

Abstract
Rapid postnatal growth and differentiation of fetal arterial smooth muscle is coordinated by a cacophony of growth factors, one of the most important of which is vascular endothelial growth factor (VEGF). In fetal arterial smooth muscle, VEGF influences both the expression and intracellular organization of contractile proteins and helps mediate hypoxic vascular remodeling. Numerous factors influence the expression of VEGF and its receptors, including chronic hypoxia, maternal food restriction, glucocorticoids, and miRNA. Continued study of the coupling between VEGF and transcription factors such as myocardin that govern smooth muscle differentiation, offers great promise for better clinical management of neonates at risk for cardiovascular dysregulation.
Keywords: contractile proteins, fetal arteries, hypoxia, smooth muscle phenotype, vascular endothelial growth factor
Introduction
One of the most impressive accomplishments of modern medicine is the ability to sustain and manage extremely small infants born very prematurely. An increasing number of these small patients are now populating Neonatal Intensive Care Units (NICU) around the world, and many survive and even prosper into early childhood and beyond. However, a major early challenge faced by many of these premature infants is cardiovascular instability, altered vascular reactivity to most drugs, and an increased tendency for brain arteries to rupture, leading to intracranial hemorrhage and hypoxic ischemic encephalopathy.1 How the immature vascular system responds to environmental stresses and injury, even if there is no immediate pathology, may also have a major influence on cardiovascular health and vitality in the adult years, as suggested by the Barker Hypothesis.2 Indeed, the very factors that predispose an infant to premature birth may also enhance the chances for coronary artery disease and cerebral stroke in later life.3 Thus, the study of early vascular development also offers unique access to the growth-related and proliferative processes that govern not only blood vessel maturation but also contribute to adult vascular pathologies such as atherosclerosis.4 And of course, improved understanding of vascular function and regulation in the neonate can only facilitate the efforts of those working to improve strategies for clinical management of NICU infants.
CONTRACTILE SPECIALIZATION IN THE FETAL VASCULATURE
A key feature of the fetal vasculature is its unique structural and functional differences from the adult vasculature. When contractile capacity is determined by exposure to potassium, which is a receptor-independent method of contraction, contractile force per unit length of artery typically increases with postnatal age in direct proportion to artery diameter and wall thickness. Correspondingly, when the potassium-induced increases in contractile force are normalized relative to the cross-sectional area of the artery wall, postnatal age has relatively little effect on receptor-independent contractile capacity.5 In contrast, postnatal age has major effects on the contractile responses to many receptor-dependent agonists. For example, the contractile potency of norepinephrine, when normalized relative to tissue contractile capacity, decrease markedly with postnatal age in an artery-specific manner (Fig. 1). For serotonin, the effect is different; postnatal maturation decreases efficacy in the common carotid but increases it slightly in the middle cerebral artery. Thus, the effects of postnatal maturation on vascular reactivity are highly dependent upon both the contractile agonist and the specific vascular bed. This heterogeneity suggests that receptor coupling to the contractile proteins is under developmental control and is finely specialized to meet the functional requirements of each vascular bed in the fetus, as in the adult. In turn, the ability of calcium to stimulate myosin light chain phosphorylation (thick filament reactivity) and the ability of phosphorylated myosin light chain to interact with actin (thin filament reactivity) is also under tight developmental control.8 The mechanisms mediating this control, however, remain uncertain but most probably involve the growth factors and trophic influences that govern smooth muscle differentiation.
FIGURE 1.

Fetal contractility is highly specialized but fully functional. The full-term fetus is fully capable of regulating arterial pressure, but the vascular component of this regulation is very different than in adults. For some contractile agonists, contractility reactivity is upregulated in the fetus compared with the adult. For norepinephrine (upper panels), potency is markedly upregulated in the fetal common carotid but is only slightly upregulated in the fetal middle cerebral. For serotonin (lower panels), efficacy is markedly upregulated in the fetal common carotid but is only slightly depressed in the fetal middle cerebral. These differences emphasize that vasoreactivity is highly specialized in an artery-specific manner in the fetal systemic circulation. All contractile responses were normalized relative to the maximum contractile response to potassium depolarization. Error bars indicate SEM for N≥6.6,7
Not surprisingly, the rapid rates of growth and differentiation typical of immature arteries also have important consequences for the specialized functions of the vascular endothelium. In addition to age-dependent changes in the endothelium’s role in angiogenesis and trophic support of vessel development, the endothelium’s critically important barrier functions improve dramatically with postnatal age as indicated by increased density of functional tight junctions and concomitant increases in expression of tight junction proteins such as occludin in certain vascular beds.9 Similarly, endothelial regulation of hemostasis is also age-dependent, such that endothelial content of pro-coagulant factors are generally greater in immature than mature arteries,10 which suggests that the thrombotic risk of many neonatal procedures, such a catheterization, may be greater than in adults. Conversely, the ability of the endothelium to promote vasodilatation may be attenuated in immature arteries11 because of a reduced abundance and enzyme-specific activity of endothelial nitric oxide synthase (Fig. 2). Equally important, the ability of shear stress produced by fluid flow across endothelial cells to stimulate nitric oxide (NO) production and release appears to be downregulated in fetal arteries compared with adult arteries.12 Although the reduced ability of immature arteries to release NO may be counterbalanced by an increased ability to produce and release vasodilator prostanoids in some,13 but not all 11 species, it is generally true that immature arteries have a greater abundance of soluble guanylate cyclase14 and thus can exhibit increased reactivity to NO15 and thereby offset the reduced NO release, characteristic of immature vascular endothelium. Caution must be exercised, however, when drawing generalizations about the effects of postnatal maturation on endothelial function, given that it is notoriously heterogeneous among different vascular beds and species.16 Correspondingly, postnatal maturation can influence this heterogeneity, as indicated by age-dependent changes in the ability of some agonists to promote endothelium-dependent vasodilatation.11 However, the growth factors and environmental influences that control endothelial function remain poorly understood, but their study remains an active and promising area of current research with numerous clinical implications.
