

Abstract
Break-induced replication (BIR) is the pathway of homologous recombination (HR) conserved from phages to eukaryotes that serves to repair DNA breaks that have only one end. BIR contributes to the repair of broken replication forks and allows telomere lengthening in the absence of telomerase. Nonallelic BIR may lead to translocations and other chromosomal rearrangements. In addition, BIR initiated at sites of microhomology can generate copy-number variations and complex chromosomal changes. The level of mutagenesis associated with DNA synthesis in BIR is significantly higher than during normal replication. These features make BIR a likely pathway to promote bursts of genetic changes that fuel cancer progression and evolution.
Multiple functions of BIR
One of the major types of spontaneous DNA damage is a single-stranded nick. When a replication fork encounters a nick, a single-ended double strand break is formed and needs to be repaired by HR. DNA single end substrates for HR can also form at chromosome ends in telomerase-deficient cells or upon missegregation of fragmented chromosomes. BIR is an efficient way to repair such breaks. BIR is initiated by invasion of a single strand into a homologous DNA molecule followed by DNA synthesis that may continue as far as the next replication fork or even to the end of the chromosome. BIR has been described in various organisms including viruses, bacteria, and eukaryotes (reviewed in [1-4]).
BIR has been examined in great detail in bacteriophage T4, where it is referred to as RDR (for recombination-dependent replication) and represents a mechanism to replicate chromosome ends [2]. The initial round of phage T4 replication is initiated by an R- loop, however lagging strand synthesis never reaches the chromosome ends. This produces 3′-ssDNA overhangs, a common problem of replication of linear chromosomes. These ssDNA substrates are recognized by HR proteins that mediate strand invasion into homologous DNA found either at the end of the same DNA molecule or within other co-infecting phage DNA molecules (Figure 1A). Following strand invasion, cycles of late bacteriophage replication produce multiple T4 chromosomes. RDR is also used in the recovery of broken replication forks.
Figure 1. Multiple functions of BIR.
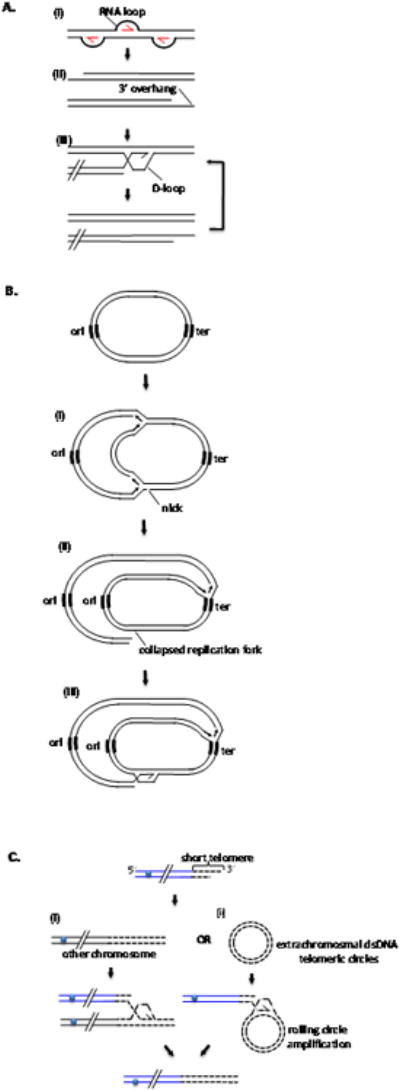
A. Replication of T4 bacteriophage. (i) Several R-loop structures mediate initial rounds of DNA replication. (ii) End-replication problem of lagging strand, collapsed replication forks or resection leave 3′ overhangs that initiate rounds of recombination mediated replication (RDR). (iii) Strand invasion occurring at homologous sequence present at the other end of the same molecule (this is possible because T4 chromosome is terminally redundant; not shown) or at internal part of co-infected molecule (shown) as T4 DNA is circularly permuted. B. Repair of broken replication forks in bacteria. (i) Replication fork runs into a nick and collapses (ii). (iii) Repair of collapsed fork by BIR. Since there is only one origin of replication (ori) extensive synthesis has to be primed from repaired replication fork to terminator (ter). C. Telomere lengthening by recombination. (i) BIR proceeds between telomeres of different chromosomes. (ii) BIR by rolling circle mechanism with extrachromosomal dsDNA telomeric circles.
In E. coli, which does not experience an end replication problem due to the circular nature of its genome, RDR functions to recover broken replication forks (Figure 1B). The recovery begins with the invasion of the sister chromatid by the broken chromosome end followed by the assembly of a new replication fork (reviewed in [5]). This is an essential process for E.coli because its replication involves just two replication forks moving in opposite directions from a single replication origin and terminating at a single locus, affording no opportunity for complete genome duplication upon replication fork failure (Figure 1B).
