

Abstract
During DNA repair by homologous recombination (HR), DNA synthesis copies information from a template DNA molecule. Multiple DNA polymerases have been implicated in repair-specific DNA synthesis1–3, but it has remained unclear whether a DNA helicase is involved in this reaction. A good candidate is Pif1, an evolutionarily conserved helicase in S. cerevisiae important for break-induced replication (BIR)4 as well as HR-dependent telomere maintenance in the absence of telomerase5 found in 10–15% of all cancers6. Pif1 plays a role in DNA synthesis across hard-to-replicate sites7, 8 and in lagging strand synthesis with Polδ9–11. Here we provide evidence that Pif1 stimulates DNA synthesis during BIR and crossover recombination. The initial steps of BIR occur normally in Pif1-deficient cells, but Polδ recruitment and DNA synthesis are decreased, resulting in premature resolution of DNA intermediates into half crossovers. Purified Pif1 protein strongly stimulates Polδ-mediated DNA synthesis from a D-loop made by the Rad51 recombinase. Importantly, Pif1 liberates the newly synthesized strand to prevent the accumulation of topological constraint and to facilitate extensive DNA synthesis via the establishment of a migrating D-loop structure. Our results uncover a novel function of Pif1 and provide insights into the mechanism of HR.
To understand how Pif1 promotes BIR, we used an established system wherein only one end of a site-specific DSB has extensive homology to the donor sequence, so that most cells (>80%) employ BIR for repair12. Following strand invasion, over 100 kb of the full length chromosome III donor is copied. Chromosomal markers provide a means to determine the frequency of BIR or alternative mechanisms by growth on selective media (Fig. 1a). BIR was evaluated in pif1-m2 cells, wherein the mutant Pif1 protein is excluded from the nucleus but retains mitochondrial function,13 or in pif1∆ cells.
Figure 1. Pif1 promotes DNA synthesis during BIR.

a. Schematic of the BIR assay. Products are distinguished by genetic markers. b. Repair outcomes in WT and indicated mutant cells. c. Quantification of Southern blot band intensities corresponding to the BIR product and template chromosome in WT and pif1∆ cells. d. Analysis of initial DNA synthesis by PCR. e. ChIP analysis of Pif1-13xMyc recruitment at the indicated loci. f. DSB recruitment of the indicated polymerases during BIR as measured by ChIP. Plotted are the mean values ±SD from at least three independent experiments.
Cells lacking Pif1 are BIR deficient and have a large increase in half crossover products (Fig. 1b) with pif1∆ showing a greater impairment, likely because the pif1-m2 protein retains residual nuclear activity13. The Pif1 helicase activity is indispensable for BIR as revealed by testing the helicase dead pif1-K264A mutant (Fig. 1b). Southern blot analysis showed loss of the template chromosome in pif1∆ consistent with an increase in half crossover products (Fig. 1c, Extended Data Fig. 1a). An examination of repair products from individual colonies revealed elevated gross chromosomal rearrangements and changes in template chromosome size, which likely stemmed from half crossovers (Extended Data Fig. 2a-d). Pif1’s role in BIR is general and highly specific, as BIR induced at a different locus is also Pif1-dependent (Extended Data Fig. 3a-e) and elimination of other 5’ – 3’ helicases, does not affect BIR (Extended Data Fig. 1b). The BIR function of Pif1 is unrelated to its known role in telomerase inhibition, as the elimination of telomerase components does not suppress the BIR defect of pif1-m2 cells (Extended Data Fig. 1c).
Extended Data Figure 1. Analysis of BIR and conversion tracts in pif1∆ mutants and crossover frequency in polymerases mutants.
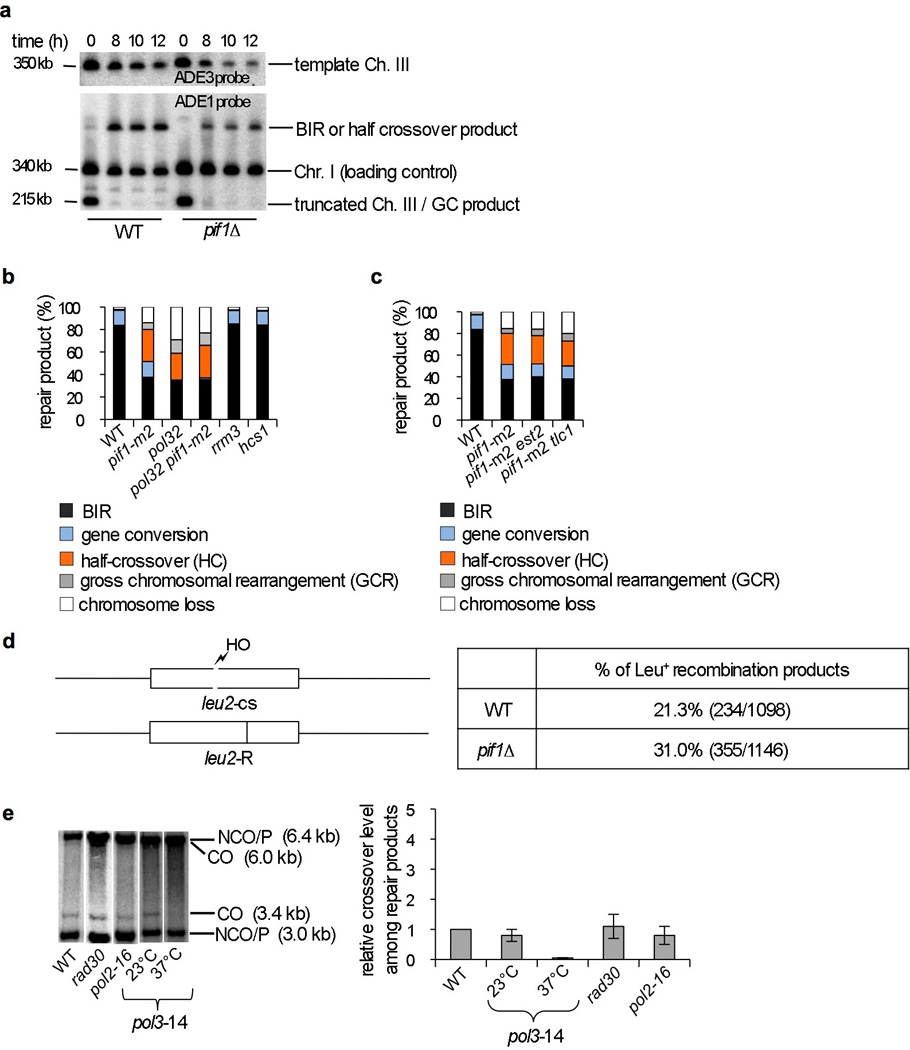
a. Southern blot analysis of BIR product formation and template chromosome maintenance. Chromosomes were separated by PFGE and a DNA probe specific for either ADE1 or ADE3 was used. Quantification is shown in Figure 1c. b,c. Analysis of DSB repair outcomes in the BIR assay of the indicated mutants. d. Schematic of allelic recombination between the leu2 alleles (left). Longer conversion tracts associated with conversion of “R” leads to formation of Leu− recombinants, while shorter conversion tracts lead to formation of Leu+ recombinants. Quantification of gene conversion events with shorter conversion tracts (Leu+) in WT and pif1∆ cells. The difference between WT and pif1∆ cells is statistically significant, p<0.0001. e. Southern blot analysis of gene conversion with and without crossing over in the indicated strains using the ectopic recombination assay shown in Figure 2a. Quantification of crossover product in the indicated mutants compared to WT that is set to 1.
Extended Data Figure 2. Analysis of recombination products in Pif1 deficient cells.
