

Abstract
No curative therapy is currently available for locally advanced or metastatic pancreatic cancer. Therefore, new therapeutic approaches must be considered. Measles virus (MV) vaccine strains have shown promising oncolytic activity against a variety of tumor entities. For specific therapy of pancreatic cancer, we generated a fully retargeted MV that enters cells exclusively through the prostate stem cell antigen (PSCA). Besides a high-membrane frequency on prostate cancer cells, this antigen is expressed on pancreatic adenocarcinoma, but not on non-neoplastic tissue. PSCA expression levels differ within heterogeneous tumor bulks and between human pancreatic cell lines, and we could show specific infection of pancreatic adenocarcinoma cell lines with both high- and low-level PSCA expression. Furthermore, we generated a fully retargeted and armed MV-PNP-anti-PSCA to express the prodrug convertase purine nucleoside phosphorylase (PNP). PNP, which activates the prodrug fludarabine effectively, enhanced the oncolytic efficacy of the virus on infected and bystander cells. Beneficial therapeutic effects were shown in a pancreatic cancer xenograft model. Moreover, in the treatment of gemcitabine-resistant pancreatic adenocarcinoma cells, no cross-resistance to both MV oncolysis and activated prodrug was detected.
Keywords: oncolysis, measles virus, prodrug-converting enzyme, single-chain antibody, pancreatic cancer
Introduction
Pancreatic cancer is the fourth most frequent cause of cancer-related deaths in the United States and the sixth in Europe.1 Owing to the aggressive neoplastic nature and lack of early specific symptoms, more than 80% of patients have locally advanced tumors and/or metastases at diagnosis.2 In addition, there is a high risk of locoregional recurrence after surgical resection.3 Gemcitabine (2′,2′-difluorodeoxycytidine, Gmc) has been the front-line chemotherapy for advanced pancreatic cancer over the past 10 years. As resistance to Gmc treatment is a major issue,4 other chemotherapeutics are explored extensively, but survival benefit has not been improved substantially. Therefore, the development of alternative therapies is urgently needed.
Currently, oncolytic viruses from different taxa are being evaluated in pre-clinical and clinical settings.5 We and others have previously shown that derivatives of the measles virus (MV) vaccine strain have potent oncolytic activity on a wide array of cancer types.6–13 Recombinant MV with ablated natural receptor recognition have been engineered,14–18 fully retargeted by specific ligands or single-chain antibodies (scFv) displayed as a fusion to the viral attachment protein H.19
For targeted therapy approaches of pancreatic cancer, a unique surface antigen is sought. Candidate targets include the prostate stem cell antigen (PSCA), a small, glycosyl-phosphatidyl-inositol-anchored cell surface protein overexpressed on prostatic adenocarcinomas and urinary bladder cancer cells.20,21 As PSCA is also displayed on pancreatic adenocarcinoma, but not on non-neoplastic tissue,22 it is an antigen for targeted therapy of pancreatic cancer,23 currently evaluated in a phase II clinical trial using the anti-PSCA monoclonal antibody AGS-1C4D4.2
To enhance oncolytic efficacy, viruses have been modified to express suicide genes, in particular prodrug-converting enzymes.24 MV oncolysis was successfully enhanced with the Escherichia coli purine nucleoside phosphorylase (PNP),7,8 which converts fludarabine (2-fluoro-araAMP) to 2-fluoroadenine with high bystander activity. 2-Fluoroadenine is highly diffusible and can be metabolized to toxic ATP analogues that inhibit DNA, RNA and protein synthesis immediately.25 In a conventional approach without PNP, fludarabine is not metabolized to 2-fluoroadenine and, therefore, is not as effective. Thus, local activation of fludarabine by MV-encoded PNP can enhance its therapeutic efficacy killing both MV-infected and non-infected cancer cells.
In this study, we have engineered a recombinant MV for the treatment of pancreatic cancer fully retargeted with a single-chain antibody conferring selective entry through PSCA.26 This virus was armed with the prodrug convertase PNP to further enhance its oncolytic potency. We show that MV-PNP-anti-PSCA can infect pancreatic adenocarcinoma cells in a receptor-specific manner, and exhibits high oncolytic efficacy in both high- and low-level PSCA-expressing cell lines. Furthermore, we show that therapeutic efficacy in a xenograft model of pancreatic cancer is enhanced significantly after systemic administration of the cognate prodrug fludarabine. Moreover, in the treatment of Gmc-resistant pancreatic adenocarcinoma cells, cross-resistance neither to MV oncolysis nor to the activated prodrug was detected.
Materials and methods
Cell culture
Vero cells (African green monkey kidney) and human pancreatic adenocarcinoma cell lines BxPC-3 and Capan-1 were purchased from American Type Culture Collection. T3M4 and MiaPaCa-2 cells were kindly provided by Zahari Raykov (DKFZ, Heidelberg, Germany). Pancreatic adenocarcinoma cell lines, except for Capan-1, were maintained in RPMI medium (Invitrogen, Darmstadt, Germany) supplemented with 10% heat-inactivated fetal bovine serum in a humidified atmosphere of 5% CO2 at 37 °C. Capan-1 cells were grown at 37 °C in Iscove's modified Dulbecco's medium containing 20% fetal bovine serum and 1% ultraglutamine (Lonza, Basel, Switzerland). The cell lines Vero-αHis (kind gift from Stephen J Russell, Mayo Clinic, Rochester, MN), Vero and 293T-PSCA were cultivated in Dulbecco's modified Eagle's medium with 10% fetal bouine serum. All cell types were routinely checked for mycoplasma contamination using a multiplex cell contamination test (DKFZ Genomics and Proteomics Core Facility).27
Construction of recombinant MV
The constructions of MV genomic cDNA plasmids were based on pMV-EGFP (designated pMeGFPNV)28 containing the MV Edmonston-B vaccine lineage strain. The coding sequence of the single-chain variable fragment against PSCA (scFv AM1)—generated previously from the monoclonal antibody 7F526—was polymerase chain reaction amplified using primers flanked by a 5′-NotI and 3′-SfiI cloning site. The NotI–SfiI fragment was inserted by exchange subcloning in pCG-HmutI-αCD20 containing the ‘blind’ H protein with CD46- and SLAM-ablating mutations,8 leading to pCG-HmutI-αPSCA. Thereof, the PSCA-retargeted PacI–SpeI fragment was isolated and exchanged in pMV-EGFP and ‘p(+)MV-PNP Hblind antiCD20’,8 resulting in the full-length genomic plasmids pMV-EGFP-anti-PSCA and pMV-PNP-anti-PSCA, respectively. The accuracy of generated plasmids was verified by single colony polymerase chain reaction, restriction digest and sequencing. All cloning steps were designed to fulfill the rule of six as a requirement for recombinant MV.29,30
Recombinant MV particles were generated from cDNA constructs according to Radecke et al.29 and fully retargeted viruses were subsequently propagated on Vero-αHis cells as described previously.19 To prepare virus stocks, Vero-αHis cells were infected at an MOI of 0.03 and incubated at 37 °C for 36 h. Viral particles were harvested by one freeze–thaw cycle and centrifugation from their cellular substrate resuspended in Opti-MEM (Invitrogen). All following infection experiments were performed with viral stocks from the third passage. For determination of the viral titers in a 96-well plate, a serial 1:10 dilution of the respective virus in 100 μl medium (Dulbecco's modified Eagle's medium + 10% fetal bovine serum) per well was carried out in octuplicates. Thereafter, 1 × 104 Vero cells in 100 μl medium were added to each well. At 48 h p.i., individual syncytia in all wells of the appropriate dilution step were counted and the viral titer (infectious units per ml) was calculated.