Eukaryotic cells are less dependent on BIR for completion of scheduled replication, as they rely on telomerase to solve the end replication problem. Moreover, eukaryotes have multiple replication origins per chromosome, eliminating the need for extensive DNA synthesis from a broken fork. Nevertheless, BIR has been preserved in eukaryotes and under certain circumstances becomes essential (reviewed in [3,4]). BIR may play an important role in the recovery of collapsed replication forks, particularly in regions where there is no replication fork coming from the opposite direction, such as at subtelomeric regions. In the yeast Saccharomyces cerevisiae and likely in most eukaryotes that lack telomerase, BIR plays an essential role in maintaining chromosome ends (Figure 1C) [6,7]. BIR can also efficiently repair double-strand breaks that are created in such a way that only one end can find homology in the genome, or when both ends can find homology, but in different places [7-9]. Several recombination assays were developed in yeast to study the mechanisms and enzymology of BIR, while in higher eukaryotes such a convenient experimental system is currently not available [7,10,11]. However, recent studies using Xenopus laevis egg extracts directly demonstrate the existence of BIR and its ability to re-start broken replication forks in vertebrates [12].
Molecular mechanism of BIR
BIR, like other HR processes, is initiated by strand invasion mediated by RecA in bacteria or Rad51 in eukaryotes to form a displacement loop (D-loop) [8,9,13,14]. However, what follows strand invasion, specifically replication fork assembly and extensive DNA synthesis, distinguishes BIR from other HR pathways. Proteins important for BIR in phage, bacteria and eukaryotes are listed in Figure 2. In bacteria, the mediator protein PriA recognizes the D-loop and with the help of several additional proteins promotes loading of the replicative helicase dnaB and primase dnaG onto the D-loop [5]. Once helicase and primase are loaded, the polymerase Pol III is recruited along with a clamp, and DNA synthesis is initiated (Figure 2B).
Figure 2. Assembly of functional replication forks during BIR.
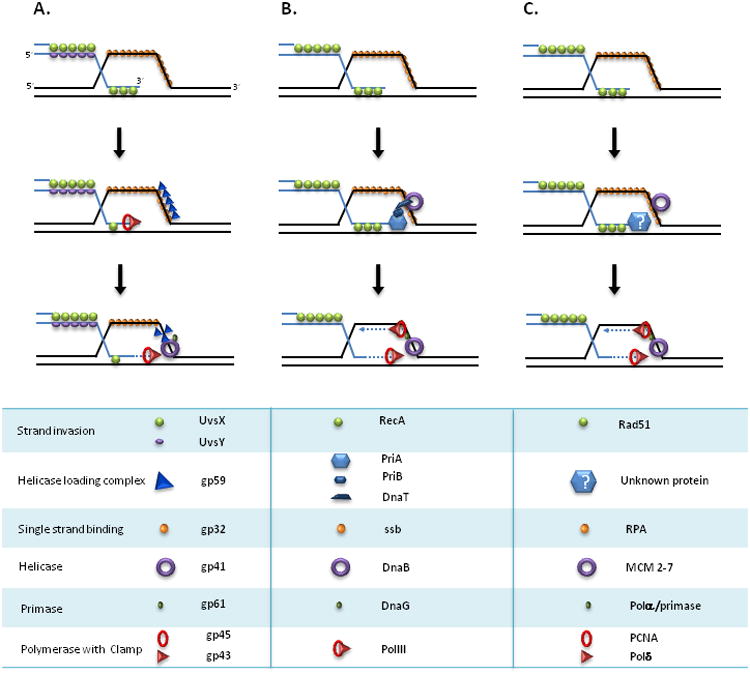
A. Initiation of RDR replication in bacteriophage T4. Formation of a D-loop by strand invasion of ssDNA bound to UvsX (RecA-homolog) and UvsY (mediator protein). Gp59 (helicase loading protein) is recruited to the D-loop coated by gp32. Gp59 recruits replicative helicase, gp41, which promotes unwinding of the parent duplex. Polymerase gp43 (together with sliding clamp (gp45) is recruited to the 3′-end of the invading strand and initiates leading strand synthesis. Recruitment of primase gp61 may initiate lagging strand synthesis (not shown). B. Initiation of RDR in E. coli. RecA-mediated strand invasion leads to the formation of a D-loop covered with SSB. PriA recognizes and binds a three-way junction formed by a D-loop and the 3′-end of the invading strand. PriA binding initiates formation of PriA-PriB-DnaT-D-loop complex, which recruits replicative helicase DnaB to the D-loop. DnaB then recruits primase DnaG. Next, Polymerase III is loaded to initiate leading and lagging strand synthesis. C. Initiation of BIR in yeast. Rad51-mediated strand invasion leads to the formation of a D-loop. Initiation of BIR involves the main replicative helicase MCM2-7(shown) and the majority of other proteins required for initiation of S-phase (not shown). The blue hexagon with a question mark represents an unknown hypothetical protein that initiates recruitment of MCMs to the D-loop (similar to PriA and gp59). DNA synthesis associated with BIR is carried out by Polδ, while the exact role of Polε remains unclear.
