

Abstract
The reductively activated nitroaromatic class of antimicrobials, which include nitroimidazole and the more metabolically labile nitrofuran antitubercular agents, have demonstrated some potential for development as therapeutics against dormant TB bacilli. In previous studies, the pharmacokinetic properties of nitrofuranyl isoxazolines were improved by incorporation of the outer ring elements of the antitubercular nitroimidazole OPC-67683. This successfully increased stability of the resulting pentacyclic nitrofuran lead compound Lee1106 (referred to herein as 9a). In the current study, we report the synthesis and antimicrobial properties of 9a and panel of 9a analogs, which were developed to increase oral bioavailability. These hybrid nitrofurans remained potent inhibitors of Mycobacterium tuberculosis with favorable selectivity indices (>150) and a narrow spectrum of activity. In vivo, the pentacyclic nitrofuran compounds showed long half-lives and high volumes of distribution. Based on pharmacokinetic testing and lack of toxicity in vivo, 9a remained the series lead. 9a exerted a lengthy post antibiotic effect and was highly active against nonreplicating M. tuberculosis grown under hypoxia. 9a showed a low potential for cross resistance to current antitubercular agents, and a mechanism of activation distinct from pre-clinical tuberculosis candidates PA-824 and OPC-67683. Together these studies show that 9a is a nanomolar inhibitor of actively growing as well as nonreplicating M. tuberculosis.
Introduction
Mycobacterium tuberculosis remains an important global pathogen that is believed to have infected a third of the world’s population and kills over a million persons annually [1]. The US Centers for Disease Control and Prevention recommended regime for drug sensitive tuberculosis (TB) infections consists of rifampin, isoniazid, pyrazinamide and ethambutol for an initial two months followed by continued four month treatment with isoniazid and rifampin. Poor patient compliance to this lengthy drug regimen can lead to patient relapse and promotes the emergence of drug-resistant strains, which have become a major treatment challenge and global health threat. Tuberculosis strains resistant to isoniazid and rifampin, two of the most powerful antituberculosis agents, are classified as multi drug-resistant (MDR TB). Extensively drug-resistant (XDR) strains, which are additionally resistant to a fluoroquinolone, and an injectable second-line drug, have been reported in over 80 countries [2]. MDR/XDR TB dramatically increases the medical burden, requiring extended treatment lengths with drugs that are less effective and show significant toxicity [3], [4]. It has been widely postulated that the key to decreasing treatment times and the spread of drug-resistant tuberculosis is the development of new therapeutics that target dormant bacilli, which are metabolically inactive cells that display phenotypic (non-genetic) resistance to most drugs [5]. Dormant bacteria are believed to reside within the nutrient and oxygen limited environment of necrotic lesions. Therefore, it is imperative that novel antitubercular agents target these persisting bacterial subpopulations [6], [7].
Nitroaromatic antibiotics, such as metronidazole and nitrofurantonin, are widely used to treat anaerobic bacterial infections (Figure 1). Based on this knowledge, researchers at the PathoGenesis Corporation developed a nitroimidazole pyran, PA-824 (Figure 1), with specific anti-tuberculosis activity against both actively growing and hypoxic tuberculosis bacilli [8]. PA-824 is structurally related to metronidazole and was selected for its specificity in terms of antitubercular activity, favorable metabolic stability, low toxicity profile, and lack of cross-resistance with other TB drugs. PA-824 has advanced into Phase II combination clinical testing by the Global Alliance for TB Drug Development [8], [9], [10], [11]. Researchers at Otsuka Pharmaceutical Company subsequently developed the nitroimidazole oxazole OPC-67683 (Figure 1), which is now being progressed to Phase III clinical trials against tuberculosis [12], [13], [14]. These studies highlight the potential for the successful development of antitubercular nitroaromatics.
Figure 1. Known nitroaromatic antimicrobial drugs and nitrofurans explored in this study.

In addition to the nitroimidazoles, other nitroaromatics belonging to nitrothiazole and nitrofuran classes have notable antitubercular activity [8], [12]. This includes our recently reported series of tetracyclic nitrofuranyl isoxazolines, which had improved solubility and bioavailability but decreased potency and metabolic stability compared to precursor nitrofurans [15], [16]. Key differences in these compounds arise from differences in their redox potential, which drive how these compounds are activated and the types of reactive products that are produced resulting in different modes of cell killing [17]. Therefore all three classes of nitroaromatics have the potential to be simultaneously developed as TB treatments, as shown in this study. Previously, incorporation of the outer ring elements of OPC-67683 into tetracyclic nitrofuranyl isoxazolines produced a novel pentacyclic hybrid nitrofuran with improved metabolic stability but limited oral bioavailability, Lee1106 (herein referred to as compound 9a) [18]. Herein, we report the synthesis and evaluation of 9a and a series of 9a-analogs with improved in vivo exposure profiles (area under the curve, AUC). Of the pentacyclic hybrid nitrofurans reported, 9a was the most metabolically stable and did not show any adverse effects in vivo. Therefore, 9a remained the most promising lead candidate and was subjected to a detailed antimicrobial characterization. 9a was highly active in two separate in vitro models of nonreplicating tuberculosis, was active in a murine model of acute tuberculosis infection, and was activated by a mechanism distinct from that required for PA-824 and OPC-67683.
Materials and Methods
Reagents and Instrumentation
All anhydrous solvents and starting materials were purchased from Aldrich Chemical Co. (Milwaukee, WI). All reagent grade solvents used for chromatography were purchased from Fisher Scientific (Suwanee, GA) and flash column chromatography silica cartridges were obtained from Biotage Inc. (Lake Forest, VA). The reactions were monitored by thin-layer chromatography (TLC) on pre-coated Merch 60 F254 silica gel plates and visualized using UV light (254 nm). A Biotage FLASH column chromatography system was used to purify mixtures. All NMR spectra were recorded on a Bruker-400 spectrometer. Chemical shifts (δ) are reported in parts per million relative to the residual solvent peak or internal standard (tetramethylsilane), and coupling constants (J) are reported in hertz (Hz). High resolution mass spectra were recorded on a Waters Xevo G2 QTOF LCMS using ESI. Purity of the products was confirmed before testing by analytical RP-HPLC on a Shimadzu HPLC system, and all final compounds had a purity of 95% or greater as determined by RP-HPLC. Gradient Conditions: solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile): 0–1.00 min 95% A, 1.00–6.00 min 0–95% B (linear gradient), 6.00–9.50 min 100% B, 9.50–9.75 min 0–95% A, 9.75–10.0 min 95% A, detection by UV at 254 nm and by ELSD. The reference compound PA-824 was synthesized in six step sequence following the procedure reported by Baker et al., starting from 2,4-dinitro, 1H-imidazole and S-glycidol [19]. OPC-67683 was synthesized from 2-chloro-4-nitro-1H-imidazole as described by Tsubouchi et al. [20].
Synthesis of tert-Butyl 4-((methylsulfonyl)oxy)piperidine-1-carboxylate 4
Et3N (10.38 mL, 74.62 mmol) was added to a stirred and cooled (0°C) solution of tert-butyl 4-hydroxypiperidine-1-carboxylate 3 (5 g, 24.87 mmol) in CH2Cl2 (50 mL), followed by methanesulfonyl chloride (2.88 mL, 37.31 mmol) drop wise and stirred at the same temperature for 1 h. The reaction was then quenched with ice-cold water (20 mL), washed with water (20 mL), dried over anhydrous Na2SO4, and concentrated under reduced pressure to afford 4 (6.52 g) in 94% yield. 1H NMR (500 MHz, CDCl3): δ 4.72–4.82 (m, 1H), 3.29–3.39 (m, 4H), 3.16 (s, 3H), 1.52–1.77 (m, 4H), 1.38 (s, 9H); LCMS: 280 (M++1).
General Procedure I, for Preparation of Compounds 5a–b
A mixture of appropriate phenol (1 mmol), 4 (1.5 mmol), K2CO3 (3 mmol) and tetra n-butyl ammonium chloride (0.2 mmol) in H2O was heated to reflux overnight. The reaction mass was then cooled to room temperature and the product was extracted with CH2Cl2 twice. The combined organic layers were washed with water, 10% NaOH, water, dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford 5a–b in 86–90% yields.
tert-Butyl 4-(4-(trifluoromethoxy)phenoxy)piperidine-1-carboxylate 5a: 4-Trifluoromethoxy phenol (3 g, 16.85 mmol), 4 (7 g, 25.28 mmol), K2CO3 (6.97 g, 50.56 mmol) and tetra n-butyl ammonium chloride (0.933 g, 3.37 mmol) were stirred in H2O and the reaction was carried out as described in general procedure I to afford 5a (5.5 g) in 90% yield. 1H NMR (500 MHz, CDCl3): δ 6.78–6.82 (m, 4H), 3.72–3.75 (m, 1H), 3.29–3.42 (m, 4H), 1.78–2.02 (m, 4H), 1.37 (s, 9H); LCMS: 362 (M++1).
tert-Butyl 4-(4-fluorophenoxy)piperidine-1-carboxylate 5b: 4-Fluorophenol (1.3 g, 11.60 mmol), 4 (4.86 g, 17.39 mmol), K2CO3 (4.81 g, 34.8 mmol) and tetra n-butyl ammonium chloride (0.645 g, 2.31 mmol) were stirred in H2O and the reaction was carried out as described in general procedure I to afford 5b (2.9 g) in 86% yield. 1H NMR (500 MHz, CDCl3): δ 7.13 (d, J = 2.8 Hz, 2H), 7.08 (d, J = 3.4 Hz, 2H), 3.66–3.70 (m, 1H), 3.29–3.39 (m, 4H), 1.75–2.00 (m, 4H), 1.38 (s, 9H); LCMS: 296 (M++1).