Establishment of Gmc-resistant pancreatic adenocarcinoma cell lines
Gmc-resistant pancreatic adenocarcinoma cell lines were established according to Angelova et al.31 with slight modifications by exposure to gradually increasing Gmc concentrations. Sensitive pancreatic adenocarcinoma cell lines were cultured for 2 weeks at their specific initially determined Gmc-threshold concentration. This concentration varied depending on the cell type (Capan-1, MiaPaCa-2: 10 nm; BxPC-3, T3M4: 25 nm). After 14 days, the cells were subcultivated and grown for 3 days in the absence of Gmc, followed by 4 days cultivation in media containing a slightly increased Gmc concentration compared with the initial threshold. The second cycle started with 3 days of cultivation without Gmc, followed by 4 days cultivation in Gmc containing media, and every second cycle the Gmc concentration was increased.
To characterize the induced Gmc resistance, we determined the half-maximal inhibitory concentration inducing a 50% reduction of viability of pretreated and of parental cell lines. Cells were grown in 96-well plates (104 cells per well) in the respective culture medium for 14 h. The cells were exposed to increasing Gmc doses (0 nm, 5 nm, 10 nm, 25 nm, 50 nm, 75 nm, 100 nm, 500 nm, 1 μm and 10 μm) and incubated for 72 h at 37 °C followed by a XTT cell viability assay.
Virus infections
Cell lines were infected with the respective MV at an MOI of 1, 0.1 or 0.01 in Opti-MEM (Invitrogen) for 2.5 h at 37 °C. At the end of the incubation period, the inoculum was removed and the cells were maintained in their standard medium. Infected cells were photographed using a fluorescence microscope (Cell Observer; Zeiss, Jena, Germany) and the software Axiovision, or subjected to cell viability assay.
Cell viability assay of infected/prodrug-converting cells
The cell proliferation kit II (XTT; Roche, Mannheim, Germany) was performed as recommended by the manufacturer. Cells were grown in 96-well plates (104 cells per well) in the respective culture medium for 14 h and infected with the respective virus at an MOI of 1. 2-Fluoro-araA (9-β-d-arabinofuranosyl-2-fluoroadenine; Sigma-Aldrich, Taufkirchen, Germany) was added 24 h after infection at a final concentration of 5 μm and cells were further incubated for 48 or 72 h. Cell viability was measured by dye absorbance at 465 nm using an automated microplate reader (SpectraMax 340PC384; Molecular Devices, Ismaning, Germany). The viability of cells treated with or without the respective agents was calculated as the mean of triplicate optical density values and expressed in relation to the untreated control samples (100% viable).
Bystander killing measurement in vitro
In a 24-well plate, 105 Vero-αHis cells were seeded, infected 6 h later at an MOI of 0.1 with MV-PNP-anti-PSCA and incubated for 36 h. Fludarabine was added to the media at a final concentration of 5 μm for 12 h. Supernatants were harvested, cleared by centrifugation and heat-inactivated at 60 °C for 30 min. Fresh pancreatic adenocarcinoma and Vero-αHis cells in 96-well plates (104 cells per well) were incubated for 72 h with different fractions of conditioned media (0.5, 0.1 and 0.01). Living cells were quantified by the XTT cell viability assay.
Subcutaneous xenograft tumor model
All animal experimental procedures were approved by the responsible animal protection officer at the German Cancer Research Center and by the regional council according to the German Animal Protection Law. Tumors were established by inoculating 6.6 × 106 BxPC-3 cells into the right flank of 6–8 weeks old, female non-obese diabetic/severe combined immunodeficient mice (Harlan, Borchen, Germany). When tumors reached an average volume of 50 μl, they were injected with 7.25 × 105 infectious units of MV-PNP-anti-PSCA in 100 μl Opti-MEM on 5 consecutive days. At 3 days after the last virus application, mice received 250 mg/kg per dose of fludarabine (Hexal, Holzkirchen, Germany) intraperitoneally on 3 consecutive days. The control animals were injected with equal volumes of Opti-MEM containing no virus and 0.9% NaCl containing no fludarabine, respectively. Tumor diameters were measured every third day (starting from the day of implantation) using a caliper and the volume was calculated by the formula (largest diameter) × (smallest diameter)2 × 0.5. The animals were euthanized by cervical dislocation when the tumor burden reached a volume of >1500 μl, if the animals were unable to drink or eat or if weight loss >20% of body weight was observed. Statistical analyses were performed using the GraphPad Prism software (version 5.04).
Results
Generation of PNP-armed and PSCA-targeted MVs
We generated recombinant MV from the Edmonston vaccine strain lineage with modifications in the attachment protein hemagglutinin (H) that ablate entry through the natural MV receptors CD46 and SLAM,17 and include the C-terminally fused single-chain antibody (AM1) against human PSCA.26 To enhance oncolytic properties, the prodrug convertase E. coli PNP gene was inserted in an additional transcription unit preceding the N gene (Figure 1, line MV-PNP-anti-PSCA). Furthermore, MV vectors coding for the reporter gene enhanced green fluorescent protein (EGFP) in combination with unmodified H (MV-EGFP), or PSCA-targeted (MV-EGFP-anti-PSCA), and CD20-targeted H (MV-EGFP-anti-CD20)8 were generated as controls.