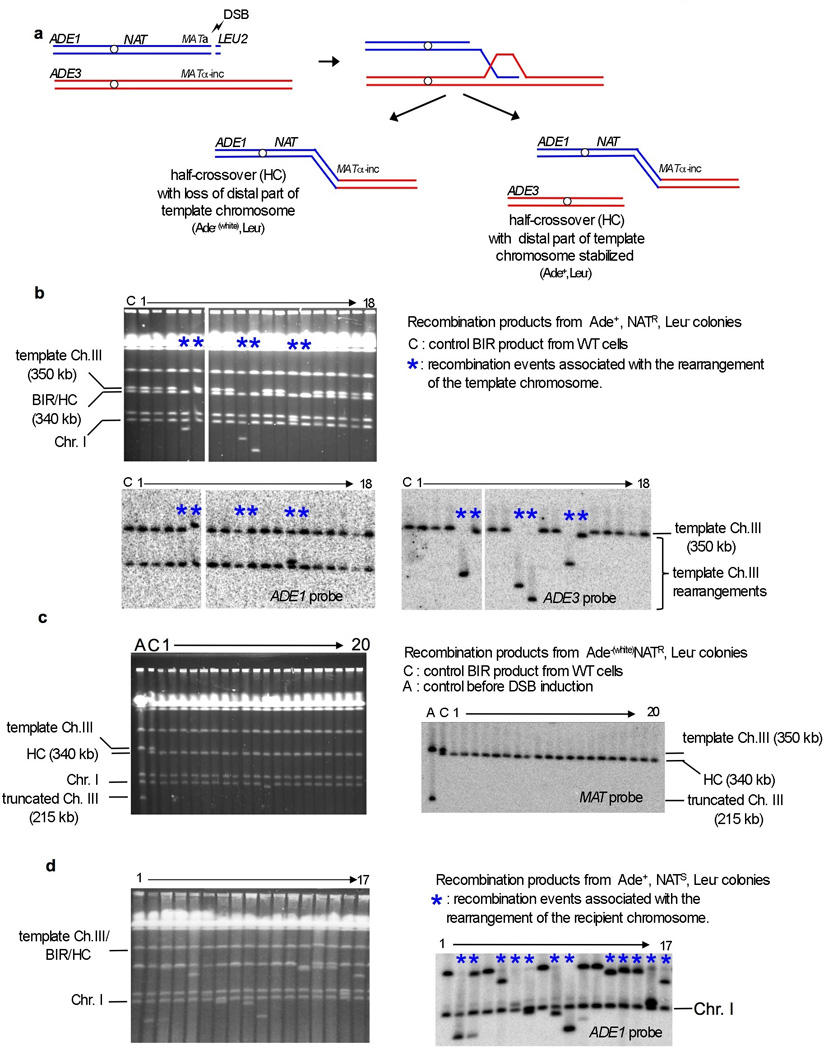
a. Illustration of the half crossover pathway where the part of the template chromosome distal to the initial invasion site is fused to the broken chromosome, with the remainder of the template chromosome either becoming stabilized (examples shown in Fig. 2b) or lost (as shown in Fig. 2c). b. Analysis of recombination products from Ade+ NATR Leu− colonies. Examples where rearrangements of the template chromosome are indicated by an asterisk. c. Analysis of half crossover recombination products from Ade− NATR Leu− colonies. d. Analysis of rare NATS Ade+ colonies.
Extended Data Figure 3. Role of Pif1 in ectopic BIR.
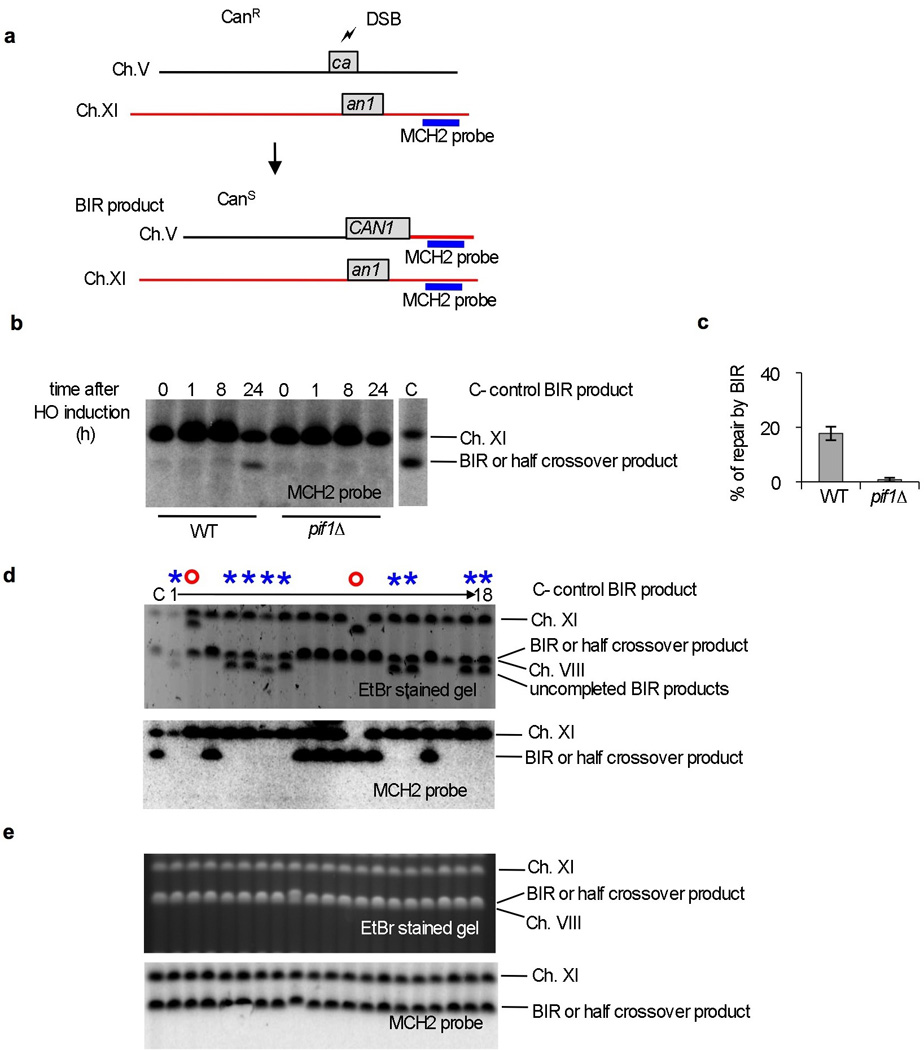
a. Schematic of the ectopic BIR assay. b. Southern blot analysis of ectopic BIR kinetics in WT and pif1∆ cells. A probe specific for the MCH2 gene located at the end of chromosome XI was used in the analysis. c. Quantification of ectopic BIR repair (CanS colonies) in WT and pif1∆ cells. d-e. CHEF analysis of rare products from canavanine sensitive colonies in pif1∆ (d) and WT cells (e). Examples where synthesis is initiated but not finished are indicated by an asterisk. In these cases, a functional CAN1 gene is formed but synthesis is abandoned resulting in shorter products. Red circles indicate major rearrangement of chromosome V or template chromosome XI.
A similar deficiency in BIR with high levels of half crossovers was observed in pol32∆ cells, which lacks the nonessential subunit of Polδ12 (Extended Data Fig. 1b), implicating Pif1 in DNA synthesis. Consistent with this deduction, strand invasion occurs normally in pif1∆ cells (Extended Data Fig. 4a-b), while DNA synthesis monitored by qPCR is decreased (Fig. 1d). In ChIP analyses we found Pif1 enrichment at the DSB and along the template molecule further implicating Pif1’s role in DNA synthesis (Fig. 1e). This Pif1 enrichment is repair-specific, as it is compromised in mutants deficient in strand invasion or extensive DNA synthesis (Extended Data Fig. 4c-d). Since Pif1 appears to affect DNA synthesis, we used ChIP to examine the initial recruitment of Polδ which is essential for BIR14, and other polymerases. Interestingly, only the recruitment of Polδ is decreased in pif1-m2 cells (Fig. 1f).
Extended Data Figure 4. Analysis of Pif1’s role in the initial steps of BIR and of Pif1 recruitment at the DSB and template.
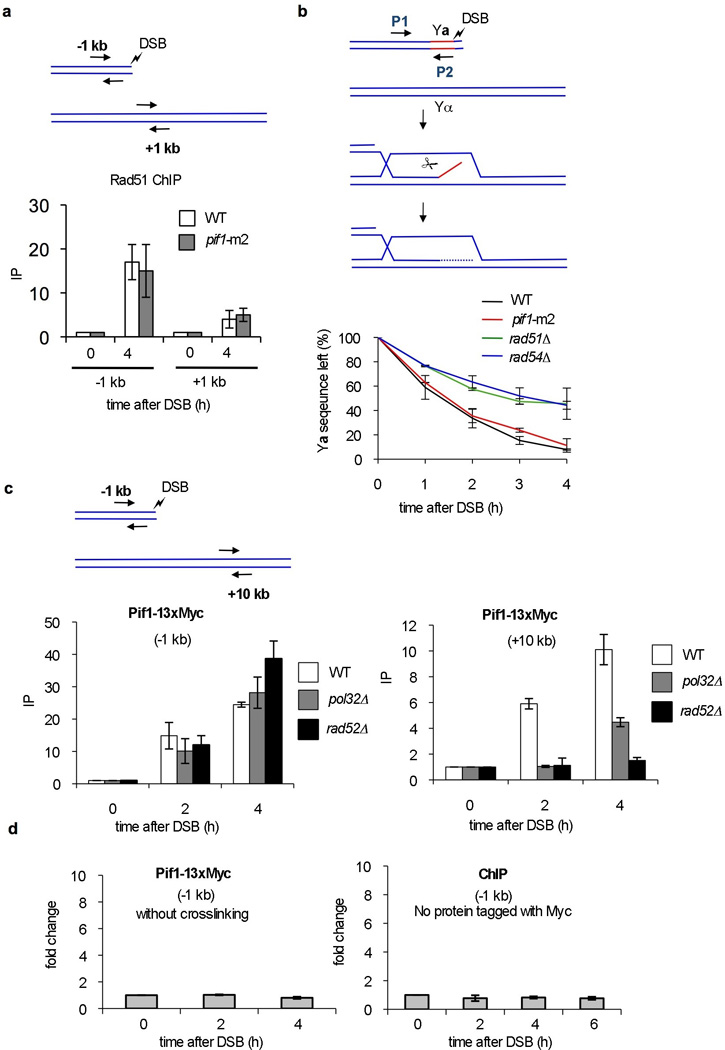
a-b. Analysis of initial strand invasion in WT and pif1∆ cells. a. Enrichment of Rad51 at the DSB site and template by ChIP analysis. b. Kinetics of removal of the non-homologous Ya tail by qPCR analysis in WT and pif1-m2 strains compared to the control strains rad51∆ and rad54∆ that are defective in strand invasion. c. Enrichment of Pif1 at the DSB and template by ChIP analysis in WT, pol32 and rad52 cells. The regions amplified by qPCR are indicated. d. Control ChIP experiments in the PIF1-13xMyc strain where crosslinking was omitted and in a strain where the Myc tag was absent.