FIGURE 2.
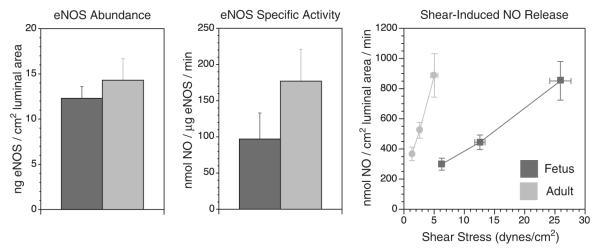
Endothelial vasodilator function is depressed in fetal arteries. Postnatal maturation is associated with increase in both the abundance and specific activity of endothelial nitric oxide synthase (eNOS). In addition, the shear stress produced by blood flow at the endothelial surface is much more potent at stimulating NO release in adult than in fetal arteries. Shown are results obtained from ovine carotid arteries and vertical error bars indicate SE for N≥6 in all cases. eNOS indicates endothelial nitric oxide synthase; NO, nitric oxide.11,12
NUMEROUS FACTORS GOVERN VASCULAR SMOOTH MUSCLE GROWTH AND DIFFERENTIATION
A broad variety of growth factors work in concert to regulate the growth and differentiation of both the smooth muscle and endothelium (Fig. 3). Many strategies have been adopted to categorize these factors into families, some of which are based on chemical characteristics (peptide and nonpeptide, heparin binding, etc.), some of which are categorized by function [fibroblast growth factor, vascular endothelial growth factor (VEGF), etc.], and others which are grouped as products of metabolism (ADP, prostaglandins, small lipids, etc.). This diversity emphasizes that both the local tissue environment and blood borne factors integrate their influences to regulate overall smooth muscle and endothelial growth, which helps explain the great diversity among cell type and vascular characteristics in different vascular beds. Equally important, this diversity is highly dynamic and changes almost continuously in response to both physiologic and to pathophysiologic perturbations.
FIGURE 3.

Numerous growth factors influence vascular smooth muscle growth and differentiation. At birth, the smooth muscle and endothelial cells that make up neonatal arteries are both shorter and narrower than their adult counterparts (left panels). Postnatal maturation can double the size and width of these cells, and the extent of dimensional change is artery-specific. Governing these major changes in cell dimensions are a broad variety of both stimulatory and inhibitory growth factors that selectively target the endothelium or the smooth muscle. One of the most important of these is VEGF, which can influence both endothelial and smooth muscle growth. Please note that in this figure, the fetal cells (left panels) are indicated by open symbols and the adult cells are indicated by closed symbols. Artery types are abbreviated as: Com, common carotid; Bas, basilar artery; PCA, posterior communicating artery; and MCA, middle cerebral artery. Error bars indicate standards for N≥5 for all measures. For morphometric details, please refer to Elliott and colleagues.17,18 For details about growth factors, please refer Hoeben and colleagues.19,20 EGF indicates epidermal growth factor; eNOS indicates endothelial nitric oxide synthase; FGF, fibroblast growth factors; NO, nitric oxide; PDGF, platelet-derived growth factor; TNF, tumor necrosis factors; VEGF, vascular endothelial growth factor.
One of the most important molecules that governs the growth and differentiation of vascular smooth muscle and endothelium is the glycoprotein VEGF.19 This growth factor was originally identified as a vascular permeability factor but was later recognized to the same molecule identified as a potent pro-angiogenic factor, first recognized in tumor biology.21,22 For many years after its discovery, VEGF was considered to act mainly through activation of tyrosine kinase receptors on microvascular endothelial cells to promote angiogenesis.19 More recent work, however, has demonstrated clearly that VEGF receptors are expressed by many different cell types and enable VEGF to exert neuroprotective23 and neurotrophic24 influences while also participating in the differentiation of skeletal25 and vascular26 muscle (Fig. 4). From this perspective, VEGF can exert direct effects on smooth muscle through VEGF receptors expressed by smooth muscle27,29,30 and can exert indirect effects on smooth muscle through VEGF receptors expressed by the vascular endothelium, which in turn can promote the release of NO and endothelin that can independently modulate smooth muscle growth and differentiation.31,32 In addition, VEGF may also promote the growth of perivascular sympathetic nerves24 that can also exert potent trophic effects on the growth and differentiation of arterial smooth muscle.33,34
FIGURE 4.