In yeast and other eukaryotes, replication fork assembly during BIR is not well understood, and the functional counterparts of PriA are yet to be identified. In BIR repair that occurs outside the context of a collapsed replication fork, initial DNA synthesis is a very slow process [8] that involves the replicative helicase Mcm2-7 (Figure 2C) and almost all of the proteins that normally start the S-phase replication fork, with the exception of Orc and Cdc6. Pol δ also plays an essential role in BIR, whereas the role of Pol ε is still under debate [7]. Thus far investigations of BIR have been limited to DNA damage checkpoint-arrested G2/M cells [8], as an experimental system to study BIR during S-phase of the cell cycle has not yet been developed. It remains unknown how cells assemble an active replication fork at a DSB in G2/M phase because cells normally tightly regulate Mcm2-7 activity to prevent unscheduled replication licensing [15] and certain components of replication are inhibited upon DNA damage [16,17]. Importantly, it has been shown that Pif1 helicase is needed for efficient BIR while it is mostly dispensable for regular replication [18]. The recent data demonstrate that Pif1 is required for the proper recruitment of Polδ to the D-loop and is capable of promoting Polδ-mediated synthesis via bubble migration (Grzegorz Ira and Patrick Sung, manuscript submitted). Further studies are needed to evaluate the respective roles of Pif1 and Mcm2-7 helicases in BIR.
Two major models were postulated to describe the nature of the BIR replication fork. In the first model, the D-loop is resolved to form a normal replication fork associated with synchronous leading and lagging strand synthesis and semi-conservative inheritance of newly-synthesized strands [7,8,19]. (Figure 3A). In the second model, BIR is carried out by a migrating D-loop (bubble), where branch migration of an unresolved HJ promotes displacement of a newly synthesized strand and therefore leads to conservative inheritance of newly synthesized strands [10], (reviewed in [3,20]) (Figure 3B, C). Testing the proposed models of BIR is the focus of ongoing experiments in model systems including bacteriophage, bacteria and budding yeast.
Figure 3. Models for BIR and associated mutagenesis.
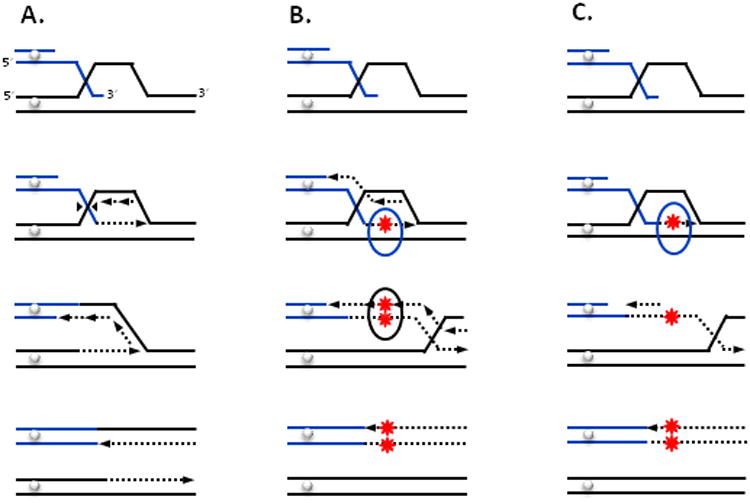
A. Single end invasion of 3′-end leads to the formation of a Holliday junction (HJ). Resolution of HJ close to the position of strand invasion leads to the establishment of a unidirectional replication fork, which carries out semi-conservative DNA synthesis. BIR proceeding in this mode is not expected to be mutagenic. B HJ remains unresolved, and BIR proceeds via D-loop migration associated with synchronous synthesis of the leading and lagging strands. Both newly synthesized strands are displaced from their templates, which leads to their conservative inheritance. C. Similar to shown in B, BIR proceeds via D-loop migration associated with conservative inheritance of newly synthesized strands. However, leading strand is synthesized first, while lagging strand synthesis is delayed and proceeds using the leading strand as a template after its displacement. Replication error (for example, nucleotide mis-incorporation; one star) remains uncorrected due to a quick dissociation of newly synthesized DNA from its template (blue oval) (B,C). Replication error (nucleotide mis-incorporation) is present in both strands of DNA (two stars) due to copying of error (C) or due to inability of MMR to discriminate between two newly synthesized strands (B, black oval).
Several in vitro studies have been undertaken to model DNA synthesis associated with BIR. Initial results from a reconstituted bacteriophage T4 recombination assay supported the D-loop migration mechanism [14]. However, these experiments were inconclusive because a protein essential for the recruitment of the replicative helicase and for initiation of lagging strand DNA synthesis was not included in the reaction [2]. Subsequent studies that included all necessary enzymes supported a model of synchronous synthesis of leading and lagging strands (see [21]). Analogous in vitro studies performed with a complete set of purified E.coli proteins also provided support for this model. Thus, it was proposed that DNA synthesis in BIR proceeds via a semi-conservative mechanism as in S-phase replication [22].
Other attempts have been made to determine the mode of BIR synthesis in vivo. Analysis of phage lambda recombination in E. coli using isotope density transfer implicated conservative segregation of newly synthesized strands [23]. To address this question in yeast, we used dynamic molecular combing coupled with FISH to analyze BIR products and found that conservative inheritance of DNA strands predominates (Malkova and Lobachev, manuscript submitted). Similarly, during gap repair that shares some similarities with the BIR pathway [24], a conservative mode of strand inheritance was observed [25].
