

Abstract
It is time for those working on oncolytic viruses to take stock of the status of the field. We now have at our disposal an array of potential therapeutic agents, and are beginning to conduct early-phase clinical trials in patients with relapsed/metastatic cancers. By drawing on lessons learned during the development of other biological therapies, such as monoclonal antibodies and targeted small molecule inhibitors, we are now in a position to chart the course of the next wave of trials that will go beyond the phase I studies of safety and feasibility. In this article we review our approach to the development of oncolytic viruses as cancer therapeutics. In doing so, we emphasise the fact that this process is modular and involves multiple iterative steps between the laboratory and the clinic. Ultimately, at least in the medium term, the future of oncolytic virotherapy lies in combination regimens with standard anti-cancer agents such as radiation and chemotherapy.
Keywords: Oncolytic virus, Reovirus, Chemotherapy, Radiotherapy, Clinical trial
1. Introduction
The clinical development of oncolytic viruses is in its infancy, and there are many obstacles to be overcome before these agents prove that they truly can make a contribution to the management of cancer patients. At present, the field suffers a serious credibility problem. Critics acknowledge the conceptual beauty of using finely crafted self-replicating molecular machines to seek and destroy locoregional or systemic deposits of cancer, but point to the fact that the host immune response will inevitably prove to be an insuperable barrier to effective systemic therapy. In addition, to many observers oncolytic virotherapy is tarred with the same brush as gene therapy, a discipline that was seen to offer much, but deliver little, in the 1980s and 1990s.
In considering the current state of oncolytic virotherapy, and as a means of counterbalancing some of the pessimism that exists, it is useful to remember lessons that we have learned from the development of other biological therapies. Many of the new agents that are now held as exemplars of the power of biomedical science to deliver therapeutic benefits had a circuitous and often rocky road to clinical approval. The first set of lessons comes from the clinical development of monoclonal antibodies [1]. Following their initial description [2] there was huge enthusiasm for their potential as specific and selective anti-cancer drugs, based on their ability to target unique tumour-associated antigens. However, the first wave of expectancy yielded to emerging despondency as it became clear that systemically administered antibodies failed to reach their targets on tumour tissue in high concentrations [3]. The reasons for this will be familiar to virotherapists: attrition through non-specific or specific adsorption in non-target tissues and immune-mediated clearance of foreign antigens (early monoclonal antibodies were frequently murine in origin) accounted for the vast majority of the injected antibody dose. The human anti-mouse antibody (HAMA) response altered the pharmacokinetics of subsequent antibody infusions and also exposed patients to the risk of immune-mediated adverse events [4]. In some quarters there was a sense that antibodies would fail to make a mark in the clinical arena. However, as scientists and clinicians wrestled with these problems, there emerged two clear strands of thought: (i) the immune response against the monoclonal antibody was seen as potentially therapeutic in its own right [5]; and (ii) efforts were made to reduce the immunogenicity of the antibody molecule by making it less visible to the immune system [6], [7]. Again, these considerations will be very familiar to those working in the field of virotherapy, and have direct parallels to the current debate on the role of the immune response in oncolytic virotherapy. On one hand, there is persuasive data showing that an intact immune system is required for oncolytic virotherapy in order to mediate optimal anti-tumoural efficacy, and there are those who espouse the view that oncolytic virotherapy is essentially a form of immunotherapy [reviewed in 8]. On the other hand, there are studies that suggest that judicious suppression of the immune response can significantly improve systemic delivery of oncolytic virotherapy with resultant enhancement of intratumoural viral replication and treatment efficacy [8], [9], [10], [11], [12].
If we review the steps that were taken to make monoclonal antibodies clinically successful, it is clear that the greatest benefit resulted from reducing their immunogenicity, rather than attempting to exploit their ability to raise an anti-antibody immune response. Studies were performed in which attempts were made to blunt the HAMA response by administering anti-T cell immunosuppressive drugs [6], [7], but these had relatively little success. It was only through the generation of chimeric murine/human or fully humanised monoclonal antibodies that these agents have been able to enter the clinic in large numbers [13]. Indeed, a number of monoclonal antibodies have been licensed for routine clinical use against malignant (and other) diseases. Clearly, corresponding “humanization” of viral proteins is not going to be possible, but there are a number of ways in which antigenic epitopes may be masked from the immune system, either by loading the viruses onto human carrier cells [14], [15] or by cloaking them in surface polymers [16].
The second set of lessons learned from the development of other biological therapies comes from the new generation of small molecules that have been generated as inhibitors of key signal transduction pathways in cancer cells. In the last 5 years, a number of these agents have demonstrated their efficacy in randomised phase II and III trials and have received approvals from regulatory agencies. However, with the exception of the very rare diseases that are reliant on defects in a single signalling pathway (e.g. gastrointestinal stromal tumour and c-Kit), many of these agents show only modest single-agent activity against established treatment-resistant cancers. Furthermore, even when they show dramatic single-agent efficacy, this is inevitably followed by the emergence of treatment resistance as a result of adaptive genetic alteration(s) in the tumour [17]. As a result of these considerations, it is now widely accepted that the future development of targeted therapies will occur as part of combination therapeutic regimens in which they are used alongside standard anti-cancer therapies such as radiotherapy and chemotherapy.
Therefore, for translational clinicians/scientists working with oncolytic virotherapy, the direction of study is abundantly clear: studies should focus on understanding how to integrate oncolytic viruses with standard agents in order to achieve therapeutic synergy and minimise normal tissue toxicity [18]. This review will describe our initial attempts to build an integrated programme of iterative preclinical and clinical research that has allowed us to evaluate the use of oncolytic viruses with cytotoxic chemotherapy and radiotherapy. In this process we have used oncolytic reovirus, but there is no reason to believe that this approach cannot be adopted with a range of other oncolytic agents.
