

Abstract
Organophosphate-induced brain damage is an irreversible neuronal injury, likely because there is no pharmacological treatment to prevent or block secondary damage processes. The presence of free glutamate (Glu) in the brain has a substantial role in the propagation and maintenance of organophosphate-induced seizures, thus contributing to the secondary brain damage. This report describes for the first time the ability of blood glutamate scavengers (BGS) oxaloacetic acid in combination with glutamate oxaloacetate transaminase to reduce the neuronal damage in an animal model of paraoxon (PO) intoxication. Our method causes a rapid decrease of blood Glu levels and creates a gradient that leads to the efflux of the excess brain Glu into the blood, thus reducing neurotoxicity. We demonstrated that BGS treatment significantly prevented the peripheral benzodiazepine receptor (PBR) density elevation, after PO exposure. Furthermore, we showed that BGS was able to rescue neurons in the piriform cortex of the treated rats. In conclusion, these results suggest that treatment with BGS has a neuroprotective effect in the PO intoxication. This is the first time that this approach is used in PO intoxication and it may be of high clinical significance for the future treatment of the secondary neurologic damage post organophosphates exposure.
Keywords: animal models, excitotoxicity, glutamate, neuroprotection, neurotransmitters
Introduction
Organophosphate compounds are highly toxic chemicals widely used as pesticides (e.g. malathion and paraoxon (PO)) and as chemical warfare nerve agents (e.g. soman, sarin). Pesticide poisoning is one of the most common poisonings worldwide, estimated at one million cases each year with several hundred thousand deaths.1 Organophosphates inactivate the enzyme acetylcholine esterase (AChE; EC 3.1.1.7), a serine protease that hydrolyzes the neurotransmitter acetylcholine. Exposure to organophosphates leads to acetylcholine accumulation that causes the overstimulation of cholinergic receptors, which consequently results in the rapid and profound excitotoxicity and dysfunction of cholinergic neurons. This effect is reactivated very slowly, if at all.2
Exposure to organophosphates damages several brain areas including the entorhinal and piriform cortex, amygdala, and hippocampus CA1/CA3 [ref 3]. Much of the brain damage does not typically occur at the time of the initial lesion, making secondary neuronal damage a major contributor to the neuronal loss.4 The degree of brain damage depends on the severity of the convulsions and is in direct relation to the increase in peripheral benzodiazepine receptors (PBR) density.5 Peripheral benzodiazepine receptors are located in the microglia and are used as reliable markers of brain damage.6 The density of PBRs increases when microglias are activated in response to tissue damage. This increase appears as early as 24 hours post systemic organophosphates exposure and lasts for at least 2 weeks post exposure.7
Glutamate (Glu) has a substantial role in the propagation and maintenance of organophosphate-induced seizures, thus contributing to the secondary brain damage.8 Glu was shown to be released in excess in the brain under soman intoxication and had a prominent role during seizures.9 Furthermore, Glu receptor antagonists in general, and N-Methyl-D-aspartate (NMDA) blockers in particular, were proposed as potential antidotes against organophosphates intoxication.10 Moreover, De Groot et al11 showed a reduction in the brain damage post soman exposure in animals treated with a competitive NMDA antagonist. N-Methyl-D-aspartate blockers were also shown to be potent protective agents against neuronal loss caused by cholinergic seizures.12
The standard treatment for organophosphates intoxication consists of pretreatment with pyridostigmine, a reversible inhibitor of AChE and anticholinergic agents such as atropine sulfate.13 In addition, it was reported that benactyzine (combined with anticholinergic and anti-NMDA properties), or benzodiazepines, could reduce some of the brain damage if administered early enough.14, 15 Yet, such a treatment failed to control the brain damage associated with excitotoxicity of Glu.9 It also failed to prevent the elevation in PBR density post organophosphate exposure.3, 16 N-Methyl-D-aspartate receptor antagonists also failed to fill their optimistic prognosis, as none of the treatments that showed preclinical promise are available to date.17, 18
The maintenance of brain extracellular Glu at levels below its excitotoxic threshold is performed not only by Glu transporters located on glia and neurons but also by those present on the anti-luminal side of the brain capillary endothelial cells.19, 20 These transporters remove the excess extracellular brain Glu into the blood stream.21 In a recent study, we have established the feasibility of accelerating the naturally occurring brain–to-blood Glu efflux by inverting the concentration gradient between the blood versus brain Glu level as a novel neuroprotective treatment.20 This was achieved by the Glu scavenging properties of the blood-resident enzyme Glu oxaloacetate transaminase (human source—hGOT) which, with oxaloacetate (OxAc), converts Glu into 2-ketoglutarate and aspartate, thereby decreasing the blood concentration of Glu. Blood glutamate scavengers (BGS) provided highly significant brain neuroprotection in rat animal models of closed head injury, ischemic stroke and glioma brain tumors, in which there is a local breach of the blood–brain barrier.22, 23, 24
In the present study, we investigated whether BGS could be used as a neuroprotective treatment in a well-established and reliable animal model of PO intoxication by inhibiting the secondary neuronal damage associated with organophosphates.
