

Abstract
The requirement that leukemic GATA1 mutations be present in cells harboring trisomy 21 led to the discovery that overexpression of ERG drives aberrant megakaryopoiesis. Given that constitutive PI3K/AKT signaling is a frequent component of hematologic malignancies and the relationship between AKT and Notch in this lineage, we studied the cross talk between AKT signaling and ERG in megakaryopoiesis. We discovered that constitutive AKT signaling is associated with a dramatic increase in apoptosis of WT MKs, but that overexpression of ERG blocks AKT-induced death. We further found that Gata1 mutations protect megakaryocytes from activated AKT-induced apoptosis. As a consequence, however, the enhanced signaling inhibits differentiation of Gata1 mutant, but not WT, megakaryocytes. Gata1 mutant cells that overexpress ERG with hyperactive AKT are characterized by diminished FOXO1/3a expression and an increased dependency on the c-Jun pathway similar to that seen in AMKL cell lines, AML with knockdown of FOXO3a, or AML with expression of myrAKT. Additionally, we found that the AKT allosteric inhibitor MK2206 caused reduced cell viability and proliferation of AMKL cell lines. The contribution of aberrant AKT signaling during the ontogeny of DS-TMD/AMKL indicates that AKT is a therapeutic target in this form of AML.
Keywords: Acute Megakaryocytic Leukemia, MK2206, AKT inhibition
Introduction
AKT expression is significantly higher in DS-AMKL than non-DS AMKL (1) leading to the model that expression of chromosome 21 genes, such as ERG, collaborate with GATA1s and additional mutations in signaling pathways, such as JAK2, JAK3, MPL, and FLT3 (2–4) as well as perturbed IGF-1 signaling, during the ontogeny of myeloid leukemia in DS. Here, we demonstrate the ability of AKT to collaborate with ERG and GATA1 during fetal derived megakaryopoiesis. We show that although constitutive AKT signaling is associated with a dramatic increase in apoptosis of megakaryocytes, overexpression of ERG blocks AKT-induced cell death. Moreover, we find that GATA1s protects megakaryocytes from AKT induced apoptosis and that activated AKT signaling inhibits differentiation of Gata1 mutant, but not wild-type, megakaryocytes. We further demonstrate that aberrant AKT signaling is compatible with ERG driven megakaryopoiesis in the Gata1 mutant background, with maintenance of AKT function over long term culturing to sustain decreased expression/nuclear localization of FOXO1/FOXO3a and up-regulation of phospho-c-Jun. (1). Finally, we show that both DS- and non-DS AMKL cell lines are sensitive to the effects of AKT inhibition mediated by the AKT allosteric inhibitor MK2206 leading to alterations in downstream signaling targets of activated AKT, increased apoptosis and decreased proliferation.
Methods
Cell culture
Isolation and manipulation of wild-type (C57Bl6) and Gata1s knock-in (Gata1Δex2) C57B/6 mice were performed as described(5). For short-term culture experiments, transduced cells were cultured in megakaryocyte expansion media composed of RPMI complete with 20% serum, TPO (20 ng/ml) and SCF (100 ng/ml) for 72 hours. For experiments with sorted hematopoietic progenitors, cells were expanded for one week in M3234 (StemCell Technologies) supplemented with IL3 (10 ng/ml), SCF (100 ng/ml), IL6 (10 ng/ml) and TPO (10 ng/ml) before being collected from the methylcellulose and then placed in RPMI complete media with 15% serum and TPO, IL3, IL6 and SCF (same concentrations as in the methylcellulose semi-solid media) for the duration of experiments. Replating experiments used 5000 fluorescently labeled retrovirally transduced cells sorted simultaneously for both GFP and mcherry and then plated in M3234 (duplicate 35 mm dishes) with IL3 (10 ng/ml), SCF (100 ng/ml), IL6 (10 ng/ml) and TPO (10 ng/ml). Colonies were enumerated between 7–9 days and 5000 cells replated for each time point as previously described (5). MK2206, 8-[4-(1-aminocyclobutyl)phenyl]-9-phenyl-1,2,4-triazolo[3,4-f] [1,6]naphthyridin-3(2H)-one hydrochloride [1:1], was generously provided by Merck. For in vitro experiments, 1 µM, 5 µM, and 10 µM stock solutions of MK2206 were formulated in DMSO and subsequently diluted in RPMI-1640 media, with the amount of DMSO added to media no greater than 0.1%. Megakaryocytic lines were grown in RPMI-1640 with 10% serum. Statistical significance of differences between the different conditions was ascertained by the Student’s t-test.