BIR promotes hyper-mutability
Deem et al. [26] determined that BIR is extremely mutagenic, as the frequency of frameshift mutations associated with BIR was 1000 times higher as compared to S-phase replication. Importantly, Pol δ, the main replicative polymerase, was responsible for the majority of mutations induced by BIR. One possible reason for the reduced fidelity of Pol δ could be the bubble-migration mechanism that drives BIR. Thus, during bubble migration, the newly synthesized DNA is quickly dissociated from its template, likely interfering with mismatch repair (MMR) that relies on the recognition of mismatches between the “old” and “new” DNA strands (Figure 3B,C). Consistently, it was observed that while MMR is active during BIR, it corrects errors less efficiently than it does during S-phase [26]. Similar model was proposed by Kuzminov to explain increased mutagenesis associated with recombination in bacteria [27].
BIR promotes structural chromosome changes
Several classes of chromosomal rearrangements including template switching, translocations and half-crossovers result from BIR. It is likely that these instabilities are promoted by frequent interruptions of DNA synthesis during BIR (Figure 4E).
Figure 4. BIR-induced chromosomal rearrangements.
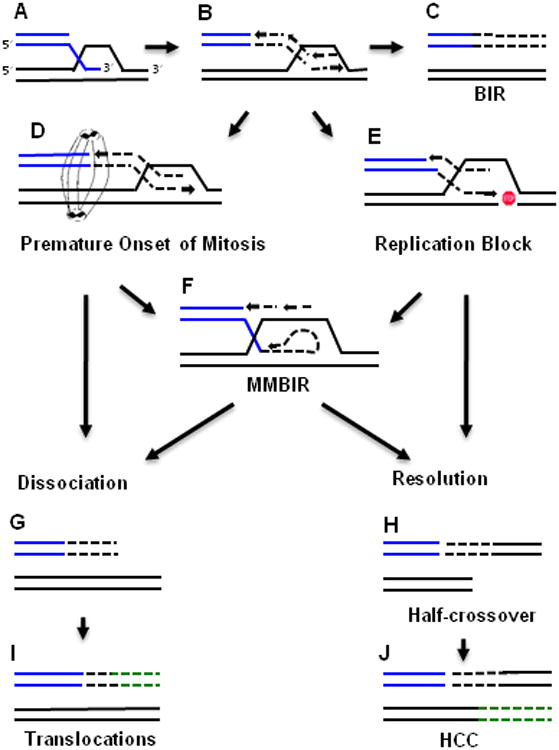
A scheme indicating proposed pathways of BIR-induced instability. A. Invasion of 3′-ssDNA into homologous DNA molecule. B. Replication via migrating bubble associated with conservative inheritance of newly synthesized strands (C). D. Premature onset of mitosis due to checkpoint failure interfering with completion of BIR. E. A pause during BIR replication (indicated by “STOP” sign). D and E can lead to one of the following: (i) F. a switch to MMBIR; (ii) G. dissociation of a newly synthesized strand; (iii) H. HJ resolution leading to a half-crossover. I. Translocation resulting from recombination with homologous sequences at ectopic position. J. Half-crossover inducing half-crossover cascades (HCC) where template gets broken and itself initiates second round of recombination. Here broken template gets stabilized by recombination with homologous sequences at ectopic position.
Template switching
The Symington lab investigated BIR in yeast diploid cells and observed frequent switches of BIR between two homologous DNA templates [10]. Switching was especially frequent close to the place of BIR initiation, but subsided after the first 10 kb of synthesis. The authors proposed that BIR is initiated by an unstable replication fork, which is later transformed into another “stabilized” form. It was suggested that template switching proceeds through multiple rounds of dissociation and re-invasion events at the beginning of BIR. However, the specific mechanisms of multiple strand invasions and D-loop displacements and transition to a stable replication fork remain unknown.
Nonrecurrent copy number change
All organisms, particularly higher eukaryotes, show differences in the copy number of some segments of the genome which is referred to as copy number variation (CNV) [15]. Non-allelic homologous recombination between low copy repeats is the mechanism for recurrent CNVs, whereas the mechanism of frequently observed nonrecurrent CNVs is not well understood. Recent studies in bacteria, yeast, worms, and humans implicate a replicative mechanism with similarities to BIR in nonrecurrent CNVs. These rearrangements are likely initiated by DSB ends such as broken replication forks or severely shortened telomere ends, and involve extensive DNA synthesis, frequent template switches and, in yeast, require Pol32, all of which are hallmarks of BIR [28-31]. The major distinction between this CNV mechanism and BIR is that 2-15 bp microhomologies mark the endpoints of these complex rearrangements, which excludes Rad51-mediated strand invasion. Based on all of these features, the mechanism was named microhomology-mediated BIR (MMBIR) (Figure 4F) (reviewed in [20]). It is currently not known under what circumstances a cell would switch from extensive homology- to microhomology-driven BIR. However, stress and loss of HR component(s) likely play important roles [20].