Materials and Methods
Materials
Paraoxon was obtained from Chem Service (West Chester, PA, USA), Glu dehydrogenase was obtained from Roche Diagnostics (Rotkreuz, Switzerland), glass-fiber filters (GF/B 2.5 cm radius) were obtained from Wathaman (Kent, UK), [3H]PK 11195 was obtained from PerkinElmer (Boston, MA, USA); all other materials were obtained from Sigma-Aldrich (St Louis, MO, USA). His-tagged version of the human glutamate oxaloacetate transaminase (hGOT) cDNA that was cloned from the human hepatoma cell line hepG2 was expressed in Escherichia coli and purified by Ni-agarose chromatography.
Animals
The experiments were conducted according to the Guidelines for the Use of Experimental Animals of the European Community approved by the Animal Care Committees of the Weizmann Institute of Science. Forty-one male, 9 to 11 weeks old, Sprague–Dawley rats (Harlan Laboratories, Rehovot, Israel), were used in this study. Weight variation of animals at the time of PO exposure was in the range of 300 to 320 g. The experiments began after at least 5 days of acclimation. During acclimation and throughout the study, the animals were housed within a limited access rodent facility, two rats in each cage, and after jugular vein cannulation, animals were housed individually in polypropylene cages (37.5 × 21 × 18 cm). The animals were provided diet ad libitum and free access to drinking water. Automatically controlled environmental conditions were set to maintain temperature at 20°C to 24°C with a relative humidity of 30% to 70%, a 12:12-hour light–dark cycle and 15 to 30 air changes/hour in the study room. On the first day of the study, the animals were randomly divided into two groups: control and treated.
Study Design
Jugular vein cannulation was performed as previously described25 and the rats were allowed to recover for 3 days. After recovery, PO was injected intramuscularly into the hind limb with a dosing of 450 μg/kg. One minute post PO challenge, atropine and Toxogonin at a dose of 0.9 mg/100 μl and 6 mg/100 μl per animal, respectively were administered intramuscularly into the thigh muscle of the contralateral hind limb. Starting 10 minutes post PO challenge, the treated animals were injected intravenously with the OxAc+hGOT infusion dissolved in saline (3.95 mg/3 ml per animal and 45 μg/animal, respectively). The control group was injected with saline at a volume of 3 ml/animal. Both groups were injected at a constant rate of 0.1 ml/minute for 30 minutes. Immediately before the treatment session and connection to the indwelling cannula, the entire infusion array, consisting of extensor polyethylene (PE 50) tubing, a swivel (Instech Solomon, Plymouth Meeting, PA, USA) syringe and infusion pump (Harvard Apparatus, Model No.02054, South Natick, MA, USA), were appropriately flushed and filled with the treatment solution dosing (OxAc+hGOT or saline). Prior and 40 minutes post PO challenge, 400 μl of blood was drawn from the jugular vein cannulation to assess blood Glu concentration and the activity of butyrylcholinesterase (BuChE). Clinical signs were monitored as described below for seven successive days. At day 7 post PO challenge, the animals were perfused and brains were used for immunohistochemistry and PBR assays.
Clinical Signs
Throughout the PO challenge, infusion, and later on, test animals were maintained within a transparent container and were observed for clinical signs with particular attention paid to the onset, intensity, and duration of characteristic and representative peripheral and central cholinomimetic manifestations. These refer mostly to dyspnea, fasciculation, salivation, and lacrimation, ataxia, paralysis, tonic–clonic convulsions, coma, and death. Recorded clinical signs parameters were graded as absent, mild, moderate, or severe. The latency until evident onset of convulsing seizures, as well as the intensity, scored by using the Racine's scale and respective time of cessation was documented.26 Clinical signs observation was carried out before PO administration and thereafter at 2.5, 5, 7.5, 10, 15, 30, 60, 120, 180 minutes, 5, 6, and 24 hours post administration and then at least once daily throughout the successive 7-day study period. Animals that did not survive the first 24 hours were excluded from the study.
Histology and Immunohistochemistry
One week post PO challenge, rats were anesthetized and perfused transcardially with chilled phosphate-buffered saline/heparin. Brains were removed and fixed in Bouin solution or in 4% formalin. Samples were processed for paraffin embedding by standard procedures. Six-micron thick coronal brain serial sections were generated with 500 μm spacing between series. Sections were stained with hematoxylin and eosin and used for immunohistochemistry. Sections were observed and captured through an optical Eclipse E800 microscope (Nikon, Tokyo, Japan) and a digital camera DMX 1200 (Nikon). ImagePro Plus software (Media Cybernetics, Silver Spring, MD, USA) was used for quantitative analysis. The number of neurons per 100 mm2 was calculated in 10 microscopic fields of each sample (objective magnification Å∼40). At least eight images in each case were used for analysis. All morphometric analyses were performed without previous knowledge of the experimental group from which the sections were obtained.