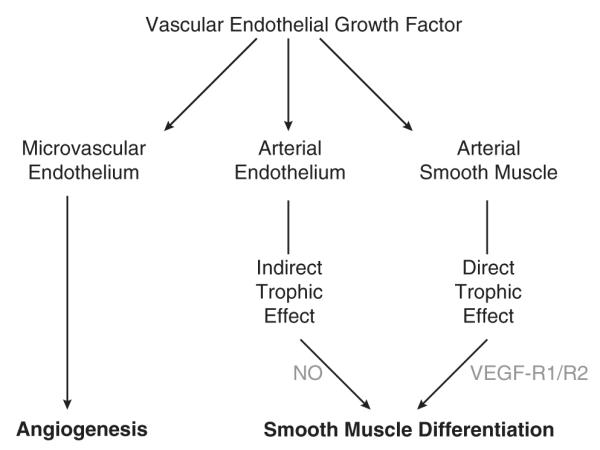
VEGF exerts trophic effects on vascular smooth muscle. The traditional view of VEGF held that this growth factor acted largely on the microvascular endothelium to promote angiogenesis.19 More recent evidence, however, has demonstrated a broad variety of effects of VEGF on many different cell types.23 Most importantly, it is now clear that VEGF can act indirectly through the vascular endothelium and directly through VEGF receptors on smooth muscle cells, to influence smooth muscle differentiation. NO indicates nitric oxide; VEGF, vascular endothelial growth factor.27,28
VEGF CONTRIBUTES TO HYPOXIC MODULATION OF SMOOTH MUSCLE PHENOTYPE
One of the most potent regulators of VEGF synthesis and release is hypoxia. When tissue oxygen tensions fall, the transcriptional coactivator HIF-1α accumulates, which allows it to combine with its constitutively expressed binding partner HIF-1β and then bind to hypoxia responsive elements in many gene promoters.35 The VEGF promoter includes hypoxia responsive elements, which allows the transcription of the VEGF gene to closely follow the severity of tissue hypoxia.36
Given that low oxygen tensions are a fundamental characteristic of the fetal circulation, it is reasonable to expect that VEGF contributes significantly toward the growth and differentiation of the fetal vasculature. To test this hypothesis, Butler et al29 first established that VEGF could influence smooth muscle phenotype in vessels other than microvascular capillaries. Using common carotid arteries organ cultured for only 24 hours in a low, physiological concentration of VEGF (3 ng/mL), these studies demonstrated significant and age-specific effects of VEGF on contractility, abundance, and intramural distribution of multiple smooth muscle contractile proteins [smooth muscle α-actin, myosin light chain kinase (MLCK), and regulatory myosin light chain]. More specifically, evidence from this study advanced the “gradient hypothesis,” which posits that the lumen-to-adventitial gradient in smooth muscle morphology and protein expression is the result of bidirectional gradients in trophic factors released from the vascular endothelium and the tissue parenchyma, respectively. Because the effects observed in organ culture occurred in the absence of any other growth factors in the culture media, the effects were unambiguously attributable to the actions of VEGF. Equally important, these studies also demonstrated that mRNA for both main VEGF receptors (Flk-1 and Flt-1) were expressed in fetal carotid arteries at abundances that were significantly greater than observed in corresponding adult arteries.
In another test of the hypothesis that VEGF contributes significantly to the growth and differentiation of the fetal vasculature, Hubbell et al30 explored the ability of VEGF to modulate smooth muscle phenotype, as indicated by the expression of smooth muscle myosin heavy chain (MHC) isoforms. In addition, these studies also examined the hypothesis that responses to chronic hypoxia should be more pronounced in fetal than adult arteries by virtue of the lower oxygen tensions in fetal arteries. The results demonstrated that chronic hypoxia increased expression of the nonmuscle isoform of MHC, and this effect was more pronounced in fetal than adult arteries. Conversely, the results also indicated that hypoxia depressed expression of the smooth muscle isoform of MHC in fetal arteries but increased its expression in adult arteries. Together, these results suggest that hypoxia transforms smooth muscle toward a more synthetic and less contractile phenotype in fetal arteries but promotes a more contractile phenotype in adult arteries. In carotid arteries organ cultured with VEGF, the fetal arteries again exhibited phenotypic transformation toward a more synthetic phenotype but only a few changes in MHC abundance were observed in adult arteries. These findings thus supported the VEGF hypothesis in fetal arteries but suggested that additional factors must be involved in adult arteries.