Translocation
When BIR is initiated by the invasion of a broken DNA end at a non-allelic position, it leads to non-reciprocal translocations (Figure 4I) [7,10,32]. Ectopic invasions occur at positions of DNA repeats, which are highly dispersed throughout the genome [8,33]. Translocations are especially frequent when BIR occurs in the absence of Rad51, but are also common in its presence [33,34].
Half-crossover (HC)
HCs result from fusions between portions of recombining donor and recipient chromosomes, while other portions of the participating chromosomes are lost (Figure 4H) [35-37]. HCs are frequently observed following initiation of BIR in Pol δ mutants that successfully undergo strand invasion but fail to initiate DNA synthesis [11,38]. This process leads to the accumulation of joint molecules that are probably processed by nucleases to form HC products. Although HCs are more frequent in mutants, they are also observed in wild-type cells [11,38]. This raises the possibility that uninterrupted DNA synthesis during BIR is important to prevent premature resolution into HC outcomes. Besides interrupted DNA synthesis, checkpoint deficiency leads to the formation of HC products likely due to the untimely onset of mitosis (Figure 4D), which interrupts BIR (Vasan and Malkova, in preparation for publication). A dangerous feature of HCs is that they lead to the breakage of a previously intact donor chromosome, which can potentially initiate cycles of HCs called half-crossover cascades (HCC) (Figure 4J).
BIR and human disease
BIR has not been systematically studied in mammals, but it is likely that several disease-associated phenomena, including alternative lengthening of telomeres, non-reciprocal translocations and complex chromosomal rearrangements, result from BIR.
Alternative lengthening of telomeres (ALT)
Unlimited proliferation of cancer cells causes shortening of telomeres with each round of replication. To counteract telomere attrition, most cancer cells up-regulate telomerase activity, but the remaining 5-15% extend telomeres via recombination. Several lines of evidence support telomere recombination in the ALT pathway (reviewed in [39]). First, a DNA tag at a single telomere is frequently copied to other chromosomal ends in ALT cells. Second, extrachromosomal linear and circular telomere fragments that can be used as templates in recombination are present in ALT cells. Third, telomeres have highly heterogeneous sizes that can be explained by recombination. Finally, telomere elongation and maintenance depends on a number of recombination proteins.
The mechanism of telomere recombination very likely resembles BIR, as there is only one end that invades and copies telomere templates which include the opposite end of the same chromosome, a sister chromatid, or an extrachromosomal telomere fragment. Indeed, rolling-circle replication-mediated telomere amplification was demonstrated in yeast and in human cell lines [40,41]. Further evidence for BIR as the mechanism of ALT comes from studies in yeast where efficient telomere recombination required Pol32 and Pif1, proteins also participating in BIR [7,18,42]. Surprisingly, the Reddel laboratory demonstrated recently that ALT functions even in normal somatic mammalian cells [43]. How cancer cells up-regulate the ALT pathway is not yet understood, but loss of the ATRX/DAXX chromatin remodeling complex that forms heterochromatin at telomeres is a common feature of most ALT cancers [44].
Chromothripsis
The occurrence of multiple genomic rearrangements within a cell is a well-accepted hallmark of cancer cells. However, the recent discovery of a new phenomenon called chromothripsis demonstrated that the complexity of chromosomal change that leads to cancer could be more intricate than previously appreciated [45,46]. The unique feature of chromothripsis is that many rearrangements are localized to a single chromosome and appear to result from one catastrophic event that initiated reshuffling of this chromosome. It was believed at first that chromothripsis was mediated by nonhomologous end-joining (NHEJ) repair events [45,46]. However, recent studies of several neurological disorders demonstrated that the illnesses were often caused by complex chromosomal rearrangements that were structurally similar to those associated with chromothripsis, and also showed similarities to changes attributed to MMBIR [47]. Since then, several other groups have published evidence supporting the role of MMBIR in the formation of complex chromosomal rearrangements and possibly in chromothripsis [48-53].
Non-reciprocal translocation (NRT)
NRTs are frequently observed in cancer cells. One well-known example is a pathway of telomere acquisition described in mammalian tumors where broken chromosomes initiate recombination with an intact donor, which in turn leads to the breakage of the donor [54]. This destabilization of the donor initiates a self-perpetuating cycle of genome-destabilizing events that results in multiple chromosomal rearrangements. Even though the exact molecular mechanism mediating NRT cycles remains undefined, it is possible that NRTs are promoted by half-crossover cascades resulting from aberrant processing of BIR intermediates (Figure 4J).
Conclusions
Break induced replication remains one of the least characterized pathways of DSB repair. Although BIR plays a positive role in repairing DSBs, it can alternatively be a dangerous source of several types of genetic instabilities. Tremendous progress in whole genome analysis revealed that BIR is likely the mechanism of multiple genomic rearrangements in all eukaryotes, including humans. To date, there is no clear understanding of how BIR can be transformed from a beneficial pathway aimed at rescuing cells into a dangerous mechanism with high destabilizing potential. In addition, it remains unclear how eukaryotic cells suppress BIR repair of two-ended breaks.