Blood Glutamate Determination Assay
Two-hundred microliters of rat blood was collected 10 minutes before and 40 minutes after PO injection. The blood was added to an ice-cold Eppendorf tube containing 200 μl 1 mol/L perchloric acid and centrifuged at 10,000 g for 10 minutes. The supernatant was collected and pH was adjusted to 7.2 with 2 mol/L carbonic acid and centrifuged again at 10,000 g for 10 minutes. The supernatant was stored at −80°C until analysis. Glutamate concentrations were measured using the fluorometric method of Graham and Aprison.27 Briefly, 60 μl duplicated blood samples were added to 90 μl solution containing 0.3 mol/L glycine, 0.25 mol/L hydrazine hydrate buffer adjusted to pH 8.6 with 0.5 M H2SO4 and 7.5 U of Glu dehydrogenase in 0.2 mmol/L nicotinamide adenine dinucleotide. After incubation for 30 to 45 minutes at room temperature, the fluorescence was measured at 460 nm with excitation at 350 nm, and its concentration was determined using a Glu standard curve in the 0 to 60 μmol/L range.
Peripheral Benzodiazepine Receptor Assay
Three groups of animals were investigated, the control and treated group of rats (1 week after PO challenge), and in addition, a group of naïve animals. The method of PBR assay was modified from Lavoieet al.28 The animals were briefly anesthetized with CO2 (less than 25 seconds) before decapitation and the brain (without cerebellum) was rapidly harvested, placed into Petri dish filled with ice-cold saline, weighted, and stored at −80°C until analysis.
After thawing of brain from −80°C, 50 mL cold Tris buffer (20 nmol/L pH 7.4) was added and the brain was manually homogenized using a dounce homogenizer (Bellco, Chicago, IL, USA). Four 5 mL tubes were filled with the homogenized brain and subjected to two rounds of centrifugation at 40,000 g for 30 minutes at 4°C. The supernatant was discarded and the pellet was re-suspended in 5 mL cold Tris buffer and after the second centrifugation in 1 mL Tris buffer. The re-suspended pellet was tested for protein content with BCA assay.29 Approximately 10 μg of protein was added to four Eppendorf tubes, which contained 500 μl of Tris buffer at room temperature. A radioactive PBR ligand [3H] PK11195 was added to all tubes at a concentration of 5 nmol/L, and unmarked PK11195 was added to two control tubes at a concentration of 20 μmol/L for the detection of unspecific binding. The tubes were incubated at room temperature for 90 minutes and then filtered with vacuum through glass-fiber filters presoaked with polyethyleneimine (0.3%). The filters were then washed five times with 4 mL cold Tris buffer and folded into a tube containing 4 mL of scintillation liquid and disintegrations per minute were counted in a β-counter. Control tubes showed fewer than 10% of unspecific binding.
Butyrylcholinesterase Activity Assay
Determination of erythrocyte AChE activity and cholinesterase status are no standard laboratory assays. However, determination of plasma BuChE (EC 3.1.1.8) is routinely used for monitoring organophosphate poisoning.30 Compared with AChE, BuChE may show different inhibition, reactivation, and aging kinetics. Nevertheless, we are using the BuChE activity assay as a marker for the inactivation of cholinesterase in combination with other clinical signs of organophosphate poisoning.
Briefly, 200 μL of rat blood was collected before and 40 minutes after PO injection. The blood was added to heparinized tubes maintained on ice. After centrifugation at 2,000 g for 10 minutes at 4°C, the supernatant was collected and an equal amount of 0.1 mol/L phosphate buffer (pH 8.0) was added. The samples were stored at −80°C until analysis.
BuChE activity was determined by the method of Ellman et al31 with some modifications. Hydrolysis rates were measured at acetylthiocholine concentration of 0.8 mmol/L in 1 mL assay final volume in a solution with 0.1 mol/L phosphate buffer, pH 7.5, and 1 mmol/L DTNB at 25°C. Fifty microliters of serum was added to the reaction mixture and preincubated for 10 minutes. The hydrolysis was monitored by determining the formation of the thiolate anion of DTNB at 436 nm for 10 minutes (intervals of 30 seconds) using a Hitachi 2001 spectrophotometer (Mho, Japan). All samples were run in triplicates. Results are presented as 'nmoles per minute per mL of plasma' using the extinction coefficient for the yellow product, according to the Beer–Lambert law.
Data Analysis
Results were analyzed by one-way analysis of variance (ANOVA) with Newman–Keuls multiple comparison test. The accepted level of significance for all tests was P<0.05 unless stated otherwise. Results are presented as mean±standard error unless stated otherwise.
Results
In this study, we investigated the effect of the BGS, OxAc, and hGOT on the neuronal damage of rats after PO exposure, a potent AChE inhibitor (see Figure 1). From 31 rats that were exposed to PO and survived up to the end of the study, 15 animals were challenged with saline and 16 were with the OxAc+hGOT infusion. Ten naïve rats were used as a standard for PBR and histology assays. Eleven rats (26.2%), six from the control group (28.6%) and five from the treated group (23.8%), died within first 24 hours and therefore were excluded from the study.
Figure 1.