Retroviral transduction
The Migr1-hERG1-IRES-GFP and Migr1-IRES-GFP constructs have been described (5, 6). MSCV-myrAKT1-IRES-GFP was provided by Thomas Mercher (6–8). Migr1-IRES-mcherry was provided by Lou Dore. ERG1 was subcloned from the Migr1-hERG1-IRES-GFP into Migr1-IRES-mcherry. All experiments used doubly transduced cells that harbor a GFP expression construct and an mcherry expression construct. Retroviral transduction was performed by spinoculation of the mcherry construct on day one and the GFP construct on day 2.
Flow cytometry, immunofluorescence and western blotting
Flow cytometry experiments were performed with a BD LSR Fortessa or BD LSRII, and sorted by a BD FacsAria II. Flowjo software (version 8.8.6, Treestar, Ashland, OR) was used to analyze FCS files. Antibodies and stains for flow include: PE-rat-anti-mouse CD41 (BD Biosciences), Dylight 649-rat-anti-mouse CD42b (GPIbα) (Emfret Analytics), anti-mouse CD117 (c-Kit) APC-efluor 780 (Ebioscience, clone 2B8), APC-Annexin V,7-AAD (BD Biosciences), phospho-AKT (Ser473) XP rabbit mAb (Alexa Fluor 647), phospho-c-jun (Ser73) XP rabbit mAb, phospho- S6 ribosomal protein (Ser235/236) rabbit mAb (Alexa Fluor 647) and anti-rabbit IgG F(ab)2 fragment Alexa Fluor 647 (Cell Signaling Technology). BrdU incorporation/detection was performed with a BD Pharmingen BrdU APC flow kit. DAPI (Sigma) was used for detection of DNA. For immunofluorescence, nuclei were stained with Hoechst 33342 (Invitrogen, Molecular Probes). FOXO1 rabbit mAb (#2880) and FOXO3a rabbit mAb (#2497) were used with the anti-rabbit IgG F(ab)2 fragment Alexa Fluor 647 as secondary antibody (Cell Signaling Technology). Immunofluoresence imaging was performed with a laser scanning confocal microscope (Nikon AR1 system). All bars shown are of 20 micron size. Anti-phospho-FOXO3a antibody purchased from Millipore, while other primary antibodies were purchased from Cell Signaling Technology and secondary antibodies from GE Healthcare.
Results
Activated AKT blocks megakaryocytic expansion in the presence of Gata1 mutation
Given that AKT signaling is required in DS-AMKL, and AMKL in general, we investigated the contribution of constitutively active AKT in primary fetal liver megakaryocytes that express the GATA-1 leukemic isoform, GATA1s, and/or overexpress the chromosome 21 gene ERG. Retroviral transduction was used to express different combinations of ERG, constitutively active AKT (myrAKT), GFP and m-cherry in murine fetal liver progenitors. The four groups included: Migr1-IRES-GFP + MIGR1-IRES-mcherry (abbreviated as Migr1); MIGR1-IRES-mcherry + MSCV-myrAKT1-IRES-GFP (abbreviated as myrAKT), Migr1-hERG1-IRES-mcherry + Migr1-IRES-GFP (abbreviated as ERG), Migr1-hERG1-IRES-mcherry + MSCV-myrAKT1-IRES-GFP (abbreviated as myrAkt+ERG). After 72 hours of culture with TPO and SCF, transduced fetal liver hematopoietic progenitors with wild-type GATA1 expression were assessed for megakaryocyte development (Figure 1A, B, E, and Supplementary Figure 1A). We found that both samples expressing ERG (ERG alone and myrAKT+ERG) demonstrated a significant increase in the CD41 single positive and CD41+CD42 double positive populations (Figure 1A, B), indicating that myrAKT does not disrupt ERG’s megakaryocyte expansion activity.
Figure 1. Short-term in vitro assessment of megakaryocyte potential demonstrates activated AKT blocks megakaryocyte maturation in the presence of Gata1 mutation.

(A) Average percentages (± SEM) for the CD41 positive, CD42 negative population for indicated wild-type sample; * p-value ≤0.04. For all graphs, black bars, MigR1; striped bars, myrAKT; grey, ERG; white, myrAKT+ERG. (B) Average percentage (± SEM) for the CD41 positive CD42 positive population for the indicated wild-type sample; * p-value ≤ 0.04. (C) Average percentage (± SEM) for the CD41 positive, CD42 negative population for indicated Gata1s knock-in sample; * p-value ≤ 0.04. (D) Average percentage (± SEM) for the CD41 positive CD42 positive population for the indicated Gata1s knock-in sample; * p-value ≤ 0.04. (E) Average (±SD) ploidy percentage of the CD41 positive population for indicated samples on the wild-type background with ≤ 8N on the left and ≥8n on the right; * p-value ≤ 0.04. (F) Average (±SD) ploidy percentage of the CD41 positive population for indicated samples on the Gata1s background with ≤ 8N on the left and ≥8n on the right; * p-value ≤ 0.04. Flow cytometry plots of CD41 versus CD42 for the wild-type and Gata1s knock-in samples shown in Supplementary Figure 1 A, B.