Besides BIR, fully processive Polδ is needed for the crossover HR pathway and promotes long conversion tracts15. Importantly, Pif1 is also needed for both processes as monitored in ectopic or allelic gene conversion assays (Fig. 2a-c; Extended Data Fig. 1d). There is no change in cell viability or repair efficiency in pif1∆ cells, but the crossover frequency decreases by half, similar to pol32∆ cells (Fig. 2c). Furthermore, the increase in crossover frequency caused by deleting the crossover suppressors Mph1 and Srs216, 17 is also dependent on Pif1 and Pol32 (Fig. 2b-c). Thus, Pif1 and Polδ are key players in crossover recombination. Indeed, the conditional depletion of Polδ but not other polymerases almost completely eliminates crossovers while slightly reducing noncrossovers (Extended Data Fig. 1e).
Figure 2. Pif1 is important for crossover recombination.

a. Schematic of the ectopic recombination assay. b. Southern blot analysis of gene conversion with and without crossovers in the indicated strains. c. Quantification of crossover frequency in ectopic recombination. d. Repair outcomes in BIR assay in WT and the indicated mutant cells. e. ChIP analysis of RPA binding during BIR at the indicated loci. f. Quantification of BIR product formation upon conditional depletion of Psf2 or Mcm4. Plotted are the mean values ±SD from at least three independent experiments.
We performed biochemical reconstitution to examine how Pif1 influences DNA synthesis in D-loops made by Rad51, RPA, and Rad5418, 19. According to a published procedure3, we loaded the polymerase clamp PCNA onto the primer end of the D-loop with RFC, then added Polδ with Pif1 (Fig. 3a). Our Pif1, RFC, PCNA, and Polδ preparations had no detectible nuclease or topoisomerase contamination (Extended Data Fig. 5a-b).
Figure 3. Pif1 promotes DNA extension at a D-loop.

a. Extension of 32P-labeled invading strand. Extension products by Polδ with Pif1 or pif1 K264A (13, 27, 40 nM) were analyzed in a native gel (upper) or denaturing gel (lower). Representative gels from three independent experiments are shown. b. Extension of unlabeled invading strand with [α-32P]-dCTP. Products were resolved in a native gel and quantified. Plotted are the mean values ±SD from three independent experiments. c. Products prepared as in lanes 1, 10, and 13 of panel a were subject to 2-D gel analysis.
Extended Data Figure 5. Quality analyses of proteins, protein requirements for DNA extension, effect on Pif1 on D-loop stability, and time course analysis of DNA extension.

a. Purified Pif1 and pif1-K264A were analyzed by SDS-PAGE and staining with Coomassie Blue. b. The plasmid DNA in all the lanes was pBluescript SK replicative form I (RFI). DNA synthesis reactions were performed with 13, 27, 40 nM Pif1 and the reaction mixtures (lanes 1–8) from the 8-min timepoint were incubated at 95°C for 2 min to disrupt the D-loop, followed by native gel electrophoresis and staining with ethidium bromide. Various other DNA forms (lane 9, plasmid DNA alone; lane 10, plasmid DNA linerized with XhoI; lane 11, plasmid DNA relaxed by calf thymus topoisomerase I; lane 12, plasmid DNA relaxed by E. coli topoisomerase I; lane 13, plasmid DNA digested with DNase I) are shown. c. DNA synthesis reactions by Polδ in conjunction with Pif1 (40 nM Pif1 and 8-min incubation) with the omission of one or more of the protein factors or dNTPs, as indicated. The reaction products were analyzed in a native gel (upper) or denaturing gel (lower). Note that a substantial portion of the D-loop was dissociated by Pif1 in the absence of PCNA, RFC, Polδ, or dNTPs (lanes 5, 7, 9, and 19). d. Time course of DNA synthesis by Polδ in conjunction with Pif1 (40 nM Pif1). The reaction products were analyzed in a native gel (upper) or a denaturing gel (lower).
We first made the D-loop with a 5’ 32P-labeled 90-mer oligonucleotide as the invading strand and pBluescript (2,961 bp) DNA as acceptor (Fig. 3a). DNA synthesis by Polδ generated DNA species that migrated above the D-loop (Fig. 3a, lanes 2 and 10). Importantly, the addition of Pif1 led to the appearance of DNA species that harbored a much larger amount of new synthesis (Fig. 3a, lanes 3–5, 11–13). Interestingly, with Pif1 present, DNA species migrating above the substrate oligonucleotide but below the D-loop were observed (indicated by the asterisk in Fig. 3a). Since Pif1 can disrupt the D-loop structure (Extended Data Fig. 5c), these DNA species likely stemmed from the release of the extended invading strand.
The helicase activity of Pif1 is required for the stimulation of Polδ-mediated DNA synthesis, as revealed by analyzing the pif1 K264A mutant13, 20 (Fig. 3a). Likewise, no DNA synthesis occurred if either Rad51 or Rad54 was absent or upon the omission of RFC, PCNA, or deoxynucleoside triphosphates (Extended Data Fig. 5c). Efficient D-loop formation and optimal DNA synthesis require RPA (Extended Data Fig. 5c), in concordance with previous observations that it promotes Rad51-mediated strand exchange19, 21 and DNA unwinding by Pif122. Importantly, in the absence of Pif1, RPA was unable to promote extensive synthesis by itself (Fig. 3 and Extended Data Fig. 5c), even when present in excess (data not shown). In addition, when either Rad51 or Rad54 was removed (Extended Data Fig. 6a), or when Rad54 was heat-deactivated after D-loop formation (Extended Data Fig. 6b), Pif1 was still able to stimulate DNA synthesis.
Extended Data Figure 6. Effect of Rad51 and/or Rad54 removal on DNA extension.

a. DNA synthesis from a deproteinized D-loop by Polδ in conjunction with Pif1 (8, 16, and 24 nM) was examined. Pif1 was at 24 nM in lane 6. The reaction products were analyzed in a native gel (left) or denaturing gel (right). b. After the D-loop reaction had proceeded for 2 min, Rad54, which is highly heat labile11, 12, was inactivated by incubation at 42°C for 20 min. DNA extension reaction and analysis were then performed by adding RPA, RFC, PCNA, Polδ and Pif1 (40 nM Pif1 and 8-min incubation). The reaction products were analyzed in a native gel (left) or denaturing gel (right). The inactivation of Rad54 was verified by examining the ATPase activity of Rad54, which decreased by ~95% compared to the unheated control (data not shown).
In another set of experiments, an unlabeled invading strand was extended with [α-32P]-dCTP present (Fig. 3b). Two-dimensional gel electrophoresis (Fig. 3c)3, 23 showed that the extended DNA species made by Polδ harbored ~200–500 nucleotides (Fig. 3c), whereas the products made by Polδ with Pif1 could reach a few thousand nucleotides (Fig. 3c, Extended Data Fig. 5d).
The effect of Pif1 is highly specific, as neither S. cerevisiae Rrm3 nor E. coli DinG, which possess 5’–3’ helicase activity, could substitute for it. Likewise, no enhancement of Polδ-mediated DNA synthesis occurred with the 3’ –5’ S. cerevisiae Mph1 helicase (Extended Data Fig. 7a). Moreover, Pif1 has no effect on E. coli DNA polymerase I Klenow fragment (Extended Data Fig. 7b).
Extended Data Figure 7. Specificity of Polδ-Pif1-mediated DNA extension.

a. DNA synthesis reactions were conducted with Polδ and Pif1, Mph1, Rrm3, or DinG (13, 27, 40, 120 nM). The reaction products from the 8-min timepoint were resolved in a native (upper) or denaturing gel (lower) (lane 1, no protein control; lane 2 D-loop formed by Rad51-Rad54, lanes 3-20, D-loop extended with Polδ and the indicated helicase). b. E. coli DNA polymerase I Klenow fragment (100 nM, from NEB) was tested for DNA extension with Pif1 (13, 27, 40 nM) with or without PCNA (200 nM) and RFC (200 nM). The reaction products from the 8-min timepoint were analyzed in a native gel.
S. cerevisiae cells lacking the POL32 gene are impaired for HR12, 14, 15, 24. The Pol32 protein interacts with PCNA and enhances the processivity of Polδ25. We observed a severely reduced level of DNA synthesis by Polδ*, which lacks Pol32 (Extended Data Fig. 8a), alone or in conjunction with Pif1 (Extended Data Fig. 8c). Purified Pol32 interacts with Polδ* (Extended Data Fig. 8b) and its addition to Polδ* led to an enhancement of DNA synthesis activity comparable to that of Polδ (Extended Data Fig. 8c).
