

Abstract
Objective:
Mitochondria-derived reactive oxygen species (ROS) play important roles in the development of cardiovascular disease highlighting the need for novel targeted therapies. This study assessed the potential therapeutic benefit of combining the mitochondria-specific antioxidant, MitoQ10, with the low-dose angiotensin receptor blocker (ARB), losartan, on attenuation of hypertension and left ventricular hypertrophy. In parallel, we investigated the impact of MitoQ10 on cardiac hypertrophy in a neonatal cardiomyocyte cell line.
Methods and results:
Eight-week-old male stroke-prone spontaneously hypertensive rats (SHRSPs, n = 8–11) were treated with low-dose losartan (2.5 mg/kg per day); MitoQ10 (500 μmol/l); a combination of MitoQ10 and losartan (M + L); or vehicle for 8 weeks. Systolic pressure and pulse pressure were significantly lower in M + L rats (167.1 ± 2.9 mmHg; 50.2 ± 2.05 mmHg) than in untreated SHRSP (206.6 ± 9 mmHg, P < 0.001; 63.7 ± 2.7 mmHg, P = 0.001) and demonstrated greater improvement than MitoQ10 or low-dose losartan alone, as measured by radiotelemetry. Left ventricular mass index was significantly reduced from 22.8 ± 0.74 to 20.1 ± 0.61 mg/mm in the combination group (P < 0.05). Picrosirius red staining showed significantly reduced cardiac fibrosis in M + L rats (0.82 ± 0.22 A.U.) compared with control (5.94 ± 1.35 A.U., P < 0.01). In H9c2 neonatal rat cardiomyocytes, MitoQ10 significantly inhibited angiotensin II mediated hypertrophy in a dose-dependent manner (500 nmol/l MitoQ10 153.7 ± 3.1 microns vs. angiotensin II 200.1 ± 3.6 microns, P < 0.001).
Conclusion:
Combining MitoQ10 and low-dose losartan provides additive therapeutic benefit, significantly attenuating development of hypertension and reducing left ventricular hypertrophy. In addition, MitoQ10 mediates a direct antihypertrophic effect on rat cardiomyocytes in vitro. MitoQ10 has potential as a novel therapeutic intervention in conjunction with current antihypertensive drugs.
Keywords: cardiac fibrosis, cardiac hypertrophy, hypertension, mitochondria, reactive oxygen species
INTRODUCTION
Accumulating evidence demonstrates that mitochondria-derived reactive oxygen species (ROS) play an important role in the development of cardiovascular disease [1,2]. Conventional antioxidants cannot impact on mitochondria-derived ROS owing to their limited accumulation within mitochondria, highlighting the need for targeted therapies. Previously, we demonstrated that oral administration of the mitochondria-targeted antioxidant MitoQ10 attenuates the development of hypertension, improves endothelial nitric oxide bioavailability and reduces cardiac hypertrophy in the stroke-prone spontaneously hypertensive rat (SHRSP) [3]. Although the MitoQ10-mediated reduction in blood pressure was modest, it may represent an important novel therapeutic agent for resistant hypertension and end-organ damage if combined with established antihypertensive drugs. Resistant hypertension remains a common problem with an estimated prevalence of 20–30%, despite a three-drug regimen [4]. This suggests existence of blood pressure elevating mechanisms, which are not fully addressed by current antihypertensive therapies. Combining a mitochondria-targeted antioxidant such as MitoQ10 with commonly used antihypertensive agents may provide additive effects against the morbidity and mortality associated with resistant hypertension.
Studies by Dai et al.[5,6] indicate a critical role of mitochondrial ROS in cardiac hypertrophy, fibrosis and failure strengthening the rationale for mitochondria-targeted drugs. Our previous studies suggest a direct effect of MitoQ10 on cardiac hypertrophy, demonstrated by a significant reduction in cardiac mass index (CMI) despite relatively small blood pressure attenuation [3]. Similarly, direct blood pressure independent effects on cardiac hypertrophy have been demonstrated with low-dose angiotensin-converting enzyme (ACE) inhibitors and angiotensin receptor blockers (ARBs) [7–9]. Combining MitoQ10 with a low-dose commonly used antihypertensive agent may achieve additional risk reduction beyond blood pressure lowering by addressing both cytosolic and mitochondrial mechanisms of ROS generation. The aims of this study were to investigate the potential complementary effects of a combined therapeutic strategy on the development of hypertension and cardiac hypertrophy using the mitochondria-targeted antioxidant MitoQ10 and a low-dose ARB, losartan. In parallel, we investigated the contribution of mitochondrial oxidative damage to cardiomyocyte hypertrophy in vitro using H9c2 neonatal cardiomyocytes. These studies suggest new therapeutic interventions for resistant hypertension and related organ damage in humans.
MATERIALS AND METHODS
An expanded Methods section is available in the Online Data Supplement.
In-vivo experimental procedures
An inbred colony of SHRSP has been maintained at the University of Glasgow since 1991. Animals were housed under 12 h light/dark cycles at ambient temperature and were maintained on normal rat chow (Rat and Mouse No. 1 maintenance diet, Special Diet Services). All studies were conducted in accordance with the Animals Scientific Procedures Act 1986. Eight-week-old male SHRSP (8–11 rats per group) were treated with low-dose losartan (2.5 mg/kg per day); MitoQ10 (500 μmol/l); a combination of MitoQ10 and losartan (M + L); or vehicle for 8 weeks. MitoQ10 was administered in drinking water, and losartan potassium (Sigma-Aldrich, Gillingham, Dorset, UK) administered daily and mixed with highly palatable baby food. SBP was measured by tail-cuff plethysmography [10,11] for the first 4 weeks of study. At 12 weeks of age, rats were implanted with radiotelemetry probes (Dataquest IV telemetry system; Data Sciences International, St Paul, Minnesota, USA) for haemodynamic measurement over the final 4 weeks of treatment [3,12]. Metabolic cages were used for the measurement of volume intake, urine output and collection of 24-h urine samples from control and treated rats at 16 weeks of age. At sacrifice, blood samples were taken by cardiac puncture, followed by measurement of heart weight, left ventricle as well as septum weight and kidney weight for the calculation of CMI, left ventricular mass index and renal mass index, respectively (CMI, LVMI, RMI corrected for tibia length). Heart apexes were fixed in 10% formalin for histological fibrosis assessment by picrosirius red staining. Aortae were taken for organ bath pharmacology and liver samples snap-frozen in liquid nitrogen.