As BIR constitutes the mechanism of telomere maintenance in telomerase-negative cancers, which often lack effective treatment, enzymes promoting BIR remain attractive targets for cancer therapy. Therefore identification and characterization of enzymes specifically needed for BIR serves as an immediate goal. In addition, while BIR induced by DSBs outside the context of a replication fork was extensively studied, the repair of single-end DSBs at collapsed replication forks in eukaryotes remains unexplored. Most importantly, BIR needs to be further investigated in mammals, where our knowledge of BIR remains elusive. In particular, it is critical to further characterize the role that BIR plays in promoting various human diseases.
Acknowledgments
AM is funded by NIH grant GM084242. GI is funded by NIH grant GM080600.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
• of special interest.
- 1.Maher RL, Branagan AM, Morrical SW. Coordination of DNA replication and recombination activities in the maintenance of genome stability. Journal of cellular biochemistry. 2011;112:2672–2682. doi: 10.1002/jcb.23211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kreuzer KN, Brister JR. Initiation of bacteriophage T4 DNA replication and replication fork dynamics: a review in the Virology Journal series on bacteriophage T4 and its relatives. Virol J. 2010;7:358. doi: 10.1186/1743-422X-7-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Llorente B, Smith CE, Symington LS. Break-induced replication: what is it and what is it for? Cell Cycle. 2008;7:859–864. doi: 10.4161/cc.7.7.5613. [DOI] [PubMed] [Google Scholar]
- 4.McEachern MJ, Haber JE. Break-induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem. 2006;75:111–135. doi: 10.1146/annurev.biochem.74.082803.133234. [DOI] [PubMed] [Google Scholar]
- 5.Marians KJ. PriA-directed replication fork restart in Escherichia coli. Trends in biochemical sciences. 2000;25:185–189. doi: 10.1016/s0968-0004(00)01565-6. [DOI] [PubMed] [Google Scholar]
- 6.Le S, Moore JK, Haber JE, Greider CW. RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics. 1999;152:143–152. doi: 10.1093/genetics/152.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lydeard JR, Jain S, Yamaguchi M, Haber JE. Break-induced replication and telomerase- independent telomere maintenance require Pol32. Nature. 2007;448:820–823. doi: 10.1038/nature06047. [DOI] [PubMed] [Google Scholar]
- 8.Malkova A, Naylor ML, Yamaguchi M, Ira G, Haber JE. RAD51-dependent break-induced replication differs in kinetics and checkpoint responses from RAD51 -mediated gene conversion. Mol Cell Biol. 2005;25:933–944. doi: 10.1128/MCB.25.3.933-944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis AP, Symington LS. RAD51-dependent break-induced replication in yeast. Mol Cell Biol. 2004;24:2344–2351. doi: 10.1128/MCB.24.6.2344-2351.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith CE, Llorente B, Symington LS. Template switching during break-induced replication. Nature. 2007;447:102–105. doi: 10.1038/nature05723. [DOI] [PubMed] [Google Scholar]
- 11.Deem A, Barker K, Vanhulle K, Downing B, Vayl A, Malkova A. Defective break-induced replication leads to half-crossovers in Saccharomyces cerevisiae. Genetics. 2008;179:1845–1860. doi: 10.1534/genetics.108.087940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12•.Hashimoto Y, Costanzo V. Studying DNA replication fork stability in Xenopus egg extract. Methods Mol Biol. 2011;745:437–445. doi: 10.1007/978-1-61779-129-1_25. This paper provides the first evidence of BI R-mediated restart of collapsed replication forks in vertebrates. The addition of Xenopus egg extracts to a nicked DNA template initiated DNA replication creating a one-ended DSB that was repaired by BIR. [DOI] [PubMed] [Google Scholar]
- 13.Asai T, Sommer S, Bailone A, Kogoma T. Homologous recombination-dependent initiation of DNA replication from DNA damage-inducible origins in Escherichia coli. Embo J. 1993;12:3287–3295. doi: 10.1002/j.1460-2075.1993.tb05998.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Formosa T, Alberts BM. DNA synthesis dependent on genetic recombination: characterization of a reaction catalyzed by purified bacteriophage T4 proteins. Cell. 1986;47:793–806. doi: 10.1016/0092-8674(86)90522-2. [DOI] [PubMed] [Google Scholar]