To examine the hypothesis that vascular differentiation involves changes in not only the abundances of contractile proteins but also in their intracellular organization, the Hubbell study30 also employed confocal microscropy to quantitate the effects of maturation, chronic hypoxia, and VEGF on colocalization among MHC isoforms and the smooth muscle α-actin cytoskeleton. For these measurements, the effects of chronic hypoxia and organ culture were closely similar in both fetal and adult arteries. Both hypoxia and VEGF decreased colocalization of nonmuscle isoform of MHC with α-actin in fetal arteries but had no effect on this colocalization in adult arteries. For the colocalization of smooth muscle isoform of MHC with α-actin, neither hypoxia nor VEGF had any effect in fetal arteries, but both treatments decreased colocalization in adult arteries. These findings emphasize on several points. First, contractile protein abundance and organization appear to be independently regulated, and contraction may be more closely dependent upon the organization than the simple abundance of contractile proteins. Second, both chronic hypoxia and VEGF transform smooth muscle phenotype in an age-dependent manner; these effects in fetal arteries are more pro-synthetic but in adult arteries are more pro-contractile. Third, VEGF can explain many of the effects of chronic hypoxia on smooth muscle phenotype and to a greater extent in fetal than adult arteries.
The hypothesis that VEGF helps mediate the effects of hypoxia on fetal vascular smooth muscle requires that VEGF is able to exert a sustained effect over relatively long periods of time. In response to chronic hypoxia, most arteries undergo major structural and functional changes (hypoxic vascular remodeling) that persist as long as hypoxic exposure continues.37 Typically, however, sustained hypoxia promotes a transient increase in VEGF that returns to baseline within 3 weeks, even in the presence of continued hypoxia.38 For VEGF to help maintain hypoxic vascular remodeling even after VEGF levels have returned to baseline, the VEGF receptor levels must be upregulated in arterial smooth muscle, as proposed to occur in pulmonary vascular endothelial cells.39 To test this hypothesis in large nonpulmonary arteries, Adeoye et al27 examined the effects of 110 days of chronic hypoxia on both VEGF and VEGF receptor levels in fetal lamb carotid arteries. These studies revealed that as suggested by previous studies, VEGF levels were not different in hypoxic and normoxic fetal arteries. In contrast, the abundances of the Flk-1 and Flt-1 receptors were both >2-fold greater in hypoxic than in normoxic fetal arteries. These results advance the hypothesis that VEGF continuously contributes to hypoxic vascular remodeling through a transient increase in VEGF levels and a sustained increase in VEGF receptors.
To further explore the hypothesis that chronic hypoxia and VEGF independently regulate contractile protein abundance and organization within the arterial smooth muscle, the Adeoye study also compared abundance and confocal colocalization for smooth muscle α-actin, MLCK, and regulatory myosin light chain. These results demonstrated that at least for these contractile proteins in fetal carotid arteries, the effects of VEGF could almost fully replicate the effects of chronic hypoxia on both protein abundance and colocalization.27 In addition, these studies again revealed that the effects of VEGF and chronic hypoxia on contractile protein abundance and colocalization were not consistently correlated (Fig. 5). In some cases, colocalization changed with no change in abundance (see MLCK-actin and MLC20-actin panels in Fig. 5). In other cases, abundance changed with no change in colocalization (see MLCK-actin panel in Fig. 5). In yet other cases, abundance and colocalization changed in opposite directions. Together, these findings emphasize that contractile protein abundance is regulated independently from its organization and distribution within the smooth muscle cell. In relation to contractile function, contractile protein organization may be a much more important determinant than simple contractile protein abundance.
FIGURE 5.