Ex-vivo experimental procedures
Organ bath pharmacology
Organ bath pharmacology was used as described previously [3,11] to test contractile responses in aorta from control and drug-treated SHRSP. Vessels (approximately 4 mm in length) were pretreated with potassium chloride (KCl, 100 mmol/l), followed by construction of cumulative concentration–response curves to phenylephrine (10 nmol/l to 10 μmol/l in the absence and presence of NG-nitro-l-arginine methyl ester (L-NAME, 100 μmol/l) to inhibit nitric oxide species (NOS). Consecutive dose–response curves were carried out in the same vessel, measured first in the absence of L-NAME, followed by two washes with Krebs’ buffer and then in the presence of L-NAME. The difference in tension in the absence and presence of L-NAME provides a measure of basal NO bioavailability and was calculated over the full dose–response curve and expressed as area under the curve (AUC). In addition, the rings were preconstricted to the EC50 of phenylephrine and a concentration–response curve for relaxation to carbachol (10 nmol/l to 10 μmol/l) was obtained to measure stimulated NO release for which AUC was calculated. Responses to phenylephrine were standardized against the initial contractile response to KCl.
Histology
Five-micrometre paraffin-embedded sections of heart apex were cut and deparaffinized with two washes in Histoclear (Fisher Scientific, Loughborough, UK) followed by rehydration through an ethanol concentration gradient. Fibrosis was assessed using picrosirius red staining (Sigma-Aldrich) specific for collagen type I and III. Sections were incubated under dark conditions in 0.1% picrosirius red solution, washed, rehydrated and cover-slip mounted. A colour threshold application was used to measure the average intensity of picrosirius red stain in five sections per heart apex from 3–8 rats per treatment group (ImageProPlus 4.1; Media Cybernetics, Marlow, UK). This involved transformation of pixel values to optical density units using the Area of Interest selection tool and Macro from the ImageProPlus software [13].
Biochemical analysis
Urine sodium, potassium, urea and total protein levels were measured by routine biochemical analysis (Biochemistry Department, Gartnavel General Hospital, Glasgow, UK). Urinary and plasma creatinine were assessed for estimated glomerular filtration rate (eGFR) by the QuantiChrom Creatinine Assay Kit [Universal Biologicals (Cambridge) Ltd, Cambridge, UK] and urinary protein concentrations assessed by bicinchoninic acid (BCA) Protein Assay (Pierce, Rockford, Illinois, USA). Lipid peroxidation in liver samples was determined by malondialdehyde (MDA) assay kit (Bioxytech LPO-586 Assay Kit; OxisResearch, Portland, Oregon, USA) according to manufacturer's instructions and normalized to protein concentration.
In-vitro treatment of cardiomyocytes with MitoQ10
In-vitro assays were conducted in the H9c2 immortalized cardiomyocyte cell line derived from rat neonatal cardiomyocytes [14,15]. Cells were pretreated with MitoQ10 or the control compound decyl triphenylphosphonium (dTPP) (10–500 nmol/l) for 18 h prior to angiotensin II (AngII) stimulation (100 nmol/l). After 96 h, cells were fixed with 2% paraformaldehyde, stained overnight with 2% crystal violet and cell size measured using ImageProPlus 4.1 software (Media Cybernetics). For each condition, 180 random cells were measured from 18 fields of view; experiments were repeated in triplicate on three independent occasions. The MitoQ10 analogue IBTP was colocalized to mitochondria using confocal microscopy as previously demonstrated (see supplementary methods) [16].
Cell Titre 96R Non-Radioactive Cell Proliferation Assay (Promega, Southampton, UK) was used according to manufacturer's instructions to test the cytotoxicity of MitoQ10 and dTPP. A spectrometer (Wallac Victor2 plate-reader; Perkin Elmer, Turku, Finland) measured absorbance of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) at 570 nm. Experiments were repeated in quadruplicate on three independent occasions.
Statistical analysis
Results are expressed as mean ± SEM. In-vivo experiments were performed with 8–11 rats per group. Repeated-measures analysis of variance (ANOVA) with Tukey pairwise comparison was used to compare radiotelemetry data. Ex-vivo comparisons were performed by one-way ANOVA with Tukey's multiple comparison test. In-vitro comparisons between groups were performed by one-way ANOVA with Bonferoni's multiple comparison test. Statistical significance was considered with P values less than 0.05.
RESULTS
Average day-time SBP of control SHRSP increased from 129.0 ± 4.9 mmHg to a maximum of 206.6 ± 9.0 mmHg over the 8-week study (Fig. 1a). MitoQ10 or low-dose losartan alone resulted in significant attenuation of SBP (MitoQ10; 185.5 ± 6.0 mmHg, P = 0.032, Los; 172.9 ± 1.7 mmHg, P = 0.004). When both drugs were used in combination, there was a highly significant reduction in SBP compared with control (167.1 ± 2.9 mmHg, P < 0.001), which was significantly lower than either treatment alone. DBP was significantly reduced by either drug alone (MitoQ10; 126.3 ± 3.7 mmHg, P = 0.032, Los; 118.2 ± 0.77 mmHg, P = 0.003) or in combination (116.3 ± 2.1 mmHg, P = 0.005) compared with control (142.6 ± 6.5 mmHg, Fig. 1b) but suggest no additive benefit of combined therapy. Pulse pressure was significantly reduced by combination treatment (50.2 ± 2.1 mmHg, P = 0.001) compared with control (63.7 ± 2.7 mmHg) and indicates additional benefit compared with either drug alone (Fig. 1c). Heart rate was significantly lower in MitoQ10-treated SHRSP (329.3 ± 3.2 bpm) than in control (351.6 ± 4.3 bpm, P = 0.012, Fig. 1d). Night-time heart rate, when the rats are most active, was significantly reduced in MitoQ10 alone and combination treatment groups (Fig. 1e) compared with control or losartan only. Tibia length, fluid excretion/intake ratio and motor activity were not significantly different between groups (Supplementary figure i).