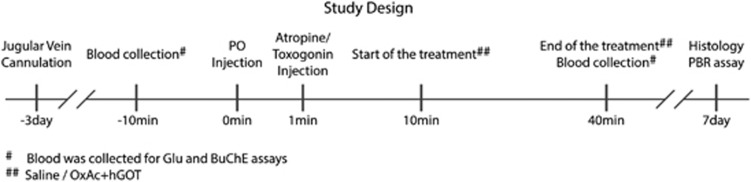
Study design. An illustration of the steps performed in the study.
Effect of Paraoxon Exposure on the Central and Peripheral Cholinergic System
In order to evaluate the action of PO as a potent AChE inhibitor, an equivalent assay verifying the activity of BuChE was performed (see Materials and Methods). We measured the control activity of BuChE (n=31 all animals before PO exposure) and found a rate of 200.9±8.6 nmoles per minute per mL of plasma using the extinction coefficient for the yellow product (nmol/minute per pmL). Upon PO injection, both groups of animals displayed more than 50% inhibition in the activity of BuChE with no significant differences between them (see Figure 2; 95.6±17 nmol/minute per pmL and 89.2±11.7 nmol/minute per pmL for saline and OxAc+hGOT groups accordingly; n=16 saline, n=15 OxAc+hGOT; One-way ANOVA with Newman–Keuls multiple comparison test, P<0.001 for both). These results validate the potency of PO as an organophosphate in our animal model, inhibiting the activity of cholinesterase's activity, in an equal manner for both the saline and OxAc+hGOT groups. Furthermore, the most important clinical sign of organophosphates exposure, convulsions, was similar in the saline and OxAc+hGOT groups (see Figure 3 for convulsions and Supplementary Figure 1 for all other clinical signs n=16 saline, n=15 OxAc+hGOT P>0.05). In conclusion, these results suggest that there is no direct effect of OxAc+hGOT on the activity of PO as an AChE inhibitor.
Figure 2.

Butyrylcholinesterase activity decreases after paraoxon (PO) challenge. The rate of activity of butyrylcholinesterase (BuChE) is significantly lower 40 minutes after PO injection for both groups as compared with before the injection (control). There is no significant difference between PO and saline and PO and OxAc+hGOT (human glutamate oxaloacetate transaminase) groups indicating no direct effect of the blood glutamate scavenger (BGS) treatment on the PO activity. Control here refers to before the PO challenge (n=31). Saline n=16, OxAc+hGOT n=15, P<0.001 for both using one-way analysis of variance with Newman–Keuls multiple comparison test, ***P<0.001. OxAc, oxaloacetate.
Figure 3.

Clinical signs are similar between control and treated groups. Seizures were quantified on a Racine's scale (0 to 5). No significant difference was observed in the control (saline–red squares; n=16) versus treated (OxAc+hGOT (human glutamate oxaloacetate transaminase)–blue triangles; n=15) groups indicating that there was no direct effect of the blood glutamate scavenger treatment on the paraoxon (PO) activity. One-way analysis of variance with Newman–Keuls multiple comparison test, P>0.05. OxAc, oxaloacetate.
In order to evaluate the peripheral blood potency the Glu scavenger hGOT in combination with OxAc to reduce blood Glu level, we monitored the Glu levels before (165±15 μmol/L Glu; n=31 control—average baseline of all treated animals 10 minutes before PO injection) and 40 minutes after OxAc+hGOT administration versus saline. As the treatment consisted of Glu scavenger injection, blood Glu levels were expected to drop after the administration of OxAc+hGOT. Indeed, a significant decrease in the blood Glu levels in the OxAc+hGOT group was observed (97±17 μmol/L) as compared with the saline group (see Figure 4; 176±12 μmol/L n=16 saline; n=15 OxAc+hGOT; One-way ANOVA with Newman–Keuls multiple comparison test, P<0.05). This result demonstrates that the administration of OxAc+hGOT in contrast to saline reduces blood Glu levels after PO exposure.
Figure 4.

Blood glutamate (Glu) levels decreases with administration of Glu scavengers. The injection of OxAc+hGOT (human glutamate oxaloacetate transaminase) (n=15) significantly reduced the blood Glu concentration in contrast to the saline (n=16) injection P<0.05 using one-way analysis of variance with Newman–Keuls multiple comparison test. We suggest that this creates a larger brain-to-blood efflux that is a critical part of the blood glutamate scavenger method. Control here refers to 10 minutes before the paraoxon (PO) challenge of all treated animals (n=31); Two other groups are 40 minutes after PO injection. *P<0.05. OxAc, oxaloacetate.
