

Abstract
PAR-2 is a G-protein coupled protease receptor whose activation in endothelial cells (ECs) is associated with increased solute permeability. VE-cadherin is an endothelial specific junction protein, which exhibits a disorganized distribution at cell junction during inflammation and is a useful indicator of endothelial barrier dysfunction. In the present study, we tested the hypothesis that PAR-2 activation mediates placenta-derived chymotrypsin-like protease (CLP)-induced endothelial junction disturbance and permeability in preeclampsia (PE). PAR-2 and VE-cadherin were examined by immunofluorescent staining. Specific CLP-induced PAR-2 activation and altered VE-cadherin distribution was assessed following depletion of protease chymotrypsin in the placental conditioned medium and after PAR-2 siRNA. VE-cadherin assembly was determined by treating cells with protease chymotrypsin and/or the specific PAR-2 agonist SLIGKV-NH2. Our results showed: 1) placental conditioned medium not only disturbed VE-cadherin distribution at cell junctions but also activated PAR-2 in ECs; 2) PAR-2 siRNA blocked the placental conditioned medium induced PAR-2 upregulation and disorganization of VE-cadherin at cell junctions; 3) PAR-2 agonist induced PAR-2 activation and VE-cadherin reorganization were dose-dependent; and 4) PAR-2 agonist could stimulate ERK1/2 activation. These results strongly suggest that proteases produced by the placenta elicit endothelial barrier dysfunction via a PAR-2 signaling regulatory mechanism in PE.
Keywords: PAR-2, VE-cadherin, protease, endothelium, placenta, preeclampsia
Introduction
Increased endothelial permeability is a hallmark of endothelial dysfunction in preeclampsia, a hypertensive and multi-system disorder in human pregnancy. In the systemic microvasculature, increased endothelial permeability leads to interstitial edema. By studying the mechanisms underlying endothelial barrier dysfunction in preeclampsia, we found that increased endothelial permeability is associated with decreased expression of endothelial-specific junction protein VE-cadherin and an altered distribution of VE-cadherin at endothelial junctions (1). We have now identified that chymotrypsin-like proteases (CLP) derived from the placenta promote endothelial inflammatory responses (2,3), disintegrate VE-cadherin binding between neighbor endothelial cells (4), and diminish endothelial barrier function (5). However, the cellular response and the mechanism of placenta-derived CLP induced endothelial barrier dysfunction in preeclampsia remain largely unknown.
Protease-activated receptors (PARs) are members of the G-protein-coupled receptor super-family that are activated by the proteolytic cleavage within the N terminal domain. PARs exist in 4 isoforms. PAR-1, -3, and -4 are cleaved and activated by thrombin while PAR-2 is cleaved and activated by trypsin, chymotrypsin, and other CLP's including chymase. PARs are now emerging as important modulators of diverse biological functions in the vascular system such as endothelial-dependent vessel contraction (6). PAR-2 activation has been reported in various cell types including endothelial cells, platelets, leukocytes, and fibroblasts and PAR-2 activation modulates numerous physiological and pathophysiological conditions (7,8). In endothelial cells, PAR-2 activation induces tissue factor expression and von Willebrand factor release from Weibel-Palade bodies (9). PAR-2 activation also promotes blood coagulation (10), leukocyte recruitment, and vascular permeability (11). Thus, PAR-2 activation represents an important hallmark of inflammatory response in vascular endothelial cells.
We previously reported that soluble factors derived from the preeclamptic placenta could promote PAR-2 activation in endothelial cells (8), and that placenta-derived CLP could disturb endothelial adhesion protein VE-cadherin spatial distribution to increase endothelial permeability (4). However, it is not known if PAR-2 activation is involved in altered VE-cadherin distribution induced by CLP-derived from preeclamptic placenta. The objective of this study intended to establish the link of placental derived CLP - endothelial dysfunction and increased endothelial permeability in preeclampsia. In the present study, we employed an in vitro cell culture model to specifically investigate and test the hypothesis that altered VE-cadherin expression and distribution induced by placenta-derived CLP is mediated through PAR-2 activation in endothelial cells and to explore the potential signaling cascade event that is involved in endothelial barrier dysfunction in preeclampsia.
Materials and Methods
Chemicals and reagents
Endothelial cell growth medium (EGM) was purchased from Lonza Walkersville, Inc. (Walkersville, MD). PAR-2 agonist SLIGKV-NH2 was purchased from Bachem (Buberdorf, Switzerland). Antibodies for PAR-2, VE-cadherin, ERK and pERK were purchased from Santa Cruz (San Diego, CA). β-actin antibody was from Sigma Chemicals (St. Louis, MO) and chymotrypsin antibody was from Abcam (Cambridge, MA). Cy3 labeled donkey anti-mouse IgG (H+L) was from Jackson Immunoresearch laboratories Inc. (Westgrove, PA). PAR-2 siRNA (sc-36188) and scrambled siRNA were purchased from Santa Cruz. Dulbecco's Modified Eagle's Medium (DMEM), horseradish peroxidase (HRP), guaiacol, hydrogen peroxide (H2O2), and protease inhibitors were from Sigma. All other reagents were obtained from Sigma unless otherwise noted.