FIGURE 1.
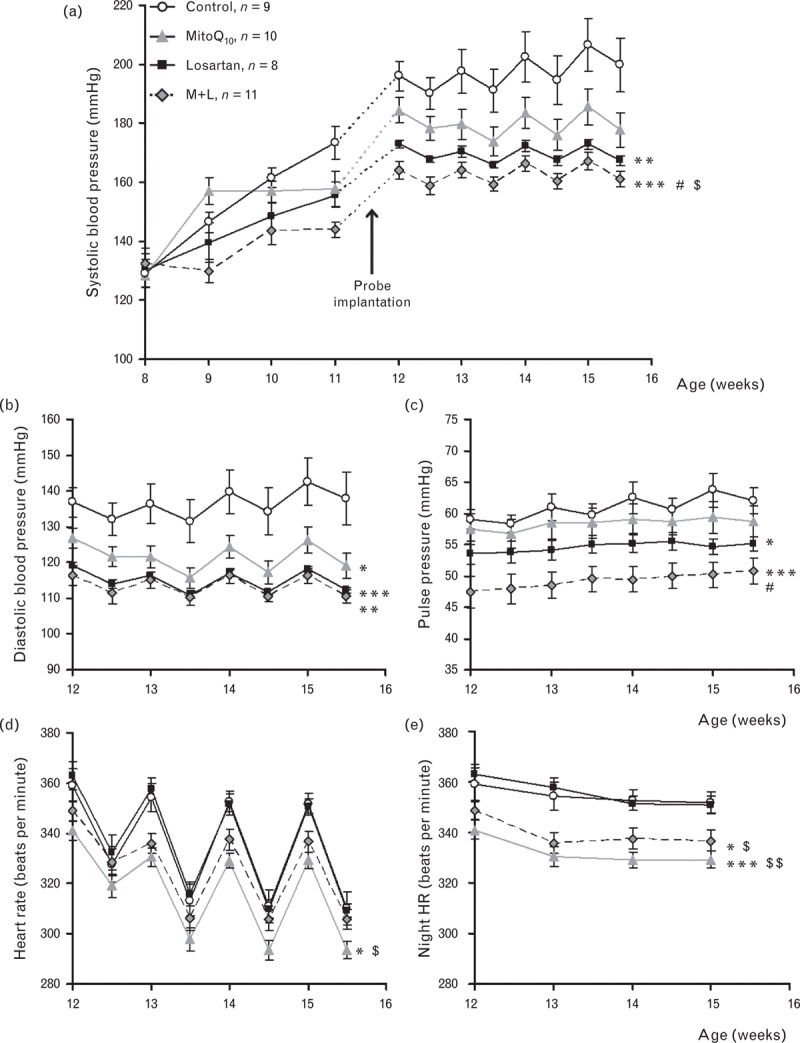
Effect of MitoQ10 treatment alone and in combination with low-dose losartan on the haemodynamic profile of SHRSP. (a) SBP measured by tail-cuff plethysmography and radiotelemetry was reduced by MitoQ10 and losartan alone, and demonstrated greatest reduction with combined therapy (∗∗P = 0.004, ∗∗∗P < 0.001 vs. control; #P = 0.006 vs. MitoQ10 and $P = 0.05 vs. losartan). (b) DBP was significantly reduced by individual and combination treatment (∗P = 0.032, ∗∗P = 0.005, ∗∗∗P = 0.003 vs. control). (c) Pulse pressure shows small but significant reduction in losartan-only treated rats, and more pronounced reduction with combination therapy (∗P = 0.025, ∗∗∗P = 0.001 vs. control; #P = 0.01 vs. MitoQ10,‡P = 0.057 vs. losartan only). (d) Heart rate measured by radiotelemetry was significantly reduced by MitoQ10 (∗P = 0.012 vs. control, $P = 0.001 vs. losartan). (e) Night-time heart rate was significantly lowered by MitoQ10 alone and by combination treatment compared with control and losartan only (∗P = 0.02 ∗∗∗P = 0.001 vs. control, $P = 0.006 $$P < 0.001 vs. losartan only; n = 8–11).
Basal nitric oxide bioavailability in aorta from control and drug-treated SHRSP is illustrated in Fig. 2a and expressed as AUC in Fig. 2b. Basal nitric oxide bioavailability was significantly improved in losartan-treated SHRSP compared with control (∗P < 0.05) and in combination treatment compared with control (∗∗P < 0.05) and MitoQ10 only (#P < 0.05). Relaxation to carbachol was not significantly different in aortic rings from control and drug-treated SHRSP (Fig. 2c). Lipid peroxidation, measured by MDA assay, was used to assess oxidative stress in liver samples from control and drug-treated SHRSP (Fig. 2d). Treatment with losartan only resulted in a significant decrease in liver MDA levels (P < 0.05). The other treatment groups demonstrated similar trends but were not significantly different to untreated controls. No significant differences were observed for basal superoxide production in aortic rings or plasma lipid peroxidation (Supplementary Figure ii).
FIGURE 2.