Effects of Glutamate Scavengers on the Central Nervous System After Exposure to Paraoxon
Our hypothesis asserts that peripheral effects of Glu scavengers cause beneficial effects by leading to the reduction in the excess brain Glu level responsible for the brain damage associated with organophosphates exposure. Previous investigations have indicated that the measurement of PBR density could be of widespread applicability in the quantification of neural tissue damage in the central nervous system as this astrocytes and microglia receptor is a known indicator of cell stress in many conditions.32, 33 Moreover, given that different areas in the brain are affected upon exposure to organophosphate (see Introduction); we sought to analyze and compare the brains of the naïve versus the two treated groups. Using a radioactive affinity assay, we observed a significantly lower concentration of PBR in the brain of the naïve animals (see Figure 5; 132±13 fmol/mg; n=5) versus the two groups that were exposed to PO, indicative of the stress caused by this challenge. Interestingly, there was a significant difference in the concentration of PBR between the two PO exposed groups, where animals treated with Glu scavengers showed a significantly lower concentration of PBR (312±47 fmol/mg versus 436±25 fmol/mg OxAc+hGOT versus saline respectively; for naïve n=4 for saline and OxAc+hGOT, One-way ANOVA with Newman–Keuls multiple comparison test, P<0.05 for saline versus OxAc+hGOT, P<0.01 for OxAc+hGOT versus naïve, P<0.001 for saline versus naïve). These results suggest that Glu scavengers were able to reduce at least part of the stress induced by exposure to PO.
Figure 5.

Whole-brain peripheral benzodiazepine receptor (PBR) concentration is lowered upon administration of glutamate (Glu) scavengers. Peripheral benzodiazepine receptor concentration is a good indicator of neuronal stress. Here we see a significant reduction in PBR concentration as a result of the BGS treatment. Although naïve animals (n=5) have the lowest concentration of PBR, there is a significant difference between saline (n=4) and OxAc+hGOT (human glutamate oxaloacetate transaminase) (n=4) treated groups 7 days after the paraoxon (PO) challenge. One-way analysis of variance with Newman–Keuls multiple comparison test; **P<0.01, ***P<0.001.
To further confirm the central protective effects of Glu scavengers, an immunohistological analysis of cell numbers in the piriform cortex, an area highly susceptible to seizure-induced neuropathology was conducted. This area was the only one in which significant morphologic damage in the saline group was found (data not shown). The same results were observed: hence naïve animals had a significantly higher number of traceable cells (see Figure 6C for a representative example; 14.8±0.95 cells/mm2) compared with the brains of OxAc+hGOT-treated animals (see Figure 6A for representative examples; 10.6±1.37 cells/mm2) and the saline groups (see Figure 6B for representative examples; 3.72±0.9 cells/mm2). Nevertheless, in the OxAc+hGOT group, there was a substantial rescue of cell numbers (n=8 for OxAc+hGOT and saline, n=5 for naïve; one-way ANOVA with Newman–Keuls multiple comparison test, P<0.001 for saline versus OxAc+hGOT and saline versus naïve, P<0.05 for OxAc+hGOT versus naïve).
Figure 6.

Cell numbers in the piriform cortex are rescued as a result of glutamate (Glu) scavengers. (A, B) Example sections from paraoxon (PO) and OxAc+hGOT (human glutamate oxaloacetate transaminase) (A) and PO and saline (B) groups of hematoxylin and eosin (H&E) staining. Left and right columns represent left and right piriform cortex accordingly. (C) Example section of the piriform cortex from a naïve animal of H&E staining. All scale bars are 100 μm. (D) Quantification of cell densities per mm2 from eight cortex sections of each group, each 6 μm width. Lower neuronal cell number indicates increased brain damage as a result of neuronal death. The amount of the rescued neurons is significantly higher in the OxAc+hGOT-treated group (n=8) as compared with the saline group (n=8), although the naïve group (n=5) had more neurons then both groups. *P<0.05, ***P<0.001. OxAc, oxaloacetate.
Discussion
To date, organophosphate-induced brain damage is an irreversible neuronal injury because no pharmacological treatment is currently available to prevent or block secondary damage processes.34, 35 The major contributors to the secondary neuronal brain damage, manifested in cell death, are thought to be calcium influx and apoptosis as a result of excessive release of extracellular Glu.36 It has been previously shown that after organophosphates intoxication, there is a release of excess Glu in the brain, which appears to cause the neurologic damage.3 Understanding secondary neuronal damage processes offer a potential therapeutic window in which progressive neural injury and neuronal cell death may be inhibited, and the extent of disability may be reduced during the first few hours after exposure to organophosphates.37, 38 In this study, we demonstrate for the first time the ability of BGS to reduce the secondary neuronal damage in an animal model of PO intoxication treated with a combination of OxAc and hGOT.
Under normal physiologic conditions, Glu, which is released into the synaptic cleft, is quickly removed by excitatory amino-acid transporters located in astrocytes and neurons. However, this widely accepted view of the homeostasis of Glu in brain fluids does not take into consideration the possible contribution of the blood–brain barrier. The maintenance of brain extracellular Glu at levels below its excitotoxic threshold is performed not only by the Glu transporters located on glia and neurons but also by those present on the anti-luminal side of the brain capillary endothelial cells.19, 20 These transporters shuttle the excess extracellular brain Glu to the blood.21 The method presented in the current study causes a rapid decrease of blood Glu levels. Based on our previous studies, we suggest that the gradient that was created after OxAc/GOT injection leads to the efflux of the excess brain Glu into the blood stream thereby reducing its potential to cause neurologic damage.