Tissue collections
Placentas from normal and preeclamptic pregnant women were obtained at the main hospital, Louisiana State University Health Sciences Center - Shreveport (LSUHSC-S), LA. Normal pregnancy was defined as a pregnancy with normal blood pressure (<140/90mmHg), negative proteinuria, and absence of obstetrical and medical complications. Preeclampsia was defined as follows: sustained systolic blood pressure of ≥ 140 mmHg or a sustained diastolic blood pressure of ≥ 90mmHg on two separate readings; proteinuria measurement of 1+ or more on dipstick, or 24 hrs urine protein with ≥ 300mg in the specimen. None of the patients had signs of infection and smokers were excluded. Tissue collections were approved by the Institutional Review Board (IRB) for Human Research at LSUHSC-S. Umbilical cords from normal placentas were used to isolate HUVECs. Placentas from preeclamptic pregnancies were used to prepare placental conditioned medium.
Endothelial cell isolation and culture
HUVECs were isolated by collagenase digestion of umbilical cord vein of placentas delivered by normal term pregnant women as previously described (12). Isolated HUVECs were cultured with EGM containing recombinant human epithelial growth factor (rhEGF), hydrocortisone, gentamicin sulfate/amphotercin-B, bovine brain extract, and 2% fetal bovine serum (FBS). Cells used for fluorescent staining were grown on glass coverslips in 24 well/plates. Cells used for protein expression by Western blot were grown in 6 well/plates or T25 flasks. First passage cells were used in all experiments.
Placental conditioned medium preparation
Placental conditioned medium was prepared by culturing villous tissue from preeclamptic placentas as previously described (2). Briefly, Placental tissue was gently separated by sterile dissection from different cotyledons, excluding chorionic and basal plates, and washed repeatedly with phosphate buffered saline (PBS) to remove blood. Villous tissue explants 500mg/well in 6 well/plates were incubated with 7 ml DMEM containing penicillin, streptomycin, and amphotericin B without serum. The incubation was carried out for 48 hours at 37C in an incubator gassed with 95% air-5% CO2 (Forma Scientific, Inc., Marietta, OH). Medium samples were collected at the end of incubation as conditioned medium (CM) and stored at -80C freezer. In general, conditioned medium was used within 6 months after preparation. Pooled conditioned medium from 2-3 placental cultures were used to treat endothelial cells in each treatment assay and conditioned medium from at least 15 placentas were used in this study.
PAR-2 siRNA transfection
Transfections were conducted using siPORT™ Lipid Transfection Agent (Ambion Inc. Austin, TX) according to the manufacturer's instructions. Briefly, 0.5 nmol of siRNA was diluted with Opti-MEM I medium without serum and antibiotics and mixed with siPORT™ Lipid Transfection Agent. Cells were transfected at 37C gassed with 95% air-5% CO2 for 4 hours and then medium was replaced with fresh EGM containing 2% FBS. Scrambled siRNA was transfected as control. Cells were treated with conditioned medium 48 hours after transfection.
Immunofluorescent staining
Endothelial PAR-2 and VE-cadherin distribution and abundance were determined by immunofluorescent staining. Briefly, confluent endothelial cells grown on glass coverslips were treated with placental conditioned medium or PAR-2 agonist. After treatment, cells were fixed with 95% ethanol, permeabilized with 50% acetone, and then stained with monoclonal antibodies against PAR-2 or VE-cadherin. Cy3 labeled donkey anti-mouse IgG (H+L) was used as the secondary antibody. Cells stained without primary antibody served as negative control. Stained cells were examined by fluorescent microscopy (Olympus IX71, Tokyo, Japan). Images were recorded using a digital camera with PictureFrame computer software (Optronics Inc., Sunnyvale, CA).
Protein expression
Endothelial VE-cadherin, ERK, and pERK abundance were examined by Western blotting. An aliquot of total cellular protein (10μg of each sample) was subjected to electrophoresis (Bio-Rad, Hercules, CA) and then transferred to Hybond-protein transfer membrane (Amersham Corp, Arlington Heights, IL). The membrane was blocked with 5% milk in phosphate buffered saline and then probed with primary antibody at 4°C overnight. Proteins were visualized using enhanced chemiluminescent (ECL) detection Kit (Amersham Corp). The membrane was stripped before being probed with β-actin antibody (used as the loading control for each sample).
Endothelial permeability assay
Endothelial permeability was determined by measuring the passage of horseradish peroxidase (HRP) across confluent endothelial monolayers grown on polycarbonate cell culture inserts as we previously described (1,13). The HRP enzymatic activity was measured by a spectrophotometer with the wavelength at 470 nm (Ultraspec 3000, Pharmacia Biotech). Data were calculated as OD470nm sample-OD470nm blank, and expressed as OD470nm for permeation of HRP across transwell filters.
