

Abstract
Background
Complement has the potential to provoke severe impairment to host tissues, as shown in autoimmune diseases where complement activation has been associated with diminished CD55 and/or CD59 expression on peripheral blood cell membranes. The aim of this study was to evaluate the presence of CD55- and/or CD59-deficient erythrocytic populations in patients with different rheumatic diseases and to investigate possible correlations with clinical or laboratory parameters.
Material/Methods
CD55 and CD59 expression was evaluated in erythrocytes of 113 patients with rheumatic diseases, 121 normal individuals, and 10 patients with paroxysmal nocturnal hemoglobinuria (PNH) using the Sephacryl gel microtyping system. Ham and sucrose tests were also performed.
Results
Interestingly, the majority of patients (104/113, 92%) demonstrated CD55- and/or CD59-deficient erythrocytes: 47 (41.6%) with concomitant deficiency of CD55 and CD59, 50 (44.2%) with isolated deficiency of CD55, and 6 (6.2%) with isolated deficiency of CD59. In normal individuals, only 2 (1%) had concomitant CD55/CD59 negativity and 3 (2%) had isolated CD55 or CD59 deficiency. All PNH patients exhibited simultaneous CD55/CD59 deficiency. Positive Ham and sucrose tests were found only in PNH patients. There was no association between the CD55- and/or CD59-deficient erythrocytes and hemocytopenias or undergoing treatment. However, CD55 expression significantly influenced hemoglobin values (F=6.092, p=0.015).
Conclusions
This study provides evidence supporting the presence of erythrocytes with CD55 and/or CD59 deficiency in patients with rheumatic diseases. Moreover, CD55 deficiency on red cells influences hemoglobin concentration. Further studies using molecular techniques will clarify the exact pathophysiological mechanisms of this deficiency.
MeSH Keywords: Autoimmune Diseases, complement regulatory proteins, Antigens, CD55, Antigens, CD59, Hemoglobinuria, Paroxysmal, Erythrocytes
Background
Paroxysmal nocturnal hemoglobinuria (PNH) is an acquired clonal hematopoietic stem cell disorder characterized by a variety of clinical manifestations, including the classic triad of chronic intravascular hemolysis, thrombotic events, and bone-marrow (BM) failure. Somatic mutation of X-linked gene PIG-A in hematopoietic stem cells, which encodes the first essential enzyme of the glycosylphosphatidylinositol (GPI)-anchor biosynthetic pathway, results in absent or decreased cell membrane expression of all surface proteins normally anchored by it – including CD55 and CD59 – in all circulating cells [1–5]. The incidence of “PNH-like” defect has been also demonstrated in many hematological diseases [2,3,6–12] and on peripheral blood cells (PBCs) of normal individuals [13–16].
Complement system (CS) represents a fundamental part, not only of the host’s innate immunity against pathogen invasion, but also of adaptive and humoral immunity [17–23]. Nevertheless, when not properly regulated, CS is recognized as having the potential to provoke severe impairment to host tissues through a process mainly mediated by autoantibodies (AAb) against specific tissue antigens or non-specific immune complexes (IC) deposited in organs, like renal glomerulus – known as damage to the “innocent bystander” [24–26]. This has been demonstrated in autoimmune diseases, but the mechanisms have not been thoroughly explained [18,27–29]. However, autoimmunity is also characterized by the loss of immunological tolerance, the over-activation of T-cell populations against specific antigenic epitopes and, generally, an enhanced and extensive overall immune response [30].
Multiple regulatory and inhibitory enzymes, either membrane-bound or soluble in plasma, known as complement regulatory proteins (Cregs), regulate the progression of complement cascade (CC) at all levels, protecting the autologous cells [31]. CD55 or DAF (decay- accelerating Factor), CD59 or MIRL (Membrane Inhibitor of Reactive Lysis), CD46 or MCP (Membrane Cofactor Protein), and CD35 or CR1 (complement receptor 1) are the 4 major membrane-bound Cregs [32]. Both CD55 and CD59 are globular GPI-anchored membrane glycoproteins [33] widely expressed in all circulating cells and most human tissues. CD55 accelerates the degradation of C3 and C5 convertases and prevents the formation of new enzymes. CD59 interferes with membrane attack complex (MAC) formation [27,34]. Indeed, the pathophysiologic consequence of GPI-anchored Cregs defect was initially observed in PNH, leading to an unusual sensitivity of abnormal red blood cells (RBCs) to complement lysis and subsequent intravascular hemolysis and hemoglobinuria.
Several studies in experimental models of autoimmune injury, such as encephalomyelitis [35–37], glomerulonephritis [38,39], vasculitis [40,41], myasthenia gravis [42], rheumatoid arthritis (RA) [43,44], and systemic lupus erythematosus (SLE) [45,46], have tried to evaluate the participation of CD55 and CD59 in the evolution of these disorders. As inflammation is not only restricted to a specific tissue, but also occurs in a systemic context, hemocytopenias may occur due to BM failure or excessive destruction of PBCs, both of which may be immune-mediated [47–54], without disregarding the role of drug-induced toxicity [55,56]. In SLE, hematological involvement is frequent [57], while anemia (mainly due to chronic disease) [56], and lymphopenia are the most common forms [55]. Lymphopenia has been related to clinical exacerbation in these patients [54]. Nevertheless, the potential role of CD55 and CD59 expression on the surface of PBCs in the pathogenesis of hemocytopenias and in disease severity has been poorly elucidated. Earlier studies have been mainly performed on PBCs in patients with SLE [58–65] and RA [66–68].
The aim of this study was to evaluate the presence of CD55 and/or CD59 antigens on erythrocytes of patients with rheumatic disorders, using the Sephacryl gel test microtyping system (SGT) [13], a semi-quantitative, inexpensive, and simple method useful in screening “PNH-like” red-cell defect, as well as to examine possible correlation with patient demographic characteristics, clinical and complete blood count (CBC) parameters, and undergoing treatment.
Material and Methods
Patients
In this study, 113 patients with rheumatic diseases (94 females, 19 males; median age: 64 years), who presented or were referred to our Department’s outpatient clinic from February 2009 to February 2013, were evaluated. The study population included 38 patients with rheumatoid arthritis (RA), 25 patients with systemic lupus erythematosus (SLE), 17 patients with Sjögren’s syndrome (SS), 7 patients with systemic sclerosis (Sc), 12 patients with vasculitis (Vsc), 2 patients with dermatomyositis (Drm), 1 patient with ankylosing spondylitis (ASp), and 11 patients with mixed connective tissue disease (MCTD). At the time of the evaluation, 86 patients underwent immunosuppressive (IS) and/or immunomodulary treatment (IM), and 27 received no treatment (N). Basic patient characteristics are shown in Table 1.
Table 1.
Basic characteristics of patients with rheumatic disorders.
| CTD (n)/disease (n) | Gender (M/F) | Age (years) (median/range) | Hb (g/dl) (mean/range) | Neu* (×109/lt) (median/range) | Lym (×109/lt) (mean/range) | PLT (×109/lt) (mean/range) | Treatment** |
|---|---|---|---|---|---|---|---|
| RA (38) | 7/31 | 70 (41–88) | 12.1 (8.0–20.0) | 4.8 (1.4–20.5) | 2.0 (0.57–4.9) | 259 (26–471) | N(3), IM(7),IS(6), C(22) |
| SLE (25) | 7/18 | 45 (18–71) | 11.7 (5.7–16.3) | 3.3 (1.2–7.4) | 1.67 (0.37–3.5) | 231 (114–532) | N(8), IM(4), IS(5), C(8) |
| SS (17) | 1/16 | 69 (43–79) | 12.7 (9.2–14.5) | 3.6 (1.6–17.5) | 1.69 (1.2–2.2) | 199 (74–347) | N(10), IM(4), IS(3) |
| Sc (7) | 0/7 | 59 (43–87) | 10.7 (7.2–13.0) | 4.75 (3.1–8.5) | 1.7 (0.96–3.1) | 250 (160–518) | N(2), IS(5) |
| Vsc (12) | 4/8 | 66.5 (48–80) | 12.9 (10.6–15.7) | 6.8 (3.7–10.9) | 0.8 (0.26–1.7) | 274 (155–393) | N(1), IS(11) |
| Drm (2) | 2/2 | 65 (54–76) | 11.1 (8.1–14.0) | 4.45 (4.3–4.6) | 1.89 (0.9–2.8) | 288 (247–328) | IM(1), IS(1) |
| ASp (1) | 0/1 | 50 | 13.9 | 5.6 | 2.78 | 222 | IM(1) |
| MCTD (11) | 0/11 | 59 (47–73) | 11.7 (8.8–13.0) | 2.0 (0.3–5.2) | 1.35 (0.46–2.5) | 207 (9–330) | N(3), IM(4), IS(1), C(3) |
| Total (113) | 19/94 | 64 (18–88) | 12.03 (5.7–20.0) | 4.3 (0.3–20.5) | 1.68 (0.26–4.9) | 222 (9–532) | N(27), IM(21), IS(32), C(33) |
Neutrophils’ distribution was not normal; so, logarithm was used for parametrics tests, wherever it was needed.
N – no treatment; IM – immunomodulary treatment; IS – immunosuppressive treatment; C – combination of IM and IS treatment. RA – rheumatoid arthritis & Still’s disease; SLE – systemic lupus erythematosus with or without antiphospholipid syndrome; SS – Sjögren syndrome; Sc – systemic sclerosis; Vsc – vasculitis; Drm – dermatomyositis; ASp – ankylosing spondylitis; MCTD – mixed connective tissue disease.
Anemia (hemoglobin<12.0 g/dl) was present in 43 (38.1%) patients, neutropenia (neutrophils <2.0×109/lt) in 14 (12.7%), lymphopenia (lymphocytes<1.0×109/lt) in 21 (18.9%), and thrombocytopenia (platelets<150×109/lt) in 13 (11.6%) patients. Cytopenias were further categorized in grades according to their severity (Grade 0: absence of cytopenia, Grade 1: mild cytopenia, Grade 2: moderate cytopenia, Grade 3: severe cytopenia, Grade 4: life-threatening cytopenia, Grade 5: death related to cytopenia) (Table 2). The National Cancer Institute (NCI), Common Terminology Criteria for Adverse Events version 3.0 (CTCAEv3.0) (Publish Date: August 9, 2006) was used for this purpose.
Table 2.