Extended Data Figure 8. Requirement for the Polδ subunit Pol32 in DNA extension, and interaction of Pif1 with PCNA.

Purified Polδ (FLAG-Pol3, GST-Pol31, Pol32), Polδ* (FLAG-Pol3, GST-Pol31) and MBP-Pol32 were analyzed by SDS-PAGE and staining with Coomassie Blue. b. Pulldown assay to examine Pol32-Polδ* interaction. c. DNA synthesis was performed with Polδ or Polδ* (20 or 40 nM) with Pif1 (40 nM). In lanes 10–13, Polδ* and Pol32 (125 nM) were preincubated on ice for 10 min before use. The reaction products from the 8-min timepoint were resolved in a native gel (left) or denaturing gel (right). d. Pulldown reactions of 6XHis-Pif1 and PCNA (left), Polδ (FLAG-Pol3, GST-Pol31, Pol32) and PCNA, Polδ and Pif1 (right).
Extensive DNA synthesis in a covalently closed DNA molecule generates topological stress that would impede polymerase movement, yet, in our reconstituted system several kilobases of DNA can be synthesized without a topoisomerase (Fig. 3c, Extended Data Fig. 5d). In fact, while topoisomerase I enhanced DNA synthesis by Polδ alone (Extended Data Fig. 9a, lanes 5 and 6)3, it had no stimulatory effect when Pif1 was present (Extended Data Fig. 9a-b). Thus, Pif1-dependent DNA synthesis may entail concomitant dissociation of the newly synthesized DNA from the 5’ side, a premise supported by our observation that Pif1 can efficiently dissociate the unmodified (Extended Data Fig. 5c) and extended D-loops (Fig. 3a).
Extended Data Figure 9. Effect of topoisomerase I in DNA extension.

a. DNA synthesis products, initiated by Polδ for 4 min, and then continued with Pif1 (13, 27, 40 nM) with and without Topo I (0.4 U/µl) for 8 min. The reaction mixtures were resolved in a native gel (top) or denaturing gel (middle). Lanes 1 and 2 contained DNA substrates only and lanes 3-12 contained D-loop made by Rad51-Rad54. An overexposed image and the scan of lanes 5 and 6 to highlight the effect of topoisomerase when Pif1 was absent are shown (bottom). b. 2-D gel analysis of the extension products. The reaction products, prepared as in lanes 5, 6, 11 and 12 of panel a, were subject to 2-D gel analysis.
We tested the hypothesis that Polδ-Pif1-mediated DNA synthesis occurs within a migrating D-loop. First, the extended D-loops were analyzed by restriction digests (Fig 4a). If the extended invading strand were released, then a significant fraction of the D-loop would be resistant to the enzymes AhdI and XmnI, which incise at 115 and 714 nucleotides from the 5’ terminus of the invading strand, respectively. D-loops were made with a 5’ 32P-labeled invading strand and extended with unlabeled deoxynucleotides (see Fig. 3a), followed by treatment with AhdI or XmnI and analysis in a denaturing gel (Fig. 4a). The D-loop extended by Polδ alone could be cleaved quantitatively by AhdI to produce a 115-nt DNA fragment (lanes 7 and 9), whereas the majority of the extended product made with Polδ-Pif1 was resistant. Little of the Polδ-extended D-loop was susceptible to XmnI, consistent with the short Polδ-alone synthesis tract (Fig. 3). A small fraction of the extended D-loop from the Polδ-Pif1 reaction was cleavable by XmnI to generate the 714-nt fragment, indicative of DNA synthesis proceeding beyond the +714 site and of the fact that much the +714 site in the extended DNA existed as ssDNA (Fig. 4a, lanes 13 and 15). Thus, in the Polδ-Pif1 reaction, Pif1 continually dissociates the extended strand from the D-loop. This ”bubble migration” mode of DNA synthesis was previously suggested for a reconstituted bacteriophage T4 system that harbors the Dda helicase26.
Figure 4. Evidence for DNA synthesis in a migrating D-loop.

a. Biochemical analysis. Products were analyzed in a denaturing gel. A representative gel from three independent experiments is shown. b. EM analysis. Micrographs of plasmid (pBluescript) DNA, Rad51-made D-loop, extension products by Polδ and Polδ-Pif1. ssDNA was decorated with T4 gp32 and appears thicker than duplex DNA. Arrows indentify the D-loop. Scale bar: 100 nm. Representative micrographs from two experiments are shown. c. Model depicting the dual role of Pif1. In BIR and crossover HR, Pif1 promotes DNA synthesis by a functional interaction with Polδ-PCNA (this work), template strand separation10, and by displacing the newly synthesized strand (this work).
We next examined the extended D-loops by electron microscopy (EM). We treated the D-loop products with the T4 gp32 protein to decorate the ssDNA region, followed by protein-DNA crosslinking with glutaraldehyde. The crosslinked nucleoprotein complexes were analyzed by EM with metal shadowing 27. Figure 4b shows typical EM images of pBluescript DNA, unextended D-loop, D-loops extended by Polδ, and D-loops extended by Polδ-Pif1. This analysis clearly showed D-loop enlargement by Polδ and that the Polδ-extended invading strand remains hybridized to its complementary strand. Importantly, the inclusion of Pif1 generated a long ssDNA tail protruding from the D-loop (Fig. 4b). Furthermore, we tested Pif1 for interaction with Polδ and PCNA in affinity pulldown reactions. Interestingly, the analysis revealed that Pif1 physically associates with PCNA but not with Polδ (Extended Data Fig. 8d).
We asked whether BIR in cells entails the formation of a canonical replication fork with the replicative helicase Mcm2-7 playing a crucial role. Several observations suggest that this is not the case. First, mutants deficient in structure-specific resolvases show only a mild BIR deficiency, suggesting that in BIR the D-loop does not need to be converted to a canonical replication fork (Fig. 2d). Second, monitoring the association of RPA with the template chromosome using ChIP, revealed that extensive ssDNA is generated during template copying in a Pif1-dependent manner. We confirmed the presence of extensive ssDNA by applying qPCR after restriction digest of DNA synthesis intermediates (Extended Data Fig. 10a-b). These results suggest that the first and complementary strands are synthesized asynchronously (Fig. 2e). Finally, conditional depletion of Mcm4 or Psf2, components of the S-phase replication fork, leads to only a mild BIR deficiency (Fig. 2f and Extended Data Fig. 10c-e). Although we cannot exclude the possibility that in wild type (WT) cells there is a switch from Pif1 to Mcm2-7-mediated synthesis, we have provided clear evidence that Pif1 can support extensive DNA synthesis in the absence of the replicative helicase.
Extended Data Figure 10. Measurement of ssDNA intermediates formed during BIR and analysis of BIR efficiency in the absence of Psf2 and Mcm4.
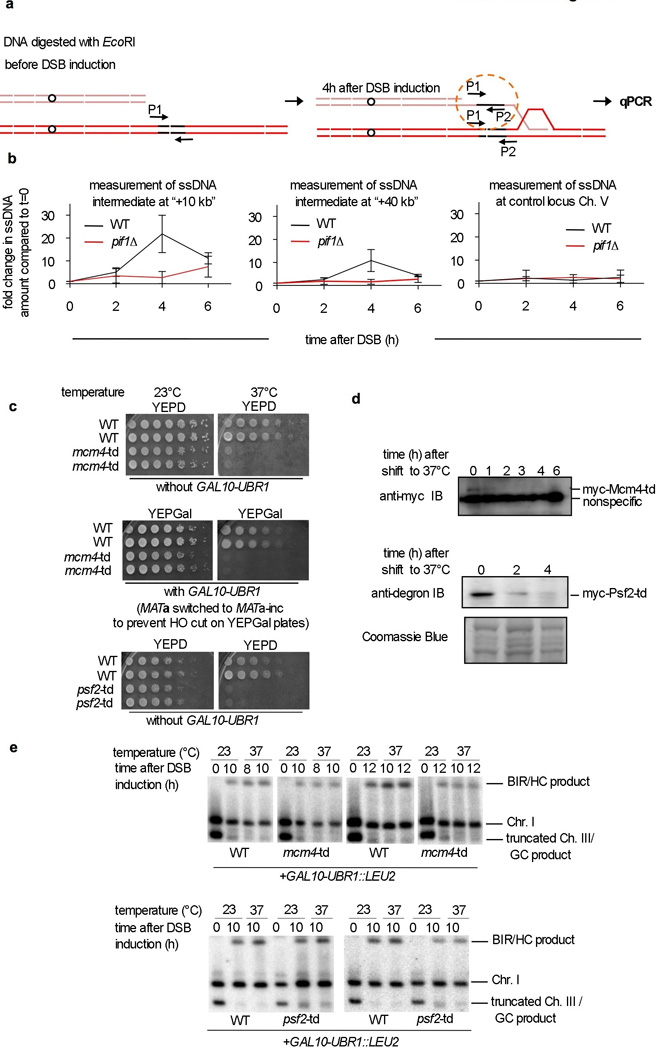
a. Schematic of the assay. b. Measurement of the relative increase of ssDNA at the indicated time after DSB induction compared to the amount of ssDNA in logarithmically growing cells (t=0). Measurement of ssDNA intermediate 10 and 40 kb from the site of strand invasion at the template chromosome and at a control locus on chromosome V which does not participate in recombination. c. An analysis of the growth of cells harboring temperature sensitive degron alleles of td-mcm4 and td-psf2. Both strains are inviable at 37°C even without overexpression of the ubiquitin ligase Ubr1. d. Western blot analysis of td-Mcm4 and td-Psf2 protein degradation. e. Southern blot analysis of the BIR assay in cells with conditional depletion of td-Mcm4 or td-Psf2. Quantification of the Southern blots is shown in Figure 2f.