Ultimately, the aim of our programme is to move oncolytic virotherapy further up on the therapeutic agenda, so that it is used earlier in the patient's treatment before his/her disease has become (multiply) resistant. Again, this goal has a firm foundation in lessons learned from the development of other biological therapies. In the last 4 years, cetuximab has been shown to enhance the therapeutic efficacy of radical radiotherapy in a randomised phase III trial, where the median overall survival was increased from 29 to 49 months [19], [20]. This result was achieved despite the fact that anti-EGFR-targeted therapies have relatively poor single-agent response rates.
2. Oncolytic reovirus as an archetype for clinical development
Reovirus (respiratory enteric orphan virus) is a member of the Reoviridae family. Reovirus is ubiquitous and has been isolated from untreated sewage, stagnant water and rivers throughout the world [21], [22]. As the name suggests, reovirus infects the respiratory and gastrointestinal tract but is not associated with a named human disease, although it can infrequently cause flu-like upper respiratory tract symptoms or a mild diarrhoeal illness. Exposure to reovirus is very common, with up to 100% of healthy human adults showing seropositivity [23], [24], [25].
The virions consist of a non-enveloped, icosahedral capsid with a double shell of proteins. The genome comprises double-stranded RNA that is split into 10 segments of three sizes, designated L, M and S, depending on size. This segmental genomic structure has thwarted attempts to generate genetically engineered reoviruses, but some progress has recently been made in this area [26]. The oncolytic nature of reovirus was first recognised when wild-type reovirus was shown to replicate in transformed, but not normal, cells [27]. Cells expressing high levels of EGFR and v-erbB (a truncated mutant of EGFR) were shown to be susceptible to reovirus infection and cytotoxicity [28], [29], [30]. Detailed analysis demonstrated that an activated Ras pathway, a signalling pathway downstream of EGFR, was a prerequisite of sensitivity to reovirus. The Ras pathway is frequently activated in cancers, either through mutation of the RAS gene or overexpression/mutational activation of EGFR [31].
Reoviral infection of normal cells leads to the activation of protein kinase R (PKR), a serine/threonine protein kinase that requires binding of double-stranded RNA and phosphorylation to become activated [32] (Fig. 1 ). PKR's main function is to act as a defence against viral infection and to contribute to the anti-proliferative response of interferon following viral infection. However, in cells harbouring an activated Ras mutation, PKR remains hypophosphorylated and in its inactive state. This allows reoviral protein synthesis to continue freely, with the result that viral replication proceeds and cell lysis occurs [33].
Fig. 1.
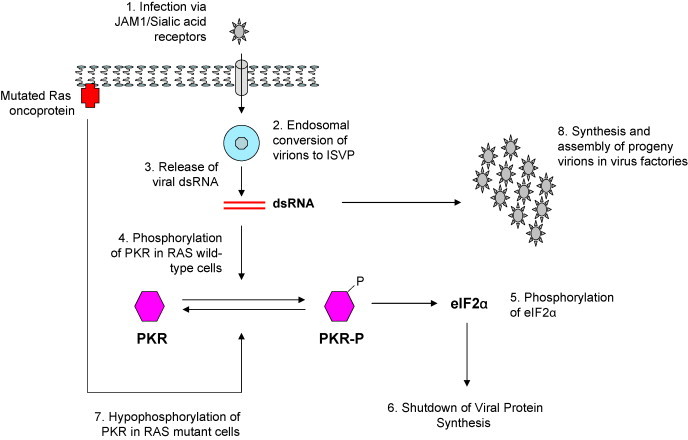
Schematic view of the infective cycle of reovirus. Initial infection occurs through interaction between the virion and junctional adhesion molecule 1 (JAM1) and sialic acid residues on the cell membrane (1). The virus is internalised in an endosomal compartment where it is partially digested to form the intermediate subviral particle (ISVP) (2). Viral double-stranded RNA (dsRNA) escapes for the endosome (3). In cells that contain normal wild-type RAS genes, the presence of dsRNA leads to phosphorylation of PKR (4) and subsequent activation of eIF2α (5). This in turn leads to shutdown of viral protein synthesis, thus aborting a productive infection (6). In contrast, in cells with mutant RAS or an activated Ras pathway, PKR remains in a hypophosphorylated form (7) and viral RNA species are able to direct synthesis and assembly of daughter virions (8).
Cell lines originating from almost all common tumour types have been found to be susceptible to reoviral oncolysis. Intratumoural and intravenous injection of reovirus has been shown to cause regression of a variety of syngeneic and xenograft tumours in immunocompetent and immunodeficient animal models, respectively [reviewed in 34].
3. Conducting clinical trials of oncolytic virotherapy
In contrast to phase I trials of small molecules or monoclonal antibodies, studies involving oncolytic viruses face a number of additional organisational, legal and ethical issues. Such considerations are extremely important, given the fact that the public perception of viruses is based on their experiences of them as the cause of disease. Recent press coverage of viral diseases such as severe acute respiratory syndrome coronavirus (SARS) and the ‘swine flu’ pandemic have added to the generally cautious view of viruses as potential therapeutics. Despite this fact, those of us who enrol patients in clinical trials can attest that there is a real willingness among patients to hear about the potential for using these ancient foes for therapeutic benefit. Indeed, it is the authors’ collective personal experience that very few patients are discouraged from taking part in such studies merely because the proposed study agent is a virus. If anything, it is the attendant safety measures that come as part of the trial design that are often viewed by patients as being too onerous to allow them to contemplate trial participation.
It is, perhaps, not surprising that the greatest degree of caution in this area comes from the regulatory authorities that oversee clinical trials of oncolytic virotherapies. These organisations, which include national, regional and local bodies, place an appropriately strong emphasis on safety considerations and the direct risks to patients, staff and the public at large. As a result, clinical studies tend to be conducted under conditions of stringent biosafety, with detailed analysis of the risk of vector shedding and environmental contamination by the study agent. Unfortunately, at the local level, there can be significant variability in the operational characteristics of the safety committees that are set up to supervise trials involving genetically modified organisms. Such local variations in rules and practices can, in fact, represent the single greatest obstacle to study initiation, enrolment and completion.