Cytopenias and their Grading (CTCAEv3.0) in patients with rheumatic disorders.
| CTD (n)/disease (n) | Anemia −/+ (n), [%] | Grade/ anemia (n), [%] | Neutropenia −/+ (n), [%] | Grade/ neutropenia | Lym/penia −/+ (n), [%] | Grade/lymphopenia | Thromb/penia −/+ (n), [%] | Grade/thrombocytopenia |
|---|---|---|---|---|---|---|---|---|
| RA (38) | (21/17) | 0: (21) [55.3] | (34/4) | 0: (34) [89.5] | (35/3) | 0: (35) [92.1] | (31/6) | 0: (31) [83.8] |
| [55.3/44.7] | 1: (8) [21.0] | [89.5/10.5] | 1: (3) [7.9] | [92.1/7.9] | 1: (2) [5.3] | [83.8/16.2] | 1: (4) [10.8] | |
| 2: (9) [23.7] | 2: (1) [2.6] | 2: (1) [2.6] | 2: (1) [2.7] | |||||
| 3: (1) [2.7] | ||||||||
|
| ||||||||
| SLE (25) | (13/12) | 0: (13) [52.0] | (19/4) | 0: (19) [82.6] | (19/5) | 0: (19) [79.2] | (21/4) | 0: (21) [84.0] |
| [52.0/48.0] | 1: (7) [28.0] | [82.6/17.4] | 1: (3) [13.0] | [79.2/20.8] | 1: (2) [8.3] | [84.0/16.0] | 1: (4) [16.0] | |
| 2: (2) [8.0] | 2: (1) [4.4] | 2: (1) [4.2] | ||||||
| 3: (2) [8.0] | 3: (2) [8.3] | |||||||
| 4: (1) [4.0] | ||||||||
|
| ||||||||
| SS (17) | (14/3) | 0: (14) [82.3] | (15/2) | 0: (15) [88.2] | (17/0) | 0: (17) [100.0] | (16/1) | 0: (16) [94.1] |
| [82.4/17.6] | 1: (1) [5.9] | [88.2/11.8] | 1: (2) [11.8] | [100.0/0.0] | [94.1/5.9] | 1: (1) [5.9] | ||
| 2: (2) [11.8] | ||||||||
|
| ||||||||
| Sc (7) | (4/3) | 0: (4) [57.1] | (6/0) | 0: (6) [100.0] | (5/1) | 0: (5) [83.3] | (7/0) | 0: (7) [100.0] |
| [57.1/42.9] | 2: (1) [14.3] | [100.0/0.0] | [83.3/16.7] | 1: (1) [16.7] | [100.0/0.0] | |||
| 3: (2) [28.6] | ||||||||
|
| ||||||||
| Vsc (12) | (9/3) | 0: (9) [75.0] | (12/0) | 0: (12) | (4/8) | 0: (4) [33.3] | (12/0) | 0: (12) [100.0] |
| [75.0/25.0] | 1: (3) [25.0] | [100.0/0.0] | [100.0] | [33.3/66.7] | 1: (2) [16.7] | [100/0.0] | ||
| 2: (3) [25.0] | ||||||||
| 3: (3) [25.0] | ||||||||
|
| ||||||||
| Drm (2) | (1/1) | 0: (1) [50.0] | (2/0) | 0: (2) [100.0] | (1/1) | 0: (1) [50.0] | (2/0) | 0: (2) [100.0] |
| [50.0/50.0] | 3: (1) [50.0] | [100.0/0.0] | [50.0/50.0] | 1: (1) [50.0] | [100.0/0.0] | |||
|
| ||||||||
| ASp (1) | (1/0) | 0: (1) [100.0] | (1/0) | 0: (1) [100.0] | (1/0) | 0: (1) [100.0] | (1/0) | 0: (1) [100.0] |
| [100.0/0.0] | [100.0/0.0] | [100.0/0.0] | [100.0/0.0] | |||||
|
| ||||||||
| MCTD | (7/4) | 0: (7) [63.6] | (7/4) | 0: (7) [63.6] | (8/3) | 0: (8) [72.7] | (9/2) | 0: (9) [81.8] |
| (11) | [63.6/36.4] | 1: (2) [18.2] | [63.6/36.4] | 1: (2) [18.2] | [72.7/27.3] | 1: (1) [9.1] | [81.8/18.2] | 1: (1) [9.1] |
| 2: (2) [18.2] | 2: (1) [9.1] | 2: (0) [0.0] | 4: (1) [9.1] | |||||
| 4: (1) [9.1] | 3: (2) [18.2] | |||||||
|
| ||||||||
| Total | (70/43) | 0: (70) [61.9] | (96/14) | 0: (96) [87.2] | 90/21 | 0: (90) [81.1] | 99/13 | 0: (99) [88.4] |
| (113) | 1: (21) [18.6] | 1: (10) [9.1] | 1: (9) [8.1] | 1: (9) [8.0] | ||||
| [62.0/38.0] | 2: (16) [14.2] | [87.3/12.7] | 2: (3) [2.7] | [81.1/18.9] | 2: (5) [4.5] | [88.4/11.6] | 2: (2) [1.8] | |
| 3: (5) [4.3] | 3: (0) [0.0] | 3: (7) [6.3] | 3: (1) [0.9] | |||||
| 4: (1) [0.9] | 4: (1) [0.9] | 4: (0) [0.0] | 4: (1) [0.9] | |||||
| 5: (0) [0.0] | 5: (0) [0.0] | 5: (0) [0.0] | 5: (0) [0.0] | |||||
RA – rheumatoid arthritis & Still’s disease; SLE – systemic lupus erythematosus with or without antiphospholipid syndrome; SS – Sjögren syndrome; Sc – systemic sclerosis; Vsc – vasculitis; Drm – dermatomyositis; ASp – ankylosing spondylitis; MCTD – mixed connective tissue disease.
One hundred and twenty-one (121) healthy blood donors of similar age and gender and 10 patients with PNH were also studied and served as control groups.
Evaluation of CD55- and/or CD59-deficient red cells
The CD55- and CD59-deficient red-cell populations were detected using a commercial kit (DiaMed-ID Micro Typing System-PNH test, DiaMed AG, Switzerland). Testing was performed within 2 hours of sampling. Venous blood in EDTA-K3 was collected and suspended in low ionic strength buffer (ID-diluent 2, modified LISS in red cell suspension) at 0.8% (v/v).
Fifty microliters of the suspension were added in microtubes, on top of the Sephacryl gel containing microbeads coated with rabbit anti-mouse immunoglobulin (DiaMed-ID Micro Typing System PNH test), at room temperature. Fifty microliters of monoclonal mouse anti-human CD55 (clone BRIC 216) or CD59 (clone MEM 43) and ID-PNH negative control (dilution buffer for anti-CD55 and anti-CD59) were added to the corresponding microtube. The microtubes were incubated at 37°C for 15 minutes, centrifuged at 126 g for 10 minutes in an ID-centrifuge, and the result was read after centrifugation.
RBCs bearing CD55 or CD59 bind to the microbeads of the gel and remain on top of the gel (positive population). In contrast, RBCs lacking CD55 or CD59 do not agglutinate, and pellet at the bottom of the microtube (negative population). When both positive and negative populations are detected, a part of the RBCs lack the corresponding (CD55 or CD59) antigen. The red cells of PNH are characterized by the absence of CD55 and CD59 populations. In preliminary experiments using different mixtures (75%, 50%, 25%, and 10%) of the patient’s red cells with compatible normal red cells, defective CD55 or CD59 populations can be detected that account for 10% or more of the red cells [13]. All the blood samples that were used for this purpose had been tested in advance for CD55 or CD59 deficiency. The presence of the individual populations was blindly scored by 2 independent observers and expressed semi-quantitatively as 100%, 75%, 50%, 25%, and 10%.
In all samples with CD55- or CD59-negative red-cell populations, Ham and sucrose lysis tests were performed.
Statistical Analysis
Statistical analysis was performed with the MedCalc statistical software package. The effect of the age on the existence of CD55- and/or CD59-deficient red-cell populations was evaluated using Spearman’s coefficient of rank correlation (rs). The impact of gender on the existence of CD55- and/or CD59-negative erythrocytic population was evaluated using the chi-square test (χ2-test). The occurrence of CD55- and/or CD59-deficient RBCs among rheumatic disorders was calculated with chi-square test (χ2-test). The relation between the presence or percentage of CD55 and/or CD59 deficiency and the incidence or severity of anemia, neutropenia, lymphopenia, and thrombocytopenia was examined with the chi-square test (χ2-test) or Fisher’s test (when there was a small number of expected frequencies, for which chi-square is not appropriate). The variation of hematological parameters (hemoglobin, neutrophils, lymphocytes, and platelets) depending on CD55 and/or CD59 red-cell expression or CD55/CD59 phenotype was studied with one-way ANOVA, 2wo-way ANOVA, t-test, and Mann-Whitney test – (when t-test was not applicable-) for unpaired measurements. The correlation between CBC parameters was examined with Pearson’s coefficient of rank correlation (rp), and the regression of hematological parameters by CD55% deficiency and CD59% deficiency was studied with rs and multiple linear regression. Although the distribution of hemoglobin (Hb), lymphocytes (Lym), and platelets (Plt) were approximately normal, neutrophil (Neu) distribution differed significantly from normal; thus, logarithm transformation was used to apply parametric tests. All p values were two-sided, the level of statistical significance was <0.05, and confidence intervals (CI) refer to 95% boundaries.
Results
Presence of “PNH-like” erythrocytic populations in patients with rheumatic disease
Interestingly, only 9 out of 113 (8.0%) patients with rheumatic disorders were not observed with “PNH-like” RBCs, CD55(+)/CD59(+). As a result, deficient erythrocytic populations for CD55 and/or CD59 were detected in 104 (92.0%) patients (92.0% vs. 8.0%, p<0.0001). CD55 deficiency was present in 97 out of 113 patients (85.8% vs. 14.2%, p<0.0001), and CD59 deficiency was present in 54 (47.8%) out of 113 patients (47.8% vs. 52.2%, p=0.707). Moreover, it was noticeable that the frequency of these populations’ appearance was statistically significantly higher (p<0.05) when compared to that of normal individuals, with the exception of Drm and ASp, in which the number of observations was very limited. There was a significant difference (δ) (δ=38.0%, p<0.0001, McNemar test) between those with CD55-negative red-cell populations and those with CD59-negative ones. Indeed, no significant correlation was found between the proportions of these antigens’ deficiency (rs=0.122, p=0.1989). The population of erythrocytic clones with negativity in CD55 and/or CD59 antigen never surpassed 25% of the total red-cell population. The most common proportion of deficiency for both GPI-anchored proteins was 10% (CD55: 88/113, 77.9%, CD59: 48/113, 42.5%). In addition, neither an isolated CD59 negativity of 25%, nor a simultaneous CD55/CD59 negativity of 25% was detected. Thus, 3 different “PNH-like” red-cell populations were present in this cohort of patients; 1 with isolated CD55 antigen deficiency (50/113, 44.2%), CD55(−)/CD59(+); 1 with isolated CD59 antigen deficiency (7/113, 6.2%), CD55(+)/CD59(−); and 1 with concomitant deficiency of CD55 and CD59 antigens (47/113, 41.6%), CD55(−)/CD59(−) (Figure 1). This difference of occurrence was statistically significant (p<0.0001).
Figure 1.

(A) Patient (RA) with isolated CD55 (DAF) red-cell deficiency (10% of his total red-cell population), CD55(−)/CD59(+). (B) Patient (SeA) with concomitant CD55 (DAF) and CD59 (MIRL) red-cell deficiency (10%/25%, of his total red-cell population, respectively), CD55(−)/CD59(−). None of these patients had positive Ham or sucrose test.
There was no significant difference among the patients of the 6 major – in this study – categories of CTD (RA, SLE, SS, Sc, Vsc, and MCTD) concerning the presence and the proportion of CD55 and/or CD59 deficiency on the surface of RBCs. The highest occurrence of “PNH-like” red-cell populations was noticed in patients with SLE (25/25, 100%), Sc (7/7, 100.0%), and Vsc (12/12. 100.0%) (Figure 2A). On the contrary, the highest frequency of normal CD55/CD59 erythrocytic phenotype was in patients with MCTD (3/11, 27.3%).
Figure 2.

(A) Patient (Vsc) with concomitant CD55 (DAF) and CD59 (MIRL) red-cell deficiency (25%/10%, of his total red-cell population, respectively). 12/12 patients with Vsc presented with “PNH-like” red-cell phenotype. However, only 7 (6.2%) patients on this study were presented with this red-cell phenotype of CD55/CD59 percentage-deficiency. (B) Patient (Sc) with concomitant CD55 (DAF) and CD59 (MIRL) red-cell deficiency (10%/10%, of his total red-cell population, respectively). 30.1% of patients were presented with this red-cell phenotype of CD55/CD59 percentage-deficiency. None of these patients had positive Ham or sucrose test, for PNH.
The most common red-cell phenotypes of CD55/CD59 percentage-deficiency were 10%/0% (44/113, 42.5%) and 10%/10% (34/113, 30.1%). The first phenotype (Figure 1A) was most apparent in patients with SS (9/17, 52.9%), RA (18/38, 47.4%), and SLE (11/25, 44%), and the second in patients with Sc (5/7, 71.4%), SLE (12/25, 48.0%), and Vsc (5/12, 41.7%) (Figure 2B).
Findings on the red-cell deficiency of CD55 and/or CD59 proteins, the percentage of deficiency, the presence of different “PNH-like” phenotypes on RBCs of patients with rheumatic diseases, including their occurrence per disease, are shown in detail in Tables 3 and 4.
Table 3.