Our results provide evidence for repair-specific Pif1-dependent DNA synthesis via a migrating D-loop (Fig. 4c) that can copy tens of kilo bases. Aside from BIR and telomere recombination, such a mechanism could function in gene conversion in fungi28 and can cause various genome rearrangements29, 30.
Methods summary
The strains listed Methods are derivatives of (i) tGI354 to study ectopic recombination (hml::ADE1 MATa-inc hmr::ADE1 ade1 leu2-3,112 lys5 trp1::hisG ura3-52 ade3::GAL::HO arg5,6::HPH::MATa) and (ii) AM1003 to study BIR (MATa-LEU2-tel/MATα-inc ade1 met13 ura3 leu2-3,112/leu2 thr4 lys5 hml::ADE1/hml::ADE3 hmr::HYG ade3::GAL-HO FS2::NAT/FS2). The DSB was induced upon expression of the HO endonuclease by adding galactose to the media. Southern blotting and probes specific for either the broken or template chromosome were used to follow the kinetics of DSB repair and for the detailed analysis of individual repair products. Protein recruitment to DSBs was studied by ChIP followed by qPCR. Pif1 and other helicases, homologous recombination proteins, and DNA replication factors were expressed either in E. coli or yeast cells and purified by a multi-step procedure to near homogeneity in each case. For the DNA synthesis reaction, a D-loop is made using Rad51, RPA, and Rad54, followed by loading of PCNA with RFC onto the 3’ end of the invading strand, and incubation with combinations of Pif1 or another helicase together with Polδ. Reaction products were analyzed by gel electrophoresis and phosphorimaging, or by electron microscopy with metal shadowing.
Methods
Media, strains and plasmids
The plasmids pVS31 (pif1-m2), pSH380-PIF1, and pSH380-pif1-K264A were from Virginia Zakian. The pif1-m2 mutation was introduced into the genome as described13. For complementation tests, we amplified PIF1 from pSH380-PIF1 and introduced it into pRS316 to create pRS316-PIF1; pif1-K264A was created by subcloning the 0.7 kb AflII/ClaI fragment from pSH380-pif1-K264A into pRS316-PIF1.
For HO induction, cells (GAL10::HO) from an overnight culture in YEPD (1% yeast extract, 2% peptone, 2% dextrose) were transferred to YEP-raffinose (1% yeast extract, 2% peptone, 2% raffinose) and incubated overnight. Galactose was added to 2% when the cell density reached ~1×107 cells/ml.
To study ectopic recombination, allelic BIR, or ectopic BIR we used tGI354, AM1003, or JRL346 strains, respectively, or their derivatives. tGI354 hml::ADE1 MATa-inc hmr::ADE1 ade1 leu2-3,112 lys5 trp1::hisG ura3-52 ade3::GAL::HO arg5,6::HPH::MATa16 and its derivatives: pif1-m2 (yWH42); pif1::KANMX (yWH1217); pol32::KANMX (yWH80); pif1-m2 pol32::KANMX (yWH198); pif1::KANMX pol32::URA3 (yWH1226); mph1::KANMX (tGI772)16, 17; pif1-m2 mph1::KANMX (yWH1043); pol32::TRP1 mph1::KANMX (yWH221); srs2::LEU2 (tGI383) 16; pif1-m2 srs2::LEU2 (yWH1072); pol32::KANMX srs2::LEU2 (yWH1076); pol2-16 (yWH1116); pol3-14 (yWH1103); rad30::KANMX (yWH1222).
AM1003 MATa-LEU2-tel/MATa-inc ade1 met13 ura3 leu2-3,112/leu2 thr4 lys5 hml::ADE1/hml::ADE3 hmr::HYG ade3::GAL-HO FS2::NAT/FS212 and its derivatives: trp1::hisG (yWH422); leu2::KANMX (yWH271); pho87::URA3 (yWH279); pif1-m2 (yWH121); pif1-m2+pRS316-PIF1 (yWH530); pif1-m2+pRS316-pif1-K264A (yWH531); pol32::KANMX (yWH321); pif1-m2 pol32::KANMX (yWH304); pif1::KANMX (yWH465); rad51::KANMX (yWH615); rad54::KANMX (yWH616); exo1::TRP1 sgs1::KANMX (yWH612); pif1-m2 tlc1::LEU2 (yWH308); pif1-m2 est2::KANMX (yWH328); hcs1::KANMX (yAP427); pif1-m2 pho87::URA3 (yWH298); POL1-13Myc-TRP1 (yWH1176); pif1-m2 POL1-13Myc-TRP1 (yWH1177); POL2-13Myc-TRP1 (yWH499); pif1-m2 POL2-13Myc-TRP1 (yWH501); POL3-13Myc-TRP1 (yWH634); pif1-m2 POL3-13Myc-TRP1 (yWH1110); RAD30-13Myc-TRP1 (yWH1179); pif1-m2 RAD30-13Myc-TRP1 (yWH1178); pif1-m2 MCM7-3HA-TRP1 (yWH1056); pif1-m2 pif1-m1-4Myc-TRP1 pol32::KANMX (yWH971); pif1-m2 pif1-m1-4Myc-TRP1 rad52::KANMX (yWH1003); ura3 ::URA3 thr4 ::THR4 (yMW 331); pif11 ::KANMX (yMW 335); MATa-LEU2::URA3-tel/MATa-inc (yMW393); CUP1::mcm4-td ::KANMX (yMW412); CUP1:: psf2-td ::KANMX (yMW467).
JRL346 mata ::HOcsDEL::hisG ura3DEL851 trp1DEL63 sup53DEL::leu2DEL ::NATMX hmlDEL ::hisG hmrDEL::ADE3 ade3 ::GAL10 ::HO can1,1-1446 ::HOcs ::HPH ::DEL AVT2 ykl215c ::LEU2 ::hisG ::can1DEL1-28914; and its derivative, pif1 ::KANMX (yGI272)
Pulsed-field gel electrophoresis (PFGE) and analysis of DSB repair products
To analyze DSB repair kinetics and products in AM1003 derivative strains, chromosomal DNA plugs were prepared and separated as described 4, followed by Southern blotting and hybridization with probes specific for either ADE1, ADE3, MCH4 or MAT4. Allelic BIR product formation was estimated as the percent of the initial uncut chromosome III. Percentage of the chromosome III template remaining during repair was measured as the normalized intensity of the band corresponding to chromosome III in each time point after break induction multiplied by 100 and divided by the intensity the band corresponding to chromosome III at time point “0”.
Chromatin immunoprecipitation (ChIP)
ChIP analyses of DNA polymerases, Rad51, and Pif1 were performed and quantified as described31. The α-Myc (9E10) antibody was from Sigma (M4439). Anti-Rfa2 antibody was from Wolf-Dietrich Heyer 32 and anti-Rad51 antibody was from our laboratory stock 31, 33. Experiments were done at least three separate times and the t-test was used to establish the statistical significance of the results. The primers used for qPCR were: MATX-F2 -1kb, (5’-GGTAGGCGAGGACATTATCTATCA-3’) and MATX-R3 -1kb, (5’-GAAGAATACCAGTTTATCTCGCATTCAAATC-3’); 5 kb RHO-F1 (5’-ATTCACTAACAATGGCTCTAGGAGTGGCG-3’), 5 kb RHO-R1 (5’-CTTGCGGATATCGTGCTACAAAATCAGTC-3’), 10 kb RHO-F1 (5’-TCTCTCCCTTTCAGCAGCTGCTCAGAG-3’), 10 kb RHO-R1 (5’-GAAGAAACACACATCCTCACACGCATATTC-3’), 30 kb RHO-F1 (5’-CTCTCATGGTTCGGACTTACTTAAAACACCC-3’), 30 kb RHO-R1 (5’-AATTCGTTGCGCTTGTGAGGACATCGG-3’), 67 kb (5’-GCTGCAGTTGCTAATAATCTG-3’) and 67 kb (5’-CGGGAGGAGTGGAAGCC-3’). These primer pairs are for sites −1, +5, +10, +30 and +67 kb from the break.
Analysis of nonhomologous DNA tail removal
To determine removal of the nonhomologous Ya tail as a measure of successful DNA strand invasion, we performed real-time PCR using genomic DNA and primers specific for the Ya tail (642 bp): MATX-F1 (5’-GTTGTTACACTCTCTGGTAACTTAGGTAAA-3’) and MATYa-R2 (5’-CAATCTCAGTACCTAGAATGTTAAACAGAG-3’) designated as P1 and P2 in Extended Data Fig. 4b.