Figure 1.
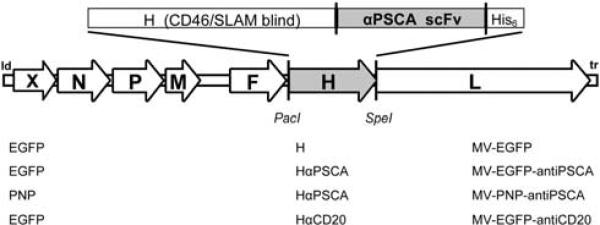
Schematic representation of the recombinant measles virus (MV) genomes. Vectors harbor either unmodified hemagglutinin (H) or fully retargeted H against prostate stem cell antigen (PSCA) and CD20 (listed at the bottom, middle), respectively, in combination with the transgene X = enhanced green fluorescent protein (EGFP) or purine nucleoside phosphorylase (PNP) (listed at the bottom, left). Corresponding names of the viruses are listed at the bottom right side.
PSCA-targeted MVs infect specifically pancreatic adenocarcinoma cells
The specificity and oncolytic potential of MV-EGFP-anti-PSCA was tested on four pancreatic adenocarcinoma cell lines, including BxPC-3 and MiaPaCa-2 with low expression levels, and Capan-1 and T3M4 with higher expression levels of the targeted antigen PSCA.21 The transgenic cell line 293T-PSCA with high ectopic expression of PSCA26 served as a control. First, to show the susceptibility of pancreatic adenocarcinoma cell lines to MV, we used a control virus expressing the reporter green fluorescent protein and the unmodified H protein, MVEGFP. This virus can bind all these cell lines through the ubiquitous protein CD46, which is expressed at high levels on many human cancer cells.11,32–35 We infected four pancreatic adenocarcinoma cell lines, as well as the producer cell line Vero-αHis19 and the standard host cell Vero (multiplicity of infection (MOI) of 1). In all these cell lines, MV-EGFP infection caused the characteristic MV cytopathic effect of syncytia formation 48 h post-infection (p.i.), followed by cell death (Figure 2a, upper panel), leading to a complete lysis of Vero and Vero-αHis cells.
Figure 2.

Susceptibility of pancreatic adenocarcinoma cell lines with high- and low-level prostate stem cell antigen (PSCA) expression to measles virus (MV) infection. (a) Four pancreatic adenocarcinoma cell lines, Vero and Vero-αHis cells were infected with the respective viruses (indicated on the right of each row) at a multiplicity of infection (MOI) of 1. Photographs were taken at 48 h post-infection (p.i.) using a fluorescence microscope (×100 magnification). Merged pictures of enhanced green fluorescent protein (EGFP) fluorescence and phase contrast are shown. (b) In total, 104 293T-PSCA, BxPC-3 and Vero cells each were infected at an MOI of 1 in quadruplicates and cell viability was determined at the indicated time points. Means and standard deviations are shown (white diamond, mock-treated cells defining 100% viability; white triangle, MV-EGFP-anti-PSCA; black triangle, MV-EGFP-anti-PSCA).
Next, the fully retargeted, CD46/SLAM-blinded MV-EGFP-anti-PSCA infected specifically cells expressing the addressed PSCA (Figure 2a, middle panel), whereas the control virus MV-EGFP-anti-CD20,8 fully retargeted against the B-cell-specific marker CD20, was not able to infect (Figure 2a, lower panel). As a control, Vero cells could be infected only with the parental virus, whereas the producer cells Vero-αHis were highly susceptible to all MV variants (fully retargeted H variants are displaying a C-terminal His hexapeptide for infection via the His-antibody pseudoreceptor on Vero-αHis cells). Apparently, in this experiment syncytia formation was profoundly reduced by PSCA targeting.
To quantify and compare the cytotoxic impact between PSCA-targeted MV and MV with native tropism, we assessed the kinetics of cell killing in a cell viability assay (XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide)) using 293T-PSCA (high-level PSCA expression), BxPC-3 (low-level PSCA expression) and Vero (no PSCA expression) after infection at an MOI of 1 with MV-EGFP and MV-EGFP-anti-PSCA, respectively (Figure 2b). Cell killing of both viruses after infection of 293T-PSCA was massive with a similar impact over the time course (cell viability 15% with MV-EGFP vs 23% with MV-EGFP-anti-PSCA 120 h p.i.). In the same experiment using BxPC-3, both viruses showed moderate cytotoxic effects at early infection times (cell viability 72% with MV-EGFP vs 91% with MV-EGFP-anti-PSCA 72 h p.i.), whereas more enhanced cell killing of MV-EGFP compared with MV-EGFP-anti-PSCA was detected in later infection phases (cell viability 40% with MV-EGFP vs 68% with MV-EGFP-anti-PSCA 120 h p.i.). As expected, on Vero cells no cell killing was provoked by MV-EGFP-anti-PSCA, but was extensive at all time points after infection with MV-EGFP.
Altogether, these data suggest that PSCA retargeting of MV to pancreatic adenocarcinoma is highly specific at the cost of approximately 30% loss in cytotoxic efficacy in vitro associated with the targeting strategy. Therefore, arming was intended to compensate for the attenuation of the PSCA-retargeted MV.
The prodrug convertase PNP enhances cytotoxicity of MV-PNP-anti-PSCA
The cytotoxic function of the PNP system was characterized in four prototypic pancreatic adenocarcinoma cell lines after infection with retargeted and armed MV-PNP-anti-PSCA at an MOI of 1. Cell viability was determined 3 days after incubation in the presence or absence of the prodrug fludarabine (final concentration 5 μm). As controls, all cell lines were either mock treated or incubated with prodrug alone (Figure 3a).
Figure 3.

Cytotoxic effect of prodrug convertase. (a) In total, 104 cells each were infected with measles virus (MV)-purine nucleoside phosphorylase (PNP)-anti-PSCA (multiplicity of infection (MOI) of 1) for 24 h and incubated with 5 μm fludarabine for 72 h, followed by a viability assay. Means and standard deviations from two experiments are shown (black, mock-treated cells defining 100% viability; white, fludarabine only; dark gray, MV-PNP-anti-PSCA; light gray, MV-PNP-anti-PSCA + fludarabine). (b) Cytotoxic efficacy of activated fludarabine in conditioned media. Vero-αHis cells were mock treated, treated with fludarabine only, infected with MV-PNP-anti-PSCA only (MOI = 0.1) or infected with MV-PNP-anti-PSCA and incubated with 5 μm fludarabine 36 h post-infection (p.i.) for 12 h. Different fractions (0.1 and 0.5) of conditioned and heat-inactivated media were added to a fresh layer of test cells. Viability was determined after 72 h by XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) assay. Means and standard deviations from three experiments are shown (black, mock-treated cells defining 100% viability; white, fludarabine only; dark gray, MV-PNP-anti-PSCA; light gray, MV-PNP-anti-PSCA + fludarabine).