CD55, CD59 antigens’ expression, percentage of deficient red-cell population for each antigen and presence of “PNH-like” red-cell phenotype in patients with rheumatic diseases.
| CTD (n)/disease (n) | CD55 (+/−)* (n), [%] | CD55%# (n), [%] | CD59 (+/−)* (n), [%] | CD59%# (n), [%] | “PNH-like” phenotype** (−/+) (n), [%] |
|---|---|---|---|---|---|
| RA (38) | (5/33) [13.2/86.8] | 0: (5) [13.2] | (22/16) [57.9/42.1] | 0: (22) [57.9] | (3/35) [7.9/92.1] |
| 10: (30) [79.0] | 10: (15) [39.5] | ||||
| Ra (37) | (4/33) [10.9/89.1] | 25: (3) [7.8] | (21/16) [56.8/43.2] | 25: (1) [2.6] | (2/35) [5.4/94.6] |
| Stl (1) | (1/0) [100.0/0.0] | (1/0) [100.0/0.0] | (1/0) [100.0/0.0] | ||
|
| |||||
| SLE (25) | (1/24) [4.0/96.0] | 0: (1) [4.0] | (12/13) [48.0/52.0] | 0: (12) [48.0] | (0/25) [0.0/100.0] |
| 10: (21) [84.0] | 10: (11) [44.0] | ||||
| SL (20) | (0/20) [0.0/100.0] | 25: (3) [12.0] | (10/10) [50.0/50.0] | 25: (2) [8.0] | (0/20) [0.0/100.0] |
| SeA (5) | (1/4) [20.0/80.0] | (2/3) [40.0/60.0] | (0/5) [0.0/100.0] | ||
|
| |||||
| SS (17) | (3/14) [17.6/82.4] | 0: (3) [17.6] | (11/6) [64.7/35.3] | 0: (11) [64.7] | (2/15) [11.8/88.2] |
| 10: (13) [76.5] | 10: (4) [23.5] | ||||
| 25: (1) [5.9] | 25: (2) [11.8] | ||||
|
| |||||
| Sc (7) | (1/6) [14.3/85.7] | 0: (1) [14.3] | (1/6) [14.3/85.7] | 0: (1) [14.3] | (0/7) [0.0/100.0] |
| 10: (6) [85.7] | 10: (6) [85.7] | ||||
| 25: (0) [0.0] | 25: (0) [0.0] | ||||
|
| |||||
| Vsc (12) | (2/10) [16.7/83.3] | 0: (2) [16.7] | (5/7) [41.7/58.3] | 0: (5) [41.7] | (0/12) [0.0/100.0] |
| 10: (8) [66.6] | 10: (7) [58.3] | ||||
| 25: (2) [16.7] | 25: (0) [0.0] | ||||
|
| |||||
| Drm (2) | (1/1) [50.0/50.0] | 0: (1) [50.0] | (1/1) [50.0/50.0] | 0: (1) [50.0] | (1/1) [50.0/50.0] |
| 10: (1) [50.0] | 10: (1) [50.0] | ||||
| 25: (0) [0.0] | 25: (0) [0.0] | ||||
|
| |||||
| ASp (1) | (0/1) [0.0/100.0] | 0: (0) [0.0] | (0/1) [0.0/100.0] | 0: (0) [0.0] | (0/1) [0.0/100.0] |
| 10: (1) [100.0] | 10: (1) [100.0] | ||||
| 25: (0) [0.0] | 25: (0) [0.0] | ||||
|
| |||||
| MCTD (11) | (3/8) [27.3/72.7] | 0: (3) [27.3] | (7/4) [63.6/36.4] | 0: (7) [63.6] | (3/8) [27.3/72.7] |
| 10: (8) [72.7] | 10: (3) [27.3] | ||||
| RSS (3) | (0/3) [0.0/100.0] | 25: (0) [0.0] | (1/2) [33.3/66.7] | 25: (1) [9.1] | (0/3) [0.0/100.0] |
| RSc (1) | (0/1) [0.0/100.0] | (1/0) [100.0/0.0] | (0/1) [0.0/100.0] | ||
| SeSS (6) | (2/4) [33.3/66.7] | (4/2) [66.7/33.3] | (2/4) [33.3/66.7] | ||
| ScSS (1) | (1/0) [100.0/0.0] | (1/0) [100.0/0.0] | (1/0) [100.0/0.0] | ||
|
| |||||
| Total (113) | (16/97) [14.2/85.8] | 0: 15 [13.6] | (59/54) [52.2/47.8] | 0: 59 [52.2] | (9/104) [8.0/92.0] |
| 10: 86 [78.2] | 10: 48 [42.5] | ||||
| 25: 9 [8.2] | 25: 6 [5.3] | ||||
(+) indicates normal expression of CD55 or CD59 molecule in patient’s red cells, (−) indicates the existence of CD55- or CD59-deficient red-cell population.
CD55% or CD59%: percentage of CD55- or CD59-deficient red-cell population in patient’s total red cells.
“PNH-like” phenotype (+): the existence of deficient red-cell populations for CD55 and/or CD59 antigen. RA – rheumatoid arthritis & Still’s disease; Ra – rheumatoid arthritis apart from Still’s disease; Stl – Still’s disease; SLE – systemic lupus erythematosus with or without antiphospholipid syndrome; SL – SLE without antiphospholipid syndrome; SeA – SLE with antiphospholipid syndrome; SS – Sjögren syndrome; Sc – systemic sclerosis, Vsc – vasculitis, Drm – dermatomyositis, ASp – ankylosing spondylitis; MCTD – mixed connective tissue disease; RSS – RA & SS; RSc – RA & Sc; SeSS – SLE & SS; ScSS – Sc & SS.
Table 4.
Types of CD55/CD59 deficient erythrocytic populations (“PNH-like” red-cell phenotypes) and percentage of concomitant CD55/CD59 red-cell deficiency in patients with rheumatic diseases.
| CTD (n)/disease (n) | CD55(−)/CD59(+) (n), [%] | CD55%/CD59% (n) | CD55(+)/CD59(−) (n), [%] | CD55%/CD59% (n) | CD55(−)/CD59(−) (n), [%] | CD55%/CD59% (n) |
|---|---|---|---|---|---|---|
| RA (38) | (19) [50.0] | 10%/0% (18) [47.4] | (2) [5.3] | 0%/10% (2) [5.3] | (14) [36.8] | 10%/10% (11) [28.9] |
| 25%/0% (1) [2.6] | 0%/25% (0) [0.0] | 25%/10% (2) [5.3] | ||||
| Ra (37) | (19) [51.4] | (2) [5.4] | (14) [37.8] | 10%/25% (1) [2.6] | ||
| 25%/25% (0) [0.0] | ||||||
| Stl (1) | (0) [0.0] | (0) [0.0] | (0) [0.0] | |||
|
| ||||||
| SLE (25) | (12) [48.0] | 10%/0% (11) [44.0] | (1) [4.0] | 0%/10% (1) [4.0] | (12) [48.0] | 10%/10% (8) [32.0] |
| 25%/0% (1) [4.0] | 0%/25% (0) [0.0] | 25%/10% (2) [8.0] | ||||
| SL (20) | (10) [50.0] | (0) [0.0] | (10) [50.0] | 10%/25% (2) [8.0] | ||
| 25%/25% (0) [0.0] | ||||||
| SeA (5) | (2) [40.0] | (1) [20.0] | (2) [40.0] | |||
|
| ||||||
| SS (17) | (9) [52.9] | 10%/0% (9) [52.9] | (1) [5.9] | 0%/10% (1) [5.9] | (5) [29.4] | 10%/10% (2) [11.8] |
| 25%/0% (0) [0.0] | 0%/25% (0) [0.0] | 25%/10% (1) [5.9] | ||||
| 10%/25% (2) [11.8] | ||||||
| 25%/25% (0) [0.0] | ||||||
|
| ||||||
| Sc (7) | (1) [14.3] | 10%/0% (1) [14.3] | (1) [14.3] | 0%/10% (1) [14.3] | (5) [71.4] | 10%/10% (5) [71.4] |
| 25%/0% (0) [0.0] | 0%/25% (0) [0.0] | 25%/10% (0) [0.0] | ||||
| 10%/25% (0) [0.0] | ||||||
| 25%/25% (0) [0.0] | ||||||
|
| ||||||
| Vsc (12) | (5) [41.7] | 10%/0% (5) [41.7] | (2) [16.6] | 0%/10% (2) [16.7] | (5) [41.7] | 10%/10% (3) [25.0] |
| 25%/0% (0) [0.0] | 0%/25% (0) [0.0] | 25%/10% (2) [16.7] | ||||
| 10%/25% (0) [0.0] | ||||||
| 25%/25% (0) [0.0] | ||||||
|
| ||||||
| Drm (2) | (0) [0.0] | 10%/0% (0) [0.0] | (0) [0.0] | 0%/10% (0) [0.0] | (1) [50.0] | 10%/10% (1) [50.0] |
| 25%/0% (0) [0.0] | 0%/25% (0) [0.0] | 25%/10% (0) [0.0] | ||||
| 10%/25% (0) [0.0] | ||||||
| 25%/25% (0) [0.0] | ||||||
|
| ||||||
| ASp (1) | (0) [0.0] | 10%/0% (0) [0.0] | (0) [0.0] | 0%/10% (0) [0.0] | (1) [100.0] | 10%/10% (1) [100.0] |
| 25%/0% (0) [0.0] | 0%/25% (0) [0.0] | 25%/10% (0) [0.0] | ||||
| 10%/25% (0) [0.0] | ||||||
| 25%/25% (0) [0.0] | ||||||
|
| ||||||
| MCTD (11) | (4) [36.4] | 10%/0% (4) [36.4] | (0) [0.0] | 0%/10% (0) [0.0] | (4) [36.4] | 10%/10% (3) [27.3] |
| 25%/0% (0) [0.0] | 0%/25% (0) [0.0] | 25%/10% (0) [5.3] | ||||
| RSS (3) | (1) [33.3] | (0) [0.0] | (2) [66.7] | 10%/25% (1) [9.1] | ||
| 25%/25% (0) [0.0] | ||||||
| RSc (1) | (1) [100.0] | (0) [0.0] | (0) [0.0] | |||
| SeSS (6) | (2) [33.3] | (0) [0.0] | (2) [33.3] | |||
| ScSS (1) | (0) [0.0] | (0) [0.0] | (0) [0.0] | |||
|
| ||||||
| Total (113) | (50) [44.2] | 10%/0% (48) [42.5] | (7) [6.2] | 0%/10% (7) [6.2] | (47) [41.6] | 10%/10% (34) [30.1] |
| 25%/0% (2) [1.7] | 0%/25% (0) [0.0] | 25%/10% (7) [6.2] | ||||
| 10%/25% (6) [5.3] | ||||||
| 25%/25% (0) [0.0] | ||||||
RA – rheumatoid arthritis & Still’s disease; Ra – rheumatoid arthritis apart from Still’s disease; Stl – Still’s disease; SLE – systemic lupus erythematosus with or without antiphospholipid syndrome; SL – SLE without antiphospholipid syndrome; SeA – SLE with antiphospholipid syndrome; SS – Sjögren syndrome; Sc – systemic sclerosis, Vsc – vasculitis; Drm – dermatomyositis; ASp – ankylosing spondylitis; MCTD – mixed connective tissue disease; RSS – RA & SS; RSc – RA & Sc; SeSS – SLE & SS; ScSS – Sc & SS.
Treatment/demographic characteristics and “PNH-like” red-cell deficiency
No difference was found regarding the presence of CD55 and/or CD59 red-cell deficiency in relation to the type of undergoing treatment, at the time of evaluation. Moreover, when patients who did not receive any treatment were compared with those who underwent IS and/or IM treatment, no significant heterogeneity was found concerning the presence of CD55- and/or CD59-deficient erythrocytic populations. Similarly, there was no significant heterogeneity with regard to age or gender of rheumatic patients.
Hemocytopenias and “PNH-like” red-cell defect
Anemia was present in 43 out of 113 patients (38%). In anemic patients, grade 1 and grade 2 of anemia were the most common forms (18.6% and 14.2%, respectively). Patients with SLE, RA, and Sc were presented more often with anemia than others. There was no significant relation between the presence and proportion of CD55 and/or CD59 deficiency on erythrocytes, and the occurrence or grade of anemia. Moreover, among the different “PNH-like” red-cell phenotypes, no significant difference relating to the presence or grade of anemia was detected.