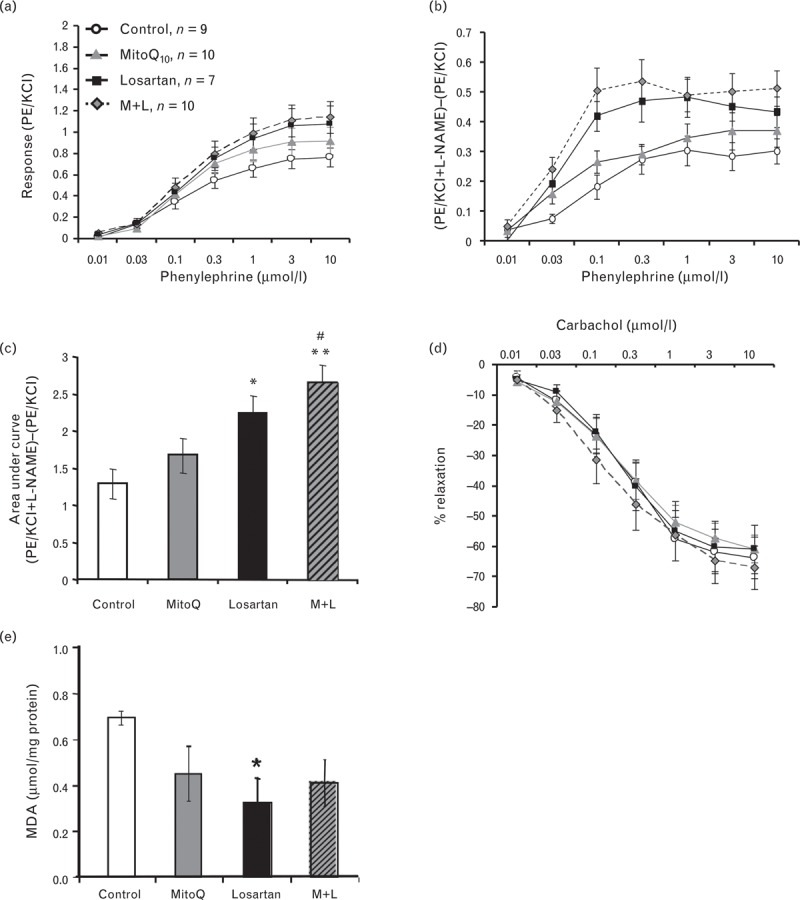
Effect of MitoQ10 alone and in combination with low-dose losartan on vascular function and oxidative stress. (a) Concentration response curves to PE stimulation. (b) Nitric oxide bioavailability measured as the difference in contraction to PE stimulation in the presence and absence of the NOS inhibitor L-NAME. (c) AUC calculated for basal NO bioavailability curves. Basal NO bioavailability was significantly increased in vessels from combination and losartan-treated rats (∗P < 0.05 and ∗∗P < 0.01 vs. control. #P < 0.05 vs. MitoQ10; n = 7–10). (d) Relaxation to carbachol was not significantly different in vessels from control and treated rats. (e) Liver MDA was significantly reduced in losartan-treated rats (∗P < 0.05, n = 5) with trends towards reduction in the MitoQ10 and combination groups.
Kidney and urine characteristics for MitoQ10, losartan and combination-treated SHRSP were assessed (Table 1). There were no significant differences in kidney mass index, eGFR or urinary levels of Na+, K+ or urea between control and treatment groups. However, total urinary protein excretion was significantly reduced in MitoQ10-treated rats (∗P < 0.05) with a similar trend in the combination therapy group.
TABLE 1.
Effect of MitoQ10 alone and in combination with low-dose losartan on renal parameters
| Control | MitoQ10 | Losartan | M + L | |
| Kidney:tibia ratio (mg/mm) | 27.1 ± 1.06 | 28.1 ± 0.90 | 27.6 ± 1.04 | 28.1 ± 0.91 |
| eGFR (ml/min per g kidney weight) | 0.25 ± 0.03 | 0.17 ± 0.02 | 0.22 ± 0.02 | 0.20 ± 0.01 |
| Urinary protein (mg/ml per 24 h) | 120.7 ± 15.4 | 75.8 ± 10.5* | 110.5 ± 8.0 | 83.6 ± 7.7 |
| Urinary Na+ (mmol/24 h) | 0.92 ± 0.21 | 0.84 ± 0.35 | 0.85 ± 0.08 | 1.07 ± 0.20 |
| Urinary K+ (mmol/24 h) | 2.06 ± 0.28 | 2.00 ± 0.31 | 2.14 ± 0.20 | 2.18 ± 0.19 |
| Urea (mmol/24 h) | 6.99 ± 0.54 | 4.92 ± 1.36 | 5.74 ± 1.26 | 6.02 ± 0.41 |
Values are means ± SEM; n = 8–11 rats per group. eGFR, estimated glomerular filtration rate.
*P < 0.05 compared with untreated control.
Combination treatment significantly reduced cardiac mass index (CMI) (M + L 27.3 ± 0.77 mg/mm, control 30.9 ± 0.98 mg/mm, P < 0.05) and LVMI (M + L 20.1 ± 0.61 mg/mm, control 22.8 ± 0.74 mg/mm; P < 0.05) compared with untreated controls (Fig. 3a, b). Cardiac fibrosis was assessed by picrosirius red staining (Fig. 3c) and collagen I and III immunohistochemistry (Supplementary Figure iii). Elevated levels of fibrillar collagen in perivascular and interstitial regions (Fig. 3c) in control hearts were significantly attenuated by combination treatment (P < 0.01) (Fig. 3d).
FIGURE 3.