Infection with MV-PNP-anti-PSCA in the presence of prodrug caused a more prominent cell death (46–84%) than without prodrug (19–47%). The presence of prodrug alone had no (BxPC-3) or only a slight influence on viability (88–78%) of Capan-1, T3M4 and MiaPaCa-2 cells after 3 days of incubation. Non-susceptible Vero control cells were not affected by the PNP/prodrug system. The differences in the mean cell viability between treatment with virus only and the combined chemovirotherapy were highly significant in the case of BxPC-3 and T3M4 (***P<0.001) and very significant for Capan-1 (**P<0.01), in contrast to MiaPaCa-2 (P>0.05). Thus, local activation of fludarabine by PNP enhances viral cytotoxicity significantly in three out of the four tested cell lines.
Efficient bystander killing of activated prodrug
To assess bystander cell killing after toxification of the prodrug, Vero-αHis cells were infected with MV-PNP-anti-PSCA at an MOI of 0.1. Fludarabine (final concentration 5 μm) was added to the medium 36 h p.i. for 12 h. As controls, Vero-αHis cells were either mock treated, infected with MV-PNP-anti-PSCA alone or incubated with prodrug only. After heat inactivation of viral particles, different fractions of conditioned media (0.1 and 0.5) were transferred to subconfluent layers of four pancreatic adenocarcinoma cell lines, and cell viability was determined after 72 h by cell viability assay (Figure 3b). In all cell lines tested, toxicity increased proportionally to the concentration of the conditioned media derived from the previously infected Vero-αHis cells incubated with fludarabine (Figure 3b, light gray columns). In contrast, cells incubated with supernatants from non-infected Vero-αHis cells, infected with MV-PNP-anti-PSCA in the absence of prodrug or incubated with prodrug only did not show significant differences in cell viability. Thus, fludarabine is converted to a toxic substance and secreted from infected cells expressing PNP. Transfer of conditioned media using a fraction of 0.1 induced toxicity in all cell lines in a broad range of 15–60%. In contrast, the higher concentrated conditioned media (fraction 0.5) had a stronger and similar impact (73–95% toxicity) on all cell lines used. Even a 10-fold media dilution is toxic on the four tested pancreatic adenocarcinoma cell lines. Therefore, arming with PNP mediates efficient prodrug toxification providing bystander killing of non-infected cells.
Prodrug-enhanced virotherapy of pancreatic cancer xenografts
Next, oncolytic efficacy of the armed and retargeted MV-PNP-anti-PSCA was characterized after subcutaneous implantation of pancreatic cancer xenografts in immunodeficient non-obese diabetic/severe combined immunodeficient mice. The low-level PSCA-expressing cell line BxPC-3 was chosen for implantation to assess therapeutic effects in pancreatic tumor patients with a low or heterogeneous surface marker expression. We examined the effect of MV-PNP-anti-PSCA on tumor growth after intratumoral infection 19 days after implantation with or without subsequent intraperitoneal administration of fludarabine (Figure 4a). Control groups were treated with carrier liquid (mock) or prodrug only and tumor diameters were measured every 3 days. Tumor growth after infection with MV-PNP-anti-PSCA alone was retarded, and this therapeutic effect was significantly enhanced by additional prodrug administration. The difference in tumor sizes between mock treatment and the chemovirotherapy was considered as significant (P<0.05) from day 48, whereas the difference in tumor sizes between MV only and the combined treatment was considered as significant (P<0.05) from day 60 after implantation. Mice were euthanized when the tumor reached a volume of 1500 μl.
Figure 4.

Oncolytic effects of the measles virus (MV)-purine nucleoside phosphorylase (PNP)-anti-PSCA/fludarabine system in non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice after intratumoral (i.t.) administration. BxPC-3 cells (6.6 × 106) were implanted into the right flank of mice (9 per group) and treatment started at a tumor volume of ca. 50 μl. Mice were injected i.t. on 5 consecutive days (days 19–23) with a dose of 7.2 × 105 cell infectious unit (ciu) of MV-PNP-anti-PSCA each. Fludarabine was given intraperitoneally (i.p.) (250 mg/kg per dose) 3 days after the last MV application on 3 consecutive days (diamond, mock-treated; square, treated with fludarabine only; triangle, MV-PNP-anti-PSCA only; circle, MV-PNP-anti-PSCA + fludarabine). (a) Tumor volume measurements starting on day 3 after subcutaneous implantation (error bars are indicating standard deviation of the mean). (b) Distribution of tumor volumes at day 66 after implantation. Each dot represents one mouse with 9 mice per group. (c) Kaplan–Meier survival curve documenting the effects of oncolytic MV-PNP-anti-PSCA and prodrug, respectively. The defined end point was >1500 μl of tumor volume.
As shown for the tumor volume distribution at day 66 after implantation (Figure 4b), treatment with MV-PNP-anti-PSCA and prodrug resulted in a significantly enhanced oncolysis compared with sole viral (P = 0.0116 by the two-sample t-test) or sole fludarabine treatment (P = 0.0045). The mean tumor size at day 66 after implantation with combined viral and prodrug therapy was less than one-third the size of mock-treated tumors (P = 0.0027 by the two-sample t-test) showing a highly significant retardation in tumor growth.
Survival benefits were estimated using the Kaplan–Meier plot with a defined end point of >1500 μl tumor volume (Figure 4c). Treatment with MV-PNP-anti-PSCA in the presence of fludarabine resulted in a significant increase of survival time compared with mock treatment or administration of prodrug alone (P<0.05 each). However, survival of mice treated with MV-PNP-anti-PSCA and prodrug therapy was moderately, but not significantly extended compared with virus treatment alone. The median survival of mock-treated animals was 75 days, whereas 50% of the mice having obtained viral and prodrug therapy survived until day 90 and until day 87 with sole virus application, respectively. Thus, the combined chemovirotherapy yielded a beneficial treatment regimen in this model of pancreatic cancer.
