A, Hansson BG. Healthy skin of many animal species harbors papillomaviruses which are closely related to their human counterparts. J Virol. 2002;76:12537–12542. doi: 10.1128/JVI.76.24.12537-12542.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker KS, et al. Metagenomic study of the viruses of African straw-coloured fruit bats: detection of a chiropteran poxvirus and isolation of a novel adenovirus. Virology. 2013;441:95–106. doi: 10.1016/j.virol.2013.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SA, Stamatakis A. Aligning short reads to reference alignments and trees. Bioinformatics. 2011;27:2068–2075. doi: 10.1093/bioinformatics/btr320. [DOI] [PubMed] [Google Scholar]
- Bernard H-U. Taxonomy and phylogeny of papillomaviruses: an overview and recent developments. Infect Genet Evol. 2013;18:357–361. doi: 10.1016/j.meegid.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Bernard H-U, Calleja-Macias IE, Dunn ST. Genome variation of human papillomavirus types: phylogenetic and medical implications. Int J Cancer. 2006;118:1071–1076. doi: 10.1002/ijc.21655. [DOI] [PubMed] [Google Scholar]
- Bernard H-U, et al. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology. 2010;401:70–79. doi: 10.1016/j.virol.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo IG, Alonso A. Mucosal human papillomaviruses encode four different E5 proteins whose chemistry and phylogeny correlate with malignant or benign growth. J Virol. 2004;78:13613–13626. doi: 10.1128/JVI.78.24.13613-13626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo IG, de Sanjosé S, Gottschling M. The clinical importance of understanding the evolution of papillomaviruses. Trends Microbiol. 2010;18:432–438. doi: 10.1016/j.tim.2010.07.008. [DOI] [PubMed] [Google Scholar]
- Brody H. Human papillomavirus. Nature. 2012;488:S1. doi: 10.1038/488S1a. [DOI] [PubMed] [Google Scholar]
- Calisher CH, Childs JE, Field HE, Holmes KV, Schountz T. Bats: important reservoir hosts of emerging viruses. Clin Microbiol Rev. 2006;19:531–545. doi: 10.1128/CMR.00017-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Chan SY, et al. Genomic diversity and evolution of papillomaviruses in rhesus monkeys. J Virol. 1997;71:4938–4943. doi: 10.1128/jvi.71.7.4938-4943.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu DKW, Poon LLM, Guan Y, Peiris JSM. Novel astroviruses in insectivorous bats. J Virol. 2008;82:9107–9114. doi: 10.1128/JVI.00857-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua KB, et al. Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect. 2002;4:145–151. doi: 10.1016/s1286-4579(01)01522-2. [DOI] [PubMed] [Google Scholar]
- de Villiers EM. Cross-roads in the classification of papillomaviruses. Virology. 2013;445:2–10. doi: 10.1016/j.virol.2013.04.023. [DOI] [PubMed] [Google Scholar]
- de Villiers EM, Fauquet C, Broker TR, Bernard HU, zur Hausen H. Classification of papillomaviruses. Virology. 2004;324:17–27. doi: 10.1016/j.virol.2004.03.033. [DOI] [PubMed] [Google Scholar]
- Doorbar J, et al. The biology and life-cycle of human papillomaviruses. Vaccine. 2012;30(Suppl 5):F55–F70. doi: 10.1016/j.vaccine.2012.06.083. [DOI] [PubMed] [Google Scholar]
- Drew BT, et al. Lost branches on the tree of life. PLoS Biol. 2013;11:e1001636. doi: 10.1371/journal.pbio.1001636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drexler JF, et al. Bats host major mammalian paramyxoviruses. Nat Commun. 2012;3:796. doi: 10.1038/ncomms1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echevarría JE, Avellón A, Juste J, Vera M, Ibáñez C. Screening of active lyssavirus infection in wild bat populations by viral RNA detection on oropharyngeal swabs. J Clin Microbiol. 2001;39:3678–3683. doi: 10.1128/JCM.39.10.3678-3683.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcón A, et al. Detection of alpha and betacoronaviruses in multiple Iberian bat species. Arch Virol. 2011;156:1883–1890. doi: 10.1007/s00705-011-1057-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field HE, Barratt PC, Hughes RJ, Shield J, Sullivan ND. A fatal case of Hendra virus infection in a horse in north Queensland: clinical and epidemiological features. Aust Vet J. 2000;78:279–280. doi: 10.1111/j.1751-0813.2000.tb11758.x. [DOI] [PubMed] [Google Scholar]
- Forslund O, Antonsson A, Nordin P, Stenquist B, Hansson BG. A broad range of human papillomavirus types detected with a general PCR method suitable for analysis of cutaneous tumours and normal skin. J Gen Virol. 80(Pt. 1999;9):2437–2443. doi: 10.1099/0022-1317-80-9-2437. [DOI] [PubMed] [Google Scholar]