Based on previous experiments in our lab and others, we suggest that the combination of a single dose of OxAc and hGOT is optimal for the PO intoxication model. This treatment shows a long lasting effect of several hours in reducing blood Glu levels,21 and omits the need for repetitive dosing. Additionally, these doses and treatment plan were also proven as beneficial in a rat model of head injury and middle cerebral artery occlusion.22, 24 It is worth mentioning that in contrast to the PO animal model presented in this study, in the above mentioned models, there is a local breach of the blood–brain barrier that could facilitate the efflux of excess Glu.
As BGS treatment takes place in the peripheral blood alongside the detrimental activity of PO, it was important to monitor PO activity in both BGS and saline treated groups to exclude a direct effect of BGS on PO activity. We demonstrate that BuChE activity and the clinical signs of both groups are similar, strongly suggesting that there is no direct effect of OxAc+hGOT treatment on the activity of PO as an AChE inhibitor. Moreover, as early as 30 minutes from OxAc+hGOT injection, the level of the blood Glu level was significantly lowered in the treated group, compared with the control group, which was injected with saline. Thus and based on previous studies showing increased brain Glu levels after organophosphate exposure,9 we suggest that a gradient is formed between the blood and brain Glu levels, a necessary condition for the central nervous system Glu efflux.
Having demonstrated the blood Glu-lowering activity of the OxAc+hGOT treatment, we investigated the ability of BGS to reduce the excitotoxic effect of excess brain Glu levels after PO intoxication.39 Testing for PBR density, which is a well-established marker of the brain neuronal damage, we show that our treatment significantly reduced the elevated whole-brain PBR density in the treated animals, as compared with the control group. To further analyze if the elevated level of PBR in treated animals manifested at a whole-brain level, an immunohistochemical analysis was performed. The results from the comparison of the treated and the naïve animals clearly demonstrated that even though the number of neurons in the naïve rats was higher than both groups, the OxAc+hGOT treatment succeeded in preventing the death of neurons in the piriform cortex, an area highly susceptible to seizure-induced neuronal damage. Interestingly, unlike previous studies3, 5, 7 where different brain regions were damaged, in our study only the neurons in the piriform cortex were affected. We do not know what the source of this selectivity is, but can hypothesize that the difference in the organophosphates used, the different model system and experimental setup might explain some of the variability.
In contrast to our expectation and other studies treating organophosphate poising,18 BGS treatment did not reduce the seizure intensity or the survival of the treated animals. We hypothesize that as our treatment is relatively moderate, a stronger dosing regimen or pretreatment could have reduced mortality or seizure intensity of the treated group and not only the secondary brain damage.
Based on data obtained from several in vivo experiments it is safe to assume that an immediate elevation of Glu level in the central nervous system is intimately associated with the neuropathology of nerve agents. Despite the efficacy of anti-Glu agents in animal models has been shown in the past by several groups, most of them failed in the human clinical trials because of severe side effects. For review, seeSolberg and Belkin.18 These were attributed to the direct effect of the agents on the central nervous system.
The clear advantage of our BGS technology is that there seem to be no direct intervention with any of the brain Glu receptors or Glu transport systems. The administration of OxAc+hGOT only temporarily affects the blood Glu levels and this creates a gradient between the levels of Glu in the blood and in the brain, causing a rapid efflux of excess Glu from the brain to the blood without affecting any other brain functions.22 As plasma Glu levels normally fluctuate by ∼50% during the circadian cycle,40 we speculate that in the transition of the BGS technology into clinical trials, no severe side effects are expected.
Conclusion
We have demonstrated that the in vivo administration of BGS is effective as a neuroprotective treatment in the PO intoxication model. This is the first time that this approach is used after PO intoxication and it may be of high clinical significance for the future treatment of the secondary neurologic damage in such pathologic conditions.
Acknowledgments
We would like to thank Tamara Berkutzki for her professional support in the histology analyses and Professor David Mirelman for his excellent advice on style and content of the manuscript.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Journal of Cerebral Blood Flow & Metabolism website (http://www.nature.com/jcbfm)
This work was supported by the Nella and Leon Benoziyo Center for Neurological Diseases and by a Grant from the US Department of Defense, DOD award number W81XWH-09-2-0004.