Efficient infection of Gmc-resistant pancreatic adenocarcinoma cell lines
As resistance to Gmc is a major issue in chemotherapy for advanced pancreatic cancer, combination with other treatments, for example, oncolytic MV may be sought. We have established a stable resistance to Gmc in four different pancreatic adenocarcinoma cell lines by exposure to gradually increasing Gmc concentrations with defined incubation periods (>200 days after beginning of expositions): BxPC-3/Gmc-100; Capan-1/Gmc-50; T3M4/Gmc-100; and MiaPaCa-2/Gmc-25 (numbers are indicating the maximum Gmc concentrations (nm) at which cells are cultivatable permanently). Even after cryopreservation of the resistant cell cultures, viability was comparable to the parental cell line and the Gmc resistance at the previous threshold concentration was stably maintained (data not shown). To characterize the induced Gmc resistance, we compared the half-maximal inhibitory concentration inducing a 50% reduction of viability of pretreated and of parental cell lines. Cells were exposed for 3 days to increasing Gmc concentrations (0–10 μm) in a dilution assay, resulting in at least 4- to 17-fold higher half-maximal inhibitory concentration values for Gmc-resistant cells compared with the naive cell line with statistical significances (Supplementary Table 1).
Next, we examined the permissiveness of Gmc-resistant pancreatic adenocarcinoma cell lines to MV infection compared with the chemotherapy-naive counterparts (Figure 5a). Cells were infected with MV-EGFP and the retargeted MV-EGFP-anti-PSCA, respectively, at an MOI of 1 and syncytia formation was analyzed 72 h p.i. To compare the infectivity of both viruses, differential viral permissiveness was expressed as the ratio between the total numbers of syncytia per well caused by both viruses (unmodified H vs retargeted H-anti-PSCA) observed on each cell type (white bars, left y axis) and additionally by estimation of the dimensions of the respective syncytia expressed as ‘nuclei per syncytium’ for each virus variant (right y axis). Taken together, each corresponding cell couple developed similar numbers of equal syncytia after infection with the respective virus. As expected, the total numbers of induced syncytia as well as their size were less and smaller after infection with PSCA-retargeted MV compared with MV with unmodified tropism. However, susceptibility to infection with both MV variants was clearly not impaired by Gmc resistance.
Figure 5.

Susceptibility of gemcitabine-resistant pancreatic adenocarcinoma cells to measles virus (MV) infection and cytotoxic efficacy of activated fludarabine. (a) Naive or gemcitabine-resistant pancreatic adenocarcinoma cells were infected with MV-enhanced green fluorescent protein (EGFP) or MV-EGFP-anti-PSCA at a multiplicity of infection (MOI) of 1 and syncytia formation was analyzed 72 h post-infection (p.i.) (means of 3 wells per cell line). Differential viral permissiveness is expressed as the ratio between numbers of syncytia per well induced by the virus variants with unmodified H vs retargeted anti-PSCA (left y axis, white bars) and sizes of the respective syncytia are indicated as nuclei per syncytium (right y axis; black triangle: MV-EGFP; white triangle, MV-EGFP-anti-PSCA). (b) Cytotoxic efficacy of activated fludarabine. Different fractions (0.01, 0.1 and 0.5) of conditioned media (identical preparation procedure as described in legend to Figure 3) were added to a fresh layer of test cells. Cell viability was determined after 72 h by XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) assay. Means and standard deviations of samples normalized to mock-treated cells from three experiments are shown (light gray bars, naive cells; light gray-shaded bars, gemcitabine-resistant cells).
Gmc resistance does not interfere with the PNP/fludarabine system
Next, the efficacy of bystander cell killing of naive and Gmc-resistant pancreatic adenocarcinoma cell lines was examined. Towards this, different dilutions of conditioned media were transferred to subconfluent cell layers, and viability was determined after 48 h by cell viability assay (Figure 5b). The assessed levels of bystander killing for both naive and Gmc-resistant cells were consistently in the same range, and toxicity increased proportionally to the concentration of the conditioned media. As expected, test cells incubated with supernatants from cells not infected, infected with MV-PNP-anti-PSCA alone or incubated with prodrug only maintained viability (controls not shown). In conclusion, the level of bystander killing mediated by the PNP/fludarabine system is not impaired by Gmc resistance, showing that there is no cross-resistance to the activated prodrug.
Discussion
In this study, we retargeted an oncolytic MV to the PSCA and armed it with the prodrug convertase PNP activating the clinically approved chemotherapeutic fludarabine locally. Therapeutic efficacy of the new MV-PNP-anti-PSCA supported by toxified fludarabine was shown in a pancreas adenocarcinoma xenograft model. Moreover, a panel of four chemoresistant pancreas adenocarcinoma cell lines remained responsive to both oncolysis mediated by MV-PNP-anti-PSCA and the toxified prodrug.