We repeated the megakaryocyte expansion assays, this time infecting Gata1s knock-in fetal liver hematopoietic progenitors with the four combinations of vectors. As expected, ERG led to a significant increase in the CD41 singly positive expressing population, and this was recapitulated by the addition of myrAKT (myrAKT+ERG), in comparison to the Migr1 control (Figure 1C). Expression of myrAKT alone as well as in combination with ERG (myrAKT+ERG) led to a striking reduction in the CD41+CD42 double positive expressing population (Figure 1D). Thus, there is a dominant inhibitory effect of AKT signaling on maturation of Gata1 mutant megakaryocytes.
ERG expression in the wild-type and Gata1s knock-in background led to a significant increase in the number of singly positive CD41 expressing cells, suggesting ERG induces different stages of megakaryopoiesis possibly depending on the specific cell type infected from the original pool of lineage negative fetal liver progenitors, or alternatively, ERG may promote expansion or survival of the less mature CD41 singly positive megakaryocytes. Upon co-expression of myrAKT with ERG, this effect is recapitulated in the presence of wild-type Gata1 and potentiated in the Gata1s knock-in background upon co-expression of myrAKT, allowing myrAKT’s effects on delaying maturation to synergize with ERG’s potential to do so as well in the Gata1s knock-in background (Figure 1A–D). Polyploidization in the wild-type Gata1 background was altered in comparison to Migr1 upon the co-expression of myrAKT+ERG, with an increase in the ≤8N and decrease in the ≥ 8N population (Figure1E). Polyploidization was also altered by myrAKT, ERG and myrAKT+ERG in the Gata1s knock-in background in comparison to the Migr1 control (Figure 1F), with a decrease in the ≥8N population for the CD41 positive population and concomitant increase in the ≤8N population. Thus, overall, activated AKT inhibits maturation of Gata1 mutant megakaryocytes and these effects are dominant in the presence of ERG.
ERG, GATA1s and myrAKT expressing cells show increased replating potential
Previous studies have shown that bone marrow cells and splenocytes expressing myrAKT cannot serial replate beyond the second round of plating due to the exhaustive effects of myrAKT(7). After sorting for GFP, mcherry double positive cells, 5000 fetal liver hematopoietic progenitors of each group were plated in methylcellulose for one week and then replated two additional times under the same conditions. We found that myrAKT expressing fetal liver progenitors in the presence of wild-type GATA1 failed to expand significantly beyond two platings (Figure 2A). In contrast, myrAKT+ERG expressing cells replated in the third round and were able to grow in culture for up to four weeks (data not shown). myrAKT samples in the Gata1s knock-in background were able to replate to the third round without any significant differences in comparison to the Migr1 control (Figure 2B). Of note, myrAKT+ERG expressing Gata1s knock-in cells showed enhanced replating activity in comparison to the Migr1 and myrAKT samples, but followed a similar trend as ERG alone under these conditions. Together, these results show that ERG has a dominant effect over constitutively active AKT.
Figure 2. Long-term culturing demonstrates enhanced survival in the presence of ERG and GATA1s for myrAKT expressing cells.

(A) Serial replating of 5000 sorted GFP, mcherry double positive wild-type GATA1 (top) and (B) Gata1s knock-in (GKI) fetal liver hematopoietic progenitors (bottom); For wild-type GATA1 samples, p-value ≤ 0.04 for all * shown in figure. For GKI, p-value ≤ 0.04 for all * shown in figure. NS=not significant. The Student’s t-test was used for determination of statistical significance between each pair of linked samples. N=4 independent experiments for the wild-type cells and N=3 for the G1KD progenitors.
Increased cell survival with expression of ERG and GATA1s during long-term in vitro culturing
As it has been reported that myrAKT overexpressing cells are susceptible to high rates of apoptosis and have severely impaired proliferation (7, 9), we analyzed cell viability of long term cultures of the double positive Migr1 control, myrAKT, ERG and myrAKT+ERG transduced wild-type and Gata1s knock-in fetal liver derived hematopoietic progenitors. In an effort to ascertain the changes in cell survival as demonstrated by colony assays, sorted cells of either genotype were expanded for one week in M3234 with TPO, IL3, IL6 and SCF, recovered and placed in RPMI with the same complement of cytokines and 15% serum for an additional 14–21 days. Annexin V staining was monitored by flow cytometry at 2–3 weeks of culture for cultures with wild-type GATA1 and out to 4 weeks for Gata1s knock-in samples due to their overall enhanced fitness and proliferative capacity as has been reported (Supplementary Figure 2 A–D) (10). Live cell counts were also recorded at the same time for all samples (Figure 3 A, B). As expected from the serial replating assays, myrAKT expression in cells of the wild-type GATA1 background exhibited a decreased percentage of live cells in comparison to all other samples as well as a significant amount of Annexin V positivity.