Initial DNA synthesis analysis
To measure initial DNA synthesis in BIR, 2 ng of genomic DNA from different times after DSB induction was amplified by PCR with the primers: P1-BIR URA3 (5’-ACCCGGGAATCTCGGTCGTAATGA-3’) and P2-Z1 distal (P1 and P2 in Fig. 1d; 5’-ATCCGTCACCACGTACTTCAGC-3’). As control, the CHA1 gene on chromosome III was amplified.
Determination of DSB repair by BIR and other mechanisms
To quantify allelic BIR, we used a disomic strain with an extra, truncated copy of chromosome III wherein the arm 100 kb distal from MATa is replaced with LEU2 followed by telomeric repeats12. In this assay, the chromosomes participating in repair are marked by either LEU2, ADE1 or ADE3 which allows determination of the repair pathway by growth on selective media (Fig. 1a)12. To avoid any contribution of Ty transposon repeats to repair, the nearest Ty1 repeats to the DSB were replaced with a NAT cassette. BIR leads to the loss of the LEU2 marker. When repair fails, ADE1, NAT and LEU2 markers are lost, and the colonies appear red due to an Ade1 deficiency. When cells repair the DSB by gene conversion using the short homology on the right side of the break all the markers are retained12. In rare cases in WT cells, part of the homologous template chromosome is lost due to a half crossover that eliminates the ADE3 marker, and colonies are Ade− and appear white.
The BIR assay was performed by plating on YEP-galactose medium and replica plating on Leu−, Ade− dropout and NAT selective media. For each strain, at least one thousand colonies were scored. The frequencies of BIR, half crossovers, gene conversion and chromosome loss were estimated based on the percentage of colonies carrying markers specific for these repair outcomes, as described above (Fig. 1a) and reported previously12. Pedigree analyses and individual product size analyses by CHEF confirmed repair by BIR in WT cells12. Since the repair by BIR occurs in G2/M cells two copies of each chromosome are present. Due to random segregation of chromosomes, half of the half crossovers (the major product in pif1∆ cells) segregate with an intact copy of the full-length chromosome III, and are genetically and structurally indistinguishable from BIR as confirmed by pedigree analysis12. Therefore, the number of Ade- white colonies scored on selective media as half crossovers represent only half of these events. To correct for this, the number of half crossovers (Ade-, white colonies) was multiplied by two, and consequently the number of BIR events was adjusted by subtracting the number of Ade-, white colonies. Second, upon analysis of the repair products by CHEF from Ade+ colonies that have lost the distal NAT marker (Extended Data Fig. 2d), we found that about half of them carried a genomic rearrangement, while the other half of the products corresponded by size to BIR, where strand invasion occurred proximal to the NAT marker. Therefore, half of the NATS colonies were scored as gross chromosomal rearrangements and the other half as BIR events. The number of BIR events in pif1∆ mutants were still likely overestimated because about a third of the Ade+ NATR Leu− colonies did not result from allelic BIR, but from a half crossover event associated with a stabilization of the part of template chromosome carrying the ADE3 marker (Extended Data Fig. 2b).
The percentage of cells that repair a DSB by ectopic BIR14 (BIR between two short homologous sequences located on heterologous chromosomes) was calculated as the number of canavanine sensitive (CanS) colonies (Extended Data Fig. 3a) formed on YEPGal plates divided by the number of all colonies formed on YEPD plates, multiplied by 100. In this BIR assay, recombination between two truncated copies of the CAN1 gene located on chromosome V and XI leads to the formation of an intact CAN1 gene resulting in canavanine sensitivity. In pif1∆ cells the number of BIR events (~1%) is likely overestimated because over half of the cells that form the full length CAN1 gene during DSB repair do not complete repair (Extended Data Fig. 3d). Additionally, some of the products that appear to be BIR can correspond to half crossovers that in this assay (haploid cells) cannot be scored (half crossovers that segregate with an intact template chromatid cannot be distinguished from BIR and the half crossovers that lose a large part of the template chromosome are inviable).
Measurements of crossover frequency, DSB repair, and viability in ectopic recombination assay
We determined the frequency of crossovers, viability and efficiency of the DSB repair as previously described16, 17.
Measurement of ssDNA formation by qPCR
The ssDNA amount was measured as previously described34 10 and 41 kb away from the DSB end and as a control on the chromosome V not participating in recombination. The primers used in this assay were: oMW 1082 10832bp-F (5’-CACTAAGTTCTTGGACAGGT);oMW 1083 10832bp-R (5’-AATACTGGTCATGAAGCCAC); oMW 1116 ChrV 41701bp-F (5’-GGCAGCCACCCTTATGGTGAGG); oMW 1117 ChrV 41701bp-R (5’-GGCCGCAAGGGCCAAGACAAGG); oMW1120 40890-F (5’-CCTTTCACCGTCTATGGGCC); oMW1121 40890-R (5’-CAATTCCTCATTTCCATCGG).
Measurement of conversion tracts
We employed an assay in which an HO break is generated within LEU2 and is repaired by allelic recombination with leu2-R. In this assay, Leu+ colonies stem from short conversion tracts and Leu− colonies from longer conversion tracts35. Cells were plated on YPE-Gal plates and then replica plated on medium lacking leucine.
Preparation of proteins
Pif1 and pif1-K264: PIF1 cDNA encoding the nuclear form of Pif1 (amino acid 40-859) was inserted in the pRSF-Duet-1 vector (Novagen) to add an N-terminal 6xHis tag. The K264A mutation was introduced by QuikChange mutagenesis (Agilent Technologies). Pif1 and pif1 K264A were expressed in E. coli Rosetta cells (Novagen), with induction by 0.1 mM isopropyl-1-thio β-D-galactopyranoside (IPTG) for 18 h at 16°C. The cell paste from a 10-L culture was resuspended and sonicated in 100 ml buffer A (20 mM K2HPO4, pH 7.5, 0.5 mM EDTA, 10% glycerol, 0.01% Igepal, 2 mM DTT) containing 100 mM KCl and protease inhibitors (aprotinin, chymostatin, leupeptin, and pepstatin A at 5 µg/ml each, 1 mM phenylmethylsulfonyl fluoride). The lysate was clarified by ultracentrifugation and loaded onto a Q Sepharose column (8-ml). The flow-through fraction was applied onto a SP-Sepharose column (8-ml) and fractionated with a 90-ml gradient of 150–660 mM KCl in buffer A. Fractions containing Pif1 were incubated with 2 ml Ni-NTA-agarose beads (Qiagen) and 10 mM imidazole for 2 h. After washing three times with 10 ml buffer A containing 1 M KCl, 1 mM ATP, 8 mM MgCl2, and 15 mM imidazole, the bound proteins were eluted with 20 ml buffer A containing 150 mM KCl and 200 mM imidazole. The protein pool was fractionated in Mono S (1-ml) with a 40-ml gradient of 150–600 mM KCl in buffer A. Pif1 was concentrated in an Ultracel-30K concentrator (Amicon) and stored at −80°C.
Polδ and Polδ*: Polδ (FLAG-Pol3, GST-Pol31 and Pol32) and Polδ* (FLAG-Pol3, GST-Pol31) were expressed in S. cerevisiae strain YRP65436. Cells from a 10-L culture were disrupted using a coffee grinder and resuspended in 100 ml buffer B (50 mM Tris HCl, pH 7.5, 10% sucrose, 1 mM EDTA, 175 mM (NH4)2SO4, 200 mM NaCl, 1 mM DTT, 0.01% Igepal, protease inhibitors as above). After ultracentrifugation, the lysate was treated with 0.277 g/ml of (NH4)2SO4. The precipitate was pelleted by centrifugation and dissolved in 100 ml buffer C (25 mM Tris HCl, pH 7.5, 0.5 mM EDTA, 10% glycerol, 0.01% Igepal,10 mM 2-mercaptoethanol and 200 mM KCl) and dialyzed against the same buffer. The protein was purified by affinity chromatography in glutathione Sepharose (GE Healthcare; 5-ml) and anti-FLAG M2 resin (Sigma; 2-ml). Protein was concentrated and stored at −80°C.
RFC: RFC (GST-RFC1, RFC2, RFC3, RFC4, RFC5) was expressed in S. cerevisiae strain YRP654 using pBJ1476 (2µ, GAL-PGK-GST-RFC1/RFC4/RFC5, LEU-2d) and pBJ1469 (2µ, GAL-PGK-RFC2/RFC3, TRP1) and purified from clarified cell lysate by (NH4)2SO4 precipitation and affinity purification using glutathione Sepharose as above.
Pol32: MBP-Pol32 (with MBP cleavable with TEV protease) was expressed in E. coli Rosetta cells harboring pMAL-POL32 with induction by 1 mM IPTG for 4 h at 37°C. The cell paste from a 500-ml culture was resuspended in 50 ml buffer D (50 mM Tris-HCl, pH 7.5, 10% sucrose, 1 mM EDTA and protease inhibitors as above), sonicated, and clarified by ultracentrifugation. Nucleic acids were removed by adding 700-µl 10% polyethyleneimine (Baker) and centrifugation. MBP-Pol32 was purified by affinity chromatography with 6 ml Amylose resin (BioLabs) and fractionation in 1-ml Source S with a 30-ml gradient of 100–500 mM KCl in buffer E (25 mM Tris-HCl, pH 7.5, 10% glycerol, 0.5 mM EDTA, 0.01% Igepal, 1 mM 2-mercaptoethanol), and in a 1-ml Macrohydroxyapatite using a 30-ml gradient of 0–300 mM KH2PO4 in buffer E. MBP-Pol32 was concentrated and stored at –80°C.