- 15.Kidd JM, Graves T, Newman TL, Fulton R, Hayden HS, Malig M, Kallicki J, Kaul R, Wilson RK, Eichler EE. A human genome structural variation sequencing resource reveals insights into mutational mechanisms. Cell. 2010;143:837–847. doi: 10.1016/j.cell.2010.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lopez-Mosqueda J, Maas NL, Jonsson ZO, Defazio-Eli LG, Wohlschlegel J, Toczyski DP. Damage-induced phosphorylation of Sld3 is important to block late origin firing. Nature. 2010;467:479–483. doi: 10.1038/nature09377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zegerman P, Diffley JF. Checkpoint-dependent inhibition of DNA replication initiation by Sld3 and Dbf4 phosphorylation. Nature. 2010;467:474–478. doi: 10.1038/nature09373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18•.Chung WH, Zhu Z, Papusha A, Malkova A, Ira G. Defective resection at DNA double- strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genet. 2010;6:e1000948. doi: 10.1371/journal.pgen.1000948. This paper demonstrates that BIR is associated with extensive resection of DSB and that Pif1 helicase is required for BIR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19•.Lydeard JR, Lipkin-Moore Z, Sheu YJ, Stillman B, Burgers PM, Haber JE. Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev. 2010;24:1133–1144. doi: 10.1101/gad.1922610. This paper demonstrated that initiation of BI R in yeast involves the majority of proteins required to begin S-phase DNA replication, including Cdc7, Cdt1, Msm2-7, Cdc45, and GINs, but does not require Cdc6 and ORC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hastings PJ, Ira G, Lupski JR. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet. 2009;5:e1000327. doi: 10.1371/journal.pgen.1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kreuzer KN. Recombination-dependent DNA replication in phage T4. Trends Biochem Sci. 2000;25:165–173. doi: 10.1016/s0968-0004(00)01559-0. [DOI] [PubMed] [Google Scholar]
- 22.Xu L, Marians KJ. PriA mediates DNA replication pathway choice at recombination intermediates. Mol Cell. 2003;11:817–826. doi: 10.1016/s1097-2765(03)00061-3. [DOI] [PubMed] [Google Scholar]
- 23.Motamedi MR, Szigety SK, Rosenberg SM. Double-strand-break repair recombination in Escherichia coli: physical evidence for a DNA replication mechanism in vivo. Genes Dev. 1999;13:2889–2903. doi: 10.1101/gad.13.21.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain S, Sugawara N, Lydeard J, Vaze M, Tanguy Le Gac N, Haber JE. A recombination execution checkpoint regulates the choice of homologous recombination pathway during DNA double-strand break repair. Genes Dev. 2009;23:291–303. doi: 10.1101/gad.1751209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ira G, Satory D, Haber JE. Conservative inheritance of newly synthesized DNA in double-strand break-induced gene conversion. Mol Cell Biol. 2006;26:9424–9429. doi: 10.1128/MCB.01654-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26•.Deem A, Keszthelyi A, Blackgrove T, Vayl A, Coffey B, Mathur R, Chabes A, Malkova A. Break-induced replication is highly inaccurate. PLoS Biol. 2011;9:e1000594. doi: 10.1371/journal.pbio.1000594. This paper demonstrated that the level of frameshift mutagenesis associated with BIR is 1000 times higher as compared to normal S-phase replication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuzminov A. Collapse and repair of replication forks in Escherichia coli. Mol Microbiol. 1995;16:373–384. doi: 10.1111/j.1365-2958.1995.tb02403.x. [DOI] [PubMed] [Google Scholar]
- 28.Lee JA, Carvalho CM, Lupski JR. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 29.Payen C, Koszul R, Dujon B, Fischer G. Segmental duplications arise from Pol32- dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet. 2008;4:e1000175. doi: 10.1371/journal.pgen.1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yatsenko SA, Hixson P, Roney EK, Scott DA, Schaaf CP, Ng YT, Palmer R, Fisher RB, Patel A, Cheung SW, et al. Human subtelomeric copy number gains suggest a DNA replication mechanism for formation: beyond breakage-fusion-bridge for telomere stabilization. Hum Genet. 2012;131:1895–1910. doi: 10.1007/s00439-012-1216-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lowden MR, Flibotte S, Moerman DG, Ahmed S. DNA synthesis generates terminal duplications that seal end-to-end chromosome fusions. Science. 2011;332:468–471. doi: 10.1126/science.1199022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bosco G, Haber JE. Chromosome break-induced DNA replication leads to nonreciprocal translocations and telomere capture. Genetics. 1998;150:1037–1047. doi: 10.1093/genetics/150.3.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vanhulle K, Lemoine FJ, Narayanan V, Downing B, Hull K, McCullough C, Bellinger M, Lobachev K, Petes TD, Malkova A. Inverted DNA repeats channel repair of distant double-strand breaks into chromatid fusions and chromosomal rearrangements. Mol Cell Biol. 2007 doi: 10.1128/MCB.01740-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Downing B, Morgan R, VanHulle K, Deem A, Malkova A. Large inverted repeats in the vicinity of a single double-strand break strongly affect repair in yeast diploids lacking Rad51. Mutat Res. 2008;645:9–18. doi: 10.1016/j.mrfmmm.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haber JE, Hearn M. Rad52-independent mitotic gene conversion in Saccharomyces cerevisiae frequently results in chromosomal loss. Genetics. 1985;111:7–22. doi: 10.1093/genetics/111.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Palgrave CJ, Lu ZH, Lowden CS, Whitelaw CB. Dynein light chain 1 (LC8) sequence is highly conserved between pig species. Anim Genet. 2011;42:337. doi: 10.1111/j.1365-2052.2010.02143.x. [DOI] [PubMed] [Google Scholar]