Statistical analysis
Data are expressed as mean ± SE and analyzed by analysis of variances (ANOVA) using StatView software (Cary, NC). Student-Newman-Keuls test was used as a post hoc test. A probability level of less than 0.05 was considered statistically significant.
Results
PAR-2 activation is associated with disruption of VE-cadherin expression at endothelial junction
Our previous study showed that disturbed VE-cadherin distribution at cell junction was detected after 2hr treatment by preeclamptic placental conditioned medium and internalization of VE-cadherin was detected after 24hr treatment (14). PAR-2 is a G-protein coupled receptor. The binding of ligands and G-proteins to receptors is an early event of cell response. To focus on the changes of VE-cadherin distribution at cell junctions, we first determined if PAR-2 activation modulates VE-cadherin expression and distribution in endothelial cells. Confluent endothelial cells were treated with preeclamptic placental conditioned medium for 30min, 1, and 2 hrs and endothelial expression of PAR-2 and VE-cadherin were determined by immunofluorescent staining. Representative images for PAR-2 and VE-cadherin expression and distribution at 2 hr treatment are shown in Figure 1A. PAR-2 expression was detected in cells treated with preeclamptic placental conditioned medium, but not in control cells. VE-cadherin expression was detected at junctional regions with a zigzag pattern in control cells, but was disrupted with gap formation at cell junctions in cells treated with preeclamptic placental conditioned medium. These findings suggest that PAR-2 activation is accompanied with altered VE-cadherin distribution at cell junctions induced by factors derived from preeclamptic placentas.
Figure 1.
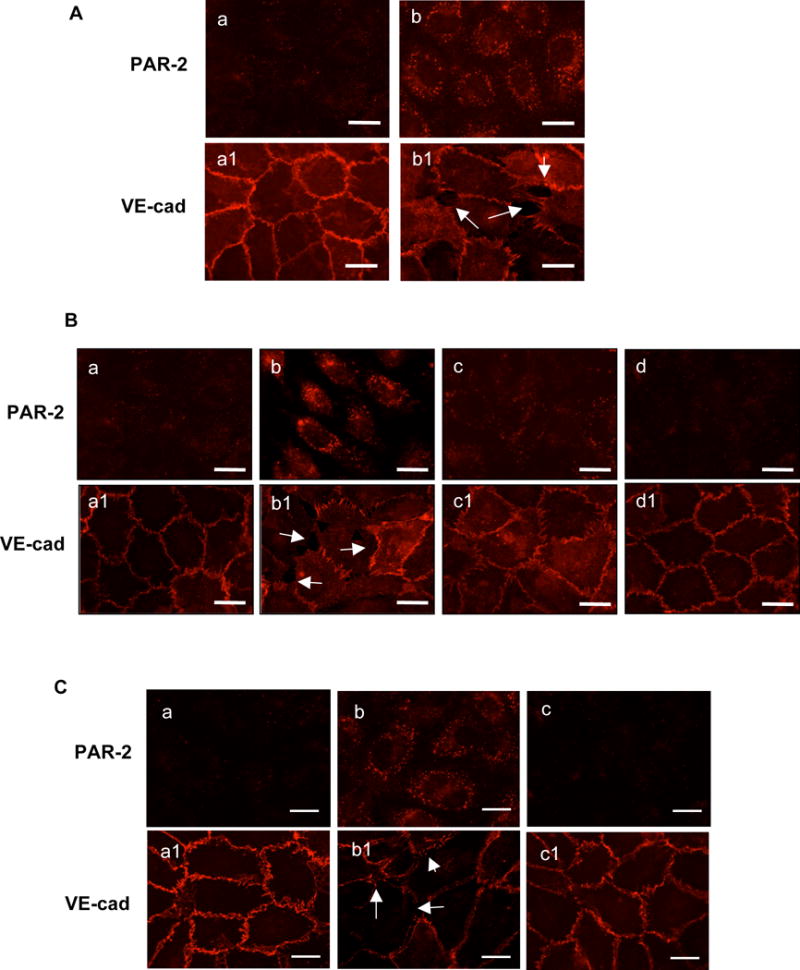
PAR-2 activation is associated with disintegration of VE-cadherin at endothelial junctions. A: PAR-2 activation and VE-cadherin disassembly induced by preeclamptic placental conditioned medium (PE-CM). a and b: PAR-2 and a1 and b1: VE-cadherin; a and a1: control cells; and b and b1: cells treated with PE-CM. B: Effects of PAR-2 siRNA on PE-CM induced PAR-2 and VE-cadherin expression in ECs. a-d: PAR-2; and a1-d1: VE-cadherin. a and a1: control cells; b and b1: cells treated with PE-CM; c and c1: cells were transfected with PAR-2 siRNA and then PE-CM; and d and d1: cells were transfected with PAR-2 siRNA only. C: PAR-2 and VE-cadherin expression in ECs treated with PE-CM with or without chymotrypsin depletion. a-c: PAR-2; and a1-c1: VE-cadherin. a and a1: control cells; b and b1: cells treated with PE-CM; and c and c1: cells treated with PE-CM after depleted with chymotrypsin. Images are representative of 3-4 independent assays in A, B, and C experiments. Bar = 25μm. Arrow shows gap formation and disturbed VE-cadherin expression at EC junctions.