General Procedure II, for Preparation of Compounds 6a–b
Compounds 5a–b were dissolved in 1∶1 CH2Cl2 and trifluoroacetic acid (TFA) and stirred at room temperature for 1 h and concentrated under reduced pressure, washed with water, 10% NaOH and water, dried over anhydrous Na2SO4 and concentrated to afford amines 6a–b in 92–95% yields.
4-(4-(Trifluoromethoxy)phenoxy)piperidine 6a: Compound 5a (5 g, 13.85 mmol) was dissolved in CH2Cl2 and TFA (1∶1, 20 mL) and the reaction was carried out as described in general procedure II to afford 6a (3.43 g) in 95% yield. 1H NMR (500 MHz, CDCl3): δ 6.84–7.02 (m, 4H), 3.68–3.71 (m, 1H), 2.68–2.78 (m, 4H), 1.72–1.98 (m, 4H); LCMS: 262 (M++1).
4-(4-Fluorophenoxy)piperidine 6b: Compound 5b (2.6 g, 8.80 mmol) was dissolved in CH2Cl2 and TFA (1∶1, 10 mL) and the reaction was carried out as described in general procedure II to afford 6b (1.58 g) in 92% yield. 1H NMR (500 MHz, CDCl3): δ 7.09–7.15 (m, 4H), 3.70–3.72 (m, 1H), 2.69–2.77 (m, 4H), 1.73–2.00 (m, 4H); LCMS: 196 (M++1).
General Procedure III, for Preparation of 7a–f
A mixture of aryl bromide (1.0 mmol), 6a (for compounds 7a–c) or 6b (for compounds 7d–f) (1.2 mmol), diacetoxypalladium (0.2 mmol), sodium butan-1-olate (2.4 mmol) and 2-(di-tert-butylphosphino)biphenyl (0.4 mmol) were stirred in anhydrous toluene and heated to 80°C for 3 h, then the reaction mass was concentrated under reduced pressure and purified by flash chromatography to afford 7a–f in 50–70% yields.
4-(4-(Trifluoromethoxy)phenoxy)-1-(4-vinylphenyl)piperidine 7a: 1-Bromo-4-vinylbenzene (1.0 g, 5.46 mmol), 6a (1.71 g, 6.55 mmol), diacetoxypalladium (0.24 g, 1.09 mmol), sodium butan-1-olate (1.26 g, 13.11 mmol) and 2-(di-tert-butylphosphino)biphenyl (0.65 g, 2.18 mmol) were stirred in anhydrous toluene and reaction was carried out as described in general procedure III to afford 7a (1.4 g) in 70% yield. 1H NMR (500 MHz, CDCl3): δ 7.32 (d, J = 8.5 Hz, 2H), 7.14 (d, J = 9.0 Hz, 2H), 6.89–6.92 (m, 4H), 6.64 (dd, J = 10.6, 17.5 Hz, 1H), 5.59 (d, J = 17.5 Hz, 1H), 5.09 (d, J = 10.9 Hz, 1H), 4.43–4.46 (m, 1H), 3.49–3.54 (m, 2H), 3.11–3.15 (m, 2H), 2.07–2.11 (m, 2H), 1.91–1.96 (m, 2H); LCMS: 364 (M++1).
2-(4-(4-(Trifluoromethoxy)phenoxy)piperidin-1-yl)-5-vinylpyridine 7b: 2-Bromo-5-vinylpyridine (1.0 g, 5.43 mmol), 6a (1.70 g, 6.52 mmol), diacetoxypalladium (0.24 g, 1.08 mmol), sodium butan-1-olate (1.25 g, 13.04 mmol) and 2-(di-tert-butylphosphino)biphenyl (0.64 g, 2.17 mmol) were stirred in anhydrous toluene and reaction was carried out as described in general procedure III to afford 7b (1.2 g) in 62.5% yield. 1H NMR (400 MHz, CDCl3): δ 8.17 (dt, J = 0.7, 2.3 Hz, 1H), 7.61 (ddd, J = 0.5, 2.5, 8.9 Hz, 1H), 7.09–7.19 (m, 2H), 6.87–6.98 (m, 2H), 6.54–6.71 (m, 2H), 5.57 (dd, J = 0.9, 17.6 Hz, 1H), 5.11 (dd, J = 0.9, 10.9 Hz, 1H), 4.51 (tt, J = 3.6, 7.3 Hz, 1H), 3.92 (ddd, J = 3.8, 7.5, 13.2 Hz, 2H), 3.48 (ddd, J = 3.6, 8.0, 13.3 Hz, 2H), 1.98–2.10 (m, 2H), 1.79–1.92 (m, 2H); LCMS: 365 (M++1).
1-(2-Fluoro-4-vinylphenyl)-4-(4-(trifluoromethoxy)phenoxy)piperidine 7c: 1-Bromo-2-fluoro-4-vinylbenzene (2.0 g, 9.95 mmol), 6a (3.12 g, 11.94 mmol), diacetoxypalladium (0.44 g, 1.99 mmol), sodium butan-1-olate (2.29 g, 23.88 mmol) and 2-(di-tert-butylphosphino)biphenyl (1.18 g, 3.98 mmol) were stirred in anhydrous toluene and reaction was carried out as described in general procedure III to afford 7c (2.1 g) in 55% yield. 1H NMR (400 MHz, CDCl3): δ 6.88–7.20 (m, 7H), 6.55–6.68 (m, 1H), 5.63 (dd, J = 0.8, 17.6 Hz, 1H), 5.19 (d, J = 10.8 Hz, 1H), 4.46 (tt, J = 3.6, 7.3 Hz, 1H), 3.34 (ddd, J = 3.3, 7.6, 11.2 Hz, 2H), 3.01 (ddd, J = 3.5, 7.8, 11.6 Hz, 2H), 2.08–2.20 (m, 2H), 1.99 (ddt, J = 3.5, 7.3, 16.5 Hz, 2H); LCMS: 382 (M++1).
4-(4-Fluorophenoxy)-1-(4-vinylphenyl)piperidine 7d: 1-Bromo-4-vinylbenzene (0.56 g, 3.06 mmol), 6b (0.71 g, 3.67 mmol), diacetoxypalladium (0.13 g, 0.61 mmol), sodium butan-1-olate (0.70 g, 7.34 mmol) and 2-(di-tert-butylphosphino)biphenyl (0.36 g, 1.22 mmol) were stirred in anhydrous toluene and reaction was carried out as described in general procedure III to afford 7d (0.5 g) in 55% yield. 1H NMR (400 MHz, CDCl3): δ 7.23–7.38 (m, 2H), 6.82–7.03 (m, 6H), 6.64 (dd, J = 17.6, 10.9 Hz, 1H), 5.59 (dt, J = 17.6, 0.8 Hz, 1H), 5.09 (dt, J = 10.9, 0.9 Hz, 1H), 4.38 (tt, J = 7.5, 3.7 Hz, 1H), 3.53 (ddd, J = 11.6, 7.2, 3.7 Hz, 2H), 3.11 (ddd, J = 12.2, 8.2, 3.5 Hz, 2H), 2.08 (ddd, J = 13.8, 7.8, 3.8 Hz, 2H), 1.91 (dtd, J = 12.0, 7.8, 3.6 Hz, 2H); LCMS: 298 (M++1).
2-(4-(4-Fluorophenoxy)piperidin-1-yl)-5-vinylpyridine 7e: 2-Bromo-5-vinylpyridine (0.2 g, 1.08 mmol), 6b (0.25 g, 1.30 mmol), diacetoxypalladium (0.04 g, 0.21 mmol), sodium butan-1-olate (0.25 g, 2.61 mmol) and 2-(di-tert-butylphosphino)biphenyl (0.13 g, 0.43 mmol) were stirred in anhydrous toluene and reaction was carried out as described in general procedure III to afford 7e (0.18 g) in 58% yield. 1H NMR (400 MHz, CDCl3): δ 8.16 (dd, J = 2.4, 0.7 Hz, 1H), 7.60 (dd, J = 8.8, 2.5 Hz, 1H), 6.94–7.01 (m, 2H), 6.85–6.91 (m, 2H), 6.54–6.70 (m, 2H), 5.56 (dt, J = 17.6, 0.7 Hz, 1H), 5.11 (dt, J = 10.9, 0.7 Hz, 1H), 4.44 (tt, J = 7.4, 3.6 Hz, 1H), 3.92 (ddd, J = 13.2, 7.3, 3.8 Hz, 2H), 3.45 (ddd, J = 13.2, 8.1, 3.6 Hz, 2H), 1.97–2.09 (m, 2H), 1.77–1.90 (m, 2H); LCMS: 299 (M++1).