- García-Pérez R, Gottschling M, Wibbelt G, Bravo IG. Multiple evolutionary origins of bat papillomaviruses. Vet Microbiol. 2013;165:51–60. doi: 10.1016/j.vetmic.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Giri I, Danos O, Yaniv M. Genomic structure of the cottontail rabbit (Shope) papillomavirus. Proc Natl Acad Sci U S A. 1985;82:1580–1584. doi: 10.1073/pnas.82.6.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschling M, Bravo IG, et al. Modular organizations of novel cetacean papillomaviruses. Mol Phylogenet Evol. 2011;59:34–42. doi: 10.1016/j.ympev.2010.12.013. [DOI] [PubMed] [Google Scholar]
- Gottschling M, Göker M, et al. Quantifying the phylodynamic forces driving papillomavirus evolution. Mol Biol Evol. 2011;28:2101–2113. doi: 10.1093/molbev/msr030. [DOI] [PubMed] [Google Scholar]
- Gottschling M, et al. Multiple evolutionary mechanisms drive papillomavirus diversification. Mol Biol Evol. 2007;24:1242–1258. doi: 10.1093/molbev/msm039. [DOI] [PubMed] [Google Scholar]
- Halpern AL. Comparison of papillomavirus and immunodeficiency virus evolutionary patterns in the context of a papillomavirus vaccine. J Clin Virol. 2000;19:43–56. doi: 10.1016/s1386-6532(00)00127-x. [DOI] [PubMed] [Google Scholar]
- Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- Ibáñez C, García-Mudarra JL, Ruedi M, Stadelmann B, Juste J. The Iberian contribution to cryptic diversity in European bats. Acta Chiropterol. 2006;8:277–297. [Google Scholar]
- Iftner A, et al. The prevalence of human papillomavirus genotypes in nonmelanoma skin cancers of nonimmunosuppressed individuals identifies high-risk genital types as possible risk factors. Cancer Res. 2003;63:7515–7519. [PubMed] [Google Scholar]
- International Union for Conservation of Nature (IUCN) Red list of threatened species. 2013 [cited 2013 Dec]. Available from: http://www.iucnredlist.org/ [Google Scholar]
- Juste J, Benda P, Garcia-Mudarra JL, Ibáñez C. Phylogeny and systematics of Old World serotine bats (genus Eptesicus, Vespertilionidae, Chiroptera): an integrative approach. Zool Scr. 2013;42:441–457. [Google Scholar]
- Kunz TH, Braun de Torrez E, Bauer D, Lobova T, Fleming TH. Ecosystem services provided by bats. Ann N Y Acad Sci. 2011;1223:1–38. doi: 10.1111/j.1749-6632.2011.06004.x. [DOI] [PubMed] [Google Scholar]
- Kurth A, et al. Novel paramyxoviruses in free-ranging European bats. PLoS One. 2012;7:e38688. doi: 10.1371/journal.pone.0038688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange CE, Tobler K, Markau T, et al. Sequence and classification of FdPV2, a papillomavirus isolated from feline Bowenoid in situ carcinomas. Vet Microbiol. 2009;137:60–65. doi: 10.1016/j.vetmic.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Lange CE, Tobler K, Ackermann M, et al. Three novel canine papillomaviruses support taxonomic clade formation. J Gen Virol. 2009;90:2615–2621. doi: 10.1099/vir.0.014498-0. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Lepage T, Blanquart S. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics. 2009;25:2286–2288. doi: 10.1093/bioinformatics/btp368. [DOI] [PubMed] [Google Scholar]
- Leroy EM, et al. Fruit bats as reservoirs of Ebola virus. Nature. 2005;438:575–576. doi: 10.1038/438575a. [DOI] [PubMed] [Google Scholar]
- Li W, et al. Bats are natural reservoirs of SARS-like coronaviruses. Science. 2005;310:676–679. doi: 10.1126/science.1118391. [DOI] [PubMed] [Google Scholar]
- Mengual-Chuliá B, García-Pérez R, Gottschling M, Nindl I, Bravo IG. Novel animal papillomavirus sequences and accurate phylogenetic placement. Mol Phylogenet Evol. 2012;65:883–891. doi: 10.1016/j.ympev.2012.08.011. [DOI] [PubMed] [Google Scholar]
- Monath TP. Ecology of Marburg and Ebola viruses: speculations and directions for future research. J Infect Dis. 1999;179(Suppl):S127–S138. doi: 10.1086/514281. [DOI] [PubMed] [Google Scholar]
- Mühldorfer K, et al. Diseases and causes of death in European bats: dynamics in disease susceptibility and infection rates. PLoS One. 2011;6:e29773. doi: 10.1371/journal.pone.0029773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munday JS, Hanlon EM, Howe L, Squires RA, French AF. Feline cutaneous viral papilloma associated with human papillomavirus type 9. Vet Pathol. 2007;44:924–927. doi: 10.1354/vp.44-6-924. [DOI] [PubMed] [Google Scholar]