Next, we determined if altered VE-cadherin distribution is associated with PAR-2 activation in cells treated with preeclamptic placental conditioned medium. Confluent endothelial cells were transfected with PAR-2 siRNA prior to placental conditioned medium treatment. We found that PAR-2 siRNA alone had no effect on VE-cadherin expression, but blocked placental conditioned medium induced PAR-2 activation and altered VE-cadherin distribution at cell junctions (Figure 1B). Control siRNA had no inhibitory effects on altered PAR-2 and VE-cadherin expressions induced by placental conditioned medium (data not shown).
PAR-2 activation induced by CLP-derived from preeclamptic placenta
We previously reported that placental-derived CLP is a likely candidate to induce endothelial activation and VE-cadherin disorganization at cell junctions (4,15). To find out if placental-derived CLP could activate PAR-2 in endothelial cells, chymotrypsin was depleted from the placental conditioned medium and then conditioned medium with or without chymotrypsin depletion were used to treat endothelial cells. Chymotrypsin depletion was carried out by immunoprecipitation as we previously described (15). Endothelial PAR-2 and VE-cadherin expressions were determined by immunofluorescent staining. Results are shown in Figure 1C. Interestingly, compared to cells treated with conditioned medium, PAR-2 expression was barely detectable in cells treated with chymotrypsin-depleted conditioned medium. This was accompanied by intact VE-cadherin distribution at the cell junction (Figure 1C). These results indicate that: 1) CLP derived from preeclamptic placentas can activate PAR-2, and 2) disturbed VE-cadherin expression at endothelial cell junctions is associated with PAR-2 activation.
PAR-2 activation-induced by chymotrypin is associated with increased endothelial permeability
To determine the specificity of CLP induced PAR-2 activation, a recombinant chymotrypsin was used. Endothelial cells were treated with chymotrypsin at a concentration of 5μg/ml for 15min, 1, 2, 6, and 24 hrs and then PAR-2 expression was determined by immunofluorescent staining. Figure 2A are representatives of PAR-2 expression in cells treated with chymotrypsin at 15min and 1hrs. Endothelial PAR-2 expression at 2, 6, and 24 hrs of treatment was similar to that at 1 hr treatment. These results indicate that PAR-2 expression was rapidly induced by chymotrypsin and this chymotrypsin-induced PAR-2 expression was time-dependent within a short period treatment.
Figure 2.
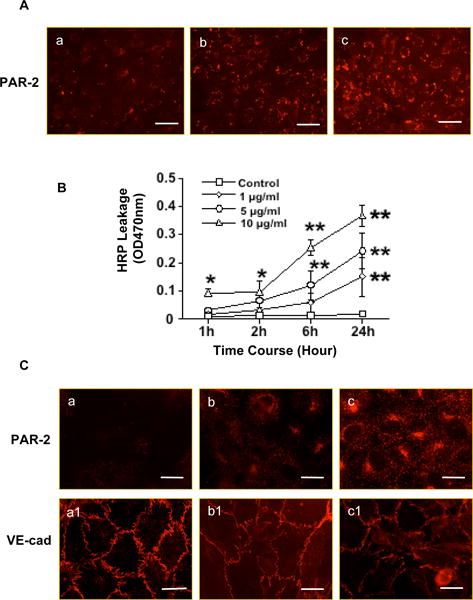
PAR-2 activation is associated with increased endothelial permeability and VE-cadherin disassembly at endothelial junctions. A: Chymotrypsin induced PAR-2 activation. a: control; b: cells were treated with chymotrypsin for 15min; and c: cells were treated with chymotrypsin for 1h. Bar = 100μm. B: Horseradish leakage in ECs treated with chymotrypsin at concentrations of 1, 5, and 10μg/ml, * p<0.05 and **p<0.01: chymotrypsin treated vs. control. Data are means from 5 independent experiments. C: Effects of PAR-2 agonist SLIGKV-NH2 on PAR-2 and VE-cadherin expression. a-c: PAR-2; and a1-c1: VE-cad. a and a1: control cells; b and b1: cells were treated with SLIGKV-NH2 at 200μM; c and c1: cells were treated with SLIGKV-NH2 at 500μM, respectively. SLIGKV-NH2 induced PAR-2 activation and VE-cadherin disintegration are in a dose-dependent manner. Bar = 25μm. Images are representative of 3-4 independent assays in the A and C experiments.
The specificity of chymotrypsin-induced endothelial permeability was further determined by horseradish leakage assay. Confluent endothelial cells grown on cell culture inserts were treated with chymotrypsin at concentrations of 1, 5, and 10μg/ml. Horseradish peroxidase was measured at 1, 2, 6, and 24hrs after chymotrypsin was added in the culture. Our results showed that chymotrypsin-induced increased endothelial permeability was both dose- and time-dependent, Figure 2B.