1-(2-Fluoro-4-vinylphenyl)-4-(4-fluorophenoxy)piperidine 7f: 1-Bromo-2-fluoro-4-vinylbenzene (0.35 g, 1.74 mmol), 6b (0.40 g, 2.08 mmol), diacetoxypalladium (0.07 g, 0.34 mmol), sodium butan-1-olate (0.40 g, 4.18 mmol) and 2-(di-tert-butylphosphino)biphenyl (0.20 g, 0.69 mmol) were stirred in anhydrous toluene and reaction was carried out as described in general procedure III to afford 7f (0.27 g) in 50% yield. 1H NMR (400 MHz, CDCl3): δ 7.04–7.14 (m, 2H), 6.56–6.66 (m, 1H), 5.62 (d, J = 17.6 Hz, 1H), 5.17 (d, J = 8.8, Hz, 1H), 4.38 (tt, J = 7.4, 3.7 Hz, 1H), 3.35 (ddd, J = 11.4, 7.3, 3.5 Hz, 2H), 2.99 (ddd, J = 11.6, 7.9, 3.5 Hz, 2H), 2.07–2.15 (m, 2H), 1.92–2.01 (m, 2H); LCMS: 316 (M++1).
General Procedure IV, for Preparation of 9a–f
At room temperature, with vigorous stirring, a solution of Et3N (1.2 mmol) in anhydrous CHCl3 was slowly added to a solution of olefin (1.0 mmol) and N-hydroxy-5-nitrofuran-2-carbimidoyl chloride 8 (1.2 mmol) in anhydrous CHCl3. The reaction mixture was stirred at room temperature for 2 h then diluted with excess CHCl3, washed with water, dried over anhydrous Na2SO4, concentrated under reduced pressure and purified by flash chromatography to afford compounds 9a–i in 50–68% yields.
3-(5-Nitrofuran-2-yl)-5-(4-(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)phenyl)-4,5-dihydroisoxazole 9a: To a solution of 7a (2.6 g, 7.16 mmol) and 8 (1.63 g, 8.59 mmol) in anhydrous CHCl3 (20 mL) was added Et3N (1.19 mL, 8.59 mmol) in CHCl3 (5 mL) and the reaction continued as described in general procedure IV to afford 2.4 g of 9a in 65% yield. 1H NMR (400 MHz, CDCl3): δ 7.39 (d, J = 3.9 Hz, 1H), 7.26 (s, 2H), 7.10–7.18 (m, 2H), 7.04 (d, J = 3.8 Hz, 1H), 6.86–7.00 (m, 4H), 5.75 (dd, J = 8.9, 11.1 Hz, 1H), 4.45 (dt, J = 3.7, 7.4 Hz, 1H), 3.75 (dd, J = 11.1, 17.2 Hz, 1H), 3.52 (ddd, J = 3.7, 7.5, 11.7 Hz, 2H), 3.40 (dd, J = 8.9, 17.2 Hz, 1H), 3.15 (ddd, J = 3.5, 7.9, 12.1 Hz, 2H), 2.04–2.14 (m, 2H), 1.87–1.99 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 155.77, 151.49, 147.84, 147.55, 142.84, 131.86, 129.35, 127.21, 122.98, 122.51, 121.82, 116.84, 116.30, 113.08, 112.39, 84.10, 72.70, 46.20, 41.15, 30.20; HRMS m/z [M+H]+ calculated for C25H22F3N3O6∶518.1539; found: 518.1533; Anal. Calculated for C25H22F3N3O6: C, 58.03; H, 4.29; N, 8.12; F, 11.01. Found: C, 58.22; H, 4.11; N, 8.24; F, 10.85.
3-(5-Nitrofuran-2-yl)-5-(6-(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)pyridin-3-yl)-4,5-dihydroisoxazole 9b: To a solution of 7b (1.2 g, 3.29 mmol) and 8 (0.75 g, 3.95 mmol) in anhydrous CHCl3 (15 mL) was added Et3N (0.55 mL, 3.95 mmol) in CHCl3 (2 mL) and the reaction continued as described in general procedure IV to afford 1.1 g of 9b in 66% yield. 1H NMR (400 MHz, CDCl3): δ 8.16 (d, J = 2.5 Hz, 1H), 7.49 (dd, J = 2.6, 8.9 Hz, 1H), 7.42 (d, J = 3.8 Hz, 1H), 7.11–7.20 (m, 2H), 7.06 (d, J = 3.9 Hz, 1H), 6.87–6.98 (m, 2H), 6.72 (d, J = 8.9 Hz, 1H), 5.74 (dd, J = 9.1, 11.0 Hz, 1H), 4.53 (tt, J = 3.5, 7.2 Hz, 1H), 3.92 (ddd, J = 3.8, 7.6, 12.0 Hz, 2H), 3.77 (dd, J = 11.1, 17.3 Hz, 1H), 3.52 (ddt, J = 3.3, 7.2, 13.8 Hz, 2H), 3.39 (dd, J = 9.1, 17.3 Hz, 1H), 2.04 (ddt, J = 3.6, 7.3, 11.5 Hz, 2H), 1.79–1.92 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 159.38, 155.77, 147.93, 147.30, 146.64, 135.51, 122.61, 116.86, 113.06, 112.52, 107.11, 82.30, 73.03, 42.23, 40.75, 30.06; HRMS m/z [M+H]+ calculated for C24H21F3N4O6∶519.1491; found: 519.1514.
5-(3-Fluoro-4-(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)phenyl)-3-(5-nitrofuran-2-yl)-4,5-dihydroisoxazole 9c: To a solution of 7c (1.8 g, 4.72 mmol) and 8 (0.98 g, 5.19 mmol) in anhydrous CHCl3 (20 mL) was added Et3N (0.72 mL, 5.19 mmol) in CHCl3 (3 mL) and the reaction continued as described in general procedure IV to afford 1.5 g of 9c in 60% yield. 1H NMR (400 MHz, CDCl3): δ 7.40 (d, J = 3.8 Hz, 1H), 6.87–7.19 (m, 8H), 5.76 (dd, J = 8.4, 11.1 Hz, 1H), 4.45 (tt, J = 3.6, 7.1 Hz, 1H), 3.79 (dd, J = 11.2, 17.2 Hz, 1H), 3.29–3.44 (m, 3H), 3.02 (t, J = 9.7 Hz, 2H), 2.08–2.18 (m, 2H), 1.94–2.03 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 155.81, 147.74, 147.16, 142.82, 140.77, 133.40, 122.50, 122.07, 119.57, 116.87, 113.96, 113.74, 113.04, 112.66, 83.09, 72.42, 47.64, 41.50, 30.74; HRMS m/z [M+H]+ calculated for C25H21F4N3O6∶536.1444; found: 536.1439.
5-(4-(4-(4-Fluorophenoxy)piperidin-1-yl)phenyl)-3-(5-nitrofuran-2-yl)-4,5-dihydroisoxazole 9d: To a solution of 7d (0.23 g, 0.77 mmol) and 8 (0.17 g, 0.92 mmol) in anhydrous CHCl3 (5 mL) was added Et3N (0.12 mL, 0.92 mmol) in CHCl3 (1 mL) and the reaction continued as described in general procedure IV to afford 0.19 g of 9d in 59% yield. 1H NMR (400 MHz, CDCl3): δ 7.39 (d, J = 3.8 Hz, 1H), 7.20–7.29 (m, 2H), 7.02–7.05 (m, 1H), 6.93–7.00 (m, 4H), 6.85–6.90 (m, 2H), 5.75 (dd, J = 11.1, 8.9 Hz, 1H), 4.39 (tt, J = 7.4, 3.6 Hz, 1H), 3.74 (dd, J = 17.2, 11.1 Hz, 1H), 3.52 (ddd, J = 11.8, 7.4, 3.7 Hz, 2H), 3.40 (dd, J = 17.2, 9.0 Hz, 1H), 3.13 (ddd, J = 12.2, 8.1, 3.5 Hz, 2H), 2.01–2.13 (m, 2H), 1.84–1.97 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 158.61, 156.24, 153.28, 152.11, 151.54, 147.84, 147.56, 129.27, 127.21, 117.56, 116.28, 116.05, 115.82, 113.08, 112.38, 84.12, 73.18, 46.25, 41.14, 30.29; HRMS m/z [M+H]+ calculated for C24H22FN3O5∶416.1621; found: 452.1622.
5-(6-(4-(4-Fluorophenoxy)piperidin-1-yl)pyridin-3-yl)-3-(5-nitrofuran-2-yl)-4,5-dihydroisoxazole 9e: To a solution of 7e (0.15 g, 0.50 mmol) and 8 (0.11 g, 0.60 mmol) in anhydrous CHCl3 (5 mL) was added Et3N (84 uL, 0.60 mmol) in CHCl3 (1 mL) and the reaction continued as described in general procedure IV to afford 0.14 g of 9e in 64% yield. 1H NMR (400 MHz, CDCl3): δ 8.16 (dt, J = 2.4, 0.7 Hz, 1H), 7.45–7.50 (m, 1H), 7.38–7.42 (m, 1H), 7.03–7.06 (m, 1H), 6.94–7.01 (m, 2H), 6.85–6.91 (m, 2H), 6.71 (dd, J = 8.9, 0.8 Hz, 1H), 5.72 (dd, J = 11.0, 9.1 Hz, 1H), 4.45 (tt, J = 7.3, 3.6 Hz, 1H), 3.92 (ddd, J = 13.5, 7.4, 3.7 Hz, 2H), 3.75 (dd, J = 17.3, 11.0 Hz, 1H), 3.32–3.55 (m, 3H), 1.96–2.07 (m, 2H), 1.77–1.90 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 159.41, 158.64, 156.26, 153.27, 147.93, 147.30, 146.63, 135.49, 122.62, 117.59, 116.07, 115.84, 113.05, 112.52, 107.11, 82.31, 73.51, 42.29, 40.74, 30.15; HRMS m/z [M+H]+ calculated for C23H21FN4O5∶453.1574; found: 453.1589.