Supplementary Material
References
- Vinay P, Shubha S, Rao S, Charmaine S, Ashwini K, Rohith V. A case of organophosphate poisoning presenting with seizure and unavailable history of parenteral suicide attempt. J Emerg Trauma Shock. 2011;4:132–265. doi: 10.4103/0974-2700.76825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos MD, Pereira EF, Aracava Y, Castro NG, Fawcett WP, Randall WR, et al. Low concentrations of pyridostigmine prevent soman-induced inhibition of GABAergic transmission in the central nervous system: involvement of muscarinic receptors. J Pharmacol Exp Ther. 2003;304:254–265. doi: 10.1124/jpet.102.043109. [DOI] [PubMed] [Google Scholar]
- Lallement G, Delamanche IS, Pernot-Marino I, Baubichon D, Denoyer M, Carpentier P, et al. Neuroprotective activity of glutamate receptor antagonists against soman-induced hippocampal damage: quantification with an omega 3 site ligand. Brain Res. 1993;618:227–237. doi: 10.1016/0006-8993(93)91270-3. [DOI] [PubMed] [Google Scholar]
- Granacher RP., Jr . Traumatic brain injury: methods for clinical and forensic neuropsychiatric assessment. CRC; 2007. [Google Scholar]
- McDonough JH, Jr., McLeod CG, Jr., Nipwoda MT. Direct microinjection of soman or VX into the amygdala produces repetitive limbic convulsions and neuropathology. Brain Res. 1987;435:123–137. doi: 10.1016/0006-8993(87)91593-9. [DOI] [PubMed] [Google Scholar]
- Weissman BA, Brandeis R, Gilat E, Cohen G, Alkalay D, Rabinovitz I, et al. Monitoring drug-induced neurodegeneration by imaging of peripheral benzodiazepine receptors. Ann. N Y Acad. Sci. 2004;1025:584–589. doi: 10.1196/annals.1316.072. [DOI] [PubMed] [Google Scholar]
- Raveh L, Brandeis R, Gilat E, Cohen G, Alkalay D, Rabinovitz I, et al. Anticholinergic and antiglutamatergic agents protect against soman-induced brain damage and cognitive dysfunction. Toxicol Sci. 2003;75:108–116. doi: 10.1093/toxsci/kfg166. [DOI] [PubMed] [Google Scholar]
- Tattersall J. Seizure activity post organophosphate exposure. Front Biosci. 2009;14:3688–3711. doi: 10.2741/3481. [DOI] [PubMed] [Google Scholar]
- Lallement G, Denoyer M, Collet A, Pernot-Marino I, Baubichon D, Monmaur P, et al. Changes in hippocampal acetylcholine and glutamate extracellular levels during soman-induced seizures: influence of septal cholinoceptive cells. Neurosci Lett. 1992;139:104–107. doi: 10.1016/0304-3940(92)90868-8. [DOI] [PubMed] [Google Scholar]
- Sparenborg S, Brennecke LH, Jaax NK, Braitman DJ. Dizocilpine (MK-801) arrests status epilepticus and prevents brain damage induced by soman. Neuropharmacology. 1992;31:357–368. doi: 10.1016/0028-3908(92)90068-z. [DOI] [PubMed] [Google Scholar]
- De Groot DMG, Bierman EPB, Van Huygenvoort A. Involvement of acetylcholine and glutamate in soman-induced brain damage. Micron and Microscopica Acta. 1990;21:247–248. [Google Scholar]
- Fujikawa DG, Daniels AH, Kim JS. The competitive NMDA receptor antagonist CGP 40116 protects against status epilepticus-induced neuronal damage. Epilepsy Res. 1994;17:207–219. doi: 10.1016/0920-1211(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Capacio BR, Shih TM. Anticonvulsant actions of anticholinergic drugs in soman poisoning. Epilepsia. 1991;32:604–615. doi: 10.1111/j.1528-1157.1991.tb04699.x. [DOI] [PubMed] [Google Scholar]
- Gilat E, Kadar T, Levy A, Rabinovitz I, Cohen G, Kapon Y, et al. Anticonvulsant treatment of sarin-induced seizures with nasal midazolam: an electrographic, behavioral, and histological study in freely moving rats. Toxicol Appl Pharmacol. 2005;209:74–85. doi: 10.1016/j.taap.2005.03.007. [DOI] [PubMed] [Google Scholar]
- McDonough JH, Jr., Zoeffel LD, McMonagle J, Copeland TL, Smith CD, Shih TM. Anticonvulsant treatment of nerve agent seizures: anticholinergics versus diazepam in soman-intoxicated guinea pigs. Epilepsy Res. 2000;38:1–14. doi: 10.1016/s0920-1211(99)00060-1. [DOI] [PubMed] [Google Scholar]
- Dematteis M, Mallaret M, Baubichon D, Pernot-Marino I, Lallement G. Evaluation of dextromethorphan and dextrorphan as a preventive treatment of soman toxicity in mice. Neurosci Lett. 1997;234:91–94. doi: 10.1016/s0304-3940(97)00682-4. [DOI] [PubMed] [Google Scholar]
- Lees KR, Dyker AG, Sharma A, Ford GA, Ardron ME, Grosset DG. Tolerability of the low-affinity, use-dependent NMDA antagonist AR-R15896AR in stroke patients: a dose-ranging study. Stroke. 2001;32:466–472. doi: 10.1161/01.str.32.2.466. [DOI] [PubMed] [Google Scholar]
- Solberg Y, Belkin M. The role of excitotoxicity in organophosphorous nerve agents central poisoning. Trends Pharmacol Sci. 1997;18:183–185. doi: 10.1016/s0165-6147(97)89540-5. [DOI] [PubMed] [Google Scholar]