Pancreatic cancer is one of the most drug-resistant type of malignancy, with extremely poor prognosis and survival rate. However, chemotherapy is currently the option of choice for the treatment of non-resectable and/or metastasized tumors using Gmc as front-line therapy ultimately not preventing a progressive disease. In this process, chemoresistance of pancreatic cancer is not fully understood and a variety of mechanisms may be involved, for example, cellular influx of Gmc by members of hENT/hCNT (human equilibrative and concentrative nucleoside transporters) nucleoside transporters36 and removal of Gmc by increased expression of efflux transporters (multidrug resistance-associated proteins).37 It has been shown that nucleoside transporter-deficient cells display resistance to Gmc38 and that deoxycytidine kinase-deficient lines exhibit a Gmc-resistant phenotype.39
Recently, new experimental therapy approaches include targeted drugs against the two known surface markers for pancreatic cancer mesothelin and PSCA.22,40,41 A phase I/II study of the mesothelin-targeted L19IL2 monoclonal antibody–cytokine fusion protein in combination with Gmc in patients with advanced pancreatic cancer is recruiting at the National Center for Tumor Diseases (Heidelberg, Germany). In parallel, a phase II clinical trial using the anti-PSCA monoclonal antibody AGS-1C4D4 at the Memorial Sloan-Kettering Cancer Center, New York, NY, USA is ongoing.2
In our experimental setting presented here, we have generated an oncolytic MV fully retargeted to PSCA. This virus, which cannot interact with the natural receptors CD46 and SLAM,17,18 infected PSCA-positive pancreatic adenocarcinoma cells specifically causing strong oncolytic effects. In vitro, this virus was tested on a panel of pancreatic adenocarcinoma cell lines with different PSCA receptor densities. It could be shown that efficient target cell infection was mediated through both high- and low-level PSCA surface expression. For in vivo studies of MV-PNP-anti-PSCA, the BxPC-3 pancreatic adenocarcinoma cell line with a very low surface expression of PSCA was chosen. In a xenograft mouse model, therapeutic efficacy of intratumorally injected MV-PNP-anti-PSCA could be shown by retarded tumor growth. By arming this virus with the prodrug convertase PNP, oncolytic effects in the above-described animal model were significantly enhanced by systemic administration of the cognate prodrug fludarabine. We could show for the first time in vivo tumor growth suppression using a retargeted MV-mediated PNP/fludarabine strategy in a pancreatic adenocarcinoma model. MV-PNP-anti-PSCA-treated tumors exposed even briefly (3 consecutive days) to fludarabine exhibited profound retardation. However, a total remission of the treated tumors could not be achieved, meaning the timing of virus administration and prodrug application may have to be adjusted to maximize therapeutic outcome. Increasing the interval between infection and prodrug administration in a chemovirotherapy approach improved therapeutic efficacy of different oncolytics because viruses can spread further in the tumor before the prodrug is added to enhance local cell killing.8,42
Fludarabine monophosphate is a nucleoside analog that is routinely used in the treatment of low-grade lymphoproliferative malignancies including chronic lymphocytic leukemia and low-grade non-Hodgkin's lymphoma, but so far has no clinical application yet in the treatment of solid tumors such as pancreatic adenocarcinoma. Nevertheless, the PNP/fludarabine system was considered previously in preclinical testing for the treatment of several solid tumors,43,44 for example, hepatocellular carcinoma, prostate cancer, bladder cancer, glioma, breast cancer and melanoma.45–52 A phase I clinical trial was initiated in 2008 (ClinicalTrials.gov identifier: NCT00625430) to evaluate the safety and tolerability of an ovine atadenovirus expressing the E. coli PNP under control of a prostate-directed promoter combined with fludarabine in prostate cancer patients.
Here we could show that various Gmc-resistant pancreatic adenocarcinoma cell lines were both still infectible with MV-PNP-anti-PSCA and sensitive to the toxified fludarabine nucleoside analog. As bystander killing of pretreated pancreatic adenocarcinoma cells was shown to the same extent as to the naive counterparts, a cross-resistance between Gmc and fludarabine could not be detected. In another in vitro study using a Gmc-resistant murine leukemic cell line, resistance to fludarabine treatment was not observed either.53 This indicates differences in the metabolism/inactivation and/or mechanism of action of Gmc and fludarabine.
In summary, we have shown that oncolytic MV can efficiently be engineered for targeted therapy of pancreatic adenocarcinoma. Arming this vector with the PNP/fludarabine system is enhancing the oncolytic effect significantly. Moreover, in the treatment of four different Gmc-resistant pancreatic adenocarcinoma cell lines, there was no cross-resistance detected against both the PSCA-retargeted MV-PNP-anti-PSCA and the virally activated prodrug. Hereby, the existing toolbox of oncolytic MV is extended by a highly specific and effective vector for pancreatic cancer therapy.
Supplementary Material
Acknowledgements
We thank Jessica Albert for her valuable technical assistance and Dr Zahari Raykov and Dr Jean Rommelaere (DKFZ) for providing the T3M4 and MiaPaCa-2 cells as well as Dr Stephen J Russell (Mayo Clinic) for the Vero-αHis cells. We also thank the Light Microscopy Facility at the German Cancer Research Center, including Manuela Brom and Felix Bestvater, for their technical support. This work was supported by the German Cancer Aid, Max Eder Grant No. 108307 (GU) and by the NIH Grant No. R01 CA139389 (RC).
Footnotes
Conflict of interest
Patent applications on which RC is an inventor have been licensed to NISCO, Mayo has an equity position in NISCO; Mayo has not yet received royalties from products developed by the company, but may receive these in the future. The other authors declare no conflict of interest.
Supplementary Information accompanies the paper on Cancer Gene Therapy website (http://www.nature.com/cgt)
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605–1617. doi: 10.1056/NEJMra0901557. [DOI] [PubMed] [Google Scholar]
- 3.Alberts SR, Gores GJ, Kim GP, Roberts LR, Kendrick ML, Rosen CB, et al. Treatment options for hepatobiliary and pancreatic cancer. Mayo Clin Proc. 2007;82:628–637. doi: 10.4065/82.5.628. [DOI] [PubMed] [Google Scholar]