5-(3-Fluoro-4-(4-(4-fluorophenoxy)piperidin-1-yl)phenyl)-3-(5-nitrofuran-2-yl)-4,5-dihydroisoxazole 9f: To a solution of 7f (0.08 g, 0.25 mmol) and 8 (0.05 g, 0.30 mmol) in anhydrous CHCl3 (2 mL) was added Et3N (42 uL, 0.30 mmol) in CHCl3 (0.5 mL) and the reaction continued as described in general procedure IV to afford 0.06 g of 9f in 55% yield. 1H NMR (400 MHz, CDCl3): δ 7.39 (dd, J = 3.9, 0.6 Hz, 1H), 7.02–7.08 (m, 3H), 6.94–7.01 (m, 3H), 6.86–6.91 (m, 2H), 5.75 (dd, J = 11.1, 8.4 Hz, 1H), 4.38 (tt, J = 7.2, 3.6 Hz, 1H), 3.79 (dd, J = 17.2, 11.2 Hz, 1H), 3.29–3.44 (m, 3H), 2.94–3.05 (m, 2H), 2.05–2.17 (m, 2H), 1.90–2.02 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 158.62, 156.86, 156.24, 154.40, 153.32, 147.73, 147.19, 140.84, 133.33, 122.04, 119.44, 117.60, 116.04, 115.81, 113.95, 113.73, 113.01, 112.60, 83.10, 72.92, 47.70, 41.51, 30.84, 13.61; HRMS m/z [M+H]+ calculated for C24H21F2N3O5∶470.1527; found: 470.1533.
MIC Determination
Minimum inhibitory concentrations (MICs) were determined according to Clinical Laboratory Standards Institute (CLSI), using microbroth dilution method in 96-well plates and read by visual inspection [21]. The lowest concentration of drug that prevented visible growth was recorded as the MIC. Liquid MICs for indicated mycobacterium were determined in Middlebrook 7H9 broth supplemented with 10% albumin–dextrose complex and 0.05% (v/v) Tween 80 after 3 days (M. abscessus, M. fortuitum), or 7 days (M. avium, M. kansasii, M. tuberculosis) unless otherwise indicated. M. ulcerans MICs were performed by the agar proportion method and results recorded after 4 weeks of incubation.
Minimum Bactericidal Concentration Determination
Liquid MICs were performed as described above with the additional step of determining the number of bacteria added to each well. Serial dilutions of bacteria from the MIC plates were prepared and spread on Middlebrook 7H11 agar supplemented with 10% oleic acid–albumin–dextrose complex. After 4 weeks of incubation at 37°C, colony forming units (CFUs) were counted and the input calculated. Following 7 days of incubation, the contents of individual MIC plate wells were resuspended and spread on drug-free 7H11 agar plates. Plates were incubated at 37°C for 4 weeks prior to enumeration of viable colonies. The MBC was recorded as the lowest treatment concentration that killed >99.9% of input bacteria.
Cytotoxicity
In vitro cytotoxicity was assessed using Vero cells (kidney epithelial cells; ATCC CCL-81) essentially as described previously, except that detection was performed using CellTiter-Glo® Luminescent Cell Viability (Promega) [22]. The concentration of treatments that produced a 50% decrease in viability (IC50 values) were computed using nonlinear regression assisted equation of log [inhibitor] vs. Response-variable slope (four parameters) symmetrical, in GraphPad prism software.
Post Antibiotic Effect (PAE)
PAE studies were performed essentially as described previously [21]. To facilitate sample handling, M. bovis strain BCG Pasteur was used as a surrogate for M. tuberculosis [21]. Briefly, a mid-log phase culture recovered from frozen stock was diluted to an OD600 of ∼0.001 and allowed to grow to early log-phase (OD600 0.2). For each experimental condition, 50 mL of cells were exposed to 9a (at 0.1, 1.0, or 10.0 µg/mL) or isoniazid (at 0.05, 0.5, or 5.0 µg/mL) for 2 hours. Drugs were subsequently removed by washing twice in pre-warmed Middlebrook 7H9 medium and treated cells resuspended in pre-warmed medium. OD600 was determined for each culture immediately following resuspension and every 24 hours thereafter until reaching growth saturation. The difference between the time required for treated cells to reach 50% of the maximum OD600 of the untreated culture and for the untreated culture to reach this density was calculated as the PAE. Average and standard deviation were calculated based on results of two biologically independent experiments.
In vitro Activity against NRP Bacteria
To evaluate the activity of compounds against dormant TB bacilli, two in vitro models were used, mimicking oxygen and nutrient starvation. These experiments were performed using M. tuberculosis strain H37Rv as described previously, with no modifications [23], [24]. Briefly, hypoxic cultures were exposed to 10 µg/mL of indicated treatments for 4 days prior to cfu enumeration. For nutrient starvation experiments, aliquots of six-week starved cultures (in PBS) were exposed to indicated treatments for one week prior to cfu enumeration.
Checkerboard Synergy Assays
Whole cell in vitro synergy assays were performed using M. tuberculosis strain H37Rv. In a 96-well assay plate, two fold serial dilutions of Drug A (ethambutol, isoniazid, linezolid, PA-824, rifampin, or streptomycin) were prepared in 100 µl of Middlebrook 7H9 media (highest and lowest concentrations in rows A and G, respectively, with no drug in row H). Using a single dip with a 200ss pintool, 0.2 µl of drug B (9a) was transferred to the assay plate columns 1 to 11 of the assay plate, with drug-free DMSO transferred to column 12. To each well of the assay plate 100 µl of mid-log phase bacteria (diluted to OD600 of 0.01) was added, and plates incubated for 7 days prior to reading MICs by visual inspection. Fractional inhibitory concentration index (FICI) scores were calculated using the formula [MIC drug B in presence of Drug A]/[MIC of drug B)+[MIC of drug A in the presence of drug B]/[MIC of drug A]. FICI scores were interpreted as follows: synergy (≤0.5), indifference (>0.5–4.0), or antagonism (>4.0) [25]. For each drug combination, FICI ranges were reported from two biologically independent experiments.
In vitro Drug Resistance Frequency
Resistance frequency determination and spontaneous mutants selection was performed by plating M. tuberculosis H37Rv in the presence of increasing super-MIC concentrations of compound as described previously [21]. Where indicated, liquid cultures were exposed to a sub-inhibitory concentration of 9a prior to selection on agar.
Pharmacokinetic Analyses
Maximum solubility, metabolic stability, and plasma protein binding assays were performed and analyzed as described previously [22]. For in vivo pharmacokinetic studies, catheterized male Sprague-Dawley rats were obtained from Harlan Bioscience (Indianapolis, IN) and maintained as previously described [22]. Test compounds were dissolved in 30% DMSO, 30% propylene glycol, 20% PEG 3000 and 20% saline and administered intravenously (IV) at 10 mg/kg or were dissolved in PEG-3000:Water (60∶40) and dosed by oral gavage at 100 mg/kg. Blood sample collection, processing, and analysis were performed exactly as described previously [22]. The pharmacokinetic parameters maximum plasma concentration (Cmax), minimum plasma concentration within 24 hours after dosing (Cmin,24h), systematic exposure (AUC0-∞), half-life (t1/2), clearance (CL), volume of distribution (Vd), fraction excreted unchanged in urine (fe), and oral bioavailability (F) were determined by standard non-compartmental analysis using the software package Phoenix WinNonlin 6.2 (Pharsight, Mountain View, CA).
In vivo Efficacy
Efficacy of 9a in an acute tuberculosis infection mouse model was performed as described previously [26], [27], [28]. Briefly, 8 week female GKO mice (C57BL/6-Ifngtm1 Ts, from Jackson Laboratories) were infected with a low dose aerosol inoculum (∼100 CFU’s per mouse) of M. tuberculosis Erdman, transformed with pFCA-LuxAB. Isoniazid was dissolved in distilled water (DI). Treatment start was 14 days after infection. 9a was assessed in various formulations and therefore prepared using several different carriers (either 5% methylcellulose in DI-H2O, 30% captisol in DI-H2O, 10% vitamin E TPGS in DI-H2O, 0.5% Tween 80 in DI-H2O, 20% non-functionalized cyclodextrin (2-hydroxypropyl-β-cyclodextrin) in DI-H2O, or cold PEG (50∶35∶15 H2O:PEG300:PG)). Groups of 5 mice per group received 9a (300 mg/kg), isoniazid (25 mg/kg), or no treatment (untreated control group) for 9 consecutive days by oral gavage prior to sacrifice. Lungs and spleens from drug treated and control groups were homogenized and bacterial loads determined by CFU enumeration to determine bacterial loads before and after the 9 day treatment period, as described previously [27]. Reduction in bacterial loads of treatment groups compared to the untreated control group was recorded as (Log10) reduction. For statistical analysis the CFU were converted to logarithms, which were then evaluated by a one-way analysis of variance and two-way analysis of variance, followed by a multiple comparison analysis of variance by a one-way Tukey test and Dunnett test (SAS Software program, Research Triangle Park, NC). Differences were considered significant at the 95% level of confidence.