When embarking on a programme of research on novel biological agents, it is important to appreciate that it will be necessary to conduct the work in modular projects with regular iterative steps between the bench and the bedside (Fig. 2 ). Indeed, such work is the epitome of true translational work, and allows lessons learned in the laboratory to shape clinical trial design directly, while analysis of clinical material in the laboratory informs ongoing preclinical experiments.
Fig. 2.
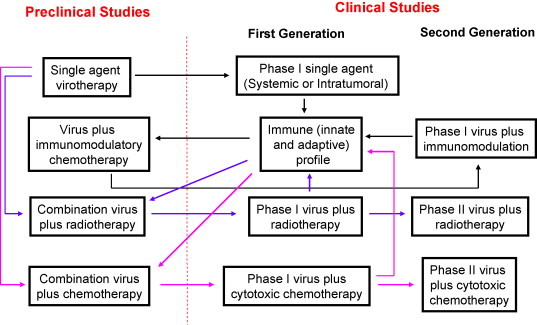
Modular and iterative nature of preclinical and clinical development of oncolytic virotherapy. Black arrows chart the course of preclinical and clinical trials involved in the use of single-agent reovirus. Specifically, data that emerged from the initial phase I study of intravenous viral administration provided essential data on the immune effects in patients. These results were used to design studies in the laboratory that ultimately provided a mechanism for attempting to modulate the NARA response with cyclophosphamide. Blue arrows show the course of studies in which reovirus is combined with radiotherapy. Once again, immune profiling has been a key component in this work. Pink arrows indicate work streams for combinations with cytotoxic chemotherapy. In this regard, phase I and II combination studies have been completed and have resulted in successful submission of a proposal for a phase III trial. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
4. Phase I studies of single-agent virotherapy
These studies are, by their very nature, designed to provide an initial platform on which a subsequent work programme can be built. As such, they represent a huge hurdle for small biotechnology companies to clear, and require significant financial investment, often with only a modest chance of achieving sufficiently eye-catching results to guarantee progression to phase II evaluation. In early examples of such trials, concerns about the perceived risks of administering live viruses to cancer patients led to regulators insisting on extraordinarily cautious dose-escalation schemes, such that trials took a very long time to reach dose levels where biological effects might have been anticipated [35]. Thankfully, the literature now contains a sufficient body of reassuring data from a number of phase I studies of oncolytic virotherapy to assuage some of the concerns of regulators. Nonetheless, it is likely that new viral agents, especially if they are first-in-family, will still have to go through relatively laborious early stage clinical development.
When we consider the clinical development of reovirus as a template for the whole development process, the first-in-man experience came with intratumoural administration. In this first study, patients with a variety of malignancies received escalating doses of intratumoural reovirus at levels ranging from a single injection of 1 × 107 plaque forming units (pfu) to three injections of 1 × 1010 pfu. The main symptoms were headaches and a flu-like illness, but no grade 3 events were reported. Best responses were as follows: complete response (1 patient), partial response (1 patient) and stable disease (8 patients) [36]. In a subsequent study that focused on patients with gliomas, 12 patients with recurrent disease were dosed at 3 dose levels (1 × 107 (n = 3), 1 × 108 (n = 6) and 1 × 109 (n = 3) tissue culture infectious dose-50 (TCID50)) by intratumoural injection. Again, the treatment was well tolerated, and grade 3 or 4 toxicities were not encountered. The median time to progression was only 4.3 weeks and only 1 patient showed disease stabilization [37].
These data on intratumoural injection (and other unpublished data on prostate cancer patients) formed the basis of a submission to conduct a first-in-man study of intravenous reovirus in patients with advanced cancers [38]. Thirty-three patients received escalating doses of reovirus from 1 × 108 for 1 day up to 3 × 1010 TCID50 for 5 days repeated 4-weekly. A total 76 cycles of reovirus were delivered, with a median number of 2 and a maximum number of 6 cycles administered. There was no dose-limiting toxicity and escalation was only limited by the limitations imposed by viral manufacture. As with the previous phase I intratumoural studies, the dominant toxicities were mild and consistent with virus infection, consisting of fever, fatigue and headache. Interestingly, these effects appeared to be independent of virus dose and treatment cycle. In view of concerns about viral excretion and environmental contamination, the study included detailed analysis of blood, urine, fecal and urinary samples by reverse transcription PCR. These analyses confirmed negativity of all pre- and post-treatment blood, urine, saliva and fecal samples for reovirus using RT-PCR screening based on 25 cycles of amplification, and this was used for decision-making regarding the patients’ suitability for discharge from the hospital. Subsequent re-analysis of the data based on 35 cycles of amplification (where the detection limit was 200 TCID50) revealed weak positive signals in the first cycle of reovirus treatment in a small number of patients at days 5 and 15. In 3 patients, we were able to obtain pre- and post-treatment biopsy samples and demonstrate recovery of replication-competent virus.
These data are important, as they confirm the ability of the virus to reach the tumour after systemic administration. Such data on tumour delivery of virus are even more impressive when viewed in light of the results of the analyses of the anti-viral immune response [39]. Significant increases in neutralising anti-reoviral antibodies (NARAs) were seen, with the peak endpoint titres reaching >1/10,000 in all but one patient. Overall, the median increase in NARA titre was 250-fold (range 9–6437). We also conducted an analysis of the dynamic changes in cell-mediated immune parameters (NK cells, CD4+ and CD8+ T cells) and these, in general, showed an immunostimulatory effect (albeit with significant patient-to-patient variability). Taken together, these data are indicative of the fact that even heavily pre-treated patients are capable of mounting dynamic immune responses during treatment with systemic virotherapy.