Neutropenia was present in 14 patients (12.7%). In neutropenic patients the most common grade was 1 (10, 9.1%). Among the rheumatic disorders, MCTD and SLE were associated more frequently with neutropenia (4/11, 36.4% and 4/25, 17.4% respectively). There was an inverse relation (p=0.0005) between the presence of “PNH-like” red-cell population and the presence of neutropenia (odds ratio, OR=0.078, 95%CI: 0.0178–0.345, p=0.0008). Moreover, when the grade of neutropenia was studied in relation to the existence (or not) of “PNH-like” RBCs, the outcome was significant (p=0.002). This result could be partially ascribed to the relation between the presence of “PNH-like” erythrocytes and the absence of neutropenia (which was described before). The fact that over 50% (4 out of 7, 57.14%) of patients with normal CD55/CD59 red-cell phenotype presented with grade 1 neutropenia (OR=12.67, 95%CI: 2.6825 to 59.812, p=0.0013), could be also responsible for this relation. No significant outcome was found concerning the severity of neutropenia in neutropenic patients. Moreover, there was no significant difference regarding the occurrence/absence or grade of neutropenia between the 3 different “PNH-like” erythrocytic phenotypes, even when only neutropenic patients were examined.
Concerning CD55 antigen, there was a significant inverse relation (p=0.0308) between its red-cell deficiency and the presence of neutropenia (OR=0.209, 95%CI: 0.058–0.749, p=0.0162). The significant heterogeneity of the severity of neutropenia among patients with different CD55 red-cell expression (p=0.0147) was attributed to high occurrence of non-neutropenia in those with CD55 negativity on RBCs (as in neutropenic patients); there was not such relation. Regarding the presence and severity of neutropenia, percentage of CD55 erythrocytic deficiency demonstrated a similar behavior. Although there was no significant difference when comparing 10% with 25% CD55 negativity, as well as 0% with 25%, the presence of 10% CD55-deficient RBCs is significantly associated with non-neutropenia (OR=0.205, 95%CI: 0.05608 to 0.7503, p=0.0167). Once again, the significant heterogeneity of neutropenia severity depending on CD55% negativity was attributed to high occurrence of non-neutropenia in patients with 10% CD55-deficient RBCs. Indeed, in neutropenic patients, such a relation was not found. CD59 expression on red-cell membrane did not generally affect the presence or grade of neutropenia, but CD59 negativity was related with absence of neutropenia in patients with normal expression of CD55 (Fisher’s test, p=0.044). This was the reason for the “significant result”, when severity of neutropenia was examined (as in neutropenic patients), such relation was not found.
Lymphopenia was present in 21 (18.9%) patients. Grade 1 was the most common form. Vsc and MCTD were the disorders with the most high frequency of lymphopenia (8/12, 66.7% and 3/11, 27.3%, respectively). Neither the presence of “PNH-like” erythrocytes nor CD55/CD59 red-cell phenotype was proved to influence the presence of lymphopenia in this cohort of patients. CD55 antigen was not proved to have an effect on presence or severity of lymphopenia. Nevertheless, “significant heterogeneity” was noted when percentage of CD55 negativity was studied in relation with severity of lymphopenia (p=0.0203). There was no clear evidence for this. It could be attributed partially to the fact that 7 out of 7 patients with lymphopenia grade 3 presented with 10% CD55 negativity on their RBCs. Yet, this hypothesis was not verified by Fisher’s test or odds ratio significance level (OR=11.47, p=0.1157). However, both the presence of CD59 deficiency and the percentage of CD59-deficient erythrocytes were not proved as statistically significant factors for the presence or grade of lymphopenia.
Thrombocytopenia was apparent in 13 patients (11.6%). Regarding the severity, grade 1 was the most frequent type (8.0%). Patients with RA and SLE presented more frequently with thrombocytopenia (6/38, 16.2% and 4/25, 16.0%, respectively). There was no heterogeneity of thrombocytopenia in relation with presence of “PNH-like” red-cell populations. However, when the severity of thrombocytopenia was studied in relation with the existence (or not) of “PNH-like” RBCs, there was a statistically significant outcome (p=0.0042). This result was attributed to the fact that 102 out of 104 patients with “PNH-like” red-cell phenotype presented with grade 0 or grade 1 thrombocytopenia (OR=17.0, 95%CI: 2.029 to 142.470, p=0.0090). In thrombocytopenic patients, there was evidence of heterogeneity when grade 1 thrombocytopenia was compared with the other scales, regarding the presence (or not) of “PNH-like” red-cell populations (p=0.0695). Similarly, while CD55/CD59 erythrocytic phenotype did not had an effect on occurrence of thrombocytopenia, the comparison of severity of thrombocytopenia between the different “PNH-like” phenotypes was significantly heterogeneous (p=0.0333). Once again, this was ascribed to the absence of moderate, severe, or life-threatening thrombocytopenia in patients with CD55- and/or CD59-deficient RBCs (OR=0.059 95%CI: 0.00702 to 0.493, p=0.009). Red-cell membrane CD55 negativity was not likely to significantly affect the presence or grade of thrombocytopenia. Percentage of CD55 negativity on erythrocyte’ surface was not proved as a significant factor. Regarding CD59 expression, there was no significant relation with the presence or severity of thrombocytopenia.
CD55 deficiency and Hb levels
There was no significant difference of Hb between patients with normal expression of CD55/CD59 antigens on their RBCs and those with deficient ones (ä=−1.3035 g/dl, p=0.0928). However, although there was only a trend regarding the influence of “PNH-like” red-cell phenotype on Hb concentration (F=2.874, p=0.093), CD55 antigen expression on RBCs was proved as a significant factor of influence on Hb variance (F=6.092, p=0.015), while percentage of CD55% negativity demonstrated a marginal impact (F=3.040, p=0.052). As a result, there was a significant difference when the arithmetic mean of Hb was compared between patients with normal expression of CD55 and those with CD55 erythrocytic negativity (δ=−1.4534 g/dl, 95%CI: −2.6202 to −0.2866 g/dl, p=0.0151). The difference of Hb between normal expression of CD55 and 10% deficiency was also significant (δ=−1.4392 g/dl, 95%CI: −2.6356 to −0.2428 g/dl, p=0.0189), while between normal expression and 25% deficiency was marginally significant (δ=−1.5924 g/dl, p=0.0508, Mann-Whitney test). No significant effect on Hb was found between 10% and 25% CD55-negative red-cell populations. Thus, a significant inverse correlation was found between CD55% deficiency on RBCs and Hb levels (rs=−0.205, p=0.0296). In contrast, CD59 expression did not have an important effect on Hb variability. CD59% deficiency was not proved as a significant factor of influence on Hb concentration (rs=+0.143, p=0.132).
Figure 3 and 4 demonstrate the scatter diagram with regression line of Hb in relation to CD55% and CD59% negativity on red-cell surface, respectively. Table 5 briefly illustrates all the outcomes.
Figure 3.

Scatter diagram with regression line between CD55% red-cell deficiency and Hb levels (g/dl), in rheumatologic patients. Regression equation: y=13.2812 + −0.1974 × + 0.005348 x2, Coefficient of determination R2=0.05237, F=3.0398, p=0.052.
Figure 4.

Scatter diagram with regression line between CD59% red-cell deficiency and Hb levels (g/dl), in rheumatologic patients. Regression equation: y=11.8458 + 0.02284 × + 0.0007997 x2, Coefficient of determination R2=0.01338, F=0.7456, p=0.477.
Table 5.
Presentation of statistic results between CD55- and/or CD59-red-cell deficiency and hematological parameters or manifestations.
| Influence factor/hematological profile | “PNH-like” red-cell phenotype | CD55 deficiency | CD55%- negativity | CD59 deficiency | CD59%- negativity | CD55-CD59 interaction# | CD55/CD59 red-cell phenotype | |
|---|---|---|---|---|---|---|---|---|
| Anemia | +/− | (NS) | (NS) | (NS) | (NS) | (NS) | (N/T) | (NS) |
|
| ||||||||
| Grade* | (NS) | (NS) | (NS) | (NS) | (NS) | (N/T) | (NS) | |
|
| ||||||||
| Hemoglobin | F=2.874 | F=6.092 | F=3.040 | (NS) | (NS) | (NS) | F=2.404 (p=0.071) | |
| (p=0.093) | δ=−1.4534 | (p=0.052) | (−/+) – (+/+) | |||||
| δ (0–10%): −1.439 | (−/+) – (+/−) | |||||||
| rs=−0.205 | (−/−) – (+/−) | |||||||
|
| ||||||||
| +/− | (StS) | (StS) | (StS) | (StS) | (StS) | (N/T) | (StS) | |
| OR=0.078 | OR=0.209 | 0–10% | CD55(+) | [Only CD55(+)] | OR=1/12.778 | |||
| OR=0.2051 | (p=0.044) | 0–10% | ||||||
| Neutro-penia | (p=0.044) | |||||||
|
| ||||||||
| Grade* | (NS) | (NS) | (NS) | (NS) | (NS) | (N/T) | (NS) | |
|
| ||||||||
| Neutrophils | F=16.077 | F=4.06 | F=2.406 | (NS) | (NS) | F=5.035 | F=5.67 | |
| δ=+2.582 | (p=0.095) | (−/+) – (+/+) | ||||||
| (+/−) – (+/+) | ||||||||
| (−/−) – (+/+) | ||||||||
|
| ||||||||
| Lympho-penia | +/− | (NS) | (NS) | (NS) | (NS) | (NS) | (N/T) | (NS) |
|
| ||||||||
| Grade* | (NS) | (NS) | (StS) ? | (NS) | (NS) | (N/T) | (NS) | |
| (p=0.0203) | ||||||||
|
| ||||||||
| (NS) | F=2.937 | F=0.156 | F=4.623 | F=2.388(p=0.097) | (NS) | F=2.713 | ||
| Lymphocytes | (p=0.089) | δ (0–10%)= | δ=+0.363 | δ (0–10%)= | (−/+) – (+/−) | |||
| δ=−0.426 | −0.455 | +0.383 | (−/+) – (−/−) | |||||
| rs=0.118 | rs=−0.157 | |||||||
|
| ||||||||
| Throm/penia | +/− | (NS) | (NS) | (NS) | (NS) | (NS) | (N/T) | (NS) |
|
| ||||||||
| Grade** | (StS) | OR=7.31 | (NS) | (NS) | (NS) | (N/T) | (StS) | |
| OR=17.0 | (p=0.0565) | OR=1/0.059 | ||||||
|
| ||||||||
| F=4.623 | F=3.797 | δ=+48.93 | (NS) | (NS) | (NS) | F=1.759 | ||
| Platelets | δ=+71.59 | (p=0.054) | (p=0.0612) | (−/+) – (+/+) | ||||
| δ=+49.23 | (−/−) – (+/+) | |||||||
| (p=0.0539) | ||||||||
Grade refers to the scale 1–5 of hemocypenias, Grade 0 is not included, apart from thrombocytopenia
in which the written odds ratios refer to the possibility of grade 0 and 1 occurrence.
CD55-CD59 interaction was calculated with Two-way ANOVA, Logarithm transformation was not conducted because there was not such choice offered by the MedCalc.? the existence of true statistically significant result and its aetiology is under question, δ=statistically significant difference that is based on antigen deficiency unless it is mentioned otherwise, (StS)=statistical significant, (NS)=non-significant, (N/T) – not tested, in bold letters whatever statistical parameter-relation is significant (p<0.05).
Correlation between Hb and different subpopulations of leucocytes in patients with concomitant CD55/CD59 deficiency
Pearson’s correlation coefficient (rp) was examined among different hematological parameters depending on either the presence or absence of “PNH-like” phenotype, or the deficiency of CD55 and/or CD59 proteins on red-cell surface. Hb was not associated, in general, either with Neu or Lym, apart from CD55(−)/CD59(−) red-cell phenotype (rp=−0.3416, p=0.0201 and rp=+0.315, p=0.0332, respectively) (Table 6). Figures 5 and 6 demonstrate the scatter diagram and regression line of these 2 correlations.