Rrm3: DNA that harbors RRM3-FLAG was cloned into the pMAL-TEV vector (BioLabs) to add an N-terminal MBP tag. Expression was in E. coli Rosetta cells with induction by 0.1 mM IPTG for 24 h at 12 °C. The cell paste from a 3.3-L culture was resuspended and sonicated in 40 ml buffer F (25 mM Tris-HCl, pH 7.5, 0.5 mM EDTA, 10% glycerol, 0.01% NP-40, 1 mM DTT, 500 mM KCl, 0.2 mM ATP, 5 mM MgCl2, and protease inhibitors as above). The lysate was clarified by ultracentrifugation and treated with 0.277 g/ml of (NH4)2SO4. The protein precipitate was pelleted by centrifugation and dissolved in 30 ml buffer F. MBP-Rrm3 was purified by two-step affinity chromatography with Amylose resin (2-ml) and anti-FLAG M2 resin (0.7-ml). Rrm3 was concentrated and stored at −80°C.
Rad51, Rad54, RPA, PCNA, and Mph1 were purified as described elsewhere17, 23, 37. DinG was from Daniel Camerini-Otero.
D-loop extension
The 32P-labeled 90-mer oligonucleotide (2.4 µM nucleotides), homologous to positions 1932-2021 of pBluescript DNA38, was incubated with Rad51 (800 nM) in buffer G (35 mM Tris-HCl, pH 7.5, 1 mM DTT, 7 mM MgCl2) containing 100 ng/µl BSA, 30 mM KCl, 2 mM ATP, an ATP-regenerating system (20 mM creatine phosphate, 30 ng/µl creatine kinase), and 100 µM each of the four dNTPs for 10 min at 37°C. This was followed by a 5-min incubation with RPA (400 nM) at 30°C, a 2 min-incubation with Rad54 (200 nM) at 23°C, and a 2-min incubation with pBluescript DNA (37 µM base pairs) at 30°C. For D-loop extension, the reaction was mixed with PCNA (200 nM) and RFC (200 nM) and incubated on ice for 2 min. Then, Polδ (100 nM) and Pif1 (13–40 nM) were added to the reaction, followed by an incubation at 15°C. Reaction mixtures were deproteinized with 0.5% SDS and 0.5 mg/ml proteinase K for 10 min at 37°C before being resolved in a native gel (0.8% agarose) in TAE buffer (40 mM Tris-acetate, pH 7.5, 0.5 mM EDTA), or in a denaturing gel (4% polyacrylamide, 7 M urea) in TBE buffer (90 mM Tris-HCl, pH 8.3, 90 mM boric acid, 2 mM EDTA), or in a 0.9% agarose gel in 50 mM NaOH, 1 mM EDTA (Extended Data Fig. 5d). Dried gels were analyzed in a phosphorimager (BioRad).
For quantification of DNA synthesis in Fig. 3b, D-loop extension was carried out as above, except that the invading strand was unlabeled and the reaction was supplemented with [α-32P]-dCTP (80 nCi/µl). The reaction products were resolved in a native gel and analyzed.
DNA extension from deproteinated D-loop
The D-loop reaction (250 µl) was performed as above with the 32P-labeled 90-mer oligonucleotide. The reaction was deproteinized with SDS and proteinase K as above. After an extraction with phenol-chloroform-isoamyl alcohol (25:24:1), the buffer was exchanged with buffer H (35 mM Tris-HCl pH 7.5, 1 mM DTT, 9.3 mM MgCl2, and 30 mM KCl) using a Zebra Spin-desalting Column (Thermo Scientific). DNA synthesis reaction was carried out with the deproteinized D-loop (equivalent to 1.2 µM nucleotides of the 32P-labeled 90-mer oligonucleotide), with 2 mM ATP, the ATP-regenerating system, 100 ng/µl BSA and 100 µM each dNTPs, 200 nM RPA, 100 nM PCNA, 100 nM RFC, 50 nM Polδ, and 8, 16, 24 nM Pif1 with an 8-min incubation.
2-D gel electrophoresis
Deproteinized reaction mixtures were run in a 0.8% agarose gel in TAE buffer. Then, lanes containing the radiolabeled species were excised and placed on top of a 0.9% agarose gel. Electrophoresis in the second dimension was done in 50 mM NaOH, 1 mM EDTA. A gel strip from the first dimension is shown above the 2-D gel.
Pulldown assay
Pol32 and Polδ*: MBP-Pol32 (5 µg) or TEV-protease treated MBP-Pol32 was incubated with Polδ* (5 µg) in 20 µl buffer I (25 mM Tris-HCl, pH 7.5, 10% glycerol, 1mM DTT, 0.01% Igepal, 150 mM KCl) for 30 min on ice, then mixed with 10 µl glutathione Sepharose for 1 h at 4°C. The resin was washed four times with 100 µl buffer I, then eluted with 20 µl 2% SDS. The supernatant (S) containing unbound proteins, final wash (W), and SDS eluate (E), 10 µl each, were analyzed by SDS-PAGE and Coomassie Blue staining.
Pif1 and PCNA: 6xHis-Pif1 (3 µg) was incubated with PCNA (3 µg) in 30 µl buffer J (25 mM Tris-HCl, pH 7.5, 0.01% Igepal, 1 mM 2-mercaptoethanol, 100 mM KCl) containing 20 mM imidazole for 30 min at 4°C, then mixed with 6 µl Ni-NTA agarose for 1 h at 4°C. The resin was washed three times with 50 µl buffer J, then eluted with 20 µl 2% SDS and analyzed as above.
Pif1 and Polδ: Polδ (9 µg) with GST-Pol31 was incubated with Pif1 (3 µg) or PCNA (3 µg) in 30 µl buffer J for 30 min at 4°C then mixed with 12 µl gluthathione Sepharose for 1 h at 4°C. The resin was washed three times with 40-µl buffer J, then eluted with 25-µl 2% SDS and analyzed as above.
Restriction digests of extended D-loops
Reactions (20 µl) were extracted with phenol-chloroform-isoamyl alcohol and DNA was precipitated with ethanol, which was dissolved in 10 µl buffer K (20 mM Tris-acetate, pH 7.9, 10 mM Mg-acetate, 50 mM K-acetate, 1 mM DTT, 100 ng/ml BSA) and incubated with AhdI (0.25 U/μl) or XmnI (1 U/μl) at 37 °C for 10 min. SDS was added to 0.5% and the DNA species were separated on a denaturing gel.
Electron Microscopy (EM)
DNA from 40 µl of reactions at a 16-min timepoint was dissolved in 20 µl buffer L (25 mM HEPES, pH 7.5, 10 mM MgCl2 and 50 mM KCl). T4 gp32 protein (NEB) was added to 1 µg/ml, and after a 10-min incubation at 25°C, crosslinking of protein to DNA was carried out using glutaraldehyde (0.6%) at 25°C for 5 min. DNA was purified in 2-ml 6% agarose beads (Agarose Bead Technologies) equilibrated with TE Buffer (10 mM Tris-HCl, pH 7.5 and 1 mM EDTA). The samples were adsorbed onto glow-charged thin carbon support in TE buffer containing 2.5 mM spermidine, dehydrated through a series of water/ethanol washes, and air dried27. The EM grids were shadowed by rotary tungsten coating at 1× 10−7 torr and examined in an FEI Tecnai 12 TEM at 40 kV. Images were captured using an Ultrascan400 scan CCD camera (Gatan Inc.). Adobe Photoshop was used to invert images.
Acknowledgements
We thank Virginia Zakian, Karim Labib, Wolf-Dietrich Heyer, James Haber, Curt Wittenberg, Robert Johnson, and Louise Prakash for the gift of antibodies, strains or plasmids, and Daniel Camerini-Otero for providing DinG. We are grateful to Jack Griffith and Smaranda Wilcox for training in metal shadowing electron microscopy. This work was funded by grants from the US National Institutes of Health (GM080600 to GI; ES007061, GM057814, ES015632 to PS, GM084242 to AM, T32 GM07526-34 to MAW), National Research Foundation of Korea, Academia Sinica National Taiwan University, and the National Science Council of Taiwan.