Ethics Statement
The pharmacokinetic study protocol was approved by Institutional Animal Care and Use Committee of the University of Tennessee Health Science Center (Protocol number 1463). The in vivo efficacy study protocol was approved by Colorado State University Institutional Animal Care and Use Committee (Protocol number 13-4263A).
Results and Discussion
Synthesis of an Expanded Set of Pentacyclic Nitrofurans
The design of the target series of new pentacyclic nitrofurans is shown in Figure 1. The nitrofuranyl and isoxazoline rings of our previous lead compounds 1, 2 and 9a were kept intact to maintain antitubercular potency, whereas the outer side chain (C-E rings) of the pentacyclic system were modified [18], [22]. Within this general scaffold, modifications were targeted to the C and E rings with substitutions aimed at blocking sites of potential metabolism and increasing solubility. The target compounds 9a–f were synthesized in a five step sequence starting from 1-Boc-4-hydroxypiperidine 3 as shown in Figure 2. Accordingly, 3 was mesylated by treating with methanesulfonyl chloride and triethylamine in dichloromethane at 0°C for 1 h to yield 4 in 93% yield which upon treatment with appropriate substituted E-ring phenols in presence of potassium carbonate and tetra n-butyl ammonium chloride in water at reflux afforded ethers 5a,b in 86–90% yields. Boc deprotection of 5a,b with trifluoroacetic acid in dichloromethane at room temperature produced the key intermediate amines 6a,b. The aryl amination reactions were carried out under Buchwald conditions by using appropriate C ring aryl bromides and secondary amines 6a,b in presence of palladium catalyst and sodium tert-butoxide in toluene at 100°C to afford 7a–f in 50–70% yields [22]. Finally, the isoxazoline ring was constructed by treating the olefins with nitrofuranyl chloroxime 8 [29] in the presence of triethylamine in chloroform at room temperature to give compounds 9a–f in 50–68% yields.
Figure 2. Synthesis of pentacyclic nitrofurans.

Reagents and conditions: a) MsCl, Et3N, CH2Cl2, RT, 2 h, 94%; b) substituted phenol, nBu4NCl, H2O, 100°C, 12 h, 86–90%; c) TFA, CH2Cl2, RT, 1 h, 92–95%; d) aryl bromide, 2-(di-tert-butylphosphino)biphenyl, NaOtBu, Pd(OAc)2, toluene, 100°C, 3 h, 50–70%; e) 8, Et3N, CHCl3, RT, 3 h, 50–68%.
Antituberculosis Activity and in vitro Metabolic Profile
All the compounds in the series maintained excellent potency against M. tuberculosis (MIC 0.001–0.02 µg/mL, corresponding to 1.9–46 nM), as shown in Table 1, which was well within their Cmax range. Mammalian cytotoxicity assays and calculation of subsequent therapeutic indices revealed very favorable selectivity, which ranged from 150–2500. Solubility of the series was low, however, with modifications yielding a ∼10 fold drop in solubility compared to our earlier series (2). Plasma protein binding remained very high (>96%) for all analogs. Compounds with a 4-fluorophenyl E ring (9d–f) showed a slight improvement in microsomal stability (t½ of 1.4–1.9 h. compared to 1.1 h for 2). The 4-trifluoromethoxyphenyl E ring series (9a–c), however, demonstrated much improved metabolic stability (t½ of 5–7 hours). Modification to the C ring (pyridyl in 9b and 9e, fluorophenyl in 9c and 9f) had negligible effects on overall properties irrespective of E ring substitution.
Table 1. In vitro antitubercular and in vitro metabolic profile.
| Compound | a M. tb. MIC(µg/mL) | bCytotoxicityIC50 (µg/mL) | cSelective Index(Cytox/MIC) | dSolubility (pH 7.4)µg/mL | eMicrosomal stability t1/2(mouse) (h) | fPlasma Proteinbinding (%) |
| 2 | 0.006 | 2.9g | 483 | 9.9 | 1.06g | 96.4g |
| 9a | 0.024 | 19.4 | 808 | 2.7h | 5.06 | 99.9h |
| 9b | 0.006 | 4.0 | 667 | 1.6 | 6.91 | 99.9 |
| 9c | 0.001 | 21.4 | 21400 | 1.9 | 7.04 | 97.6 |
| 9d | 0.012 | 14.7 | 1225 | 0.7 | 1.43 | 99.1 |
| 9e | 0.002 | 7.5 | 3750 | 2.2 | 1.93 | 99.8 |
| 9f | 0.012 | 8.0 | 667 | 0.8 | 1.74 | 99.8 |
| OPC-67683 | 0.01 | 107.1 | 10710 | 1.22 | 27.25 | 99.8 |
| PA-824 | 0.39 | 69.4 | 178 | 8.5 | 8.12 | 92.9 |
Structures, inhibitory activity, and in vitro PK properties of experimental and reference nitrofurans.
The lowest concentration of drug that prevented visible growth.
Cytotoxicity against Vero epithelial cells using MTT assay.
Selectivity for tuberculosis.
Maximum solubility calculated by µSol Evolution Software.
Metabolic stability performed with mouse liver microsomes.
Plasma protein binding assays performed using mouse plasma.
Indicates values reported in reference.
Indicates values reported in reference.
Spectrum of Antimicrobial Activity
Compounds 9a–f were weakly active or inactive against a panel of gram-positive and gram-negative pathogens (Table S1) except for 9f, which had moderate activity against most gram-positive pathogens and an efflux pump deficient strain of E. coli. This activity is consistent to that observed for other similar nitrofurans we generated in prior series [21], [22]. Compound 9a was further tested against non-tuberculosis disease causing mycobacteria (Table 2). Compound 9a was moderately active against non-tuberculosis mycobacteria (NTM) M. abscessus, M. kansasii and M. fortuitum and highly active against M. ulcerans with an MIC lower than the plasma concentrations maintained over 24 hours after administration, with a Cmax of 0.85 µg/mL and a Cmin,24h of 0.17 µg/mL. PA-824 was moderately active against M. kansasii and M. ulcerans but inactive against other NTM species tested, in agreement with previously reported studies [30], [31]. The activity for 9a against M. abscessus is notable since this difficult to treat mycobacterium is intrinsically resistant to most anti-mycobacterial agents [32]. These differences in the spectrum of activity between the nitrofurans and the nitroimidazole PA-824 is consistent with differences in the redox potential for these two compound classes, with the lower redox potential of nitroimidazoles requiring specific enzymes for activation, whilst nitrofurans have a higher redox potential and may be more easily activated by a wider spectrum of enzymes [17], [33].
Table 2. Susceptibility of non-tuberculosis mycobacteria.
| Minimum Inhibitory Concentration (µg/mL) | |||||
| Compound | a M. abscessus | a M. avium | a M. kansasii | a M. fortuitum | b M. ulcerans |
| 9a | 12.50 | 200 | 3.13–6.25 | 6.25–12.50 | 0.02–0.05 |
| Amikacin | 3.13–6.25 | 12.5 | 6.25–12.5 | 0.78 | ND |
| Rifampin | 50.00 | 6.3 | 0.39 | 1.56 | 0.1–0.4 |
| PA-824 | >200 | >200 | 3.1–12.5 | >200 | 3.1–25 |
Susceptibility of non-tuberculosis mycobacteria to nitrofurans and reference drugs.
MICs determined by macrobroth dilution and visual inspection of plates.
MIC determined by agar proportion method. Values reported are the range of at least two biologically independent experiments. Species tested included M. abscessus (ATCC 19977); M. avium (ATCC 25291); M. kansasii (ATCC 12478); M. fortuitum (ATCC 6841); and M. ulcerans (ATCC 35840).
In vivo Pharmacokinetic Profiling
The pharmacokinetic profiles of 9b–c were evaluated in rats and compared to the published data for 9a using similar formulations (Table 3). After intravenous administration of 10 mg/kg 9b–c, compounds showed biexponential concentration-time profiles similar to 9a, but elimination half-lives (t½ 2.5–5.6 h) significantly lower than 9a (t½ 10.3 h). The shorter half-lives are a consequence of a reduced volume of distribution for compounds 9b–c (Vd 1.53–1.69 L/kg) compared to 9a (Vd 6.72 L/kg), while clearance (CL) remained similar for all three compounds. The fraction of dose excreted unchanged by the kidneys (fe) was negligible for all compounds. Their relatively large volumes of distribution and non-renal elimination are probably a reflection of their high lipophilicity (clogP 4.6–5.8). The oral bioavailability of compounds 9b and 9c was poor (F 8.0%, 0.83%), possibly a reflection of poor solubility as had already been noted for 9a which also has limited oral bioavailability (F 4.6%).Compounds 9a and 9b were non-toxic by intravenous and oral dosing in rats. Central nervous system toxicity (seizures) was observed for compound 9c in both administration modes. Compound 9a was therefore advanced for in vivo efficacy testing due to its longer in vivo half-life and higher volume of distribution as compared to 9b.