PAR-2 agonist induced PAR-2 activation and altered VE-cadherin distribution
The specificity of PAR-2 activation associated with altered VE-cadherin distribution in endothelial cells was further accessed by treating endothelial cells with the PAR-2 agonist (SLIGKV-NH2) at concentrations of 200μM and 500μM for 15 and 30min, and 1, 2, and 6 hrs. Figure 2C shows representative PAR-2 and VE-cadherin expressions in cells treated with PAR-2 agonist for 2 hrs. PAR-2 expression induced by SLIGKV-NH2 was dose dependent. The increased PAR-2 expression was associated with a remarkable change in VE-cadherin distribution at cell junctions.
PAR-2 agonist triggers ERK phosphorylation in endothelial cells
We previously found that endothelial activation induced by placenta-derived CLP was associated with ERK (extracellular-signal-regulated kinase) activation (15). To elucidate if PAR-2 is associated with ERK activation, ERK phosphorylation (pERK) was determined in cells treated with PAR-2 agonist SLIGKV-NH2. Since ERK activation is an early event, confluent endothelial cells were treated with PAR-2 agonist for 15 and 30min with or without pretreatment with PD98059 for 1 hr. Interestingly, we found that pERK was expressed at 15min but not 30min after PAR-2 agonist treatment and this PAR-2 agonist induced ERK phosphorylation event could be blocked by pretreatment of cells with PD98059, indicating the involvement of ERK1/2 signal transduction pathway during PAR-2 activation in endothelial cells (Figure 3A).
Figure 3.
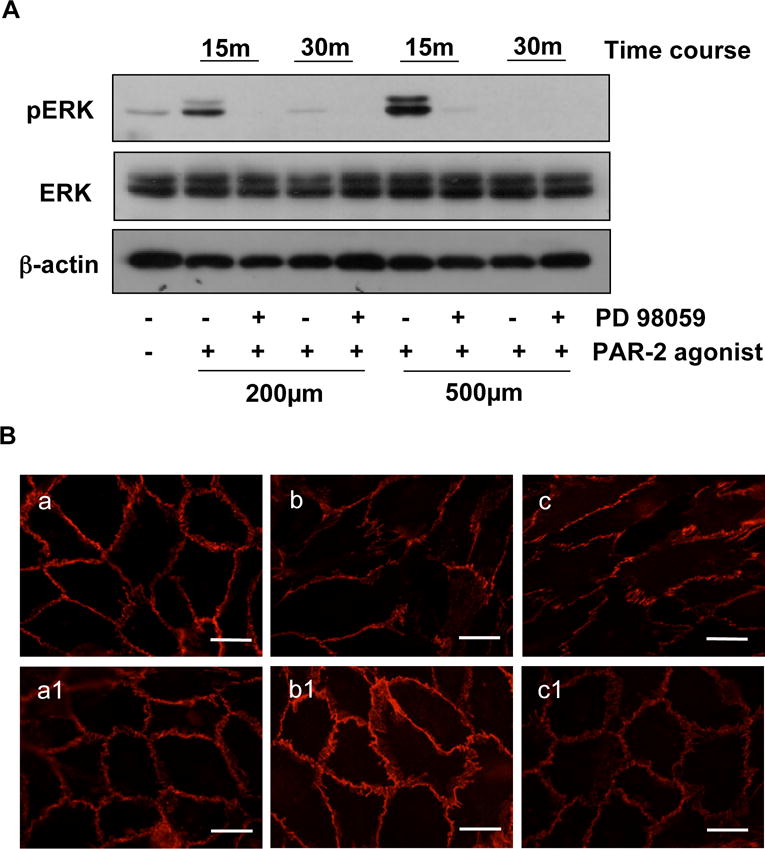
PAR-2 agonist triggers MAPK/ERK1/2 phosphorylation in endothelial cells. A: ERK, and pERK expressions in cells treated with concentrations of SLIGKV-NH2 at 200μM and 500μM in the presence or absence of PD98059. β-actin expression was determined as internal control for each sample. pERK expression was detected in cells treated with SLIGKV-NH2 at 15 min, but not at 30min, indicating that ERK phosphorylation induced by PAR-2 agonist was an early event of the cell response. B: VE-cadherin expression in ECs treated with SLIGKV-NH2 in the presence (a1, b1, and c1) or absence of PD98059 (a, b, and c). a and a1: control; b and b1: SLIGKV-NH2 200μM; and c and c1: SLIGKV-NH2 500μM, respectively. Bar = 25μm. Blots and images are representatives from 3 independent experiments.
To determine if ERK1/2 activation is associated with altered VE-cadherin expression, we examined VE-cadherin expression by Western blot and VE-cadherin distribution by immunofluorescent staining in cells treated with the PAR-2 agonist SLIGKV-NH2. We found that there was no difference in total protein expression for VE-cadherin in cells treated with PAR-2 agonist at 15 and 30min and 1 and 2hrs in the presence or absence of PD98059 (data not shown). However, cells pretreated with PD98059 showed an inhibition of PAR-2 agonist induced altered VE-cadherin distribution at cell junctions at 2 hrs of treatment (Figure 3B). These observations suggest that ERK phosphorylation is an early event of the signaling cascade in PAR-2 activation and altered VE-cadherin distribution in endothelial cells.