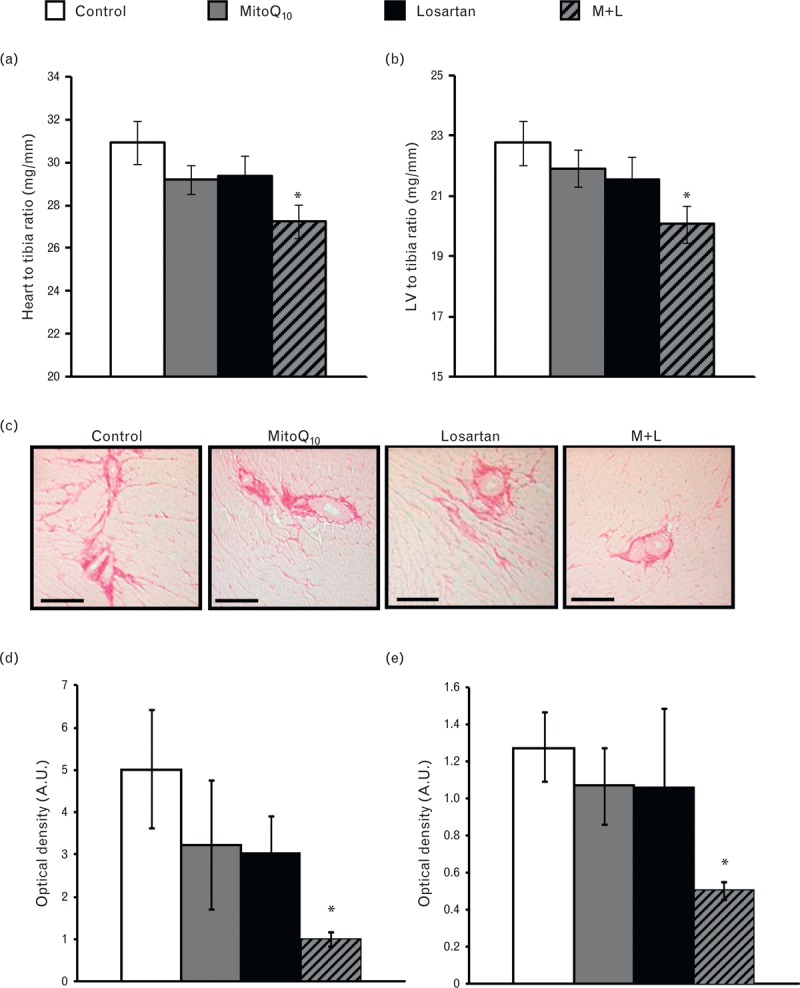
Effect of MitoQ10 alone and in combination with low-dose losartan on cardiac and left ventricular hypertrophy and fibrosis. (a) CMI and (b) LVMI were significantly reduced in combination-treated SHRSP compared with control (n = 8–11, ∗P < 0.05 vs. control). Single-drug therapy had no significant effect on CMI or LVMI. (c) Representative images of perivascular and interstitial cardiac fibrosis (magnification ×10, scale bar = 100 μm) analysed with picrosirius red staining and quantified using ImageProPlus. (d) Perivascular and (e) interstitial fibrosis was significantly reduced in combination-treated SHRSP compared with controls (∗P < 0.05 vs. control, n = 3–8).
Exposure of H9c2 cardiomyocytes to AngII increased cell size from 159.2 ± 3.3 to 200.1 ± 3.6 μm (P < 0.001) (Fig. 4a), and this was prevented by MitoQ10 but not by the control compound dTPP (Fig. 4b). MitoQ10 had no effect on cell size in unstimulated H9c2 cells and cell viability was not affected by MitoQ10 or dTPP (Supplementary Figure iv). Immunofluorescence to detect the MitoQ10 analogue IBTP in cardiomyocytes demonstrated uptake and colocalization to mitochondria during hypertrophy (Fig. 4c).
FIGURE 4.
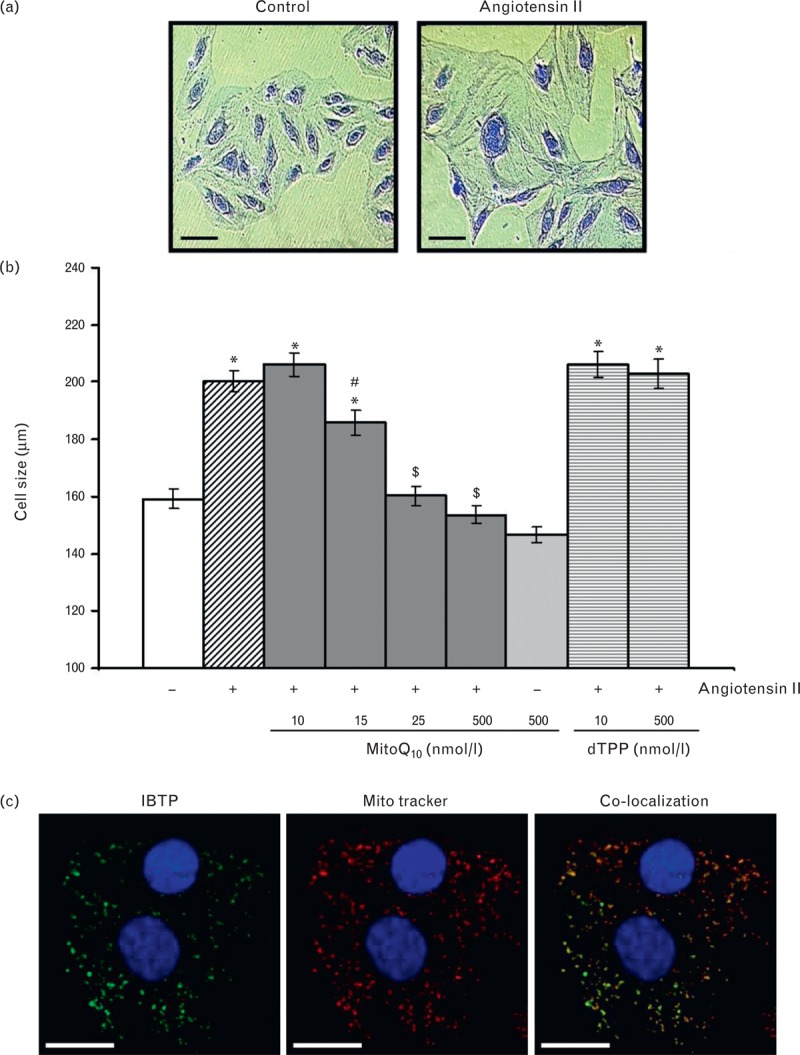
Effect of MitoQ10 on AngII-induced cardiomyocyte hypertrophy. (a) Representative images of control and AngII-stimulated H9c2 cardiomyocytes after 96 h incubation and crystal violet staining (magnification ×10, scale bar = 100 μm). (b) MitoQ10 prevented development of AngII-stimulated hypertrophy in a dose-dependent manner (∗P < 0.001 vs. control, #P < 0.05 and $P < 0.001 vs. 10 nmol/l MitoQ10), whereas dTPP had no effect. (c) The MitoQ10 analogue, IBTP, was detected in cells after 96 h, co-localized with MitoTracker Red. Nuclei were counterstained with DAPI (magnification ×63, scale bar = 50 μm).