- Cohen-Kashi-Malina K, Cooper I, Teichberg VI. Mechanisms of glutamate efflux at the blood-brain barrier: involvement of glial cells. J Cereb Blood Flow Metab. 2012;32:177–189. doi: 10.1038/jcbfm.2011.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichberg VI, Cohen-Kashi-Malina K, Cooper I, Zlotnik A. Homeostasis of glutamate in brain fluids: an accelerated brain-to-blood efflux of excess glutamate is produced by blood glutamate scavenging and offers protection from neuropathologies. Neuroscience. 2009;158:301–308. doi: 10.1016/j.neuroscience.2008.02.075. [DOI] [PubMed] [Google Scholar]
- Gottlieb M, Wang Y, Teichberg VI. Blood-mediated scavenging of cerebrospinal fluid glutamate. J Neurochem. 2003;87:119–126. doi: 10.1046/j.1471-4159.2003.01972.x. [DOI] [PubMed] [Google Scholar]
- Campos F, Sobrino T, Ramos-Cabrer P, Argibay B, Agulla J, Pérez-Mato M, et al. Neuroprotection by glutamate oxaloacetate transaminase in ischemic stroke: an experimental study. J Cereb Blood Flow Metab. 2011;31:1378–1386. doi: 10.1038/jcbfm.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruban A, Berkutzki T, Cooper I, Mohar B, Teichberg VI. Blood glutamate scavengers prolong the survival of rats and mice with brain-implanted gliomas. Invest New Drugs. 2012;30:2226–2235. doi: 10.1007/s10637-012-9794-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnik A, Gruenbaum SE, Artru AA, Rozet I, Dubilet M, Tkachov S, et al. The neuroprotective effects of oxaloacetate in closed head injury in rats is mediated by its blood glutamate scavenging activity. J Neurosurg Anesth. 2009;21:235–241. doi: 10.1097/ANA.0b013e3181a2bf0b. [DOI] [PubMed] [Google Scholar]
- Harms PG, Ojeda SR. A rapid and simple procedure for chronic cannulation of the rat jugular vein. J Appl Physiol. 1974;36:391–392. doi: 10.1152/jappl.1974.36.3.391. [DOI] [PubMed] [Google Scholar]
- Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- Graham LT, Jr., Aprison MH. Fluorometric determination of aspartate, glutamate, and gamma-aminobutyrate in nerve tissue using enzymic methods. Anal Biochem. 1966;15:487–497. doi: 10.1016/0003-2697(66)90110-2. [DOI] [PubMed] [Google Scholar]
- Lavoie J, Layrargues GP, Butterworth RF. Increased densities of peripheral-type benzodiazepine receptors in brain autopsy samples from cirrhotic patients with hepatic encephalopathy. Hepatology. 1990;11:874–878. doi: 10.1002/hep.1840110524. [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, et al. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Jun D, Musilova L, Kuca K, Kassa J, Bajgar J. Potency of several oximes to reactivate human acetylcholinesterase and butyrylcholinesterase inhibited by paraoxon in vitro. Chem Biol Interact. 2008;175:421–424. doi: 10.1016/j.cbi.2008.05.004. [DOI] [PubMed] [Google Scholar]
- Ellman GL, Courtney KD, Andres V, jr, Featherstone RM.A new and rapid colorimetric determination of acetylcholinesterase activity Biochem Pharmacol 19617IN8191–95. [DOI] [PubMed] [Google Scholar]
- Papadopoulos V, Lecanu L, Brown RC, Han Z, Yao ZX. Peripheral-type benzodiazepine receptor in neurosteroid biosynthesis, neuropathology and neurological disorders. Neuroscience. 2006;138:749–756. doi: 10.1016/j.neuroscience.2005.05.063. [DOI] [PubMed] [Google Scholar]
- Stephenson DT, Schober DA, Smalstig EB, Mincy RE, Gehlert DR, Clemens JA. Peripheral benzodiazepine receptors are colocalized with activated microglia following transient global forebrain ischemia in the rat. J Neurosci. 1995;15:5263–5274. doi: 10.1523/JNEUROSCI.15-07-05263.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande LS, Carter DS, Blair RE, DeLorenzo RJ. Development of a prolonged calcium plateau in hippocampal neurons in rats surviving status epilepticus induced by the organophosphate diisopropylfluorophosphate. Toxicol Sci. 2010;116:623–631. doi: 10.1093/toxsci/kfq157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadka AB, Andrews BT. Neurotrauma: Evidence-Based Answers To Common Questions. Thieme; 2005. [Google Scholar]
- Siesjö Bk, Siesjö P. Mechanisms of secondary brain injury. Eur J Anaesth. 1996;13:247–268. [PubMed] [Google Scholar]
- Andrews B. Intensive Care in Neurosurgery. Thieme; 2002. [Google Scholar]
- Armin SS, Colohan ART, Zhang JH. Traumatic subarachnoid hemorrhage: our current understanding and its evolution over the past half century. Neurol Res. 2006;28:445–452. doi: 10.1179/016164106X115053. [DOI] [PubMed] [Google Scholar]
- Kozhemyakin M, Rajasekaran K, Kapur J. Central cholinesterase inhibition enhances glutamatergic synaptic transmission. J Neurophysiol. 2010;103:1748–1757. doi: 10.1152/jn.00949.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai P-J, Huang P-C. Circadian variations in plasma and erythrocyte glutamate concentrations in adult men consuming a diet with and without added monosodium glutamate. J Nutr. 2000;130:1002S–1004S. doi: 10.1093/jn/130.4.1002S. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.