Footnotes
Author contributions
M.W. and Y.K. contributed equally to this work. M.W. performed genetic and physical analyses of BIR in pif1∆ cells, td-mcm4 and td-psf2; analyzed conversion tracts and tested ssDNA formation during BIR; Y.K. carried out the reconstitution of the repair synthesis reaction and other biochemical experiments; Y.X. performed the EM analyses; W.H.C. tested initial DNA synthesis by qPCR, analyzed polymerases and Rad51 recruitment by CHIP and tested crossover frequencies; P.C. provided essential help in protein expression and purification; H.N. provided key reagents for the study; X.C. analyzed Pif1 and RPA recruitment by ChIP; A.M. provided key reagents; R. M. tested the role of resolvases in BIR; G.I., P.S., M.W. and Y.K. designed the experiments, discussed the data and wrote the manuscript.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Maloisel L, Fabre F, Gangloff S. DNA polymerase delta is preferentially recruited during homologous recombination to promote heteroduplex DNA extension. Mol Cell Biol. 2008;28:1373–1382. doi: 10.1128/MCB.01651-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, et al. Role of DNA replication proteins in double-strand break-induced recombination in Saccharomyces cerevisiae. Mol Cell Biol. 2004;24:6891–6899. doi: 10.1128/MCB.24.16.6891-6899.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li X, Stith CM, Burgers PM, Heyer WD. PCNA is required for initiation of recombination-associated DNA synthesis by DNA polymerase delta. Mol Cell. 2009;36:704–713. doi: 10.1016/j.molcel.2009.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung WH, Zhu Z, Papusha A, Malkova A, Ira G. Defective resection at DNA double-strand breaks leads to de novo telomere formation and enhances gene targeting. PLoS Genet. 2010;6:e1000948. doi: 10.1371/journal.pgen.1000948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dewar JM, Lydall D. Pif1- and Exo1-dependent nucleases coordinate checkpoint activation following telomere uncapping. EMBO J. 2010;29:4020–4034. doi: 10.1038/emboj.2010.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cesare AJ, Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. 2010;11:319–330. doi: 10.1038/nrg2763. [DOI] [PubMed] [Google Scholar]
- 7.Paeschke K, Capra JA, Zakian VA. DNA Replication through G-Quadruplex Motifs Is Promoted by the Saccharomyces cerevisiae Pif1 DNA Helicase. Cell. 2011;145:678–691. doi: 10.1016/j.cell.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabouri N, McDonald KR, Webb CJ, Cristea IM, Zakian VA. DNA replication through hard-to-replicate sites, including both highly transcribed RNA Pol II and Pol III genes, requires the S. pombe Pfh1 helicase. Genes Dev. 2012;26:581–593. doi: 10.1101/gad.184697.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Budd ME, Reis CC, Smith S, Myung K, Campbell JL. Evidence suggesting that Pif1 helicase functions in DNA replication with the Dna2 helicase/nuclease and DNA polymerase delta. Mol Cell Biol. 2006;26:2490–2500. doi: 10.1128/MCB.26.7.2490-2500.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pike JE, Burgers PM, Campbell JL, Bambara RA. Pif1 helicase lengthens some Okazaki fragment flaps necessitating Dna2 nuclease/helicase action in the two-nuclease processing pathway. J Biol Chem. 2009;284:25170–25180. doi: 10.1074/jbc.M109.023325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rossi ML, et al. Pif1 helicase directs eukaryotic Okazaki fragments toward the two-nuclease cleavage pathway for primer removal. J Biol Chem. 2008;283:27483–27493. doi: 10.1074/jbc.M804550200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deem A, et al. Defective break-induced replication leads to half-crossovers in Saccharomyces cerevisiae. Genetics. 2008;179:1845–1860. doi: 10.1534/genetics.108.087940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schulz VP, Zakian VA. The saccharomyces PIF1 DNA helicase inhibits telomere elongation and de novo telomere formation. Cell. 1994;76:145–155. doi: 10.1016/0092-8674(94)90179-1. [DOI] [PubMed] [Google Scholar]
- 14.Lydeard JR, Jain S, Yamaguchi M, Haber JE. Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature. 2007;448:820–823. doi: 10.1038/nature06047. [DOI] [PubMed] [Google Scholar]
- 15.Ho CK, Mazon G, Lam AF, Symington LS. Mus81 and Yen1 promote reciprocal exchange during mitotic recombination to maintain genome integrity in budding yeast. Mol Cell. 2010;40:988–1000. doi: 10.1016/j.molcel.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ira G, Malkova A, Liberi G, Foiani M, Haber JE. Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell. 2003;115:401–411. doi: 10.1016/s0092-8674(03)00886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prakash R, et al. Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev. 2009;23:67–79. doi: 10.1101/gad.1737809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petukhova G, Stratton S, Sung P. Catalysis of homologous DNA pairing by yeast Rad51 and Rad54 proteins. Nature. 1998;393:91–94. doi: 10.1038/30037. [DOI] [PubMed] [Google Scholar]
- 19.Van Komen S, Petukhova G, Sigurdsson S, Sung P. Functional cross-talk among Rad51, Rad54, and replication protein A in heteroduplex DNA joint formation. J Biol Chem. 2002;277:43578–43587. doi: 10.1074/jbc.M205864200. [DOI] [PubMed] [Google Scholar]
- 20.Bochman ML, Sabouri N, Zakian VA. Unwinding the functions of the Pif1 family helicases. DNA Repair (Amst) 2010;9:237–249. doi: 10.1016/j.dnarep.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sugiyama T, Zaitseva EM, Kowalczykowski SC. A single-stranded DNA-binding protein is needed for efficient presynaptic complex formation by the Saccharomyces cerevisiae Rad51 protein. J Biol Chem. 1997;272:7940–7945. doi: 10.1074/jbc.272.12.7940. [DOI] [PubMed] [Google Scholar]
- 22.Boule JB, Zakian VA. The yeast Pif1p DNA helicase preferentially unwinds RNA DNA substrates. Nucleic Acids Res. 2007;35:5809–5818. doi: 10.1093/nar/gkm613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sebesta M, Burkovics P, Haracska L, Krejci L. Reconstitution of DNA repair synthesis in vitro and the role of polymerase and helicase activities. DNA Repair (Amst) 2011;10:567–576. doi: 10.1016/j.dnarep.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith CE, Lam AF, Symington LS. Aberrant double-strand break repair resulting in half crossovers in mutants defective for Rad51 or the DNA polymerase delta complex. Mol Cell Biol. 2009;29:1432–1441. doi: 10.1128/MCB.01469-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burgers PM, Gerik KJ. Structure and processivity of two forms of Saccharomyces cerevisiae DNA polymerase delta. J Biol Chem. 1998;273:19756–19762. doi: 10.1074/jbc.273.31.19756. [DOI] [PubMed] [Google Scholar]
- 26.Formosa T, Alberts BM. DNA synthesis dependent on genetic recombination: characterization of a reaction catalyzed by purified bacteriophage T4 proteins. Cell. 1986;47:793–806. doi: 10.1016/0092-8674(86)90522-2. [DOI] [PubMed] [Google Scholar]
- 27.Griffith JD, Christiansen G. Electron microscope visualization of chromatin and other DNA-protein complexes. Annu Rev Biophys Bioeng. 1978;7:19–35. doi: 10.1146/annurev.bb.07.060178.000315. [DOI] [PubMed] [Google Scholar]
- 28.Ferguson DO, Holloman WK. Recombinational repair of gaps in DNA is asymmetric in Ustilago maydis and can be explained by a migrating D-loop model. Proc Natl Acad Sci U S A. 1996;93:5419–5424. doi: 10.1073/pnas.93.11.5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hastings PJ, Lupski JR, Rosenberg SM, Ira G. Mechanisms of change in gene copy number. Nat Rev Genet. 2009;10:551–564. doi: 10.1038/nrg2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malkova A, Ira G. Break-induced replication: functions and molecular mechanism. Curr Opin Genet Dev. 2013;23:271–279. doi: 10.1016/j.gde.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sugawara N, Wang X, Haber JE. In vivo roles of Rad52, Rad54, and Rad55 proteins in Rad51-mediated recombination. Mol Cell. 2003;12:209–219. doi: 10.1016/s1097-2765(03)00269-7. [DOI] [PubMed] [Google Scholar]
- 32.Janke R, et al. A truncated DNA-damage-signaling response is activated after DSB formation in the G1 phase of Saccharomyces cerevisiae. Nucleic Acids Res. 2010;38:2302–2313. doi: 10.1093/nar/gkp1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Busygina V, et al. Hed1 regulates Rad51-mediated recombination via a novel mechanism. Genes Dev. 2008;22:786–795. doi: 10.1101/gad.1638708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zierhut C, Diffley JF. Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. Embo J. 2008;27:1875–1885. doi: 10.1038/emboj.2008.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malkova A, Klein F, Leung WY, Haber JE. HO endonuclease-induced recombination in yeast meiosis resembles Spo11- induced events. Proc Natl Acad Sci U S A. 2000;97:14500–14505. doi: 10.1073/pnas.97.26.14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Acharya N, Klassen R, Johnson RE, Prakash L, Prakash S. PCNA binding domains in all three subunits of yeast DNA polymerase delta modulate its function in DNA replication. Proc Natl Acad Sci U S A. 2011;108:17927–17932. doi: 10.1073/pnas.1109981108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Komen S, Macris M, Sehorn MG, Sung P. Purification and assays of Saccharomyces cerevisiae homologous recombination proteins. Methods Enzymol. 2006;408:445–463. doi: 10.1016/S0076-6879(06)08028-1. [DOI] [PubMed] [Google Scholar]
- 38.Raschle M, Van Komen S, Chi P, Ellenberger T, Sung P. Multiple interactions with the Rad51 recombinase govern the homologous recombination function of Rad54. J Biol Chem. 2004;279:51973–51980. doi: 10.1074/jbc.M410101200. [DOI] [PubMed] [Google Scholar]