5. Improving systemic delivery of reovirus through immunomodulation
Having demonstrated a significant humoral anti-viral immune response following intravenous administration of reovirus in the phase I study, subsequent preclinical studies were performed as part of an iterative process to improve systemic oncolytic virotherapy. We examined intravenous delivery of multiple doses of reovirus in immunocompetent C57Bl/6 mice bearing subcutaneous B16 tumours, and showed that only very low levels of virus were able to reach the tumour. Furthermore, in therapeutic experiments the activity of intravenous reovirus was only modest (at best) against B16 as lung metastases or subcutaneous tumours. In light of previous studies that had reported improved activity from combining the immunosuppressive agent cyclophosphamide with oncolytic virotherapy [9], [10], [11], we tested this approach in our model [12]. By combining intravenous reovirus with high-dose cyclophosphamide (150 mg/kg for 3 days), many of the barriers to effective intratumoural delivery and replication were overcome. In biodistribution studies, viral titers of between 107 and 108 pfu per mg were recovered from B16 tumours. In therapeutic studies this was associated with marked tumour regressions in the first 2 weeks of therapy, but this was followed by the death of the animals. Unfortunately, and surprisingly, it was found that titerable virus could also be recovered from the circulation and a number of normal tissues, most notably from the heart. Levels of recovery of reovirus from cardiac tissues were consistent with reoviral replication within cardiac myocytes, and histological evaluation demonstrated severe non-suppurative myocarditis. It emerged that these toxicities were occurring due to the absence of circulating NARA in mice treated with high-dose cyclophosphamide. Further studies of systemic reovirus administration in B6.129S2-Igh-6tm1Cgn/j mice which lack mature B cells and fail to make anti-viral antibodies confirmed that similar severe toxicities occurred and were due to viral replication in systemic organs.
Rather than seeing this as a no-go point for the clinical development of immunosuppressive therapy given with reovirus, we decided to modify the dose schedule of cyclophosphamide in an attempt to derive a clinically applicable regimen. Accordingly, we restructured the protocol such that the drug was given 1 day before intravenous reovirus, and the whole so-called “metronomic regimen” consisted of 3 repeats of this therapy, each separated by 6 days. This regimen was associated with very high levels of intratumoural viral delivery, replication (∼107 pfu/mg tumour) and tumour cures. Importantly, the NARA response was preserved (albeit at an attenuated level) and systemic toxicity was mild and did not cause death in any of the animals.
These data show that neutralising antibodies against oncolytic viruses have the capacity to play a dual role: on one hand, they can be viewed as an obstacle, in that they hinder repeated injections of virus, but, on the other hand, they represent a safeguard that neutralises virus released from vigorous intratumoural replication, thus preventing it from causing viral toxicity in normal tissues. As a direct consequence of the initial phase I study of intravenous reovirus and the subsequent preclinical studies designed to modulate immune response to the virus, we have initiated a phase I dose-escalation study of cyclophosphamide combined with the recommended phase II dose of reovirus (Fig. 3 ). This study has a unique endpoint, based on evidence of modulation of the NARA response, and recruitment is proceeding (Harrington KJ, personal communication).
Fig. 3.
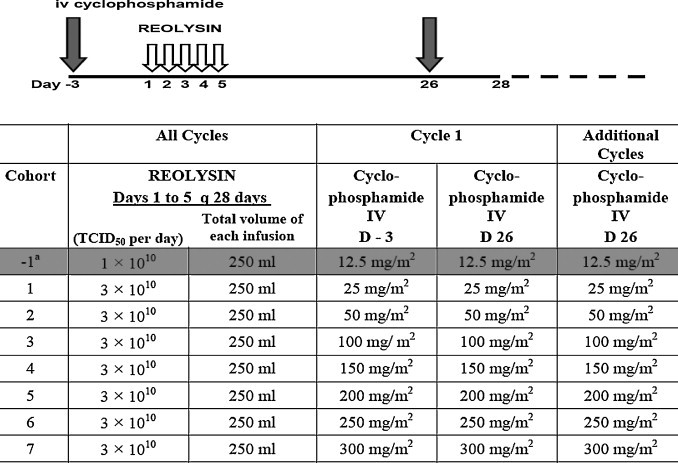
Scheme of delivery of cyclophosphamide and reovirus (REOLYSIN) in phase I study of immunomodulatory therapy. Cyclophosphamide is delivered 3 days before a 5-day cycle of reovirus with cycles repeated each 4 weeks. The −1 dose level was written into the protocol in case dose-limiting toxicity was encountered in cohort 1, but was not needed during study conduct.
6. Reovirus combined with standard therapies
Data from the phase I studies of reovirus, administered both by intratumoural and intravenous routes, and the results of previous studies of oncolytic viruses as single-agent therapies, convincingly demonstrate that they are not likely to have significant impact as stand-alone therapies. However, there are sound mechanistic reasons to believe that oncolytic viruses can be combined with standard anti-cancer agents for clinical benefit, in much the same way as monoclonal antibodies and small molecule inhibitors [40], [41].
7. Reovirus plus radiotherapy
In considering potential partner therapies to use alongside oncolytic reovirus, we were able to conceive of a number of potentially positive theoretical interactions with ionising radiation. First, radiation may increase the ability of reovirus to infect tumour cells by affecting the expression of cell surface receptors. A similar effect has been observed in adenovirus infection after therapeutic irradiation [42], [43]. Second, cells with mutated Ras will be relatively radioresistant, but should be sensitive to reovirus-induced oncolysis. Third, the stress response in irradiated cells involves increased signaling through the EGFR-Ras pathway, and this, too, should enhance reoviral replication/cytotoxicity. Fourth, radiation-induced alterations in the proportions of tumour cells at different phases of the cell cycle may render them more permissive to reovirus infection and/or replication. Fifth, in the in vivo setting, radiation may reduce the high tumour interstitial pressure that is a significant barrier to effective intratumoural spread of biological particles such as viruses [3].