Table 6.
Correlation (rp) of hematological parameters in patients with different expression of CD55 and/or CD59 antigens on RBCs.
| Parameters/ patients | Hb/Neu* | Hb/Lym | Hb/Plt | Neu*/Lym | Neu*/Plt | Lym/Plt |
|---|---|---|---|---|---|---|
| “PNH −like” phenotype | ||||||
| (−) | +0.3770 | −0.110 | −0.220 | +0.643 | +0.7848 | +0.690 |
| (p=0.0856) | (p=0.0211) | (p=0.0580) | ||||
| (+) | −0.06814 | +0.060 | −0.160 | −0.02848 | +0.2515 | −0.039 |
| (p=0.0120) | ||||||
|
| ||||||
| CD55 | ||||||
| (+) | +0.4114 | −0.165 | −0.131 | +0.1165 | +0.6269 | +0.439 |
| (p=0.0158) | ||||||
| (−) | −0.1092 | +0.110 | −0.159 | +0.001273 | +0.2714 | −0.055 |
| (p=0.0085) | ||||||
|
| ||||||
| CD59 | ||||||
| (+) | +0.03188 | −0.016 | −0.233 | +0.2898 | +0.4428 | −0.050 |
| (p=0.077) | (p=0.032) | (p=0.0007) | ||||
| (−) | −0.2321 | +0.220 | −0.120 | −0.457 | +0.224 | −0.088 |
| (p=0.0007) | ||||||
|
| ||||||
| CD55/CD59 | ||||||
| (+/+) | +0.3770 | −0.110 | −0.220 | +0.6428 | +0.7848 | +0.690 |
| (p=0.0211) | (p=0.0580) | |||||
| (−/+) | +0.06279 | +0.051 | −0.183 | +0.2077 | +0.3121 | −0.043 |
| (p=0.0327) | ||||||
| (+/−) | +0.4649 | −0.173 | +0.018 | −0.9226 | −0.09592 | +0.290 |
| (p=0.0088) | ||||||
| (−/−) | −0.3416 | +0.315 | −0.121 | −0.4223 | +0.2023 | −0.122 |
| (p=0.0201) | (p=0.0332) | (p=0.0035) | ||||
|
| ||||||
| Total | −0.04563 | +0.034 | −0.187 | +0.04645 | +0.3473 | + 0.009 |
| (p=0.0487) | (p=0.0487) | |||||
(−) indicates the absence of a characteristic, when (+) indicates the presence of it.
Due to the fact that distribution of neutrophils was significantly different from normal one, logarithm transformation was used. Statistically significant results (p<0.05) were written in bold letters, while p-value is given next to them.
Figure 5.

Scatter diagram with regression line between neutrophils’ total population (×106/lt) and Hb levels (g/dl) in patients with concomitant CD55/CD59 deficiency. Regression equation: Log(y)=4.0642 + −0.03645 x, Coefficient of determination R2=0.1167, F=5.8137, p=0.020.
Figure 6.

Scatter diagram with regression line between Hb levels (g/dl) and lymphocytes’ total population (×106/lt) in patients with concomitant CD55/CD59 deficiency. Regression equation: y=10.9051 + 0.0008468 x, Coefficient of determination R2=0.09902, F=4.836, p=0.033.
Further findings on the correlation between different CBC parameters regarding the erythrocytic deficiency of CD55 and/or CD59 proteins are depicted in Table 6.
Control groups
Among the 121 normal individuals, 2 (1.6%) had RBCs with concomitant deficiency for CD55 and CD59, while 3 (2.4%) had erythrocytes with isolated CD55 or CD59 deficiency; these erythrocytes accounted for no more than 10% of the total red-cell population. All patients with PNH had a simultaneous CD55 and CD59 deficiency (Figure 7). Positive Ham and sucrose tests were found only in patients with PNH. Results are shown in Table 7.
Figure 7.
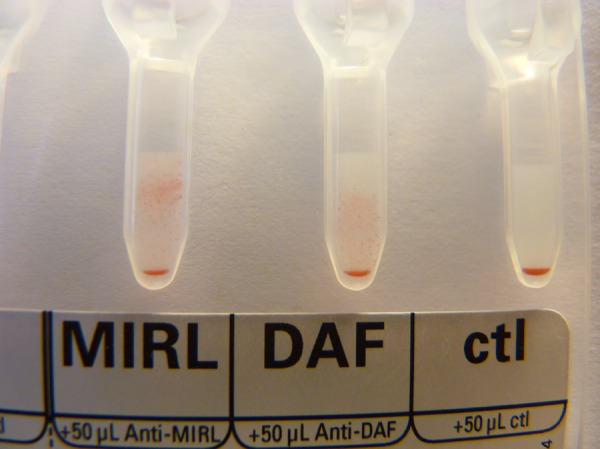
PNH patient: >75% simultaneous absence of CD55 (DAF) and CD59 (MIRL). This patient had a positive Ham and sucrose test.
Table 7.
The incidence of “PNH-like” red-cell phenotypes and the frequency of CD55/CD59 concomitant percentage-deficiency on RBCs in control groups.
| Control groups | CD55(−)/CD59(+) (n), [%] | CD55%/CD59% (n) | CD55(+)/CD59(−) (n), [%] | CD55%/CD59% (n) | CD55(−)/CD59(−) (n), [%] | CD55%/CD59% (n) |
|---|---|---|---|---|---|---|
| Normal individuals (121) | (2) [1.6] | 10%/0% (2) [1.6] | (1) [0.8] | 0%/10% (1) [0.8] | (2) [1.6] | 10%/10% (2) [1.6] |
| PNH Patients (10) | (0) [0.0] | (0) [0.0] | (0) [0.0] | (0) [0.0] | (10) [100.0] | 25%/10% (3) [30.0] 25%/25% (3) [30.0] 75%/50% (1) [10.0] 75%/75% (1) [10.0] 100%/100% (2) [20.0] |
Discussion
The fundamental role of CS in autoimmunity is clearly reflected in the fact that genetic deficiency of C1q or C4 – and other complement components – is a major predisposing factor for severe SLE [23], while S allele of CR1 has been genetically linked with this disease [69]. The main potential mechanism includes the release of self-antigens due to failure in the regulation of complement response to apoptosis [70,71], as well as the formation of IC between AAb and self-antigens against tissues, leading to activation primarily of the classic pathway [27,72]. B- and T-cell hyperactivity is also present [30]. Despite the recognized contribution of CS in autologous tissues impairment and cellular damage in patients with rheumatic diseases, the role of Cregs such as DAF and MIRL in modulating – in a systemic context – the severity of that injury remains rather obscure. However, since rare GPI-negative stem cells with confirmed PIG-A mutations have been described to occur even in BM of normal individuals, but without proliferating in competition with normal ones [73], case-reports in the literature of autoimmune disorders (such as SLE and Sc) associated with PNH development [74–77] enhance the hypothesis of a possible immune-mediated BM failure in these patients [78], in which GPI-deficient stem cells escape from cytotoxic T-cell – mediated attack, proliferate and dominate in BM [2].
Although there are few publications evaluating the expression of CD55 and CD59 on PBCs in patients with SLE, RA and SS [58–68], we believe the present study is the first to demonstrate the presence of diminished expression of CD55 and/or CD59 proteins on RBCs – “PNH-like” red-cell populations – in patients with Sjögren syndrome (15/17, 88.2%), systemic sclerosis (7/7, 100%), vasculitis (12/12, 100%), dermatomyositis (1/2), ankylosing spondylitis (1/1), and mixed connective tissue disease (8/11, 72.7%). Furthermore, incidence per disease in these populations was significantly higher when compared to normal individuals, with the exception of Drm and ASp. Interestingly, only 9 rheumatologic patients did not have “PNH-like” red-cell populations (9/113, 8.0%), using the SGT. The size of defect for each molecule never surpassed 25% of the total erythrocytic population. The 2 most common types of concomitant CD55/CD59 percentage-deficiency were 10%/0% (48/113, 42.5%) and 10%/10% (34/113, 30.1%). Yet, remarkably, the appearance of CD55-negative RBCs (97/113, 85.8%) was significantly more frequent than CD59-negative ones (54/113, 47.8%), and there was no correlation between diminished expression of these 2 Cregs. So, which possible mechanisms mediate that loss of expression?
Since 1981, when Miyakawa et al. initially managed to demonstrate the diminished expression of Cregs, particularly that of CR1 (C3b receptor), on red-cell membrane of patients with SLE [79], several flow-cytometric and immunofluorescence studies have evaluated the presence of DAF and MIRL on erythrocytes in autoimmune diseases like SLE [59,60,63–65] and RA [67,68], without determining a specific pattern of the expression of these antigens. Primarily performed on a small sample of patients, they revealed a significant deficiency of these molecules – either jointly or separately – on RBCs, although without association with complement activity, the presence of anemia or antiphospholipid antibodies (aPLA) and clinical status [61,63–65,68].
Recently, in a large-sample clinical study (100 SLE patients) on Cregs expression on PBCs’ surface, Alegretti et al. [65] showed only a diminished expression of CD59 on RBCs, which did not seem to be associated with anemia (Hb <11gr/dl) or autoimmune hemolytic anemia (AIHA), in contrast to Richaud-Patin et al. [60]. Moreover, there was a significant decrease in red-cell membrane–bound MIRL in patients with SLE nephritis, compared to the results of Arora et al. [59]. It is important to mention that the number of patients in this study was far greater than in the previous ones. In addition, among Cregs expression on red-cell surface, only that of CR1 was associated with complement activation, while none were related with disease activity [65]. In our study, besides identifying the significant increased occurrence of CD55- and CD59-negative RBCs in patients with SLE [with (SeA) or without (SL) antiphospholipid syndrome] compared to normal individuals, no difference was found between SL and SeA patients with regard to deficiency of these antigens. Moreover, our data show that this loss of expression was unrelated to the presence of anemia.
Concerning the expression of CD55 and CD59 on leucocytes in SLE patients, it has been shown that under circumstances of hemocytopenias, the deficiency of these surface antigens was greater, while an inverse correlation between their expression and complement activation was noted. Moreover, in lymphopenic patients there was a higher percentage and titers of AAb (such as anti-SSA anti-dsDNA), which remarkably were unrelated to CD55 and CD59 deficiency on lymphocytes [58,61,63,65]. Lymphopenia not only constitutes the most common hematological manifestation of SLE [56], but it also has been related with clinical exacerbation [61]. Although it has been widely accepted the involvement of anti-lymphocyte AAb in its appearance (especially IgM cryoglobulins), their pathogenic significance, and their correlation with disease activity, remain controversial [53,54,61]. In addition to functioning as complement inhibitors, CD55 and CD59 also act as signal transducers or ligands to specific cell receptors of the immune system, thus participating in lymphocyte and macrophage activation and proliferation, in modulation of antigen-presenting cells (APC) and in regulation of cytokines and proinflammatory molecule secretion [19,26,28]. Our study failed to demonstrate a significant relation between CD55 and CD59 expression on RBCs and the presence of other cell-lineages hemocytopenias. There is scientific evidence of no association between the size of “PNH-like” deficiency and the presence of marked cytopenias [80]. However, a point of interest for further investigation, revealed in our study, was the description of significant positive correlation between Hb and Lym, only in patients with concomitant DAF and MIRL deficiency, as there is no such evidence in the scientific literature.
Interestingly, to the best of our knowledge, this is the first description of correlation between Hb and proportion of CD55 deficiency on RBCs in patients with rheumatic diseases. Although DAF and MIRL deficiency on erythrocytes seems irrelevant to the presence of anemia, our data revealed a significant difference of approximately −1.45 g/dl in Hb between patients with CD55-deficient RBCs and those with normal ones. Although the pathogenesis of these patients’ anemia was mainly considered as a result of antibody-induced damage of erythrocytes in the past, evidence to date indicates that anemia of chronic disease (ACD) – a mild to moderate normocytic-hypochromic anemia – is the most common form in patients with autoimmune disorders. Inhibiting action of inflammatory cytokines, such as interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), interferon-α, and AAb against EPO (anti-EPO), plays a crucial role in ACD pathogenesis. However, ACD often coexists with anemia caused by other relatively frequent mechanisms, like iron deficiency anemia (IDA), AIHA, anemia of chronic renal insufficiency, and cyclophosphamide-induced myelotoxicity. The prevalence of AIHA is often overestimated because of a positive direct antiglobulin test (DAT) (warm type IgG anti-erythrocyte AAb) without real hemolysis, while deficiency of GPI-anchored proteins might be responsible for rare episodes of DAT-negative hemolytic anemia by increasing the susceptibility to complement-mediated lysis [56]. However, T-cell mediated BM failure is an additional factor that might contribute to anemia in autoimmunity [56,78].