Discussion
We previously reported that placental-derived factors could diminish endothelial barrier function and increased endothelial permeability through disorganized adhesion junction protein VE-cadherin (5). We also found that CLP produced by preeclamptic placentas is a likely mediator that produces this harmful effect against endothelial cells (4). However, the mechanism by which CLP induced endothelial barrier dysfunction is largely unknown. Serine proteases can induce PAR-2 activation and CLP is a potent serine protease in the vasculature. Several studies have shown that PAR-2 activation plays a role in endothelial permeability (11, 16). In the present study, we sought to determine if PAR-2 activation mediates CLP induced alterations of VE-cadherin distribution and endothelial barrier dysfunction during preeclampsia. Our key findings include the following: 1) CLP produced/released by the placenta promotes PAR-2 activation in endothelial cells, which is accompanied by altered VE-cadherin distribution and gap formation at cell junctions; 2) a selective PAR-2 agonist not only induces VE-cadherin disorganization but also mitogen-activated protein kinase (MAPK/ERK1/2) phosphorylation, suggesting that MAPK/ERK1/2 activation might be an early event in endothelial cells that is involved in the process of PAR-2 activation-mediated endothelial barrier dysfunction; and 3) PD98059, a MAPK blocker, blocks PAR-2 agonist-induced MAPK/ERK1/2 activation and VE-cadherin redistribution, indicating that MAPK/ERK1/2 activation could be an upstream signaling event associated with endothelial junction disturbances.
PAR-2 is a G-protein coupled protease activated receptor expressed by vascular, intestinal, and airway cells which mediates inflammatory and proliferative responses associated with tissue injury (17). Like other PARs, PAR-2 is irreversibly activated through proteolytic cleavage of its N terminus by serine proteases. This cleavage creates a new extracellular N terminus, which serves as tethered ligand that activates and initiates downstream intracellular signaling events. Mounting evidence has shown that not only PAR-1, but also PAR-2, activation results in endothelial injury and increased endothelial permeability (11,16,18,19). For example, Dömötör et al have found that PAR-2 activation is associated with endothelial injury in brain microvasculature (20); others observed that PAR-2 activation increases endothelial and epithelial permeability of proteins in the lung (11,19). In the present study, we found that PAR-2 mediated CLP-induced endothelial barrier disturbance and PAR-2 agonist induced ERK phosphorylation, which indicate that PAR-2 activation could represent an indicator of increased endothelial inflammatory response in preeclampsia.
VE-cadherin assembly controls endothelial junction integrity and regulates endothelial solute permeability. PAR-2 activation has now been linked to VE-cadherin disassembly and gap formation at cell junctions clearly shows that PAR-2 activation controls barrier integrity in endothelial cells. Using the PAR-2 agonist SLIGKV-NH2, we specifically examined downstream effects of PAR-2 activation in endothelial cells and found that SLIGKV-NH2 not only induced PAR-2 activation but also robust MAPK/ERK1/2 phosphorylation. These results are consistent with previously reports that ERK1/2 phosphorylation could be induced by either PAR-1 or PAR-2 receptor activation and ERK1/2 phosphorylation interferes downstream of PAR-2 activation signaling events (21,22). In addition, we also found that PD98059 (a specific inhibitor of MAPK) could block SLIGKV-NH2-induced VE-cadherin disassembly at cell junctions, but did not affect the amount of VE-cadherin protein expressed by endothelial cells. These results suggest that phosphorylation of ERK1/2 is involved in PAR-2 activation induced barrier integrity in endothelial cells. MAPK/ERK1/2 activation is a downstream event of PAR-2 activation. PAR-2 could function as a scaffold to recruit and activate ERK1/2 as well as other effecter proteins.
PAR-2 exerts diverse effects on cellular function. As a cell surface sensor of proteases, PAR-2 endows the cell with the ability to respond to the rapidly changing proteolytic microenvironments during inflammation. Currently, PAR-2 is considered a therapeutic target to improve protease-induced cell dysfunction. Animal studies have shown that PAR-2 activation could contribute to several early events in the inflammatory reaction, including leukocyte rolling/adherence/recruitment (23). Over-stimulation of PAR-2 could increase microvascular permeability, granulocyte infiltration and severe edema (24). Conversely, PAR-2 deficiency has been shown to reduce cardiac ischemia/reperfusion injury in mice and protects against impaired heart function compared with wild-type littermates at least 4 weeks after ischemia/reperfusion injury (25). Our finding of PAR-2 activation associated with altered VE-cadherin distribution at cell junction induced by placental conditioned medium together with specific PAR-2 agonist induced altered VE-cadhein expression provides evidence linking PAR-2 activation with endothelial activation in preeclampsia.