- 4.Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 5.Vaha-Koskela MJ, Heikkila JE, Hinkkanen AE. Oncolytic viruses in cancer therapy. Cancer Lett. 2007;254:178–216. doi: 10.1016/j.canlet.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blechacz B, Splinter PL, Greiner S, Myers R, Peng KW, Federspiel MJ, et al. Engineered measles virus as a novel oncolytic viral therapy system for hepatocellular carcinoma. Hepatology. 2006;44:1465–1477. doi: 10.1002/hep.21437. [DOI] [PubMed] [Google Scholar]
- 7.Ungerechts G, Springfeld C, Frenzke ME, Lampe J, Parker WB, Sorscher EJ, et al. An immunocompetent murine model for oncolysis with an armed and targeted measles virus. Mol Ther. 2007;15:1991–1997. doi: 10.1038/sj.mt.6300291. [DOI] [PubMed] [Google Scholar]
- 8.Ungerechts G, Springfeld C, Frenzke ME, Lampe J, Johnston PB, Parker WB, et al. Lymphoma chemovirotherapy: CD20-targeted and convertase-armed measles virus can synergize with fludarabine. Cancer Res. 2007;67:10939–10947. doi: 10.1158/0008-5472.CAN-07-1252. [DOI] [PubMed] [Google Scholar]
- 9.Allen C, Paraskevakou G, Iankov I, Giannini C, Schroeder M, Sarkaria J, et al. Interleukin-13 displaying retargeted oncolytic measles virus strains have significant activity against gliomas with improved specificity. Mol Ther. 2008;16:1556–1564. doi: 10.1038/mt.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu C, Hasegawa K, Russell SJ, Sadelain M, Peng KW. Prostate-specific membrane antigen retargeted measles virotherapy for the treatment of prostate cancer. Prostate. 2009;69:1128–1141. doi: 10.1002/pros.20962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Msaouel P, Iankov ID, Allen C, Morris JC, von Messling V, Cattaneo R, et al. Engineered measles virus as a novel oncolytic therapy against prostate cancer. Prostate. 2009;69:82–91. doi: 10.1002/pros.20857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H, Peng KW, Dingli D, Kratzke RA, Russell SJ. Oncolytic measles viruses encoding interferon beta and the thyroidal sodium iodide symporter gene for mesothelioma virotherapy. Cancer Gene Ther. 2010;17:550–558. doi: 10.1038/cgt.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ungerechts G, Frenzke ME, Yaiw KC, Miest T, Johnston PB, Cattaneo R. Mantle cell lymphoma salvage regimen: synergy between a reprogrammed oncolytic virus and two chemotherapeutics. Gene Therapy. 2010;17:1506–1516. doi: 10.1038/gt.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dorig RE, Marcil A, Chopra A, Richardson CD. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell. 1993;75:295–305. doi: 10.1016/0092-8674(93)80071-l. [DOI] [PubMed] [Google Scholar]
- 15.Naniche D, Varior-Krishnan G, Cervoni F, Wild TF, Rossi B, Rabourdin-Combe C, et al. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J Virol. 1993;67:6025–6032. doi: 10.1128/jvi.67.10.6025-6032.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tatsuo H, Ono N, Tanaka K, Yanagi Y. SLAM (CDw150) is a cellular receptor for measles virus. Nature. 2000;406:893–897. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 17.Vongpunsawad S, Oezgun N, Braun W, Cattaneo R. Selectively receptor-blind measles viruses: identification of residues necessary for SLAM- or CD46-induced fusion and their localization on a new hemagglutinin structural model. J Virol. 2004;78:302–313. doi: 10.1128/JVI.78.1.302-313.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakamura T, Peng KW, Vongpunsawad S, Harvey M, Mizuguchi H, Hayakawa T, et al. Antibody-targeted cell fusion. Nat Biotechnol. 2004;22:331–336. doi: 10.1038/nbt942. [DOI] [PubMed] [Google Scholar]
- 19.Nakamura T, Peng KW, Harvey M, Greiner S, Lorimer IA, James CD, et al. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat Biotechnol. 2005;23:209–214. doi: 10.1038/nbt1060. [DOI] [PubMed] [Google Scholar]
- 20.Reiter RE, Gu Z, Watabe T, Thomas G, Szigeti K, Davis E, et al. Prostate stem cell antigen: a cell surface marker overexpressed in prostate cancer. Proc Natl Acad Sci USA. 1998;95:1735–1740. doi: 10.1073/pnas.95.4.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng L, Reiter RE, Jin Y, Sharon H, Wieder J, Lane TF, et al. Immunocytochemical analysis of prostate stem cell antigen as adjunct marker for detection of urothelial transitional cell carcinoma in voided urine specimens. J Urol. 2003;169:2094–2100. doi: 10.1097/01.ju.0000064929.43602.17. [DOI] [PubMed] [Google Scholar]
- 22.Argani P, Rosty C, Reiter RE, Wilentz RE, Murugesan SR, Leach SD, et al. Discovery of new markers of cancer through serial analysis of gene expression: prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. 2001;61:4320–4324. [PubMed] [Google Scholar]
- 23.Wente MN, Jain A, Kono E, Berberat PO, Giese T, Reber HA, et al. Prostate stem cell antigen is a putative target for immunotherapy in pancreatic cancer. Pancreas. 2005;31:119–125. doi: 10.1097/01.mpa.0000173459.81193.4d. [DOI] [PubMed] [Google Scholar]
- 24.Hermiston TW, Kuhn I. Armed therapeutic viruses: strategies and challenges to arming oncolytic viruses with therapeutic genes. Cancer Gene Ther. 2002;9:1022–1035. doi: 10.1038/sj.cgt.7700542. [DOI] [PubMed] [Google Scholar]
- 25.Parker WB, Allan PW, Shaddix SC, Rose LM, Speegle HF, Gillespie GY, et al. Metabolism and metabolic actions of 6-methylpurine and 2-fluoroadenine in human cells. Biochem Pharmacol. 1998;55:1673–1681. doi: 10.1016/s0006-2952(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 26.Morgenroth A, Cartellieri M, Schmitz M, Gunes S, Weigle B, Bachmann M, et al. Targeting of tumor cells expressing the prostate stem cell antigen (PSCA) using genetically engineered T-cells. Prostate. 2007;67:1121–1131. doi: 10.1002/pros.20608. [DOI] [PubMed] [Google Scholar]
- 27.Schmitt M, Pawlita M. High-throughput detection and multiplex identification of cell contaminations. Nucleic Acids Res. 2009;37:e119. doi: 10.1093/nar/gkp581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duprex WP, McQuaid S, Hangartner L, Billeter MA, Rima BK. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. J Virol. 1999;73:9568–9575. doi: 10.1128/jvi.73.11.9568-9575.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Radecke F, Spielhofer P, Schneider H, Kaelin K, Huber M, Dotsch C, et al. Rescue of measles viruses from cloned DNA. EMBO J. 1995;14:5773–5784. doi: 10.1002/j.1460-2075.1995.tb00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Calain P, Roux L. The rule of six, a basic feature for efficient replication of Sendai virus defective interfering RNA. J Virol. 1993;67:4822–4830. doi: 10.1128/jvi.67.8.4822-4830.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Angelova AL, Aprahamian M, Grekova SP, Hajri A, Leuchs B, Giese NA, et al. Improvement of gemcitabine-based therapy of pancreatic carcinoma by means of oncolytic parvovirus H-1PV. Clin Cancer Res. 2009;15:511–519. doi: 10.1158/1078-0432.CCR-08-1088. [DOI] [PubMed] [Google Scholar]