Therefore, we have completed in vitro and in vivo preclinical studies in which we have evaluated the combination of reovirus and radiation in a range of tumour cell lines [44]. We initially demonstrated that reovirus is not significantly inactivated, even by large single fractions of X-ray irradiation up to 25 Gy. There was some minor reovirus cytotoxicity at higher radiation doses (50 and 100 Gy), but such radiation doses are not clinically relevant. Thereafter, we showed that there was enhanced cytotoxicity when radiation was combined with reovirus across a broad range of multiplicities of infection (MOI) (0.001–10) in a panel of 11 tumour cell lines. The effect of the combination treatment was most marked at low MOI and in cell lines that showed only modest susceptibility to reovirus cytotoxicity in the absence of radiation. We proceeded to examine the nature of the co-operative interaction between radiation and reovirus using the combination index methodology of Chou and Talalay [45]. Strong synergism was demonstrated for tumour cells exposed to 3, 5 and 10 Gy radiation and reovirus at MOI between 0.001 and 1. Contrary to our initial hypothesis that radiation may alter the signalling environment within the cell to make it more conducive to reoviral replication, there was no evidence of enhanced replication of reovirus in irradiated cell lines. The cellular mechanism underlying the observed increased cytotoxicity of the combination therapy was through an increase in apoptosis. Finally, the combination therapy was tested in three separate animal models, including both immunodeficient and immunocompetent systems. For all three models there was evidence that intratumoural injections of reovirus were able to significantly enhance the therapeutic efficacy of fractionated courses of radiation.
These studies formed the basis of our conducting a dose-escalation phase I study to determine the safety and feasibility of combining intratumoural reovirus and palliative radiotherapy in patients with advanced cancer [46]. The secondary goals included assessing viral biodistribution and excretion (blood, urine, stool, sputum), reoviral replication in tumours and anti-viral immune responses. Eligible patients included those with measurable disease that was amenable to palliative radiotherapy. In order to maximise the information that would be available from the study, we chose to adopt a novel two-stage design (Fig. 4 ). In the first stage, patients received radiotherapy at a dose of 20 Gy in 5 fractions, plus two intratumoural injections of reovirus at doses between 108 and 1010 TCID50. In the second stage, the radiotherapy dose was increased to 36 Gy in 12 fractions and the patients received two, four or six doses of reovirus at a stable dose of 1010 TCID50. We recruited 23 patients with a variety of solid tumours. Reassuringly, dose-limiting toxicity was not seen in any of the cohorts and the combination treatment was not associated with exacerbation of the acute radiation reaction. As with the phase I study of intravenous reovirus, we conducted a rigorous programme to check for shedding of the virus from injected patients. Reverse transcription polymerase chain reaction (RT-PCR) studies of blood, urine, stool and sputum were negative in all cases. Of the patients treated in the 20 Gy radiation group, 2 of 7 evaluable patients had a partial response (PR) and 5 had stable disease (SD). In the group of patients treated to 36 Gy in 12 fractions, 5 of 7 evaluable patients had a PR and 2 had SD. This study has shown that the combination of intratumoural reovirus and radiotherapy is well tolerated and shows a favourable toxicity profile. The absence of vector shedding from injected patients means that this approach can easily be applied to an out-patient setting. However, the patient population that was studied precluded any formal analysis of the potential efficacy of the approach, and this should be evaluated in patients with newly diagnosed cancers who are receiving radiotherapy with curative intent.
Fig. 4.
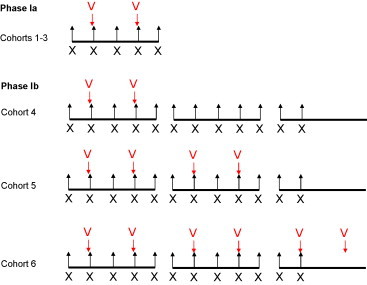
Trial design for the phase I study of reovirus in combination with radiotherapy. In phase Ia, patients in cohorts 1–3 received palliative radiotherapy to a dose of 20 Gy in 5 fractions (X = 4 Gy per fraction), with escalating doses of virus (V) delivered on days 2 and 4. In cohort 1, virus doses were 1 × 108 TCID50. In cohort 2, virus doses were 1 × 109 TCID50, and in cohort 3 doses were 1 × 1010 TCID50 per injection. In phase Ib, patients received palliative radiotherapy to a dose of 36 Gy in 12 fractions (X = 3 Gy per fraction) with increasing numbers of virus administrations. Each virus dose (V) was at the 1 × 1010 TCID50 dose level. Patients in cohort 4 received 2 injections, those in cohort 5 received 4 injections and those in cohort 6 received 6 injections.
8. Reovirus and chemotherapy
A number of combinations of chemotherapy and oncolytic viruses have already been evaluated preclinically, with many of the studies confirming marked anti-tumour effects without significant additional toxicity [reviewed in 41]. Relatively early in the development of ONYX-015 it was shown to enhance clinical efficacy in combination with systemic cisplatin and 5-fluorouracil when compared to chemotherapy alone in patients with head and neck cancers [47]. Therefore, as a prelude to conducting clinical studies, we have evaluated combinations of reovirus with cytotoxic chemotherapy in a number of cell lines (including melanomas, prostate cancers and head and neck cancers).
Initial studies have demonstrated synergistic activity when combining chemotherapy with reovirus in malignant melanoma cell lines [48]. The addition of cisplatin (10 or 100 μM) significantly enhanced tumour kill compared to reovirus alone across a range of MOIs (0.01–1), and subsequent isobolographic analyses [45] confirmed that the interaction was synergistic, particularly for cisplatin and paclitaxel. The mechanism of enhanced cell kill was through an increase in apoptosis, and in vivo studies confirmed that there were greater levels of viral replication in tumours recovered from animals that received the combination therapy. We have also completed similar studies in prostate and head and neck cancer cell lines. In the former, taxanes have been shown to be synergistic with reovirus [Pandha HS, personal communication], and in the latter, both platins and taxanes have been shown to exert synergistic effects [Harrington KJ, personal communication].