- Munday JS, Knight CG. Amplification of feline sarcoid-associated papillomavirus DNA sequences from bovine skin. Vet Dermatol. 2010;21:341–344. doi: 10.1111/j.1365-3164.2010.00872.x. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, et al. Molecular phylogenetics and the origins of placental mammals. Nature. 2001;409:614–618. doi: 10.1038/35054550. [DOI] [PubMed] [Google Scholar]
- Nafz J, et al. A novel rodent papillomavirus isolated from anogenital lesions in its natural host. Virology. 2008;374:186–197. doi: 10.1016/j.virol.2007.12.012. [DOI] [PubMed] [Google Scholar]
- Nasir L, Campo MS. Bovine papillomaviruses: their role in the aetiology of cutaneous tumours of bovids and equids. Vet Dermatol. 2008;19:243–254. doi: 10.1111/j.1365-3164.2008.00683.x. [DOI] [PubMed] [Google Scholar]
- Plummer M, Vaccarella S, Franceschi S. Multiple human papillomavirus infections: the exception or the rule? J Infect Dis. 2011;203:891–893. doi: 10.1093/infdis/jiq146. [DOI] [PubMed] [Google Scholar]
- Rector A, et al. Genetic characterization of the first chiropteran papillomavirus, isolated from a basosquamous carcinoma in an Egyptian fruit bat: the Rousettus aegyptiacus papillomavirus type 1. Vet Microbiol. 2006;117:267–275. doi: 10.1016/j.vetmic.2006.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles-Sikisaka R, et al. Evidence of recombination and positive selection in cetacean papillomaviruses. Virology. 2012;427:189–197. doi: 10.1016/j.virol.2012.01.039. [DOI] [PubMed] [Google Scholar]
- Sakakibara N, Chen D, McBride AA. Papillomaviruses use recombination-dependent replication to vegetatively amplify their genomes in differentiated cells. PLoS Pathog. 2013;9:e1003321. doi: 10.1371/journal.ppat.1003321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaratunga H, Searle JW, Hudson N. Non-rabies Lyssavirus human encephalitis from fruit bats: Australian bat Lyssavirus (pteropid Lyssavirus) infection. Neuropathol Appl Neurobiol. 1998;24:331–335. doi: 10.1046/j.1365-2990.1998.00129.x. [DOI] [PubMed] [Google Scholar]
- Scase T, et al. Equus caballus papillomavirus-2 (EcPV-2): an infectious cause for equine genital cancer? Equine Vet J. 2010;42:738–745. doi: 10.1111/j.2042-3306.2010.00311.x. [DOI] [PubMed] [Google Scholar]
- Schulz E, et al. Genomic characterization of the first insectivoran papillomavirus reveals an unusually long, second non-coding region and indicates a close relationship to Betapapillomavirus. J Gen Virol. 2009;90:626–633. doi: 10.1099/vir.0.008011-0. [DOI] [PubMed] [Google Scholar]
- Simmons NB. The case for chiropteran monophyly. Am Mus Novitat. 1994;3103:1–54. [Google Scholar]
- Simmons NB. Evolution. An Eocene big bang for bats. Science. 2005;307:527–528. doi: 10.1126/science.1108871. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Tse H, et al. Identification of a novel bat papillomavirus by metagenomics. PLoS One. 2012;7:e43986. doi: 10.1371/journal.pone.0043986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner D. The vampire bat, a field study in behavior and ecology. Baltimore (MD): London Johns Hopkins University Press; 1975. [Google Scholar]
- VV.AA. 2010. Nombre de los murcielagos en España. Revista “Barbastella (Journal of the Spanish Asociation for Conservation and Study of Bats)” 4:15.
- van Dyk E, et al. Detection of bovine papillomavirus DNA in sarcoid-affected and healthy free-roaming zebra (Equus zebra) populations in South Africa. J Virol Methods. 2009;158:141–151. doi: 10.1016/j.jviromet.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Van Ranst M, Kaplan JB, Sundberg JP, Burk RD. Molecular evolution of papillomaviruses. In: Gibbs A, Calisher CH, Garcia-Arena F, editors. Molecular basis of virus evolution. Cambridge: Cambridge University Press; 1995. pp. 455–476. [Google Scholar]
- Varsani A, et al. Evidence of ancient papillomavirus recombination. J Gen Virol. 2006;87:2527–2531. doi: 10.1099/vir.0.81917-0. [DOI] [PubMed] [Google Scholar]
- Woolford L, et al. A novel virus detected in papillomas and carcinomas of the endangered western barred bandicoot (Perameles bougainville) exhibits genomic features of both the Papillomaviridae and Polyomaviridae. J Virol. 2007;81:13280–13290. doi: 10.1128/JVI.01662-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J Virol. 2012;86:10999–11012. doi: 10.1128/JVI.01394-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur Hausen H. The search for infectious causes of human cancers: where and why. Virology. 2009;392:1–10. doi: 10.1016/j.virol.2009.06.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.