Hypoxia and VEGF differentially influence contractile protein abundance and colocalization. Numerous studies of smooth muscle cell function have focused on the relative abundances of key contractile proteins such as smooth muscle α-actin, myosin light chain kinase, and regulatory myosin light chain (MLC20). A common view was that as abundances of these proteins increased, so would their integration into the contractile apparatus. Recent work, however, suggests that contractile protein abundance and its integration with other contractile proteins may be independently regulated. Shown here are measurements of abundance and colocalization made within the same artery segments under control conditions, after organ culture with VEGF, or after acclimatization to chronic hypoxia. As evident from these data, the relations between abundance and colocalization are not tightly coupled, vary with developmental age, and are highly protein specific. Error bars indicate SEM for N≥5 in all cases. For details, please see. VEGF indicates vascular endothelial growth factor.27,30
MODULATORS AND MEDIATORS OF VEGF’S EFFECTS
Aside from the unknown mechanisms through which VEGF may influence contractile protein organization and distribution within the fetal vascular smooth muscle cell, many other aspects of VEGF biology also remain highly uncertain. Numerous physiological factors can increase or decrease the levels of VEGF and/or its receptors independent of hypoxia. For example, maternal food restriction has been shown to depress angiogenesis in the neonatal offspring, which in turn was associated with decreased expression of VEGF.40 How maternal food restriction may reduce VEGF expression is uncertain, but one hypothesis is that maternal food restriction may increase maternal glucocorticoid levels,41 which in turn may depress VEGF expression.42,43 Other studies, however, suggest at best a minor role for glucocorticoids in the fetal vascular effects of maternal food restriction.44,45 As an alternative to the glucocorticoid hypothesis, it has recently been proposed that the depressant effects of maternal food restriction on VEGF and angiogenesis in offspring are secondary to altered expression of miRNA molecules.46,47 Correspondingly, maternal food restriction has been shown to significantly alter numerous miRNA species with the potential to influence angiogenesis and vascular development in a highly age-dependent manner.46 Although the precise molecular mechanisms that couple maternal food restriction to altered patterns of miRNA expression remain unclear and under active investigation, ongoing studies of vascular miRNA metabolism offer great promise in elucidating how environmental influences such as hypoxia and food restriction can lead to altered vascular structure and function in the fetus.47–49
Independent of the many factors that govern the synthesis and expression of VEGF are the multiple pathways that mediate its intracellular effects. VEGF receptors belong to the tyrosine kinase family of cell surface receptors and are subject to regulation by diverse physiological and pathophysiological influences, particularly those that involve the Ets family of transcription factors and/or those that increase intracellular cAMP.50 Activated VEGF receptors can couple to intracellular pathways through activation of small G proteins and MAP kinases, through activation of phospholipase Cγ, through the PI3K-Akt pathway, and through other less common pathways.19,28 These cytosolic pathways, in turn, typically converge on a relatively small number of transcription factors intimately involved in regulation of endothelial and smooth muscle phenotype.4,51 In this context for smooth muscle, the transcription factors of greatest importance are ternary complex factor, serum response factor, Elk-1, and myocardin.52,53 Indeed, the discovery of myocardin and its related transcription factors has in many ways revolutionized understanding of the control of smooth muscle differentiation and phenotype (Fig. 6). How VEGF couples to activation of myocardin and the other nuclear factors that govern smooth muscle differentiation remains unknown but it is a highly active area of current research, certain to provide many new insights into how fetal vascular differentiation is regulated, how it is influenced by environmental and potentially epigenetic influences, and how these process vary between immature and mature arteries.
FIGURE 6.

Myocardin-like transcription factors regulate smooth muscle phenotype. Understanding of the regulation of smooth muscle phenotype was advanced significantly by the discovery that the coactivator myocardin participates in the activation of transcription for a majority of genes that code for contractile proteins in smooth muscle.54 This discovery brought together the roles of ternary complex factor, serum response factor, and myocardin in a hierarchy that was consistent with known patterns of smooth muscle phenotype control obtained from studies of wound repair, atherosclerosis, tumor vasculogenesis, and Alzheimer disease. The discovery of myocardin has thus helped unify these fields and stimulated many new insights into how both physiological and pathologic stimuli influence smooth muscle differentiation. For details, please see.52,53
Conclusions and Overview
The extra-pulmonary fetal vasculature is highly specialized for life in utero, and as such, exhibits major structural and functional differences when compared with mature adult arteries. Fetal arteries are often more sensitive to contractile agonists but exhibit depressed endothelial function, relative to adult arteries. The smooth muscle and endothelial cells of term fetal arteries are typically less than half the length and width of corresponding adult cells but grow very rapidly during postnatal life. This rapid growth involves major changes in both vessel structure and function that are highly specific for each vascular bed. These many changes are coordinated by a virtual cacophony of growth factors released locally by parenchymal tissues and remotely into the circulating blood stream. One factor that appears to be particularly important in fetal vascular development is VEGF, once thought to control mainly microcirculatory angiogenesis. Recent evidence, however, demonstrates that VEGF serves many functions in multiple nonendothelial cell types including large artery smooth muscle. In this latter cell type, VEGF independently influences both the expression and the intracellular organization of contractile proteins in a highly age-specific manner. VEGF contributes significantly to hypoxic vascular remodeling and in the fetal vasculature, can replicate many of the structural and functional effects of chronic hypoxia. Numerous factors can influence the expression of VEGF and its receptors, including chronic hypoxia, maternal food restriction, glucocorticoids, and miRNA. Similarly, multiple mechanisms influences the mechanisms that couple VEGF receptors to activation of cytosolic kinases and transcription factors involved in control of smooth muscle phenotype and differentiation. Continued study of this coupling, particularly that involving the transcription factor myocardin, offers great promise for better understanding of the processes that regulate fetal vascular maturation and how these processes may be manipulated therapeutically for better clinical management of neonates at risk for cardiovascular dysregulation.
Acknowledgments
W.J.P. was supported by NIH grant numbers HL54120, HD31266, HL64867, NS076945, and the Loma Linda University School of Medicine. O.K. was supported by Harbor-UCLA Medical Center.