- 37.Malkova A, Ivanov EL, Haber JE. Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc Natl Acad Sci U S A. 1996;93:7131–7136. doi: 10.1073/pnas.93.14.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith CE, Lam AF, Symington LS. Aberrant double-strand break repair resulting in half crossovers in mutants defective for Rad51 or the DNA polymerase delta complex. Mol Cell Biol. 2009;29:1432–1441. doi: 10.1128/MCB.01469-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nature reviews Genetics. 2010;11:319–330. doi: 10.1038/nrg2763. [DOI] [PubMed] [Google Scholar]
- 40.Henson JD, Cao Y, Huschtscha LI, Chang AC, Au AY, Pickett HA, Reddel RR. DNA C- circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat Biotechnol. 2009;27:1181–1185. doi: 10.1038/nbt.1587. [DOI] [PubMed] [Google Scholar]
- 41.Tomaska L, Nosek J, Kramara J, Griffith JD. Telomeric circles: universal players in telomere maintenance? Nat Struct Mol Biol. 2009;16:1010–1015. doi: 10.1038/nsmb.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dewar JM, Lydall D. Pif1- and Exo1-dependent nucleases coordinate checkpoint activation following telomere uncapping. EMBO J. 2010;29:4020–4034. doi: 10.1038/emboj.2010.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43•.Neumann AA, Watson CM, Noble JR, Pickett HA, Tam PP, Reddel RR. Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev. 2013;27:18–23. doi: 10.1101/gad.205062.112. This paper provided the first evidence of alternative telomere lengthening (ALT) occuring in normal mammalian cells. Authors proposed that ALT activity is a normal part of telomere biology in mammalian cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44•.Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, Bettegowda C, Rodriguez FJ, Eberhart CG, Hebbar S, et al. Altered telomeres in tumors with ATRX and DAXX mutations. Science. 2011;333:425. doi: 10.1126/science.1207313. This paper demonstrates that the loss of chromatin remodeling protein ATRX activates an alternative telomere maintenance in tumors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47•.Liu P, Erez A, Nagamani SC, Dhar SU, Kolodziejska KE, Dharmadhikari AV, Cooper ML, Wiszniewska J, Zhang F, Withers MA, et al. Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell. 2011;146:889–903. doi: 10.1016/j.cell.2011.07.042. This paper demonstrated that complex genomic rearrangements (CGRs) in humans, similar to those associated with cancer chromothripsis, are created by a replication mechanism. The authors proposed that varyous CGRs, including those observed in chromothripsis, are produced by microhomology-mediated BIR (MMBIR) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vissers LE, Bhatt SS, Janssen IM, Xia Z, Lalani SR, Pfundt R, Derwinska K, de Vries BB, Gilissen C, Hoischen A, et al. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Human molecular genetics. 2009;18:3579–3593. doi: 10.1093/hmg/ddp306. [DOI] [PubMed] [Google Scholar]
- 49.Motobayashi M, Nishimura-Tadaki A, Inaba Y, Kosho T, Miyatake S, Niimi T, Nishimura T, Wakui K, Fukushima Y, Matsumoto N, et al. Neurodevelopmental features in 2q23.1 microdeletion syndrome: report of a new patient with intractable seizures and review of literature. American journal of medical genetics Part A. 2012;158A:861–868. doi: 10.1002/ajmg.a.35235. [DOI] [PubMed] [Google Scholar]
- 50.Bondurand N, Fouquet V, Baral V, Lecerf L, Loundon N, Goossens M, Duriez B, Labrune P, Pingault V. Alu-mediated deletion of SOX10 regulatory elements in Waardenburg syndrome type 4. European journal of human genetics : EJHG. 2012 doi: 10.1038/ejhg.2012.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang F, Khajavi M, Connolly AM, Towne CF, Batish SD, Lupski JR. The DNA replication FoSTeS/MMBIR mechanism can generate genomic, genic and exonic complex rearrangements in humans. Nature genetics. 2009;41:849–853. doi: 10.1038/ng.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lawson AR, Hindley GF, Forshew T, Tatevossian RG, Jamie GA, Kelly GP, Neale GA, Ma J, Jones TA, Ellison DW, et al. RAF gene fusion breakpoints in pediatric brain tumors are characterized by significant enrichment of sequence microhomology. Genome Res. 2011;21:505–514. doi: 10.1101/gr.115782.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marechal A, Parent JS, Veronneau-Lafortune F, Joyeux A, Lang BF, Brisson N. Whirly proteins maintain plastid genome stability in Arabidopsis. Proceedings of the National Academy of Sciences of the United States of America; 2009; pp. 14693–14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sabatier L, Ricoul M, Pottier G, Murnane JP. The loss of a single telomere can result in instability of multiple chromosomes in a human tumor cell line. Mol Cancer Res. 2005;3:139–150. doi: 10.1158/1541-7786.MCR-04-0194. [DOI] [PubMed] [Google Scholar]