To explain the changes in the expression of these molecules on the red-cell membrane, several hypotheses could be proposed. First of all, the diminished expression of CD55 and CD59 proteins on RBCs might be due to either the impaired synthesis of the GPI anchor or the abnormal coupling of the protein to the membrane on red-cell precursors [60]. Other researchers have excluded this possibility, since different patterns of diminished expression of Cregs were observed on each cell type, strongly suggesting the participation of different lineage-specific physiopathology processes [65]. Nevertheless, similar variability has been described in the pattern of expression of GPI-anchored proteins (GPI-APs) explored among different hematopoietic cell-lineage subpopulations in patients with PNH and normal individuals [80,81]. Moreover, Hernandez-Campo et al. reported that DAF and MIRL expression is highly variable among different cell-lineage populations, while the latter shows higher amounts on RBCs. Thus, this parameter may partially contribute to the significant difference between CD55 and CD59 deficiency on RBCs in our cohort of patients [80].
Several studies have described up-regulation of CD55 and CD59 in tissues and nucleated cells that are exposed to complement-sustained activation, pro-inflammatory molecules, and cytokines such as C-reactive protein (C-RP), TNF-α, IL-1 [59,62,82–84]. This has been considered as the effect of an adaptive response against chronic – but not acute – inflammatory state, associated with prolonged systematic complement activation [85]. However, the biological function of these molecules varies, and is associated with the cell-membrane in which they are expressed. Unlike leucocytes, endothelial cells, and other nucleated cells, red-cell membrane proteome changes very rarely, being quite constant towards the cell activation state [62]. Thus, we hypothesized that this deficiency on RBCs is an adaptive phenomenon that occurs due to consumption of the GPI-anchored Cregs on the erythrocytic membrane, when trying to prevent complement-mediated lysis, as down-regulators of CS [67]. Previous studies generally did not demonstrate any correlation between complement activation and expression of these antigens on red-cell membrane. Moreover, in this study, “PNH-like” RBCs never exceeded 25% of the total erythrocytic population, while the most common proportion of deficiency for both was 10%. There is scientific evidence that phenotypic alterations (mostly up-regulation) of GPI-APs on both PNH cells and normal residual PBCs of PNH patients compared to those of healthy donors, may sometimes occur independently to complement activation, affected not only by genetic abnormalities (PIG-A gene mutations), but also the surrounding microenvironment [81].
Since no correlation was found among the presence or titers of several aPLA or anti-nuclear antibodies (ANA) and CD55 or CD59 expression on PBC membrane, nor with clinical status, several authors have suggested the existence of yet undetectable AAb or IC against specific cell self-antigens, which might hinder the binding of the proteins to the GPI anchorage or shed the GPI-protein complexes from the cell surface [60–63,65]. This was doubted by the experimental evidence, since Arora et al. observed no alteration in the expression of DAF and MIRL after incubating normal erythrocytes with RA patient sera whose RBCs exhibited significantly diminished expression of CD55 and CD59 [67]. In addition, the authors proposed as the cause, the spontaneous vesiculation that occurred to erythrocytes incubated with MAC, whose vesicles contained DAF, MIRL, and CR1[86,87]. Or perhaps a proteolytic cleavage is generated in relation to activation of complement on erythrocyte surface by enzymatic activity, as in the phosphatidylinositol-specific phospholipase C and D [60,67,88]. As such enzymes are specific for phosphatidylinositol, the latter hypothesis could not be corroborated by our results, due to lack of correlation between CD55 and CD59 deficiency on RBCs, as well as the significant difference between their loss of expression on red-cell membrane.
In the majority of these cases, normal individuals have “PNH-like” clones in a very small proportion, and PIG-A mutations characteristic for PNH have been identified [89]. Indeed, we found that 5 normal individuals had deficient red-cell populations for CD55 and/or CD59. Similarly, the existence of “PNH-like” clones has been described in a very small proportion of cells prior to selection in their favor by anti-CD52 (Campath-1H) administration in patients with chronic lymphocytic leukemia [90]. According to the “dual pathogenesis model” [1,2], findings in BM biopsies in SLE patients with hemocytopenias – without undergoing any immunosuppressive treatment – [48,49] and the presence of AAb against BM progenitor cells in rare cases of SLE patients with aplastic anemia (AA) [50,51], enhance the concept of a possible primary immune-mediated hematopoietic failure syndrome. Thus, it suggests the presence of an immunoregulatory selection in favor of GPI-defective red-cell clones to proliferate preferentially on a microenvironment of BM autoimmunity attack, compared to normal ones, and become detectable with our methodology [91,92].
None of our patients with rheumatic disease showed clinical or laboratory signs of hemolysis, and the Ham and sucrose lysis tests were negative. This is possibly due to the small proportion of erythrocytes with concomitantly reduced expression of CD55/CD59. Furthermore, isolated CD55 or CD59 deficiency is not able to produce homologous hemolysis [93].
According to the International PNH Interest Group, subclinical PNH (PNH-sc) is defined by the presence of a small population of GPI-deficient blood cells detected in patients with different types of BM failure, who do not demonstrate clinical or laboratory signs of hemolysis, [94]. Notably, 8.0% of our patients did not demonstrate “PNH-like” phenotype on RBCs using SGT; the presence of PNH-sc patients who may have less than 1% PNH-type cells, which can be detected only by high-sensitivity flow cytometric assay, has been reported in the scientific literature. Furthermore, as this assay failed to demonstrate the presence of “PNH-like” PBC clones in 43 patients with SLE, an increase in the proportion of these clones should be considered as a characteristic of BM failure [78].
The acquired CR1 loss of expression on erythrocytes [95] is envisaged by many researchers to contribute significantly to the pathophysiology of many autoimmune disorders such as SLE and RA [96,97], as well as to reflect the disease activity and inflammatory state. Moreover, it plays an important role in the erythrocyte-mediated C3 and C4 clearance of IC from the circulation [59,98–100]. Its expression on RBCs was not assessed, but in previous studies it was significantly associated with complement activation. Moreover, disease activity, complement activation, and inflammatory state were not taken into consideration. On the other hand, the use of immunosuppressive and immunoregulatory drugs may have had an impact on our outcomes, being a limiting factor in our study. Despite no statistically significant relation between GPI-anchored Cregs and treatment, heterogeneous and multiple treatments perplexed the manifestation of clear association between specific drugs and Cregs deficiency and/or hematological parameters. Undoubtedly, SGT has the disadvantage of not detecting small PNH erythrocytic clones (<2%), as well as not evaluating the presence of CD55 and/or CD59 deficiency in other hematopoietic cell-lineages, compared to flow cytometry. However, previous studies have proven it is a useful and fairly sensitive test that can be used for screening to identify the presence of PNH red-cell populations [91,92].
Although our study did not sufficiently demonstrate, the clinical significance of the CD55 and/or CD59 deficiency on RBCs in rheumatologic patients, the presence of “PNH-like” red-cell phenotype in small proportions (mainly less than 25%), that seems to be independent from the development of anemia or other hemocytopenias, as well as the absence of clinical or laboratory signs of hemolysis, supports the hypothesis that these populations may preexist in a BM failure microenvironment owing to autoimmunity. These populations may be offered a proliferative advantage that makes them detectable with SGT [91,92]. Furthermore, the disclosed unprecedented association between CD55 red-cell expression and Hb levels in rheumatologic patients of this study may reflect the effect of autoimmunity on erythropoiesis, due to an immune-mediated BM failure. Deeper understanding of the pathophysiologic mechanisms seems fundamental for the development of novel therapeutic approaches regarding the hematological involvement in rheumatic disorders. In patients with other types of BM failure such as myelodysplastic syndromes or AA, the detection of PNH-like clones is predictive of the response to immunosuppressive therapy [2,3,6,78].
Conclusions
Our study provides evidence supporting the presence of “PNH-like” erythrocytic populations in patients with rheumatic diseases. Any hypothesis about the deficiency of CD55 and/or CD59 on RBCs in patients with rheumatic disorders, jointly or on its own, could be valid, and it is equally valid to believe that the process by which expression of Cregs on PBCs’ membranes is regulated remains rather obscure. However, we provided sufficient scientific evidence that supports the pre-existence of small populations of CD55- and/or CD59-deficient clones in BM, which acquire a survival advantage to proliferate against normal hematopoietic tissue and become detectable. Nevertheless, the semi-quantitative method used for the detection of CD55 and/or CD59 absence on erythrocytic membrane has the disadvantage of not demonstrating this abnormality in other cell types and not estimating these populations quantitatively. Previous studies have shown that the results obtained by SGT are comparable with those obtained by flow cytometry, proving that it is a useful screening tool for the detection of such populations, as it is fairly sensitive and easy to perform [91,92]. This study revealed an unprecedented relation between Hb levels and DAF expression on RBCs, which may reflect the contribution of autoimmunity to impaired erythropoiesis through an immune-mediated BM failure. Moreover, an unusual association between Hb levels and lymphocytic population, as well as between Hb levels and neutrophilic population, was detected only in rheumatologic patients with concomitant deficiency of CD55 and CD59. Further research using flow cytometry and other molecular techniques is required to clarify the deeper pathophysiologic processes. This could be fundamental not only for the comprehension of their role in inflammation and autoimmunity, but also for the development of novel therapies for hematological manifestations in autoimmune disorders.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Source of support: Departmental sources
References
- 1.Rotoli B, Luzzato L. Paroxysmal nocturnal hemoglobinuria. Semin Hematol. 1989;26:201–7. [PubMed] [Google Scholar]
- 2.Meletis J, Terpos E. Recent insights into the pathophysiology of paroxysmal nocturnal hemoglobinuria. Med Sci Monit. 2003;9(7):161–72. [PubMed] [Google Scholar]