DISCUSSION
This study demonstrates that administration of MitoQ10 provides complementary therapeutic benefit to an established antihypertensive agent. Our data suggest that MitoQ10 targets elements of the hypertensive and hypertrophic processes involving mitochondrial oxidative damage that are not fully addressed by current antihypertensive drugs. Furthermore, our in-vitro studies demonstrate that MitoQ10 has a direct antihypertrophic action in cardiomyocytes indicating that its in-vivo action may be partly independent of blood pressure lowering.
We have previously demonstrated that the mitochondria-specific antioxidant MitoQ10 has important antihypertensive action in young SHRSP [3] emphasizing a key role for mitochondrial-oxidative stress in the development of hypertension. Other in-vivo studies [17–21] have shown that MitoQ10 is effective against mitochondrial oxidative damage in rodent models of sepsis, cardiac reperfusion, metabolic syndrome and diabetic nephrophathy. Together, these findings highlight the importance of targeted antioxidant therapy in a range of diseases affected by mitochondrial oxidative damage.
Several sources of ROS may contribute to the development of cardiovascular disease, including NADPH oxidase, xanthine oxidase, uncoupled nitric oxide synthase (NOS) and the mitochondrial electron transport chain [1,22–24]. There is a complex interplay between these different ROS and the precise contributions to the underlying disease mechanisms remain obscure. There is evidence for ROS-induced ROS production, with mitochondria being both stimulated by NADPH oxidases and also acting as the initiating stimulus for further ROS generation [2,5,24–26], creating a potential feed-forward cycle of ROS production. In the present study, MitoQ10 was tested in combination with a low-dose commonly prescribed ARB. This combined therapy demonstrated significant additive haemodynamic benefit, antihypertrophic and antifibrotic action when compared with control or single therapies. Only DBP failed to show additive reduction during combined versus single therapy, suggesting that MitoQ10 and losartan share a common antihypertensive mechanism for diastolic pressure. The lack of an additive effect on DBP is potentially beneficial, as evidence suggests that therapeutically induced diastolic hypotension can be a risk factor for increased coronary events [27–29]. In line with the nonadditive effects on DBP, we also observe similar significant equivalent improvements in aortic nitric oxide bioavailability in the combination and losartan-only group. The significant reduction in heart rate by MitoQ10 treatment, either alone or in combination with losartan, during the active (night-time) period, was not observed with single losartan therapy. MitoQ10 may alter heart rate by reducing oxidative stress within the rostral ventolateral medulla (RVLM), as mitochondrial-derived ROS has been shown to mediate sympathoexcitation within this brain stem cardiovascular control centre [30]. We also demonstrate reduced urinary protein levels in MitoQ10 and combination-treated rats indicating that mitochondria-specific antioxidants may also play a role in reducing end-organ damage in the kidney.
Increased ROS production stimulates myocardial fibrosis and is associated with reduced cardiac function [31]. This may be due to the accumulation of perivascular collagen compressing coronary arterioles and the accumulation of fibrillar collagen in the myocardial interstitium increasing wall stiffness and impairing cardiac systolic function [32]. Significant cardiac fibrosis has previously been reported in male SHRSP by 16 weeks of age [33,34], and in the current study, we demonstrate prevention of cardiac fibrosis by combination treatment. The design of the current study does not allow direct mechanistic assessment of the antifibrotic action of MitoQ10. This would require investigation in alternative models of cardiac fibrosis such as the transverse aortic constriction (TAC) model or the renal hypertensive rat (RHR), permitting pre and postsurgery assessment of MitoQ10 action. However, we suggest that the significant reduction in collagen deposition by combination therapy is likely to be due to decreased oxidative damage. Losartan lowers blood pressure and decreases left ventricular hypertrophy by blocking AT1 receptors, and reducing AngII-dependent activation of NADPH oxidases, [35,36]. This, in turn, can reduce mitochondrial-derived ROS production [24,26] in addition to the direct mitochondrial antioxidant potential of MitoQ10. Our data suggest that combining a mitochondria-targeted antioxidant with losartan may provide superior antihypertensive, antihypertrophic and antifibrotic action by interrupting the vicious cycle of ROS production and oxidative damage.
Although this study was unable to demonstrate direct MitoQ10-induced improvement in ROS, this was not entirely unexpected considering that O2−, the primary ROS produced by the mitochondria, is not readily diffusible across mitochondrial membranes and is largely dismutated to H2O2[37]. Moreover, it is well recognized that measurement of reactive molecules in biological environments is inherently challenging due to their short lifespan and the limited selectivity of detection systems [38–40].
Cardiac hypertrophy is an adaptive response to increased blood pressure, and as a result, practically all antihypertensive agents reduce LVMI, emphasizing the importance of haemodynamic load in the pathogenic process. However, convincing evidence from studies with low-dose ACE inhibitors and ARBs also demonstrate blood pressure independent attenuation of left ventricular hypertrophy [7–9]. The beneficial action of MitoQ10 on cardiac hypertrophy despite modest blood pressure reduction suggests a direct role for mitochondrial oxidative stress in the hypertrophic process rather than simply a secondary effect of blood pressure reduction [3]. In support of this, H9c2 cardiomyocytes, a well established model of hypertrophy [14,15,41], showed dose-dependent inhibition of hypertrophy by MitoQ10. These data suggest a potentially important therapeutic role for MitoQ10 in heart disease independent of its haemodynamic effects.
The current preclinical study has investigated the beneficial action of MitoQ10 during the developmental of hypertension and left ventricular hypertrophy. The next stage will be to determine the effects of mitochondria-specific antioxidant efficacy in the more clinically relevant setting of established hypertension. These future studies will provide important information for potential clinical trials of human resistant hypertension, as MitoQ10 was proven well tolerated in two phase II clinical trials [42,43].
Clinical perspectives
Oxidative stress and mitochondrial dysfunction are increasingly implicated in cardiovascular disease and yet this is a neglected aspect in current treatment strategies. We demonstrate that MitoQ10 is a potential complementary therapeutic intervention to current antihypertensive agents, particularly in the treatment of haemodynamic changes and end-organ damage associated with resistant hypertension.
ACKNOWLEDGEMENTS
British Heart Foundation Chair (CH98001) and Programme grant funding (RG/07/005), EU Sixth Framework Programme Integrated Project (LSHG_CT 2005-019015 EURATools), EU Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement (HEALTH-F4–2010–241504 EURATRANS) awarded to A.F.D. BHF PhD studentship (FS/07/029/23022) and project grant (PG/11/82/29136) awarded to D.G. and A.F.D.