Footnotes
The authors declare that they have nothing to disclose.
References
- 1.Cunningham S, Symon AG, Elton RA, et al. Intra-arterial blood pressure reference ranges, death and morbidity in very low birthweight infants during the first seven days of life. Early Hum Dev. 1999;56:151–165. doi: 10.1016/s0378-3782(99)00038-9. [DOI] [PubMed] [Google Scholar]
- 2.Barker DJ. Adult consequences of fetal growth restriction. Clin Obstet Gynecol. 2006;49:270–283. doi: 10.1097/00003081-200606000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Martyn CN, Barker DJ, Osmond C. Mothers’ pelvic size, fetal growth, and death from stroke and coronary heart disease in men in the UK. Lancet. 1996;348:1264–1268. doi: 10.1016/s0140-6736(96)04257-2. [DOI] [PubMed] [Google Scholar]
- 4.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 5.Pearce WJ, Hull AD, Long DM, et al. Developmental changes in ovine cerebral artery composition and reactivity. Am J Physiol. 1991;261(pt 2):R458–R465. doi: 10.1152/ajpregu.1991.261.2.R458. [DOI] [PubMed] [Google Scholar]
- 6.Teng GQ, Williams J, Zhang L, et al. Effects of maturation, artery size, and chronic hypoxia on 5-HT receptor type in ovine cranial arteries. Am J Physiol. 1998;275(pt 2):R742–R753. doi: 10.1152/ajpregu.1998.275.3.R742. [DOI] [PubMed] [Google Scholar]
- 7.Longo LD, Ueno N, Zhao Y, et al. Developmental changes in alpha 1-adrenergic receptors, IP3 responses, and NE-induced contraction in cerebral arteries. Am J Physiol. 1996;271(pt 2):H2313–H2319. doi: 10.1152/ajpheart.1996.271.6.H2313. [DOI] [PubMed] [Google Scholar]
- 8.Nauli SM, Williams JM, Gerthoffer WT, et al. Chronic hypoxia modulates relations among calcium, myosin light chain phosphorylation, and force differently in fetal and adult ovine basilar arteries. J Appl Physiol. 2005;99:120–127. doi: 10.1152/japplphysiol.01131.2004. [DOI] [PubMed] [Google Scholar]
- 9.Rubin LL, Staddon JM. The cell biology of the blood-brain barrier. Annu Rev Neurosci. 1999;22:11–28. doi: 10.1146/annurev.neuro.22.1.11. [DOI] [PubMed] [Google Scholar]
- 10.Van Cott EM, Grabowski EF. Vascular hemostasis in flowing blood in children. Semin Thromb Hemost. 1998;24:583–590. doi: 10.1055/s-2007-996058. [DOI] [PubMed] [Google Scholar]
- 11.Williams JM, Hull AD, Pearce WJ. Maturational modulation of endothelium-dependent vasodilatation in ovine cerebral arteries. Am J Physiol Regul Integr Comp Physiol. 2005;288:R149–R157. doi: 10.1152/ajpregu.00427.2004. [DOI] [PubMed] [Google Scholar]
- 12.White CR, Hamade MW, Siami K, et al. Maturation enhances fluid shear-induced activation of eNOS in perfused ovine carotid arteries. Am J Physiol Heart Circ Physiol. 2005;289:H2220–H2227. doi: 10.1152/ajpheart.01013.2004. [DOI] [PubMed] [Google Scholar]
- 13.Parfenova H, Massie V, Leffler CW. Developmental changes in endothelium-derived vasorelaxant factors in cerebral circulation. Am J Physiol Heart Circ Physiol. 2000;278:H780–H788. doi: 10.1152/ajpheart.2000.278.3.H780. [DOI] [PubMed] [Google Scholar]
- 14.White CR, Hao X, Pearce WJ. Maturational differences in soluble guanylate cyclase activity in ovine carotid and cerebral arteries. Pediatr Res. 2000;47:369–375. doi: 10.1203/00006450-200003000-00014. [DOI] [PubMed] [Google Scholar]
- 15.Nauli SM, White CR, Hull AD, et al. Maturation alters cyclic nucleotide and relaxation responses to nitric oxide donors in ovine cerebral arteries. Biol Neonate. 2003;83:123–135. doi: 10.1159/000067959. [DOI] [PubMed] [Google Scholar]
- 16.Vanhoutte PM, Miller VM. Heterogeneity of endothelium-dependent responses in mammalian blood vessels. J Cardiovasc Pharmacol. 1985;7(suppl 3):S12–S23. doi: 10.1097/00005344-198500073-00002. [DOI] [PubMed] [Google Scholar]
- 17.Elliott CF, Pearce WJ. Effects of maturation on cell water, protein, and DNA content in ovine cerebral arteries. J Appl Physiol. 1995;79:831–837. doi: 10.1152/jappl.1995.79.3.831. [DOI] [PubMed] [Google Scholar]
- 18.Williams JM, Pearce WJ. Age-dependent modulation of endothelium-dependent vasodilatation by chronic hypoxia in ovine cranial arteries. J Appl Physiol. 2006;100:225–232. doi: 10.1152/japplphysiol.00221.2005. [DOI] [PubMed] [Google Scholar]