- 3.Meletis J, Terpos E. Paroxysmal nocturnal haemoglobinuria: clinical presentation and association with other haematological disorders. Haema. 2001;4:79–88. [Google Scholar]
- 4.Luzzatto L. Paroxysmal nocturnal hemoglobinuria: an acquired X-linked genetic disease with somatic-cell mosaicism. Curr Opin Genet Dev. 2006;16:317–22. doi: 10.1016/j.gde.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 5.Tomita M. Biochemical background of paroxysmal nocturnal hemoglobinuria. Biochim Biophys Acta. 1999;1455:269–86. doi: 10.1016/s0925-4439(99)00068-x. [DOI] [PubMed] [Google Scholar]
- 6.Meletis J, Terpos E, Samarkos M, et al. Detection of CD55 and/or CD59 deficient red cell populations in patients with aplastic anaemia, myelodysplastic syndromes and myeloproliferative disorders. Haematologia. 2001;31:7–16. doi: 10.1163/15685590151092643. [DOI] [PubMed] [Google Scholar]
- 7.Meletis J, Terpos E, Samarkos M, et al. Detection of CD55- and/or CD59- deficient red cell population in patients with lymphoproliferative syndromes. Hematol J. 2001;2:33–37. doi: 10.1038/sj.thj.6200077. [DOI] [PubMed] [Google Scholar]
- 8.Meletis J, Terpos E, Samarkos M, et al. Red cells with paroxysmal nocturnal hemoglobinuria- phenotype with acute leukemia. Hematology. 2002;7:69–74. doi: 10.1080/10245330290028560. [DOI] [PubMed] [Google Scholar]
- 9.Meletis J, Terpos E, Samarkos M, et al. Detection of CD55- and/or CD59- deficient red cell populations in patients with plasma cell dyscrasias. Int J Hematol. 2002;75:40–44. doi: 10.1007/BF02981977. [DOI] [PubMed] [Google Scholar]
- 10.Meletis J, Terpos E, Mavrogianni D, et al. Aplastic anaemia with large “PNH-like” red cell population. Haema. 2003;6:103–6. [Google Scholar]
- 11.Terpos E, Samarkos M, Meletis J, et al. Unusual association between increased bone resorption and presence of paroxysmal nocturnal hemoglobinuria phenotype in multiple myeloma. Int J Hematol. 2003;78:344–48. doi: 10.1007/BF02983560. [DOI] [PubMed] [Google Scholar]
- 12.Terpos E, Sarantopoulos A, Meletis J, et al. Reduction of CD55 and/or CD59 in red blood cells of patients with HIV infection. Med Sci Monit. 2008;14(5):276–80. [PubMed] [Google Scholar]
- 13.Meletis J, Michali E, Samarkos M, et al. Detection of “PNH red cell” populations in hematological disorders using the sephacryl gel test micro typing system. Leuk Lymphoma. 1997;28:177–82. doi: 10.3109/10428199709058344. [DOI] [PubMed] [Google Scholar]
- 14.Kwong YL, Lee CP, Chan TK, Chan LC. Flow cytometric measurement of glycosylphosphatidyl-inositol-linked surface proteins on blood cells of patients with paroxysmal nocturnal hemoglobinuria. Am J Clin Pathol. 1994;102:30–35. doi: 10.1093/ajcp/102.1.30. [DOI] [PubMed] [Google Scholar]
- 15.Fujioka S, Yamada T. Decay-accelerating factor and CD59 expression in peripheral blood cells in aplastic anaemia and report of a case of paroxysmal nocturnal haemoglobinuria secondary to aplastic anaemia. Br J Haematol. 1993;83:660–62. doi: 10.1111/j.1365-2141.1993.tb04707.x. [DOI] [PubMed] [Google Scholar]
- 16.Oelschlaegel U, Besson I, Arnoulet C, et al. A standardized flow cytometric method for screening paroxysmal nocturnal haemoglobinuria (PNH) measuring CD55 and CD59 expression on erythrocytes and granulocytes. Clin Lab Haematol. 2001;23:81–90. doi: 10.1046/j.1365-2257.2001.00357.x. [DOI] [PubMed] [Google Scholar]
- 17.Song WC, Sarrias MR, Lambris JD. Complement and innate immunity. Immunopharmacology. 2000;49:187–98. doi: 10.1016/s0162-3109(00)80303-3. [DOI] [PubMed] [Google Scholar]
- 18.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–66. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 19.Dunkelberger JR, Song WC. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20:34–50. doi: 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- 20.Ogden CA, Elkon KB. Role of complement and other innate immune mechanisms in the removal of apoptotic cells. Curr Dir Autoimmun. 2006;9:120–42. doi: 10.1159/000090776. [DOI] [PubMed] [Google Scholar]
- 21.Molina H, Holers VM, Li B, et al. Markedly impaired humoral immune response in mice deficient in complement receptors 1 and 2. Proc Natl Acad Sci USA. 1996;93:3357–61. doi: 10.1073/pnas.93.8.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaya Z, Afanasyeva M, Wang Y, et al. Contribution of the innate immune system to autoimmune myocarditis: a role for complement. Nat Immunol. 2001;2:739–45. doi: 10.1038/90686. [DOI] [PubMed] [Google Scholar]
- 23.Carroll MC. The role of complement in B cell activation and tolerance. Adv Immunol. 2000;74:61–88. doi: 10.1016/s0065-2776(08)60908-6. [DOI] [PubMed] [Google Scholar]
- 24.Welch TR. Complement in glomerulonephritis. Nat Genet. 2002;31:333–34. doi: 10.1038/ng933. [DOI] [PubMed] [Google Scholar]
- 25.Storch MK, Piddlesden S, Haltia M, et al. Multiple Sclerosis: in situ evidence of antibody- and complement- mediated demyelination. Ann Neurol. 1998;43:465–71. doi: 10.1002/ana.410430409. [DOI] [PubMed] [Google Scholar]
- 26.Song WC. Complement regulatory proteins and autoimmunity. Autoimmunity. 2006;39:403–10. doi: 10.1080/08916930600739647. [DOI] [PubMed] [Google Scholar]
- 27.Walport MJ. Complement. Second of two parts. N Eng J Med. 2001;344:1140–44. doi: 10.1056/NEJM200104123441506. [DOI] [PubMed] [Google Scholar]
- 28.Kim DD, Song WC. Membrane complement regulatory proteins. Clin Immunol. 2006;118:127–36. doi: 10.1016/j.clim.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 29.Song WC. Membrane complement regulatory proteins in autoimmune and inflammatory tissue injury. Curr Dir Autoimmun. 2004;7:181–99. doi: 10.1159/000075693. [DOI] [PubMed] [Google Scholar]
- 30.Brodziak A, Ziółko E, Muc-Wierzgoń M, et al. The role of human endogenous retroviruses in the pathogenesis of autoimmune diseases. Med Sci Monit. 2012;18(6):RA80–88. doi: 10.12659/MSM.882892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9:729–40. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 32.Meri S, Jarva H. Complement regulation. Vox Sang. 1998;74:291–302. doi: 10.1111/j.1423-0410.1998.tb05434.x. [DOI] [PubMed] [Google Scholar]
- 33.Frank MM. Complement in the pathophysiology of human disease. N Engl J Med. 1987;316:1525–30. doi: 10.1056/NEJM198706113162407. [DOI] [PubMed] [Google Scholar]
- 34.Lublin DM, Atkinson JP. Decay accelerating factor: biochemistry, molecular biology and function. Ann Rev Immunol. 1987;7:35–38. doi: 10.1146/annurev.iy.07.040189.000343. [DOI] [PubMed] [Google Scholar]
- 35.Mead RJ, Neal JW, Griffiths MR, et al. Deficiency of the complement regulator CD59a enhances disease severity, demyelination and axonal injury in murine acute experimental allergic encephalomyelitis. Lab Invest. 2004;84:21–28. doi: 10.1038/labinvest.3700015. [DOI] [PubMed] [Google Scholar]
- 36.Liu J, Miwa T, Hilliard B, et al. The complement inhibitory protein DAF (CD55) suppresses T cell immunity in vivo. J Exp Med. 2005;201:567–77. doi: 10.1084/jem.20040863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mead RJ, Singhrao SK, Neal JW, et al. The membrane attack complex of complement causes severe demyelination associated with acute axonal injury. J Immunol. 2002;168:458–65. doi: 10.4049/jimmunol.168.1.458. [DOI] [PubMed] [Google Scholar]
- 38.Matsuo S, Nishikage H, Yoshida F, et al. Role of CD59 in experimental glomerulonephritis in rats. Kidney Int. 1994;46:191–200. doi: 10.1038/ki.1994.259. [DOI] [PubMed] [Google Scholar]
- 39.Sogabe H, Nangaku M, Ishibashi Y, et al. Increased susceptibility of decay-accelarating factor deficient mice to anti-glomerular basement membrane glomerulonephritis. J Immunol. 2001;167:2791–97. doi: 10.4049/jimmunol.167.5.2791. [DOI] [PubMed] [Google Scholar]
- 40.Xiao H, Schreiber A, Heeringa P, et al. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmatic autoantibodies. Am J Pathol. 2007;170:52–64. doi: 10.2353/ajpath.2007.060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lidington EA, Haskard DO, Mason JC. Induction of decay-accelerating factor by thrombin through a protease-activated receptor 1 and protein kinase C–dependent pathway protects vascular endothelial cells from complement-mediated injury. Blood. 2000;96:2784–92. [PubMed] [Google Scholar]
- 42.Kaminski HJ, Kusner LL, Richmonds C, et al. Deficiency of decay accelerating factor and CD59 leads to crisis in experimental myasthenia. Exp Neurol. 2006;202:287–93. doi: 10.1016/j.expneurol.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 43.Williams AS, Mizuno M, Richards PJ, et al. Deletion of the gene encoding CD59a in mice increases disease severity in a murine model of rheumatoid arthritis. Arthritis Rheum. 2004;50:3035–44. doi: 10.1002/art.20478. [DOI] [PubMed] [Google Scholar]
- 44.Hoek RM, de Launay D, Kop EN, et al. Deletion of either CD55 or CD97 ameliorates arthritis in mouse models. Arthritis Rheum. 2010;62:1036–42. doi: 10.1002/art.27347. [DOI] [PubMed] [Google Scholar]
- 45.Miwa T, Maldonado MA, Zhou L, et al. Deletion of decay-accelerating factor (CD55) exacerbates autoimmune disease development in MRL/1pr mice. Am J Pathol. 2002;161:1077–86. doi: 10.1016/S0002-9440(10)64268-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miwa T, Maldonado MA, Zhou L, et al. Absence of CD59 exacerbates systemic autoimmunity in MRL/1pr mice. J Immunol. 2012;189:5434–41. doi: 10.4049/jimmunol.1201621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keeling DM, Isenberg DA. Haematological manifestations of systemic lupus erythematosus. Blood Rev. 1993;7:199–207. doi: 10.1016/0268-960x(93)90006-p. [DOI] [PubMed] [Google Scholar]
- 48.Pereira RM, Velloso ER, Menezes Y, et al. Bone marrow findings in systemic lupus erythematosus in patients with peripheral cytopenias. Clin Rheumatol. 1998;17:219–22. doi: 10.1007/BF01451051. [DOI] [PubMed] [Google Scholar]
- 49.Feng CS, Ng MH, Szeto RS, Li EK. Bone marrow findings in lupus patients with pancytopenia. Pathology. 1991;23:5–7. doi: 10.3109/00313029109061430. [DOI] [PubMed] [Google Scholar]
- 50.Baily FA, Lilly M, Bertoli LF, Ball GV. An antibody that inhibits in vitro bone marrow proliferation in a patient with systemic lupus erythematosus and aplastic anemia. Arthritis Rheum. 1989;32:901–5. [PubMed] [Google Scholar]
- 51.Brooks BJ, Jr, Broxmeyer HE, Bryan CF, Leech SH. Serum inhibitor in systemic lupus erythematosus associated with aplastic anemia. Arch Intern Med. 1984;144:1474–77. [PubMed] [Google Scholar]
- 52.Voulgarelis M, Kokori SIG, Ioannidis JPA, et al. Anaemia in systemic lupus erythematosus: aetiological profile and the role of erythropoietin. Ann Rheum Dis. 2000;59:217–22. doi: 10.1136/ard.59.3.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Winfield JB, Fernsten PD, Czyzyk JK. Anti- lymphocyte autoantibodies in systemic lupus erythematosus. Trans Am Clin Climatol Assoc. 1997;108:127–35. [PMC free article] [PubMed] [Google Scholar]
- 54.Wenzel J, Gerdsen R, Uerlich M, et al. Lymphocytopenia in lupus erythematosus: close in vivo association to autoantibodies targeting nuclear antigens. Br J Dermatol. 2004;150:994–98. doi: 10.1111/j.1365-2133.2004.05848.x. [DOI] [PubMed] [Google Scholar]
- 55.Hepburn AL, Narat S, Mason JC. The management of peripheral blood cytopenias in systemic lupus erythematosus. Rheumatology (Oxford) 2010;49:2243–54. doi: 10.1093/rheumatology/keq269. [DOI] [PubMed] [Google Scholar]