Based on the data from the phase I study of intravenous reovirus and the in vitro data on the synergistic interactions between reovirus, cisplatin and paclitaxel, we designed a phase I study in which all three agents were combined in patients with locally advanced disease. In view of concerns about potential viral toxicity in patients receiving full-dose cytotoxic chemotherapy, the virus was again administered according to a dose-escalation scheme. However, the starting dose was reduced by only one log from the recommended single-agent phase II dose. Thus, in the initial phase I study, cohorts of 3 patients received escalating doses (3 × 109, 1 × 1010, 3 × 1010 TCID50) of reovirus on days 1–5, in combination with carboplatin (area under the curve = 5) and paclitaxel (175 mg/m2 over 3 h), which were administered on day 1 of a 3-weekly schedule. Patients with a variety of advanced cancers which were not amenable to curative treatment, or which were refractory to standard therapy, were treated. Having completed the dose-escalation phase I component of the study, a phase II study in patients with head and neck cancers was performed. In the dose-escalation portion of the study, there were no dose-limiting toxicities. In the 19 patients with head and neck cancer who received at least two cycles of treatment and were thus evaluable for response, PR was seen in 8 patients (42%), SD in 6 (32%) and PD in 5 (26%). Most of these patients had head and neck cancers that were refractory to previous platinum-based chemotherapy. A confirmatory US-based study in the same patient group is currently recruiting patients. These findings have prompted a successful submission to the Federal Drug Administration for a Special Protocol Agreement to test the combination of carboplatin, paclitaxel and reovirus versus carboplatin, paclitaxel and placebo in a randomised phase III trial in patients with platinum-refractory relapsed/metastatic head and neck cancer (Fig. 5 ).
Fig. 5.

Clinical trial design for the randomised phase III trial of carboplatin, paclitaxel and reovirus or placebo in patients with relapsed/metastatic, platin-refractory head and neck cancer (SCCHN, squamous cell cancer of the head and neck; AUC, area under the curve; CR, complete response; PR, partial response; SD, stable disease).
9. Conclusions
In this review we have summarised current progress in the clinical development of oncolytic reovirus. Reovirus has been used for the purpose of illustration because, at present, it represents the agent that has progressed the furthest along the track to clinical assessment. This progress has been backed up by an extensive preclinical and clinical package, in which the agent has been assessed as a stand-alone therapeutic and in combination regimens with radiotherapy or chemotherapy. At all stages, progress has been facilitated and accelerated by pursuing a modular and iterative approach, such that lessons learned in the laboratory can be applied in the clinic and vice versa. We believe that this model will be useful to other oncolytic viral therapies as they move forward in the clinic beyond the phase I setting.
Biographies

Kevin Harrington is Reader in Biological Cancer Therapies and Honorary Consultant in Clinical Oncology at the Royal Marsden Hospital. Having graduated from St Bartholomew's Hospital Medical School, he trained in clinical oncology at The Royal Postgraduate Medical School, Hammersmith Hospital and The Royal Marsden Hospital. He completed his PhD at Hammersmith Hospital and undertook post-doctoral research at the Mayo Clinic. He returned to the UK in 2001 to combine a clinical practice in head and neck cancer and melanoma with his role as Team Leader in the Targeted Therapy Team, The Institute of Cancer Research, London. His research interests include combining standard anti-cancer therapies with novel biologically targeted agents.

Richard Vile received his BA in biochemistry from Oxford University and his PhD in Viral Vectors from The Institute for Cancer Research, London, followed by post-doctoral training in Viral Vectors and Immunotherapy at St. Mary's Hospital Medical School, London, and a fellowship in Biology of Metastasis at Imperial Cancer Research Fund, London. Professor Vile is currently a consultant in the Molecular Medicine Program and Dept. of Immunology, Mayo Clinic. He is a member of the NIH Recombinant DNA Advisory Committee. His primary research interests are centered on the development of gene, viro and immuno-therapies for cancer. In addition, a long term research interest focuses on defining how the immune system can be re-educated to recognise and react against tumour-associated antigens.

Alan Melcher is a Cancer Research UK Senior Clinical Research Fellow and Professor of Clinical Oncology and Biotherapy at the Leeds Institute of Molecular Medicine. Having qualified at Oxford University, he trained in clinical oncology in Cardiff, London and Leeds. Having completed a PhD at the Hammersmith Hospital in London, he continued post-doctoral research at the Mayo Clinic, Minnesota, USA, before returning to the UK in 2000. He currently combines a clinical practice treating melanoma with chemotherapy, radiotherapy and biotherapies with a laboratory-based preclinical and clinical research programme focused on novel immune effector cells and oncolytic viruses for the treatment of cancer.

John Chester is a senior lecturer at the Leeds Institute of Molecular Medicine, University of Leeds, and an honorary consultant in Medical Oncology at the St. James's Institute of Oncology, also in Leeds. His main clinical interests are in systemic therapy for bladder and head/neck cancers, including cytotoxic chemotherapy, virus-mediated gene therapy and molecularly targeted agents, including monoclonal antibodies, tyrosine kinase and mTOR inhibitors. His research interests include the clinical application of oncolytic viral therapies, early-phase clinical trials of novel agents and prediction of response to systemic therapies, particularly neoadjuvant chemotherapy.

Hardev Pandha is Professor of Medical Oncology and head of the Oncology Department, Postgraduate Medical School, University of Surrey, in the UK. He qualified from the University of Birmingham Medical School and trained in internal medicine and medical oncology. He completed his PhD and specialist training with the Imperial Cancer Research Fund group at Hammersmith hospital, The Royal Marsden Hospital and at St George's Hospital Medical School. He combines a clinical practice focusing on urological malignancies with his laboratory program focusing on the combination of oncolytic viruses with conventional anti-cancer agents and cancer vaccines.