Microvascular endothelial permeability is increased in women with preeclampsia. Numerous investigators including our group have found that placenta-derived toxic factors play a significant role in inducing endothelial activation/dysfunction in preeclampsia (26-28). In the present study, we showed that PAR-2 activation might contribute to the increased endothelial permeability and barrier dysfunction in preeclampsia. Although we cannot determine if CLP-derived from the placenta could directly mediate PAR-2 activation to increase endothelial permeability in the systemic microvasculature during preeclampsia, the findings of increased CLP expression in systemic vessel endothelium (29) and the presence of leukocyte infiltration around microvessels (30) support the notion that CLP-PAR-2 activation may contribute to the altered endothelial barrier function and increased endothelial permeability in preeclampsia.
Acknowledgments
This study was supported in part by grants from National Institute of Health grants RO1 NHLBI (HL65997) and NICHD (HD36822) to Y.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang Y, Gu Y, Granger DN, Roberts JM, Alexander JS. Endothelial junctional protein redistribution and increased monolayer permeability in HUVECs isolated during preeclampsia. Am J Obstet Gynecol. 2002;186:214–220. doi: 10.1067/mob.2002.119638. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Adair CD, Weeks JW, Lewis DF, Alexander JS. Increased neutrophil-endothelial adhesion induced by placental factors is mediated by platelet-activating factor in preeclampsia. J Soc Gynecol Investig. 1999;6:136–141. doi: 10.1016/s1071-5576(99)00004-0. [DOI] [PubMed] [Google Scholar]
- 3.Wang Y, Zhang Y, Lewis DF, Gu Y, Li H, Granger DN, Alexander JS. Protease chymotrypsin mediates the endothelial expression of P- and E-selectin, but not ICAM and VCAM, induced by placental trophoblasts from preeclamptic pregnancies. Placenta. 2003;24:851–861. doi: 10.1016/s0143-4004(03)00132-2. [DOI] [PubMed] [Google Scholar]
- 4.Gu Y, Lewis DF, Alexander JS, Wang Y. Placenta-derived chymotrypsin-like protease (CLP) disturbs endothelial junctional structure in preeclampsia. Reprod Sci. 2008;16(5):479–488. doi: 10.1177/1933719108329818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Lewis DF, Gu Y, Zhang Y, Alexander JS, Granger DN. Placental trophoblast-derived factors diminish endothelial barrier function. J Clin Endocrinol Metabol. 2004;89:2421–2428. doi: 10.1210/jc.2003-031707. [DOI] [PubMed] [Google Scholar]
- 6.Saifeddine M, Roy SS, Al-Ani B, Triggle CR, Hollenberg MD. Endothelium-dependent contractile actions of proteinase-activated receptor-2-activating peptides in human umbilical vein: release of a contracting factor via a novel receptor. Bri J Pharmacol. 2000;125:1445–1454. doi: 10.1038/sj.bjp.0702213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Howells GL, Macey MG, Chinni C, Hou L, Fox MT, Harriott P, Stone SR. Proteinase-activated receptor-2: expression by human neutrophils. J Cell Science. 1997;110:881–887. doi: 10.1242/jcs.110.7.881. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Gu Y, Lucas MJ. Expression of thrombin receptors in endothelial cells and neutrophils from normal and preeclamptic pregnancies. J Clin Endocrinol Metabol. 2002;87:3728–3734. doi: 10.1210/jcem.87.8.8727. [DOI] [PubMed] [Google Scholar]
- 9.Langer F, Morys-Wortmann C, Kusters B, Storck J. Endothelial protease-activated receptor-2 induces tissue factor expression and von Willebrand factor release. Bri J Haematol. 1999;105:542–550. [PubMed] [Google Scholar]
- 10.Alm AK, Norstrom E, Sundelin J, Nystedt S. Stimulation of proteinase activated receptor-2 causes endothelial cells to promote blood coagulation in vitro. Thromb Haemost. 1999;81:984–988. [PubMed] [Google Scholar]
- 11.Itoh Y, Sendo T, Oishi R. Physiology and pathophysiology of proteinase-activated receptors (PARs): role of tryptase/PAR-2 in vascular endothelial barrier function. J Pharmacol Sci. 2005;97:14–19. doi: 10.1254/jphs.fmj04005x3. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Adair CD, Coe L, Weeks JW, Lewis DF, Alexander JS. Activation of endothelial cells in preeclampsia: Increased neutrophil-endothelial adhesion correlates with up-regulation of adhesion molecule P-selectin in human umbilical vein endothelial cells isolated from preeclampsia. J Soc Gynecol Investig. 1998;5:237–243. doi: 10.1016/s1071-5576(98)00023-9. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Alexander JS. Analysis of endothelial barrier function in vitro. Methods Mol Biol. 2011;763:253–264. doi: 10.1007/978-1-61779-191-8_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao J, Gu Y, Fan R, Groome LJ, Wang Y. Factors Derived From Preeclamptic Placentas Perturb Polarity Protein PARD-3 Expression and Distribution in Endothelial Cells. Reprod Sci. 2011;18(2):164–171. doi: 10.1177/1933719110382920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu Y, Liu C, Alexander JS, Groome LJ, Wang Y. Chymotrypsin-like protease (chymase) mediates endothelial activation by factors derived from preeclamptic placentas. Reprod Sci. 2009;16(9):905–913. doi: 10.1177/1933719109337333. 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klarenbach SW, Chipiuk A, Nelson RC, Hollenberg MD, Murray AG. Differential actions of PAR2 and PAR1 in stimulating human endothelial cell exocytosis and permeability: the role of Rho-GTPases. Circ Res. 2003;92(3):272–278. doi: 10.1161/01.res.0000057386.15390.a3. [DOI] [PubMed] [Google Scholar]
- 17.Traynelis SF, Trejo J. Protease-activated receptor signaling: new roles and regulatory mechanisms. Curr Opin Hematol. 2007;14(3):230–235. doi: 10.1097/MOH.0b013e3280dce568. [DOI] [PubMed] [Google Scholar]
- 18.Su X, Camerer E, Hamilton JR, Coughlin SR, Matthay MA. Protease-activated receptor-2 activation induces acute lung inflammation by neuropeptide-dependent mechanisms. J Immunol. 2005;175(4):2598–2605. doi: 10.4049/jimmunol.175.4.2598. [DOI] [PubMed] [Google Scholar]
- 19.Feistritzer C, Lenta R, Riewald M. Protease-activated receptors-1 and -2 can mediate endothelial barrier protection: role in factor Xa signaling. J Thromb Haemost. 2005;3(12):2798–2805. doi: 10.1111/j.1538-7836.2005.01610.x. [DOI] [PubMed] [Google Scholar]
- 20.Dömötör E, Bartha K, Machovich R, Adam-Vizi V. Protease-activated receptor-2 (PAR-2) in brain microvascular endothelium and its regulation by plasmin and elastase. J Neurochem. 2002;80(5):746–754. doi: 10.1046/j.0022-3042.2002.00759.x. [DOI] [PubMed] [Google Scholar]
- 21.McCoy KL, Traynelis SF, Hepler JR. PAR1 and PAR2 couple to overlapping and distinct sets of G proteins and linked signaling pathways to differentially regulate cell physiology. Mol Pharmacol. 2010;77(6):1005–1015. doi: 10.1124/mol.109.062018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou B, Zhou H, Ling S, Guo D, Yan Y, Zhou F, Wu Y. Activation of PAR2 or/and TLR4 promotes SW620 cell proliferation and migration via phosphorylation of ERK1/2. Oncol Rep. 2011;25(2):503–511. doi: 10.3892/or.2010.1077. [DOI] [PubMed] [Google Scholar]
- 23.Vergnolle N. Proteinase-activated receptor-2-activating peptides induce leukocyte rolling, adhesion, and extravasation in vivo. J Immunol. 1999;163:5064–5069. [PubMed] [Google Scholar]
- 24.Cenac N, Coelho AM, Nguyen C, Compton S, Andrade-Gordon P, MacNaughton WK, Wallace JL, Hollenberg MD, Bunnett NW, Garcia-Villar R. Induction of intestinal inflammation in mouse by activation of proteinase-activated receptor-2. Am J Pathol. 2002;161(5):1903–1915. doi: 10.1016/S0002-9440(10)64466-5. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antoniak S, Rojas M, Spring D, Bullard TA, Verrier ED, Blaxall BC, Mackman N, Pawlinski R. Protease-activated receptor 2 deficiency reduces cardiac ischemia/reperfusion injury. Arterioscler Thromb Vasc Biol. 2010;30(11):2136–2142. doi: 10.1161/ATVBAHA.110.213280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts JM, Hubel CA. The two stage model of preeclampsia: variations on the theme. Placenta. 2009;30(Suppl A):S32–37. doi: 10.1016/j.placenta.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cockell AP, Learmont JG, Smarason AK, Redman CWG, Sargent IL, Poston L. Human placental syncytiotrophoblast microvillous membranes impair maternal vascular endothelial function. Bri J Obstet Gynaecol. 1997;104:235–240. doi: 10.1111/j.1471-0528.1997.tb11052.x. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Lewis DF, Alexander JS, Granger DN. Endothelial barrier function in preeclampsia. Front Biosci. 2007;12:2412–2424. doi: 10.2741/2243. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Gu Y, Lewis DF, Alexander JS, Granger DN. Elevated plasma chymotrypsin-like protease (chymase) activity in women with preeclampsia. Hypertens Pregn. 2010;29(3):253–261. doi: 10.3109/10641950802001842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cadden KA, Walsh SW. Neutrophils, but not lymphocytes or monocytes, infiltrate maternal systemic vasculature in women with preeclampsia. Hypertens Pregnan. 2008;27(4):396–405. doi: 10.1080/10641950801958067. [DOI] [PMC free article] [PubMed] [Google Scholar]