Conflicts of interest
M.P.M. is on the scientific advisory board of, and holds shares in, Antipodean Pharmaceuticals Inc, which is commercializing MitoQ10.
Supplementary Material
Reviewers’ Summary Evaluations
Referee 1
The manuscript explores the efficacy of MitoQ10 as a combination therapy with an angiotensin receptor blocker in reducing cardiac fibrosis with hypertension. Although, there are several antihypertensives that can be used clinically to prolong survival in hypertension induced heart failure, there is still a need to find better therapeutic molecules. One of the possible targets is reducing the reactive oxygen species generated by mitochondria thus limiting cardiac fibrosis and the consequent heart failure. Among several antioxidants tested so far, none has been shown to be effective. Something like MitoQ10 may provide a benefit as combination therapy, provided the animal experiments are confirmed in humans.
Referee 2
There is convincing evidence that oxidative stress is involved in the pathogenesis of hypertension but data from clinical trials with antioxidants are inconsistent. Targeting the enzymes responsible for reactive oxygen species (ROS) generation could be an effective strategy. Mitochondria is an important source of ROS at cardiovascular level. This study shows that the combination of the mitochondria-targeted antioxidant MitoQ10 and losartan provides additive effects, attenuating hypertension and left ventricular hypertrophy of stroke-prone spontaneously hypertensive rats. Thus, mitochondria-targeted antioxidants would provide complementary therapeutic benefit to the inhibitors of renin-angiotensin-system in the treatment of hypertension. However, the clinically relevance of the observed effects in this work are unknown.
Footnotes
Correspondence to Dr Delyth Graham, Institute of Cardiovascular and Medical Sciences, BHF Glasgow Cardiovascular Research Centre, University of Glasgow, 126 University Place, Glasgow G12 8TA, UK. Tel: +44 141 330 2524; fax: +44 330 6997; e-mail: Delyth.Graham@glasgow.ac.uk
Abbreviations: A.U., arbitrary units; ACE, angiotensin-converting enzyme; AngII, angiotensin II; ARB, angiotensin receptor blocker; AUC, area under curve; CMI, cardiac mass index; De, external diameter; Di, internal diameter; dTPP, decyl triphenylphosphonium; eGFR, estimated glomerular filtration rate; KCl, potassium chloride; L-NAME, NG-nitro-l-arginine methyl ester; LVMI, left ventricular mass index; M+L, MitoQ10 and losartan; MRA, mesenteric resistance artery; NOS, nitric oxide synthase; ROS, reactive oxygen species; SHRSP, stroke prone spontaneously hypertensive rat
REFERENCES
- 1.Gutierrez J, Ballinger SW, Darley-Usmar VM, Landar A. Free radicals, mitochondria, and oxidised lipids. The emerging role in signal transduction in vascular cells. Circ Res 2006; 99:924–932 [DOI] [PubMed] [Google Scholar]
- 2.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, et al. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 2010; 107:106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graham D, Huynh NN, Hamilton CA, Beattie E, Smith RA, Cochemé HM, et al. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension 2009; 54:322–328 [DOI] [PubMed] [Google Scholar]
- 4.Calhoun DA, Jones D, Textor S, Goff DC, Murphy TP, Toto RD, et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2008; 51:1403–1419 [DOI] [PubMed] [Google Scholar]
- 5.Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintrón M, Chen T, et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res 2011; 108:837–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dai DF, Chen T, Szeto H, Nieves-Cintron M, Kutyavin V, Santana LF, Rabinovitch PS. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol 2011; 58:73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roman MJ, Alderman MH, Pickering TG, Pini R, Keating JO, Sealey JE, Devereux RB. Differential effects of angiotensin converting enzyme inhibition and diuretic therapy on reductions in ambulatory blood pressure, left ventricular mass, and vascular hypertrophy. Am J Hypertens 1998; 11:387–396 [DOI] [PubMed] [Google Scholar]
- 8.London GM, Pannier B, Guerin AP, Marchais SJ, Safar ME, Cuche JL. Cardiac hypertrophy, aortic compliance, peripheral resistance, and wave reflection in end-stage renal disease. Comparative effects of ACE inhibition and calcium channel blockade. Circulation 1994; 90:2786–2796 [DOI] [PubMed] [Google Scholar]
- 9.Yasunari K, Maeda K, Watanabe T, Nakamura M, Yoshikawa J, Asada A. Comparative effects of valsartan versus amlodipine on left ventricular mass and reactive oxygen species formation by monocytes in hypertensive patients with left ventricular hypertrophy. J Am Coll Cardiol 2004; 43:2116–2123 [DOI] [PubMed] [Google Scholar]
- 10.Evans AL, Brown W, Kenyon CJ, Maxted KJ, Smith DC. Improved system for measuring systolic blood pressure in the conscious rat. Med Biol Eng Compt 1994; 32:101–102 [DOI] [PubMed] [Google Scholar]
- 11.Graham D, Hamilton C, Beattie E, Spiers A, Dominiczak AF. Comparison of the effects of omapatrilat and irbesartan/hydrochlorothiazide on endothelial function and cardiac hypertrophy in the stroke-prone spontaneously hypertensive rat: sex differences. J Hypertens 2004; 22:329–337 [DOI] [PubMed] [Google Scholar]
- 12.Jeffs B, Negrin CD, Graham D, Clark JS, Anderson NH, Gauguier D, Dominiczak AF. Applicability of a ‘speed’ congenic strategy to dissect blood pressure quantitative trait loci on rat chromosome 2. Hypertension 2000; 35:179–187 [DOI] [PubMed] [Google Scholar]
- 13.Yang G, Li L, Volk A, Emmell E, Petley T, Giles-Komar J, et al. Therapeutic dosing with antiinterleukin-13 monoclonal antibody inhibits asthma progression in mice. J Pharmacol Exp Ther 2005; 313:8–15 [DOI] [PubMed] [Google Scholar]
- 14.Qin F, Patel R, Yan C, Liu W. NADPH oxidase is involved in angiotensin II-induced apoptosis in H9C2 cardiac muscle cells: effects of apocynin. Free Radic Biol Med 2006; 40:236–246 [DOI] [PubMed] [Google Scholar]