References
- 1.Harrington K.J., Epenetos A.A. Recent developments in radioimmunotherapy. Clin Oncol (R Coll Radiol) 1994;6:391–398. doi: 10.1016/s0936-6555(05)80193-1. [DOI] [PubMed] [Google Scholar]
- 2.Kohler G., Milstein C. Continuous culture of fused cells secreting antibodies of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 3.Jain R.K. Physiological barriers to delivery of monoclonal antibodies and other macromolecules in tumours. Cancer Res. 1990;50(Suppl.):814s–819s. [PubMed] [Google Scholar]
- 4.Courtenay-Luck N.S., Epenetos A.A., Moore R., Larche M., Pectasides D., Dhokia B. Development of primary and secondary immune responses to mouse monoclonal antibodies used in the diagnosis and therapy of malignant neoplasms. Cancer Res. 1986;46:6489–6493. [PubMed] [Google Scholar]
- 5.Forni L., Coutinho A., Köhler G., Jerne N.K. IgM antibodies induce the production of antibodies of the same specificity. Proc Natl Acad Sci USA. 1980;77:1125–1128. doi: 10.1073/pnas.77.2.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ledermann J.A., Begent R.H., Bagshawe K.D. Cyclosporin A prevents the anti-murine antibody response to a monoclonal anti-tumour antibody in rabbits. Br J Cancer. 1988;58:562–566. doi: 10.1038/bjc.1988.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ledermann J.A., Begent R.H., Massof C., Kelly A.M., Adam T., Bagshawe K.D. A phase-I study of repeated therapy with radiolabelled antibody to carcinoembryonic antigen using intermittent or continuous administration of cyclosporin A to suppress the immune response. Int J Cancer. 1991;47:659–664. doi: 10.1002/ijc.2910470505. [DOI] [PubMed] [Google Scholar]
- 8.Prestwich R.J., Errington F., Diaz R.M., Pandha H., Harrington K.J., Melcher A.A. The case of oncolytic viruses versus the immune system: waiting on the judgment of Solomon. Hum Gene Ther. 2009;20:1119–1132. doi: 10.1089/hum.2009.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ikeda K., Ichikawa T., Wakimoto H., Silver J.S., Deisboeck T.S., Finkelstein D. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat Med. 1999;5:881–887. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- 10.Kambara H., Saeki Y., Chiocca E.A. Cyclophosphamide allows for in vivo dose reduction of a potent oncolytic virus. Cancer Res. 2005;65:11255–11258. doi: 10.1158/0008-5472.CAN-05-2278. [DOI] [PubMed] [Google Scholar]
- 11.Thomas M.A., Spencer J.F., Toth K., Sagartz J.E., Phillips N.J., Wold W.S. Immunosuppression enhances oncolytic adenovirus replication and antitumor efficacy in the Syrian hamster model. Mol Ther. 2008;16:1665–1673. doi: 10.1038/mt.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qiao J., Wang H., Kottke T., White C., Twigger K., Diaz R.M. Cyclophosphamide facilitates anti-tumor efficacy against subcutaneous tumors following intravenous delivery of reovirus. Clin Cancer Res. 2008;14:259–269. doi: 10.1158/1078-0432.CCR-07-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Argyriou A.A., Kalofonos H.P. Recent advances relating to the clinical application of naked monoclonal antibodies in solid tumors. Mol Med. 2009;15:183–191. doi: 10.2119/molmed.2009.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harrington K., Alvarez-Vallina L., Crittenden M., Gough M., Chong H., Diaz R.M. Cells as vehicles for cancer gene therapy: the missing link between targeted vectors and systemic delivery? Hum Gene Ther. 2002;13:1263–1280. doi: 10.1089/104303402760128504. [DOI] [PubMed] [Google Scholar]
- 15.Willmon C., Harrington K., Kottke T., Prestwich R., Melcher A., Vile R. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol Ther. 2009;17:1667–1676. doi: 10.1038/mt.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fisher K.D., Seymour L.W. HPMA copolymers for masking and retargeting of therapeutic viruses. Adv Drug Deliv Rev. 2010;62:240–245. doi: 10.1016/j.addr.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Van Glabbeke M., Verweij J., Casali P.G., Le Cesne A., Hohenberger P., Ray-Coquard I. Initial and late resistance to imatinib in advanced gastrointestinal stromal tumors are predicted by different prognostic factors: a European Organisation for Research and Treatment of Cancer-Italian Sarcoma Group-Australasian Gastrointestinal Trials Group study. J Clin Oncol. 2005;23:5795–5804. doi: 10.1200/JCO.2005.11.601. [DOI] [PubMed] [Google Scholar]
- 18.Ottolino-Perry K., Diallo J.S., Lichty B.D., Bell J.C., McCart J.A. Intelligent design: combination therapy with oncolytic viruses. Mol Ther. 2010;18:251–263. doi: 10.1038/mt.2009.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonner J.A., Harari P.M., Giralt J., Azarnia N., Shin D.M., Cohen R.B. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 20.Bonner J.A., Harari P.M., Giralt J., Cohen R.B., Jones C.U., Sur R.K. Radiotherapy plus cetuximab for locoregionally advanced head and neck cancer: 5-year survival data from a phase 3 randomised trial, and relation between cetuximab-induced rash and survival. Lancet Oncol. 2010;11:21–28. doi: 10.1016/S1470-2045(09)70311-0. [DOI] [PubMed] [Google Scholar]
- 21.Adams D.J., Ridinger D.N., Spendlove R.S., Barnett B.B. Protamine precipitation of two reovirus particle types from polluted waters. Appl Environ Microbiol. 1982;44:589–596. doi: 10.1128/aem.44.3.589-596.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ridinger D.N., Spendlove R.S., Barnett B.B., George D.B., Roth J.C. Evaluation of cell lines and immunofluorescence and plaque assay procedures for quantifying reoviruses in sewage. Appl Environ Microbiol. 1982;43:740–746. doi: 10.1128/aem.43.4.740-746.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosen L., Evans H.E., Spickard A. Reovirus infections in human volunteers. Am J Hyg. 1963;77:29–37. doi: 10.1093/oxfordjournals.aje.a120293. [DOI] [PubMed] [Google Scholar]