- 19.Hoeben A, Landuyt B, Highley MS, et al. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev. 2004;56:549–580. doi: 10.1124/pr.56.4.3. [DOI] [PubMed] [Google Scholar]
- 20.Rocha SF, Adams RH. Molecular differentiation and specialization of vascular beds. Angiogenesis. 2009;12:139–147. doi: 10.1007/s10456-009-9132-x. [DOI] [PubMed] [Google Scholar]
- 21.Leung DW, Cachianes G, Kuang WJ, et al. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246:1306–1309. doi: 10.1126/science.2479986. [DOI] [PubMed] [Google Scholar]
- 22.Ferrara N, Houck KA, Jakeman LB, et al. The vascular endothelial growth factor family of polypeptides. J Cell Biochem. 1991;47:211–218. doi: 10.1002/jcb.240470305. [DOI] [PubMed] [Google Scholar]
- 23.Storkebaum E, Lambrechts D, Carmeliet P. VEGF: once regarded as a specific angiogenic factor, now implicated in neuroprotection. Bio-essays. 2004;26:943–954. doi: 10.1002/bies.20092. [DOI] [PubMed] [Google Scholar]
- 24.Marko SB, Damon DH. VEGF promotes vascular sympathetic innervation. Am J Physiol Heart Circ Physiol. 2008;294:H2646–H2652. doi: 10.1152/ajpheart.00291.2008. [DOI] [PubMed] [Google Scholar]
- 25.Bryan BA, Walshe TE, Mitchell DC, et al. Coordinated vascular endothelial growth factor expression and signaling during skeletal myogenic differentiation. Mol Biol Cell. 2008;19:994–1006. doi: 10.1091/mbc.E07-09-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grosskreutz CL, Anand-Apte B, Duplaa C, et al. Vascular endothelial growth factor-induced migration of vascular smooth muscle cells in vitro. Microvasc Res. 1999;58:128–136. doi: 10.1006/mvre.1999.2171. [DOI] [PubMed] [Google Scholar]
- 27.Adeoye OO, Butler SM, Hubbell MC, et al. Contribution of increased VEGF receptors to hypoxic changes in fetal ovine carotid artery contractile proteins. Am J Physiol Cell Physiol. 2013;304:C656–C665. doi: 10.1152/ajpcell.00110.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olsson AK, Dimberg A, Kreuger J, et al. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 29.Butler SM, Abrassart JM, Hubbell MC, et al. Contributions of VEGF to age-dependent trans-mural gradients in contractile protein expression in ovine carotid arteries. Am J Physiol Cell Physiol. 2011;301:C653–C666. doi: 10.1152/ajpcell.00413.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hubbell MC, Semotiuk AJ, Thorpe RB, et al. Chronic hypoxia and VEGF differentially modulate abundance and organization of myosin heavy chain isoforms in fetal and adult ovine arteries. Am J Physiol Cell Physiol. 2012;303:C1090–C1103. doi: 10.1152/ajpcell.00408.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmad S, Hewett PW, Wang P, et al. Direct evidence for endothelial vascular endothelial growth factor receptor-1 function in nitric oxide-mediated angiogenesis. Circ Res. 2006;99:715–722. doi: 10.1161/01.RES.0000243989.46006.b9. [DOI] [PubMed] [Google Scholar]
- 32.Ribatti D, Conconi MT, Nussdorfer GG. Non-classic endogenous novel [corrected] regulators of angiogenesis. Pharmacol Rev. 2007;59:185–205. doi: 10.1124/pr.59.2.3. [DOI] [PubMed] [Google Scholar]
- 33.Chalothorn D, Zhang H, Clayton JA, et al. Catecholamines augment collateral vessel growth and angiogenesis in hindlimb ischemia. Am J Physiol Heart Circ Physiol. 2005;289:H947–H959. doi: 10.1152/ajpheart.00952.2004. [DOI] [PubMed] [Google Scholar]
- 34.Baumbach GL, Heistad DD, Siems JE. Effect of sympathetic nerves on composition and distensibility of cerebral arterioles in rats. J Physiol. 1989 Sep;416:123–140. doi: 10.1113/jphysiol.1989.sp017753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Semenza GL. O2-regulated gene expression: transcriptional control of cardiorespiratory physiology by HIF-1. J Appl Physiol. 2004;96:1173–1177. doi: 10.1152/japplphysiol.00770.2003. discussion 0-2. [DOI] [PubMed] [Google Scholar]
- 36.Germain S, Monnot C, Muller L, et al. Hypoxia-driven angiogenesis: role of tip cells and extracellular matrix scaffolding. Curr Opin Hematol. 2010;17:245–251. doi: 10.1097/MOH.0b013e32833865b9. [DOI] [PubMed] [Google Scholar]