Table 3. In vivo pharmacokinetic parameters.
| After IV administration of 10 mg/kg | After oral administration of 100 mg/kg | |||||||
| Compound | t½ (h) | CL (L/h/kg) | Vd (L/kg) | fe (%) | Cmax (mg/L) | Cmin,24 h (mg/L) | AUC0-∞ (mg h/L) | F (%) |
| 9a Ref | 10.3 (1.4) | 0.46 (0.07) | 6.72 (1.10) | 0.04 (0.01) | 0.85 (0.80) | 0.17 (0.16) | 10.2 (9.1) | 4.56 (4.06) |
| 9b | 2.48 (0.62) | 0.42 (0.15) | 1.53 (0.36) | 0.01 (0.00) | 2.31 (0.62) | 0.11 (0.11) | 15.1 (7.1) | 8.00 (3.93) |
| 9c | 5.60 (0.45) | 0.41 (0.06) | 1.69 (0.27) | 0.02 (0.00) | 0.15 (0.06) | 0.02 (0.02) | 1.61 (0.83) | 0.83 (0.57) |
Pharmacokinetic analysis of experimental compounds in rats.
Values represent means (% coefficient of variation).
Abbreviations: t½: half life; CL: clearance; Vd: volume of distribution; fe: fraction excreted unchanged in urine; Cmax: maximum plasma concentration; Cmin,24h: minimum plasma concentration within 24 hours after dosing; AUC0-∞: systematic exposure; F: oral bioavailability.
In vivo Efficacy in a Mouse Model of Acute Tuberculosis Infection
9a was next evaluated for in vivo efficacy using a mouse model of acute tuberculosis infection [26]. Given the poor solubility of 9a, six different formulations were employed in the acute GKO mouse infection model. In each formulation, 9a was well tolerated with daily oral administration at 300 mg/kg and 9 days of treatment provided a statistically significant reduction (P<0.001 by Tukey Test) in bacillary loads in both the lungs and spleen (Figure 3, Table S2). The differences in efficacy between different 9a formulation groups were not statistically significant. A control drug isoniazid was included in the experiment (data not shown), which gave an expected a 3.5 and 4.6 log reduction of the bacterial load in lungs and spleen, respectively, versus the untreated controls. Although 9a did not provide superior protection compared to rapid acting control isoniazid, the reduction it produced in lungs was 91–97% in 9 days. This marks a substantial improvement over precursor tetracyclic nitrofuranyl isoxazolines, which were not active in vivo [15].
Figure 3. Murine model of acute tuberculosis infection.
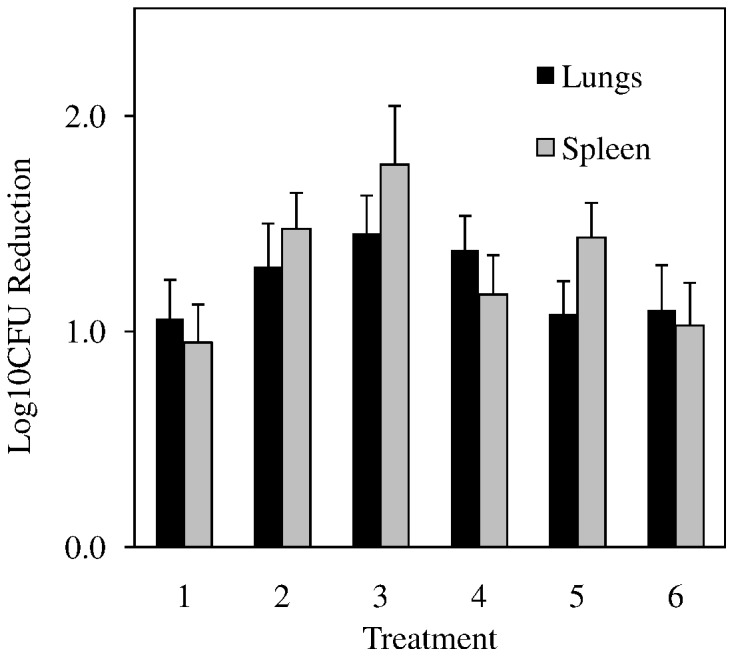
Log10 reduction provided by compound 9a in lungs (black bars) and spleen (grey bars) after 9 days of daily oral administration of 300 mg/kg was determined by calculating the difference between bacillary loads in organs from the untreated group and 9a dissolved in (1) 0.5% methylcellulose in DI-H2O (2) 30% captisol in DI-H2O (3) 10% vitamin E TPGS in DI-H2O (4) 0.5% Tween 80 in DI-H2O (5) 20% cyclodextrin in DI-H2O or (6) cold PEG (50∶35∶15 H2O:PEG300:PG). Error bars indicate SEM within treatment groups of 5–7 mice per group.
Post Antibiotic Effect and in vitro Synergy with Frontline Agents
Post antibiotic effect (PAE), defined as the duration of growth retardation following the removal of an antibiotic from the environment of the organism, is an important parameter for early consideration of potential dosing and was determined for compound 9a [34]. Pulse exposure to 9a yielded a dose-responsive PAE, with compound concentrations of 0.01, 1.0, and 10.0 µg/mL yielding PAE’s of 21.7+/−1.2 h, 105.5+/−19.8 h, and 178.4+/−8.8 h, respectively. The PAE produced by treatment with 9a at 1 µg/mL (105.5 h) was superior to that produced by exposure to 5 µg/mL isoniazid (71.8 h). The long PAE of compound 9a is highly favorable for the further development of this antitubercular compound series.
An important characteristic for novel tuberculosis therapeutics is compatibility with antitubercular agents in the clinic and late stage development, since combination therapy is applied to reduce the development of acquired drug-resistance. A preliminary investigation of 9a’s potential for co-administration was, therefore, conducted using the checkerboard approach (Table 4) [35]. In this commonly used approach to access in vitro synergy, serial dilutions of two agents are prepared in opposing directions across an MIC assay plate and the fractional inhibitory concentration index (FICI) is calculated using the formula [MIC drug B in presence of Drug A]/[MIC of drug B)+[MIC of drug A in the presence of drug B]/[MIC of drug A]. No synergy or antagonism was seen with frontline drugs ethambutol and isoniazid, nor with linezolid or streptomycin (FICI scores 0.5–4.0). However, 9a synergized with the frontline treatment rifampin and nitroimidazole PA-824 (FICI <0.5). Interestingly, in the presence of 9a, MICs of PA-824 and rifampin both decreased ∼10 fold. The favorable synergy predicted for 9a with rifampin, as well as the lack of unfavorable interactions predicted with other antitubercular drugs, suggests that 9a may complement current tuberculosis treatment regimes.
Table 4. In vitro Interactions with Antitubercular Drugs.
| Treatment | Drug Class | MIC Alone(µg/mL) | MIC in presence of9a (µg/mL) | 9a MIC in combination(µg/mL) | aFICI with 9a | Type ofInteraction |
| Rifampin | Rifamycin | 0.02–0.05 | 0.003 | 0.003–0.01 | 0.38 | Synergy |
| Isoniazid | Other | 0.02 | 0.01–0.02 | 0.02–0.0008 | 0.52–2.00 | Indifference |
| Ethambutol | Antimycobacterial | 1.6 | 0.8 | 0.01 | 0.75–1.00 | Indifference |
| Linezolid | Ooxazolidinone | 1.6 | 0.8 | 0.01–0.02 | 1.00 | Indifference |
| PA-824 | Nitroimidazole | 0.2–0.8 | 0.01–0.02 | 0.003–0.006 | 0.13–0.28 | Synergy |
| Streptomycin | Aminoglycoside | 1.6 | 0.4–0.8 | 0.025–0.006 | 0.75 | Indifference |
In vitro interactions determined by checkerboard assays.
Ex-vivo synergy assays were performed to determine if 9a displayed synergy (FICI ≤0.5), indifference (FICI >0.5–4.0) or antagonism (FICI >4.0) with a panel of anti-tuberculosis agents.
Minimum Bactericidal Concentration Determinations
The minimum bactericidal concentration (MBC) of 9a was determined (alongside controls); the results are shown in Table 5. The number of viable bacteria in individual wells of MIC plates was enumerated to determine the minimum bactericidal concentration (MBC), defined as the treatment concentration that killed >99.9% of the inoculum. Streptomycin was cidal (MBC/MIC ≤4), as anticipated, whereas all nitroaromatics compounds investigated were static (MBC/MIC >4). These observations are also consistent with nitrofurans being known to be static at low concentrations but cidal at higher concentrations.
Table 5. MIC and MBCs for select nitrofurans and controls.
| Treatment | MBC | Classification |
| 2 | – | – |
| 9a | 3.13–6.25 | Static |
| OPC-67683 | >3 | Static |
| PA-824 | >6 | Static |
| Streptomycin | 3.13–6.25 | Cidal |
Treatments were considered cidal when the MBC99.9 for M. tuberculosis H37Rv was less than 4X the MIC.