- 24.Tai J.H., Williams J.V., Edwards K.M., Wright P.F., Crowe J.E., Jr., Dermody T.S. Prevalence of reovirus-specific antibodies in young children in Nashville, Tennessee. J Infect Dis. 2005;191:1221–1224. doi: 10.1086/428911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosen L., Hovis J.F., Mastrota F.M., Bell J.A., Huebner R.J. Observations on a newly recognized virus (Abney) of the reovirus family. Am J Hyg. 1960;71:258–265. doi: 10.1093/oxfordjournals.aje.a120109. [DOI] [PubMed] [Google Scholar]
- 26.van den Wollenberg D.J., van den Hengel S.K., Dautzenberg I.J., Cramer S.J., Kranenburg O., Hoeben R.C. A strategy for genetic modification of the spike-encoding segment of human reovirus T3D for reovirus targeting. Gene Ther. 2008;15:1567–1578. doi: 10.1038/gt.2008.118. [DOI] [PubMed] [Google Scholar]
- 27.Hashiro G., Loh P.C., Yau J.T. The preferential cytotoxicity of reovirus for certain transformed cell lines. Arch Virol. 1977;54:307–315. doi: 10.1007/BF01314776. [DOI] [PubMed] [Google Scholar]
- 28.Strong J.E., Lee P.W. The v-erbB oncogene confers enhanced cellular susceptibility to reovirus infection. J Virol. 1996;70:612–616. doi: 10.1128/jvi.70.1.612-616.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coffey M.C., Strong J.E., Forsyth P.A., Lee P.W. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282:1332–1334. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- 30.Strong J.E., Coffey M.C., Tang D., Sabinin P., Lee P.W. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17:3351–3362. doi: 10.1093/emboj/17.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bos J.L. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 32.Vorburger S.A., Pataer A., Swisher S.G., Hunt K.K. Genetically targeted cancer therapy: tumor destruction by PKR activation. Am J Pharmacogenomics. 2004;4:189–198. doi: 10.2165/00129785-200404030-00006. [DOI] [PubMed] [Google Scholar]
- 33.Meurs E., Chong K., Galabru J., Thomas N.S., Kerr I.M., Williams B.R. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell. 1990;62:379–390. doi: 10.1016/0092-8674(90)90374-n. [DOI] [PubMed] [Google Scholar]
- 34.Comins C., Heinemann L., Harrington K., Melcher A., De Bono J., Pandha H. Reovirus: viral therapy for cancer “as nature intended”. Clin Oncol (R Coll Radiol) 2008;20:548–554. doi: 10.1016/j.clon.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 35.Galanis E., Hartmann L.C., Cliby W.A., Long H.J., Peethambaram P.P., Barrette B.A. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 2010;70:875–882. doi: 10.1158/0008-5472.CAN-09-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morris D.G., Forsyth P.A., Paterson A.H. A phase I clinical trial evaluating intralesional reolysin (reovirus) in histologically confirmed malignancies. Proc Am Soc Clin Oncol. 2002 [Google Scholar]
- 37.Forsyth P., Roldán G., George D., Wallace C., Palmer C.A., Morris D. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008;16:627–632. doi: 10.1038/sj.mt.6300403. [DOI] [PubMed] [Google Scholar]
- 38.Vidal L., Pandha H.S., Yap T.A., White C.L., Twigger K., Vile R.G. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res. 2008;14:7127–7137. doi: 10.1158/1078-0432.CCR-08-0524. [DOI] [PubMed] [Google Scholar]
- 39.White C.L., Twigger K.R., Vidal L., De Bono J.S., Coffey M., Heinemann L. Characterisation of the innate, humoral and cellular immune response to intravenous oncolytic reovirus (Dearing Type 3) during a phase I clinical trial. Gene Ther. 2008;15:911–920. doi: 10.1038/gt.2008.21. [DOI] [PubMed] [Google Scholar]
- 40.Harrington K.J., Melcher A., Vassaux G., Pandha H.S., Vile R.G. Exploiting synergies between radiation and oncolytic viruses. Curr Opin Mol Ther. 2008;10:362–370. [PubMed] [Google Scholar]
- 41.Kumar S., Gao L., Yeagy B., Reid T. Virus combinations and chemotherapy for the treatment of human cancers. Curr Opin Mol Ther. 2008;10:371–379. [PubMed] [Google Scholar]
- 42.Zhang M., Li S., Li J., Ensminger W.D., Lawrence T.S. Ionizing radiation increases adenovirus uptake and improves transgene expression in intrahepatic colon cancer xenografts. Mol Ther. 2003;8:21–28. doi: 10.1016/s1525-0016(03)00143-6. [DOI] [PubMed] [Google Scholar]
- 43.Qian J., Yang J., Dragovic A.F., Abu-Isa E., Lawrence T.S., Zhang M. Ionizing radiation-induced adenovirus infection is mediated by Dynamin 2. Cancer Res. 2005;65:5493–5497. doi: 10.1158/0008-5472.CAN-04-4526. [DOI] [PubMed] [Google Scholar]
- 44.Twigger K., Vidal L., White C.L., De Bono J.S., Bhide S., Coffey M. Enhanced in vitro and in vivo cytotoxicity of combined reovirus and radiotherapy. Clin Cancer Res. 2008;14:912–923. doi: 10.1158/1078-0432.CCR-07-1400. [DOI] [PubMed] [Google Scholar]
- 45.Chou T.C., Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 46.Vidal L., Twigger K., White C.L., Ahmed M., Pandha H.S., Nutting C.M. Phase I trial of intratumoral administration of reovirus type 3 (Reolysin) in combination with radiation in patients with advanced malignancies. Proc Am Assoc Cancer Res. 2005 [Google Scholar]
- 47.Khuri F.R., Nemunaitis J., Ganly I., Arseneau J., Tannock I.F., Romel L. A controlled trial of intratumoral ONYX-015, a selectively-replicating adenovirus, in combination with cisplatin and 5-fluorouracil in patients with recurrent head and neck cancer. Nat Med. 2000;6:879–885. doi: 10.1038/78638. [DOI] [PubMed] [Google Scholar]
- 48.Pandha H.S., Heinemann L., Simpson G.R., Melcher A., Prestwich R., Errington F. Synergistic effects of oncolytic reovirus and cisplatin chemotherapy in murine malignant melanoma. Clin Cancer Res. 2009;15:6158–6166. doi: 10.1158/1078-0432.CCR-09-0796. [DOI] [PubMed] [Google Scholar]