- 56.Giannouli S, Voulgarelis M, Ziakas PD, Tzioufas AG. Anemia in systemic lupus erythematosus: from pathophysiology to clinical assessment. Ann Rheum Dis. 2006;65:144–48. doi: 10.1136/ard.2005.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nossent JC, Swaak AJ. Prevalence and significance of haematological abnormalities in patients with systemic lupus erythematosus. Q J Med. 1991;80:605–12. [PubMed] [Google Scholar]
- 58.Tsunoda S, Kawano M, Koni I, et al. Diminished expression of CD59 on activated CD8+ T cells undergoing apoptosis in systemic lupus erythematosus and Sjögren’s syndrome. Scand J Immunol. 2000;51:293–99. doi: 10.1046/j.1365-3083.2000.00674.x. [DOI] [PubMed] [Google Scholar]
- 59.Arora M, Arora R, Tiwari SC, et al. Expression of complement regulatory proteins in diffuse proliferative glomerulonephritis. Lupus. 2000;9:127–31. doi: 10.1191/096120300678828154. [DOI] [PubMed] [Google Scholar]
- 60.Richaud-Patin Y, Perez-Romano B, Carrillo-Maravilla E, et al. Deficiency of red cell bound CD55 and CD59 in patients with systemic lupus erythematosus patients. Immunol Lett. 2003;88:95–99. doi: 10.1016/s0165-2478(03)00066-x. [DOI] [PubMed] [Google Scholar]
- 61.Ruiz-Arguelles A, Llorente L. The role of complement regulatory proteins (CD55 and CD59) in the pathogenesis of autoimmune hemocytopenias. Autoimmunity. 2006;6:155–61. doi: 10.1016/j.autrev.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 62.Garcia-Valladares I, Atisha-Fregoso Y, Richaud-Patin Y, et al. Diminished expression of complement regulatory proteins (CD55 and CD59) in lymphocytes from systemic lupus erythematosus patients with lymphopenia. Lupus. 2006;15:600–5. doi: 10.1177/0961203306071916. [DOI] [PubMed] [Google Scholar]
- 63.Ruiz-Delgado GJ, Vazquez-Garza E, Mendez-Ramirez N, Gomez-Almaguer D. Abnormalities in the expression of CD55 and CD59 surface molecules on peripheral blood cells are not specific to paroxysmal nocturnal hemoglobinuria. Hematology. 2009;14:33–37. doi: 10.1179/102453309X385089. [DOI] [PubMed] [Google Scholar]
- 64.Alegretti AP, Mucenic T, Merzoni J, et al. Expression of CD55 and CD59 on peripheral blood cells from systemic lupus erythematosus (SLE) patients. Cell Immunol. 2010;265:127–32. doi: 10.1016/j.cellimm.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 65.Alegretti AP, Schneider L, Piccoli AK, et al. Diminished expression of complement regulatory proteins on peripheral blood cells from systemic lupus erythematosus patients. Clin Dev Immunol. 2012;2012:725684. doi: 10.1155/2012/725684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jones J, Laffafian I, Cooper AM, et al. Expression of complement regulatory molecules and other surface markers on neutrophils from synovial fluid and blood of patients with rheumatoid arthritis. Br J Rheumatol. 1994;33:707–12. doi: 10.1093/rheumatology/33.8.707. [DOI] [PubMed] [Google Scholar]
- 67.Arora M, Kumar A, Das SN, Srivastava LM. Complement-regulatory protein expression and activation of complement cascade on erythrocytes from patients with rheumatoid arthritis (RA) Clin Exp Immunol. 1998;111:102–6. doi: 10.1046/j.1365-2249.1998.00449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pourazar AA, Andalib AR, Oreizy F, et al. A flowcytometry study of CD55 and CD59 expression on erythrocytes in rheumatoid arthritis patients. IJI. 2005;2:91–96. [Google Scholar]
- 69.Cornillet P, Gredy P, Pennaforte JL, et al. Increased frequency of the long (S) allotype of CR1 (the C3b/C4b receptor, CD35) in patients with systemic lupus erythematosus. Clin Exp Immunol. 1992;89:22–25. doi: 10.1111/j.1365-2249.1992.tb06871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Munoz LE, Lauber K, Schiller M, et al. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Reumatol. 2010;6:280–89. doi: 10.1038/nrrheum.2010.46. [DOI] [PubMed] [Google Scholar]
- 71.Nauta AJ, Raashou-Jensen N, Roos A, et al. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur J Immunol. 2003;33:2853–63. doi: 10.1002/eji.200323888. [DOI] [PubMed] [Google Scholar]
- 72.Ferreira V, Pangburn MK, Cortes C. Complement control protein factor H: the good, the bad and the inadequate. Mol Immunol. 2010;47:2187–97. doi: 10.1016/j.molimm.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wyrick- Glatzel J, MacDonald JK, Chen JJ. Paroxysmal Nocturnal Hemoglobinemia: A molecular definition of the clinical biology of the disorder. Labmed. 2006;37:237–43. [Google Scholar]
- 74.Tooze JA, Marsh JC, Wickham N, et al. Response of aplastic anemia and scleroderma to cyclosporine. Br J Haematol. 1993;85:829–31. doi: 10.1111/j.1365-2141.1993.tb03236.x. [DOI] [PubMed] [Google Scholar]
- 75.Cozzi F, Botsios C, Ostuni P, et al. Adult Henoch-Schönlein purpura with glomerulonephritis and paroxysmal nocturnal haemoglobinuria: an uncommon association. Clin Rheumatol. 2002;21:408–10. doi: 10.1007/s100670200109. [DOI] [PubMed] [Google Scholar]
- 76.Gupta A, Al Fulaij R, Gupta RK, et al. Development of paroxysmal nocturnal haemoglobinuria in systemic lupus erythematosus: un unusual cause of portal vein thrombosis. Lupus. 2009;18:743–46. doi: 10.1177/0961203308100558. [DOI] [PubMed] [Google Scholar]
- 77.Nakamura N, Sugawara T, Shirato K, et al. Paroxysmal nocturnal hemoglobinuria in systemic lupus erythematosus: a case report. J Med Cases. 2011;5:550–52. doi: 10.1186/1752-1947-5-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nakao S, Sugimori C, Yamazaki H. Clinical significance of a small population of Paroxysmal Nocturnal hemoglobinuria- type cells in management of bone marrow failure. Int J Hematol. 2006;84:118–22. doi: 10.1532/IJH97.06077. [DOI] [PubMed] [Google Scholar]
- 79.Miyakawa Y, Yamada A, Kosaka K, et al. Defective immune-adherence (C3b) receptor on erythrocytes from patients with systemic lupus erythematosus. Lancet. 1981;8245:493–97. doi: 10.1016/s0140-6736(81)90882-5. [DOI] [PubMed] [Google Scholar]
- 80.Hernandez-Campo PM, Almeida J, Sanchez ML, et al. Normal patterns of expression of glycosylphosphatidylinositol- anchored proteins on different subsets of peripheral blood cells: A frame of reference for the diagnosis of paroxysmal nocturnal hemoglobinuria. Cytometry B Clin Cytom. 2006;70:71–81. doi: 10.1002/cyto.b.20087. [DOI] [PubMed] [Google Scholar]
- 81.Hernandez-Campo PM, Almeida J, Acevedo MJ, et al. Detailed immunophenotypic characterization of different major and minor subsets of peripheral blood cells in patients with paroxysmal nocturnal hemoglobinuria. Transfusion. 2008;48:1403–14. doi: 10.1111/j.1537-2995.2008.01686.x. [DOI] [PubMed] [Google Scholar]
- 82.Cuida M, Legler DW, Eidsheim M, Jonsson R. Complement regulatory proteins in the salivary glands and saliva of Sjögren’s syndrome patients and healthy subjects. Clin Exp Rheumatol. 1997;15:615–23. [PubMed] [Google Scholar]
- 83.Li SH, Szmitko PE, Weisel RD, et al. C-reactive protein upregulates complement-inhibitory factors in endothelial cells. Circulation. 2004;109:833–36. doi: 10.1161/01.CIR.0000117087.27524.0E. [DOI] [PubMed] [Google Scholar]
- 84.Moutabarrik A, Nakanishi I, Namiki M, et al. Cytokine-mediated regulation of the surface expression of complement regulatory proteins, CD46 (MCP), CD55 (DAF), and CD59 on human vascular endothelial cells. Lymphokine Cytok Res. 1993;12:167–72. [PubMed] [Google Scholar]
- 85.van Beek J, van Meurs M, ‘t Hart BA, et al. Decay- accelerating factor (CD55) is expressed by neurons in response to chronic but not acute autoimmune central nervous system inflammation associated with complement activation. J Immunol. 2005;174:2353–65. doi: 10.4049/jimmunol.174.4.2353. [DOI] [PubMed] [Google Scholar]
- 86.Ripoche J, Sim RB. Loss of complement receptor 1 (CR1) on ageing of erythrocytes. Studies of proteolytic release of the receptor. Biochem J. 1986;235:815–21. doi: 10.1042/bj2350815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Iida K, Whitlow MB, Nussenzweig V. Membrane vesiculation protects erythrocytes from destruction by complement. J Immunol. 1991;147:2638–42. [PubMed] [Google Scholar]
- 88.Pascual M, Lutz HU, Steiger G, Stammler P, Schifferli JA. Release of vesicles enriched in complement receptor 1 from human erythrocytes. J Immunol. 1993;151:397–404. [PubMed] [Google Scholar]
- 89.Araten DJ, Nafa K, Pakdeesuwan K, Luzzato L. Clonal populations of hematopoietic cells with paroxysmal nocturnal hemoglobinuria genotype and phenotype are present in normal individuals. Proc Natl Acad Sci USA. 1999;96:5209–14. doi: 10.1073/pnas.96.9.5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rawstron AC, Rollinson SJ, Richards S, et al. The PNH phenotype cells that emerge in most patients after CAMPATH-1H therapy are present prior to treatment. Br J Haematol. 1999;107:148–53. doi: 10.1046/j.1365-2141.1999.01676.x. [DOI] [PubMed] [Google Scholar]
- 91.Nathalang O, Chuansumrit A, Prayoonwiwat W. Rapid screening of PNH red cell populations using the gel test. Southeast Asian J Trop Med Public Health. 2003;34:887–90. [PubMed] [Google Scholar]
- 92.Gupta R, Pandey P, Choudry R, et al. A prospective comparison of four techniques for diagnosis of paroxysmal nocturnal hemoglobinuria. Int J Lab Hematol. 2007;29:119–26. doi: 10.1111/j.1751-553X.2006.00838.x. [DOI] [PubMed] [Google Scholar]
- 93.Sun X, Funk CD, Deng C, Sahu A, Lambris JD, Song WC. Role of decay- accelerating factor in regulating complement activation on the erythrocyte surface as revealed by gene targeting. Proc Natl Acad Sci USA. 1999;96:628–33. doi: 10.1073/pnas.96.2.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Parker C, Omine M, Richards S, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106:3699–3709. doi: 10.1182/blood-2005-04-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lach-Trifilieff E, Marfurt J, Schwarz S, et al. Complement receptor 1 (CD35) on human reticulocytes: Normal expression in systemic lupus erythematosus and HIV-infected patients. J Immunol. 1999;162:7549–54. [PubMed] [Google Scholar]
- 96.Ross GD, Yount WJ, Walport MJ, et al. Disease-associated loss of erythrocyte complement receptors (CR1, C3b receptors) in patients with systemic lupus erythematosus and other diseases involving autoantibodies and/or complement activation. J Immunol. 1985;135:2005–14. [PubMed] [Google Scholar]
- 97.Cohen JHM, Lutz HU, Pennaforte JL, et al. Peripheral catabolism of CR1 (the C3b receptor, C35) on erythrocytes from healthy individuals and patients with systemic lupus erythematosus (SLE) Clin Exp Immunol. 1992;87:422–28. doi: 10.1111/j.1365-2249.1992.tb03013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Holme E, Fyfe A, Zoma A, et al. Decreased C3b receptors (CR1) on erythrocytes from patients with systemic lupus erythematosus. Clin Exp Immunol. 1986;63:41–48. [PMC free article] [PubMed] [Google Scholar]
- 99.Birmingham DJ, Gavit KF, McCarty SM, et al. Consumption of erythrocyte CR1 (CD35) is associated with protection against systemic lupus erythematosus renal flare. Clin Exp Immunol. 2005;143:274–80. doi: 10.1111/j.1365-2249.2005.02983.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Arora V, Mondal AM, Grover R, et al. Modulation of CR1 transcript in systemic lupus erythematosus (SLE) by IFN-gamma and immune complex. Mol Immunol. 2007;44:1722–28. doi: 10.1016/j.molimm.2006.07.300. [DOI] [PubMed] [Google Scholar]