Activity against Multiple Models of Tuberculosis
Several in vitro models have been proposed to mimic the physiological state of persistent tuberculosis infections in vivo. The most commonly used model of tuberculosis persistence is based upon the assumption that the host immune system sequesters bacilli into granulomas, where they persist at low oxygen levels. Activity of 9a was assessed in the Rapid Anaerobic Dormancy (RAD) model which uses rapid oxygen depletion to mimic the Non-Replicating-Persister 2 (NRP-2) conditions described by Voskuil. [24], [36]. As anticipated, NRP-2 bacilli were sensitive to treatment with rifampin but resistant to isoniazid (Table 6). In this in vitro model of a dormant tuberculosis, 9a and PA-824 were both highly active, killing >99.9% of hypoxic bacilli at equivalent concentrations (10 µg/mL). In addition to oxygen deprivation, nutrient starvation is also proposed as an important environmental inducer of persistent tubercular populations. A simple model of starvation-induced persistence reported by Betts et al. [23]was adopted to further assess the anti-dormancy activity of nitrofuran 9a (Figure 4). Mid-log and nutrient-starved cultures were treated with test compound (9a or isoniazid at 1 µg/mL) or DMSO drug carrier for 7 days. As anticipated, isoniazid treatment killed >95% of actively growing M. tuberculosis culture but was 7 fold less active against nutrient starved bacteria. 9a produced an 80% reduction in viability of both actively growing and nutrient starved bacilli. The sustained activity of compound 9a in two separate models of biologically relevant, drug-refractory M. tuberculosis populations suggests it may serve as a valuable, treatment-shortening addition to the existing anti-tuberculosis regimen.
Table 6. In vitro activity against NRP bacteria grown under hypoxic conditions.
| Compound | Concentration | % Growth vs. Control |
| DMSO (carrier) | 1% | 100% |
| Isoniazid | 10 µg/mL | 29.1–49.1% |
| Rifampin | 10 µg/mL | <0.1% |
| PA-824 | 10 µg/mL | <0.1% |
| 9a | 10 µg/mL | <0.1% |
Viability of hypoxic M. tuberculosis cultures was assessed using the rapid anaerobic dormancy model.
Figure 4. Nutrient starvation model of nonreplicating persisters.
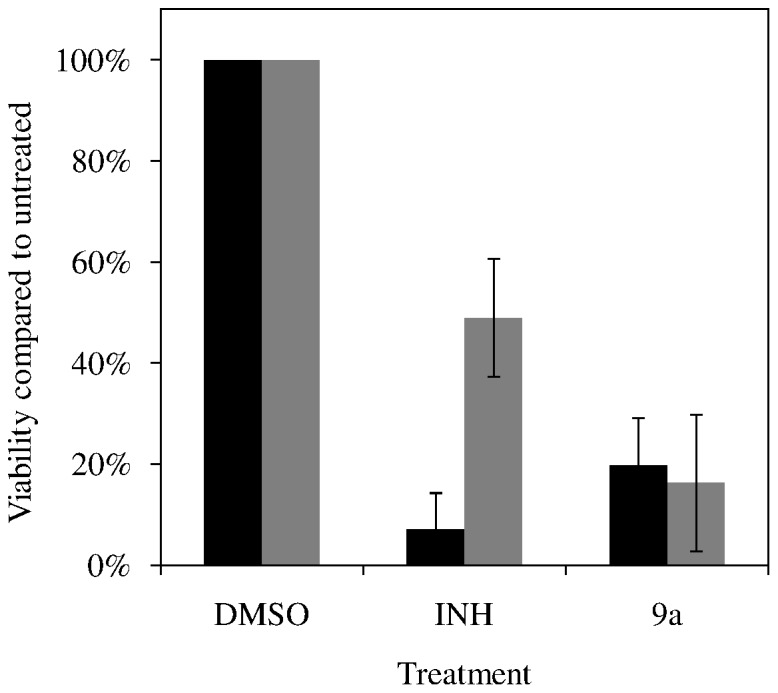
The viability of mid-log phase (black bars) or nutrient-starved (gray bars) after exposure to DMSO carrier (1% v/v), 1 µg/mL of isoniazid (INH), or 1 µg/mL of 9a. Averaged results and SEM from two biologically independent experiments are presented.
Mechanism of Action Studies
All nitroaromatic antibiotics are prodrugs for which bioreduction by the pathogen is required to generate the active antimicrobial metabolites. The bioreduction of PA-824 is well studied and is mediated by the deazaflavin-dependent nitroreductase (Ddn) in M. tuberculosis, with cellular bioreduction ultimately producing toxic nitric oxide [37], [38]. The F420-dependent glucose 6-phosphate dehydrogenase (FGD1), which is required in the recycling of F420 cofactor, is responsible for transferring electrons to Ddn (encoded by Rv3547). Thus mutations in genes encoding Ddn, FGD1 and F420 biosynthesis confer resistance to PA-824 and cross resistance to OPC-67683, which is activated in a similar manner [39]. A collection of PA-824 resistant mutants each harboring a mutation in a single gene in the PA-824 activation pathway (FGD1, F420, or Ddn) were screened against compound 9a for cross resistance (Table 7). All these mutants remained sensitive to 9a, suggesting that 9a is primarily activated by a different pathway and is unlikely to be affected by primary resistance mechanisms of bicyclic nitroimidazoles.
Table 7. Mechanism of activation and primary resistance.
| Genotype | MIC (µg/ml) | ||||
| Strain | FGD1 | F420 | Ddn | PA-824 | 9a |
| H37Rv-wt | + | + | + | 0.1 | 0.025 |
| H37Rv-T3 | − | + | + | 100 | 0.1 |
| H37Rv-5A1 | + | − | + | 50 | 0.05 |
| H37Rv-14A1 | + | + | − | 50 | 0.1 |
Activity of nitrofurans against M. tuberculosis H37Rv mutants deficient in enzymes required for bioreductive activation of PA-824.
To assess M. tuberculosis’s potential to become resistant to lead 9a, the frequency at which spontaneous resistance arises was determined by growing the susceptible M. tuberculosis strain H37Rv on agar containing 9a. Resistant clones arose at a frequency of 3×10−7 in the presence of 2–4×MIC of 9a. Repeated efforts to generate resistant clones at higher concentrations of 9a were unsuccessful, suggesting the potential for development of resistance is very low, possibly attributable to multiple mechanisms of activation or the induction of alternative modes of action at higher concentrations of compound. This appears consistent with 9a being bactericidal at higher concentrations. Pre-treatment of cultures with sub-inhibitory concentrations of 9a permitted selection of resistant colonies on agar plates containing 0.05–0.4 µg/mL of 9a. All 9a resistant clones assessed exhibited elevated 9a MIC (>3.13 µg/mL) but retained sensitivity to ethambutol, linezolid, streptomycin, rifampin, ofloxacin, and isoniazid (Table 8). All but a single 9a resistant clone remained sensitive to PA-824 and OPC-67683 (Table 8) further supporting the hypothesis that 9a is primarily activated by a unique biological pathway.
Table 8. Cross Resistance Profiles for Compounds 9a Resistant Clones.
| Clone # | Parent | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| Selection Conc. (µg/mL) | – | 0.05 | 0.05 | 0.1 | 0.1 | 0.2 | 0.2 | 0.2 | 0.4 | 0.4 |
| 2 | S | R | R | R | R | R | R | R | R | R |
| 9a | S | R | R | R | R | R | R | R | R | R |
| PA-824 | S | S | S | S | S | R | S | S | S | S |
| OPC 67683 | S | S | S | S | S | R | S | S | S | S |
Abbreviations: S, susceptible; R, resistant.
Conclusions
Given the success of nitroaromatic antibiotics in treating anaerobic bacterial infections and the current lack of suitable therapeutics to treat persistent tuberculosis infections, we report here the synthesis and antimicrobial characterization of pentacyclic nitrofuran analogs that incorporate the outer ring elements of OPC-67683. These pentacyclic nitrofurans displayed a favorable in vitro selectivity index and selective inhibitory activity against mycobacteria. The MIC of the series lead 9a (Lee1106) was 46 nM, comparable to that of OPC-67683 (19 nM), and is 20-fold more potent than PA-824 (>1000 nM). Compound 9a was active in in vitro assays against nonreplicating bacteria suggesting its potential for treatment of chronic tuberculosis infections. 9a exerted a lengthy post antibiotic effect, similar to isoniazid, and was synergistic with the frontline drug rifampin. Mechanism of actions studies showed that the three genes required for activation of PA-824 and OPC-67683 are dispensable for 9a antitubercular activity, indicating a low potential for cross resistance to other nitroimidazole antitubercular agents. This finding also shows that despite the sharing of common outer ring structural features between 9a and OPC-67683, the key difference in their nitroaromatic head groups leads to the use of different mechanisms of bioreductive activation. This likely arises from significant differences in the redox potential between nitrofurans and nitroimidazoles, with the latter compounds having a lower redox potential which means that more specific enzymes are required to transfer electrons to the nitro-group. Conversely, the high redox potential for nitrofurans makes these molecules easier to be reduced and different enzymes may be involved [17], [40]. Compound 9a was well tolerated in a murine model of acute tuberculosis infection, and showed significant in vivo efficacy in both lungs and spleens with >90% reduction in bacillary load after 9 days of treatment. In summary, the studies presented here show significant promise for development of the nitrofuran series as antituberculous agents. Further studies to improve metabolic stability, solubility and bioavailability are warranted in order to enhance the in vivo efficacy of the series.
Supporting Information
Spectrum of Antimicrobial Activity.
(DOCX)
Assessment of Efficacy in an in vivo Murine Model of Acute Tuberculosis Infection.