- 32.Anderson BD, Nakamura T, Russell SJ, Peng KW. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004;64:4919–4926. doi: 10.1158/0008-5472.CAN-04-0884. [DOI] [PubMed] [Google Scholar]
- 33.Phuong LK, Allen C, Peng KW, Giannini C, Greiner S, TenEyck CJ, et al. Use of a vaccine strain of measles virus genetically engineered to produce carcinoembryonic antigen as a novel therapeutic agent against glioblastoma multi-forme. Cancer Res. 2003;63:2462–2469. [PubMed] [Google Scholar]
- 34.Peng KW, TenEyck CJ, Galanis E, Kalli KR, Hartmann LC, Russell SJ. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res. 2002;62:4656–4662. [PubMed] [Google Scholar]
- 35.McDonald CJ, Erlichman C, Ingle JN, Rosales GA, Allen C, Greiner SM, et al. A measles virus vaccine strain derivative as a novel oncolytic agent against breast cancer. Breast Cancer Res Treat. 2006;99:177–184. doi: 10.1007/s10549-006-9200-5. [DOI] [PubMed] [Google Scholar]
- 36.Giovannetti E, Mey V, Nannizzi S, Pasqualetti G, Del Tacca M, Danesi R. Pharmacogenetics of anticancer drug sensitivity in pancreatic cancer. Mol Cancer Ther. 2006;5:1387–1395. doi: 10.1158/1535-7163.MCT-06-0004. [DOI] [PubMed] [Google Scholar]
- 37.Konig J, Hartel M, Nies AT, Martignoni ME, Guo J, Buchler MW, et al. Expression and localization of human multidrug resistance protein (ABCC) family members in pancreatic carcinoma. Int J Cancer. 2005;115:359–367. doi: 10.1002/ijc.20831. [DOI] [PubMed] [Google Scholar]
- 38.Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res. 1998;58:4349–4357. [PubMed] [Google Scholar]
- 39.Dumontet C, Fabianowska-Majewska K, Mantincic D, Callet Bauchu E, Tigaud I, Gandhi V, et al. Common resistance mechanisms to deoxynucleoside analogues in variants of the human erythroleukaemic line K562. Br J Haematol. 1999;106:78–85. doi: 10.1046/j.1365-2141.1999.01509.x. [DOI] [PubMed] [Google Scholar]
- 40.McCarthy DM, Maitra A, Argani P, Rader AE, Faigel DO, Van Heek NT, et al. Novel markers of pancreatic adeno-carcinoma in fine-needle aspiration: mesothelin and prostate stem cell antigen labeling increases accuracy in cytologically borderline cases. Appl Immunohistochem Mol Morphol. 2003;11:238–243. doi: 10.1097/00129039-200309000-00006. [DOI] [PubMed] [Google Scholar]
- 41.Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin Cancer Res. 2001;7:3862–3868. [PubMed] [Google Scholar]
- 42.Seo E, Abei M, Wakayama M, Fukuda K, Ugai H, Murata T, et al. Effective gene therapy of biliary tract cancers by a conditionally replicative adenovirus expressing uracil phosphoribosyltransferase: significance of timing of 5-fluorouracil administration. Cancer Res. 2005;65:546–552. [PubMed] [Google Scholar]
- 43.Zhang Y, Parker WB, Sorscher EJ, Ealick SE. PNP anticancer gene therapy. Curr Top Med Chem. 2005;5:1259–1274. doi: 10.2174/156802605774463105. [DOI] [PubMed] [Google Scholar]
- 44.Hebrard C, Dumontet C, Jordheim LP. Development of gene therapy in association with clinically used cytotoxic deoxynucleoside analogues. Cancer Gene Ther. 2009;16:541–550. doi: 10.1038/cgt.2009.25. [DOI] [PubMed] [Google Scholar]
- 45.Mohr L, Shankara S, Yoon SK, Krohne TU, Geissler M, Roberts B, et al. Gene therapy of hepatocellular carcinoma in vitro and in vivo in nude mice by adenoviral transfer of the Escherichia coli purine nucleoside phosphorylase gene. Hepatology. 2000;31:606–614. doi: 10.1002/hep.510310310. [DOI] [PubMed] [Google Scholar]
- 46.Cai X, Zhou J, Chang Y, Sun X, Li P, Lin J. Targeting gene therapy for hepatocarcinoma cells with the E. coli purine nucleoside phosphorylase suicide gene system directed by a chimeric alpha-fetoprotein promoter. Cancer Lett. 2008;264:71–82. doi: 10.1016/j.canlet.2008.01.027. [DOI] [PubMed] [Google Scholar]
- 47.Wang XY, Martiniello-Wilks R, Shaw JM, Ho T, Coulston N, Cooke-Yarborough C, et al. Preclinical evaluation of a prostate-targeted gene-directed enzyme prodrug therapy delivered by ovine atadenovirus. Gene Therapy. 2004;11:1559–1567. doi: 10.1038/sj.gt.3302308. [DOI] [PubMed] [Google Scholar]
- 48.Martiniello-Wilks R, Wang XY, Voeks DJ, Dane A, Shaw JM, Mortensen E, et al. Purine nucleoside phosphorylase and fludarabine phosphate gene-directed enzyme prodrug therapy suppresses primary tumour growth and pseudo-metastases in a mouse model of prostate cancer. J Gene Med. 2004;6:1343–1357. doi: 10.1002/jgm.629. [DOI] [PubMed] [Google Scholar]
- 49.Kikuchi E, Menendez S, Ozu C, Ohori M, Cordon-Cardo C, Logg CR, et al. Delivery of replication-competent retrovirus expressing Escherichia coli purine nucleoside phosphorylase increases the metabolism of the prodrug, fludarabine phosphate and suppresses the growth of bladder tumor xenografts. Cancer Gene Ther. 2007;14:279–286. doi: 10.1038/sj.cgt.7701013. [DOI] [PubMed] [Google Scholar]
- 50.Hong JS, Waud WR, Levasseur DN, Townes TM, Wen H, McPherson SA, et al. Excellent in vivo bystander activity of fludarabine phosphate against human glioma xenografts that express the Escherichia coli purine nucleoside phosphorylase gene. Cancer Res. 2004;64:6610–6615. doi: 10.1158/0008-5472.CAN-04-0012. [DOI] [PubMed] [Google Scholar]
- 51.Lockett LJ, Molloy PL, Russell PJ, Both GW. Relative efficiency of tumor cell killing in vitro by two enzyme–prodrug systems delivered by identical adenovirus vectors. Clin Cancer Res. 1997;3:2075–2080. [PubMed] [Google Scholar]
- 52.Hughes BW, Wells AH, Bebok Z, Gadi VK, Garver RI, Jr, Parker WB, et al. Bystander killing of melanoma cells using the human tyrosinase promoter to express the Escherichia coli purine nucleoside phosphorylase gene. Cancer Res. 1995;55:3339–3345. [PubMed] [Google Scholar]
- 53.Jordheim LP, Cros E, Gouy MH, Galmarini CM, Peyrottes S, Mackey J, et al. Characterization of a gemcitabine-resistant murine leukemic cell line: reversion of in vitro resistance by a mononucleotide prodrug. Clin Cancer Res. 2004;10:5614–5621. doi: 10.1158/1078-0432.CCR-04-0506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.