- 15.Flores-Muñoz M, Smith NJ, Haggerty C, Milligan G, Nicklin SA. Angiotensin1-9 antagonises pro-hypertrophic signalling in cardiomyocytes via the angiotensin type 2 receptor. J Physiol 2011; 589:939–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ross MF, Prime TA, Abakumova I, James AM, Porteous CM, Smith RA, Murphy MP. Rapid and extensive uptake and activation of hydrophobic triphenylphosphonium cations within cells. Biochem J 2008; 411:633–645 [DOI] [PubMed] [Google Scholar]
- 17.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RAJ, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J 2005; 19:1088–1095 [DOI] [PubMed] [Google Scholar]
- 18.Lowes DA, Thottakam BM, Webster NR, Murphy MP, Galley HF. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide peptidoglycan model of sepsis. Free Radic Biol Med 2008; 45:1559–1565 [DOI] [PubMed] [Google Scholar]
- 19.Chandran K, Aggarwal D, Migrino RQ, Joseph J, McAllister D, Konorev EA, et al. Doxorubicin inactivates myocardial cytochrome c oxidase in rats: cardioprotection by Mito-Q. Biophys J 2009; 96:1388–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mercer JR, Yu E, Figg N, Cheng KK, Prime TA, Griffin JL, et al. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM + /−/ApoE−/− mice. Free Radic Biol Med 2012; 52:841–849 [DOI] [PubMed] [Google Scholar]
- 21.Chacko BK, Reily C, Srivastava A, Johnson MS, Ye Y, Ulasova E, et al. Prevention of diabetic nephropathy in Ins2(+/)− (AkitaJ) mice by the mitochondria-targeted therapy MitoQ. Biochem J 2010; 432:9–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 2003; 111:1201–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 2000; 87:840–844 [DOI] [PubMed] [Google Scholar]
- 24.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res 2008; 102:488–496 [DOI] [PubMed] [Google Scholar]
- 25.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (Ros-induced) Ros release. A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 2000; 192:1001–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang GX, Lu XM, Kimura S, Nishiyama A. Role of mitochondria in angiotensin II-induced reactive oxygen species and mitogen-activated protein kinase activation. Cardiovasc Res 2007; 76:204–212 [DOI] [PubMed] [Google Scholar]
- 27.D’Agostino RB, Belanger AJ, Kannel WB, Cruickshank JM. Relation of low diastolic blood pressure to coronary heart disease death in presence of myocardial infarction: the Framingham Study. BMJ 1991; 303:385–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Messerli FH, Mancia G, Conti CR, Hewkin AC, Kupfer S, Champion A, et al. Dogma disputed: can aggressively lowering blood pressure in hypertensive patients with coronary artery disease be dangerous? Ann Intern Med 2006; 144:884–893 [DOI] [PubMed] [Google Scholar]
- 29.Osher E, Stern N. Diastolic pressure in type 2 diabetes: can target systolic pressure be reached without ‘diastolic hypotension’? Diabetes Care 2008; 31 Suppl 2:S249–S254 [DOI] [PubMed] [Google Scholar]
- 30.Nozoe M, Hirooka Y, Koga Y, Araki S, Konno S, Kishi T, et al. Mitochondria-derived reactive oxygen species mediate sympathoexcitation inducedby angiotensin II in the rostral ventrolateral medulla. J Hypertens 2008; 26:2176–2184 [DOI] [PubMed] [Google Scholar]
- 31.Zhang GX, Kimura S, Nishigama A, Shokoji T, Rahman M, Yao L, et al. Cardiac oxidative stress in acute and chronic isoproterenol-induced rats. Cardiovasc Res 2005; 65:230–238 [DOI] [PubMed] [Google Scholar]
- 32.Jalil JE, Doering CW, Janicki JS, Pick R, Shroff SG, Weber KT. Fibrillar collagen and myocardial stiffness in the intact hypertrophied rat left ventricle. Circ Res 1989; 64:1041–1050 [DOI] [PubMed] [Google Scholar]
- 33.Flores-Munoz M, Work LM, Douglas K, Denby L, Dominiczak AF, Graham D, Nicklin SA. Angiotensin-(1-9) attenuates cardiac fibrosis in the stroke-prone spontaneously hypertensive rat via the angiotensin type 2 receptor. Hypertension 2012; 59:300–307 [DOI] [PubMed] [Google Scholar]
- 34.Nakamura T, Yamamoto E, Kataoka K, Yamashita T, Tokutomi Y, Dong YF, et al. Beneficial effects of pioglitazone on hypertensive cardiovascular injury are enhanced by combination with candesartan. Hypertension 2008; 51:296–301 [DOI] [PubMed] [Google Scholar]
- 35.Rajagopalan S, Kurz S, Münzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest 1996; 97:1916–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakagami H, Takemoto M, Liao JK. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J Mol Cell Cardiol 2003; 35:851–859 [DOI] [PubMed] [Google Scholar]
- 37.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 2009; 417:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Halliwell B, Whiteman M. Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br J Pharmacol 2004; 142:231–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winterbourn CC. Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol 2008; 4:278–287 [DOI] [PubMed] [Google Scholar]
- 40.Murphy MP, Holmgren A, Larsson NG, Halliwell B, Chang CJ, Kalyanaraman B, et al. Unraveling the biological roles of reactive oxygenspecies. Cell Metab 2011; 13:361–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Watkins SJ, Borthwick GM, Arthur HM. The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. In Vitro Cell Dev Biol Anim 2011; 47:125–131 [DOI] [PubMed] [Google Scholar]
- 42.Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, et al. The mitochondria-targeted antioxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int 2010; 30:1019–1026 [DOI] [PubMed] [Google Scholar]
- 43.Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, et al. A double-blind placebo-controlled study to assess the mitochondria-targeted antioxidant as a disease-modifying therapy in Parkinson's disease. Mov Disord 2010; 25:1670–1674 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.