(DOCX)
Acknowledgments
We are grateful to Dr. Clifton E. Barry III for generously providing the PA-824 resistant mutants used in this study.
Funding Statement
This study was supported by the National Institutes of Health grant AI062415 and the American Lebanese Syrian Associated Charities (ALSAC), St. Jude Children’s Research Hospital. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.WHO (2012) Global Tuberculosis Report 2012. Geneva, Switzerland: WHO.
- 2. Koenig R (2008) Drug-resistant tuberculosis. In South Africa, XDR TB and HIV prove a deadly combination. Science 319: 894–897. [DOI] [PubMed] [Google Scholar]
- 3.Lafontaine D, Slavuski A, Vezhnina N, Sheyanenko O (2004) Treatment of multidrug-resistant tuberculosis in Russian prisons. Lancet: 246–247. [DOI] [PubMed]
- 4. Van der Walt M, Lancaster J, Odendaal R, Davis JG, Shean K, et al. (2013) Serious treatment related adverse drug reactions amongst anti-retroviral naive MDR-TB patients. PLoS One 8: e58817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Esmail H, Barry CE 3rd, Wilkinson RJ (2012) Understanding latent tuberculosis: the key to improved diagnostic and novel treatment strategies. Drug Discov Today 17: 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boshoff HI, Barry CE 3rd (2005) Tuberculosis - metabolism and respiration in the absence of growth. Nat Rev Microbiol 3: 70–80. [DOI] [PubMed] [Google Scholar]
- 7. Lenaerts AJ, Hoff D, Aly S, Ehlers S, Andries K, et al. (2007) Location of persisting mycobacteria in a Guinea pig model of tuberculosis revealed by r207910. Antimicrob Agents Chemother 51: 3338–3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, et al. (2000) A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405: 962–966. [DOI] [PubMed] [Google Scholar]
- 9. Barry CE 3rd, Boshoff HI, Dowd CS (2004) Prospects for clinical introduction of nitroimidazole antibiotics for the treatment of tuberculosis. Curr Pharm Des 10: 3239–3262. [DOI] [PubMed] [Google Scholar]
- 10. Diacon AH, Dawson R, du Bois J, Narunsky K, Venter A, et al. (2012) Phase II dose-ranging trial of the early bactericidal activity of PA-824. Antimicrob Agents Chemother 56: 3027–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Diacon AH, Donald PR, Pym A, Grobusch M, Patientia RF, et al. (2012) Randomized pilot trial of eight weeks of bedaquiline (TMC207) treatment for multidrug-resistant tuberculosis: long-term outcome, tolerability, and effect on emergence of drug resistance. Antimicrob Agents Chemother 56: 3271–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Matsumoto M, Hashizume H, Tomishige T, Kawasaki M, Tsubouchi H, et al. (2006) OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med 3: e466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sasaki H, Haraguchi Y, Itotani M, Kuroda H, Hashizume H, et al. (2006) Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles. J Med Chem 49: 7854–7860. [DOI] [PubMed] [Google Scholar]
- 14. Gler MT, Skripconoka V, Sanchez-Garavito E, Xiao H, Cabrera-Rivero JL, et al. (2012) Delamanid for multidrug-resistant pulmonary tuberculosis. N Engl J Med 366: 2151–2160. [DOI] [PubMed] [Google Scholar]
- 15. Tangallapally RP, Sun D, Rakesh, Budha N, Lee RE, et al. (2007) Discovery of novel isoxazolines as anti-tuberculosis agents. Bioorg Med Chem Lett 17: 6638–6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Carvalho LP, Lin G, Jiang X, Nathan C (2009) Nitazoxanide kills replicating and nonreplicating Mycobacterium tuberculosis and evades resistance. J Med Chem 52: 5789–5792. [DOI] [PubMed] [Google Scholar]
- 17. Sisson G, Goodwin A, Raudonikiene A, Hughes NJ, Mukhopadhyay AK, et al. (2002) Enzymes associated with reductive activation and action of nitazoxanide, nitrofurans, and metronidazole in Helicobacter pylori. Antimicrob Agents Chemother 46: 2116–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Budha NR, Mehrotra N, Tangallapally R, Rakesh, Qi J, et al. (2008) Pharmacokinetically-guided lead optimization of nitrofuranylamide anti-tuberculosis agents. AAPS J 10: 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baker WR, Cai S, Keeler EL (1997) Preparation of bicyclic nitroimidazole antibacterial compounds. WO9701562A1.
- 20.Tsubouchi H, Sasaki H, Kuroda H, Itotani M, Hasegawa T, et al. (2003) S. 2,3-Dihydro-6-nitroimidazo[2,1-b]oxazoles. WO2004033463A1.
- 21. Hurdle JG, Lee RB, Budha NR, Carson EI, Qi J, et al. (2008) A microbiological assessment of novel nitrofuranylamides as anti-tuberculosis agents. J Antimicrob Chemother 62: 1037–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rakesh, Bruhn D, Madhura DB, Maddox M, Lee RB, et al. (2012) Antitubercular nitrofuran isoxazolines with improved pharmacokinetic properties. Bioorg Med Chem 20: 6063–6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K (2002) Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol 43: 717–731. [DOI] [PubMed] [Google Scholar]
- 24. Honaker RW, Dhiman RK, Narayanasamy P, Crick DC, Voskuil MI (2010) DosS responds to a reduced electron transport system to induce the Mycobacterium tuberculosis DosR regulon. J Bacteriol 192: 6447–6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ramon-Garcia S, Ng C, Anderson H, Chao JD, Zheng X, et al. (2011) Synergistic drug combinations for tuberculosis therapy identified by a novel high-throughput screen. Antimicrob Agents Chemother 55: 3861–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lenaerts AJ, Gruppo V, Brooks JV, Orme IM (2003) Rapid in vivo screening of experimental drugs for tuberculosis using gamma interferon gene-disrupted mice. Antimicrob Agents Chemother 47: 783–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lenaerts AJ, Gruppo V, Marietta KS, Johnson CM, Driscoll DK, et al. (2005) Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrob Agents Chemother 49: 2294–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vicente E, Villar R, Burguete A, Solano B, Perez-Silanes S, et al. (2008) Efficacy of quinoxaline-2-carboxylate 1,4-di-N-oxide derivatives in experimental tuberculosis. Antimicrob Agents Chemother 52: 3321–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hamper BC, Leschinsky KL, Massey SS, Bell CL, Brannigan LH, et al. (1995) Synthesis and Herbicidal Activity of 3-Aryl-5-(haloalkyl)-4-isoxazolecarboxamides and Their Derivatives. J Agric Food Chem 43: 219–228. [Google Scholar]
- 30. Ashtekar DR, Costa-Perira R, Nagrajan K, Vishvanathan N, Bhatt AD, et al. (1993) In vitro and in vivo activities of the nitroimidazole CGI 17341 against Mycobacterium tuberculosis. Antimicrob Agents Chemother 37: 183–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ji B, Lefrancois S, Robert J, Chauffour A, Truffot C, et al. (2006) In vitro and in vivo activities of rifampin, streptomycin, amikacin, moxifloxacin, R207910, linezolid, and PA-824 against Mycobacterium ulcerans. Antimicrob Agents Chemother 50: 1921–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Griffith DE, Aksamit TR (2012) Therapy of refractory nontuberculous mycobacterial lung disease. Curr Opin Infect Dis 25: 218–227. [DOI] [PubMed] [Google Scholar]
- 33. Smith MA, Edwards DI (1995) Redox potential and oxygen concentration as factors in the susceptibility of Helicobacter pylori to nitroheterocyclic drugs. J Antimicrob Chemother 35: 751–764. [DOI] [PubMed] [Google Scholar]
- 34. Craig WA, Vogelman B (1987) The postantibiotic effect. Ann Intern Med 106: 900–902. [DOI] [PubMed] [Google Scholar]
- 35. Parsley TL, Provonchee RB, Glicksman C, Zinner SH (1977) Synergistic activity of trimethoprim and amikacin against gram-negative bacilli. Antimicrob Agents Chemother 12: 349–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Leistikow RL, Morton RA, Bartek IL, Frimpong I, Wagner K, et al. (2010) The Mycobacterium tuberculosis DosR regulon assists in metabolic homeostasis and enables rapid recovery from nonrespiring dormancy. J Bacteriol 192: 1662–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cellitti SE, Shaffer J, Jones DH, Mukherjee T, Gurumurthy M, et al. (2012) Structure of Ddn, the deazaflavin-dependent nitroreductase from Mycobacterium tuberculosis involved in bioreductive activation of PA-824. Structure 20: 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Singh R, Manjunatha U, Boshoff HI, Ha YH, Niyomrattanakit P, et al. (2008) PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 322: 1392–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gurumurthy M, Mukherjee T, Dowd CS, Singh R, Niyomrattanakit P, et al. (2012) Substrate specificity of the deazaflavin-dependent nitroreductase from Mycobacterium tuberculosis responsible for the bioreductive activation of bicyclic nitroimidazoles. FEBS J 279: 113–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Murugasu-Oei B, Dick T (2000) Bactericidal activity of nitrofurans against growing and dormant Mycobacterium bovis BCG. J Antimicrob Chemother 46: 917–919. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spectrum of Antimicrobial Activity.
(DOCX)
Assessment of Efficacy in an in vivo Murine Model of Acute Tuberculosis Infection.
(DOCX)