- 37.Longo LD, Hull AD, Long DM, et al. Cerebrovascular adaptations to high-altitude hypoxemia in fetal and adult sheep. Am J Physiol. 1993;264(pt 2):R65–R72. doi: 10.1152/ajpregu.1993.264.1.R65. [DOI] [PubMed] [Google Scholar]
- 38.Chavez JC, Agani F, Pichiule P, et al. Expression of hypoxia-inducible factor-1alpha in the brain of rats during chronic hypoxia. J Appl Physiol. 2000;89:1937–1942. doi: 10.1152/jappl.2000.89.5.1937. [DOI] [PubMed] [Google Scholar]
- 39.Tuder RM, Flook BE, Voelkel NF. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. J Clin Invest. 1995;95:1798–1807. doi: 10.1172/JCI117858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khorram O, Khorram N, Momeni M, et al. Maternal undernutrition inhibits angiogenesis in the offspring: a potential mechanism of programmed hypertension. Am J Physiol Regul Integr Comp Physiol. 2007;293:R745–R753. doi: 10.1152/ajpregu.00131.2007. [DOI] [PubMed] [Google Scholar]
- 41.Belkacemi L, Jelks A, Chen CH, et al. Altered placental development in undernourished rats: role of maternal glucocorticoids. Reprod Biol Endocrinol. 2011;9:105. doi: 10.1186/1477-7827-9-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vinukonda G, Dummula K, Malik S, et al. Effect of prenatal glucocorticoids on cerebral vasculature of the developing brain. Stroke. 2010;41:1766–1773. doi: 10.1161/STROKEAHA.110.588400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen YJ, Wang T, Liu DS, et al. Effect of budesonide on TGF-beta1-enhanced VEGF production by lung fibroblasts. Cell Biol Int. 2010;34:777–782. doi: 10.1042/CBI20090083. [DOI] [PubMed] [Google Scholar]
- 44.Lesage J, Hahn D, Leonhardt M, et al. Maternal undernutrition during late gestation-induced intrauterine growth restriction in the rat is associated with impaired placental GLUT3 expression, but does not correlate with endogenous corticosterone levels. J Endocrinol. 2002;174:37–43. doi: 10.1677/joe.0.1740037. [DOI] [PubMed] [Google Scholar]
- 45.Cottrell EC, Holmes MC, Livingstone DE, et al. Reconciling the nutritional and glucocorticoid hypotheses of fetal programming. FASEB J. 2012;26:1866–1874. doi: 10.1096/fj.12-203489. [DOI] [PubMed] [Google Scholar]
- 46.Khorram O, Han G, Bagherpour R, et al. The effect of maternal undernutrition on vascular expression of micro and messenger RNA in newborn and aging offspring. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1366–R1374. doi: 10.1152/ajpregu.00704.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dang LT, Lawson ND, Fish JE. MicroRNA control of vascular endothelial growth factor signaling output during vascular development. Arterioscler Thromb Vasc Biol. 2013;33:193–200. doi: 10.1161/ATVBAHA.112.300142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McDonald RA, Hata A, MacLean MR, et al. MicroRNA and vascular remodelling in acute vascular injury and pulmonary vascular remodelling. Cardiovasc Res. 2012;93:594–604. doi: 10.1093/cvr/cvr299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei Y, Schober A, Weber C. Pathogenic arterial remodeling: the good and bad in microRNAs. Am J Physiol Heart Circ Physiol. 2013;304:H1050–H1059. doi: 10.1152/ajpheart.00267.2012. [DOI] [PubMed] [Google Scholar]
- 50.Shibuya M, Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angio-genesis and lymphangiogenesis. Exp Cell Res. 2006;312:549–560. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 51.Ruhrberg C. Growing and shaping the vascular tree: multiple roles for VEGF. Bioessays. 2003;25:1052–1060. doi: 10.1002/bies.10351. [DOI] [PubMed] [Google Scholar]
- 52.Parmacek MS. Myocardin-related transcription factors: critical coactivators regulating cardiovascular development and adaptation. Circ Res. 2007;100:633–644. doi: 10.1161/01.RES.0000259563.61091.e8. [DOI] [PubMed] [Google Scholar]
- 53.Wamhoff BR, Bowles DK, Owens GK. Excitation-transcription coupling in arterial smooth muscle. Circ Res. 2006;98:868–878. doi: 10.1161/01.RES.0000216596.73005.3c. [DOI] [PubMed] [Google Scholar]
- 54.Chen J, Kitchen CM, Streb JW, et al. Myocardin: a component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol. 2002;34:1345–1356. doi: 10.1006/jmcc.2002.2086. [DOI] [PubMed] [Google Scholar]