Figure 3. Assessment of cell viability demonstrates survival advantage for myrAKT expressing cells due to ERG or GATA1s expression.
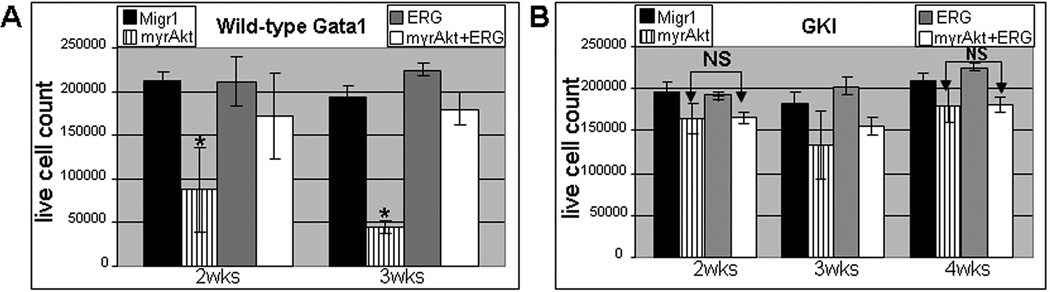
(A) Enumeration of live cells from liquid cultures in the wild-type GATA1 samples reveal a decrease in cell number for myrAKT(*) expressing cells compared to all other samples with a p-value ≤ 0.04 over time course. (B) Live cell counting on the Gata1s knock-in background reveals no significant difference (NS) in cell number between myrAKT and myrAKT+ERG samples at 2 and 4 weeks. Both samples are significantly different from Migr1 and ERG at 2 and 4 weeks with a p-value ≤ 0.03. See Supplementary Figure 2 for Annexin V staining assessed by flow cytometry over same time course for both wild-type and Gata1s knock-in samples.
MyrAKT+ERG samples also demonstrated a decrease in the percentage of live cells with time and an increase in Annexin V staining in comparison to both Migr1 and ERG samples (Figure 3A and Supplementary Figure 2A, C), suggesting that while ERG overexpression provides a potent survival signal, its function can not completely override apoptotic signal produced by continuous oncogenic AKT activity (5, 9, 11–13).
Assessment of cell viability was next assayed for the Gata1s knock-in cells infected with Migr1, myrAKT, ERG and myrAKT+ERG. Expression of myrAKT in the presence of GATA1s led to a significantly improved live cell count in which there is no significant difference between the myrAKT expressing cells and the myrAKT+ERG expressing cells, in contrast to what was observed in the wild-type background (Figure 3 A, B and Supplementary Figure 2 A, B) as well as a reduction in Annexin V positivity. Note that Gata1s knock-in cells could be cultured on average out to a minimum of 5 weeks and as long as 7 weeks (data not shown), due to their innate capacity for hyperproliferation during the fetal stage (10). Both the myrAKT and myrAKT+ERG samples on the Gata1s knock-in background were not significantly different over the time course, yet both were significantly different from ERG and Migr1 for both live cell counts and Annexin V positivity. The addition of ERG, the presence of GATA1s or the combination appears to provide a sufficient signal to overcome the apoptotic signaling from excessive AKT activity.
ERG expression sustains long-term growth in combination with myrAKT
We next evaluated the expression of c-Kit, CD41 and CD42 in double positive Migr1, myrAKT, ERG and myrAKT+ERG transduced wild-type and Gata1s knock-in fetal liver hematopoietic cells after long-term culturing. None of the samples showed consistent and/or detectable expression of CD42, suggesting that differentiated cells do not persist under these conditions (data not shown). For the Gata1s knock-in samples, myrAKT+ERG yielded the highest overall total percentage of CD41 positive c-Kit double positive cells by 4 weeks (Figure 4B). Notably, myrAKT+ERG also maintained the largest percentage of CD41 singly positive cells (loss of c-Kit and hence most differentiated population shown) during the time course (Figure 4 A, B). This similar phenomenon of enhanced maturation in the presence of myrAKT had been reported in another system with an increase in myeloid maturation (increase in CD11b) in cells doubly transduced with myrAKT and MLL-AF9 compared to control (9). This result suggests that myrAKT+ERG CD41 positive cells benefited from enhanced survival and/or proliferation and provides evidence of potent crosstalk between ERG and increased AKT activity in the presence of GATA1s. MyrAKT and ERG samples on the Gata1s knock-in background were not statistically different from each other for CD41 positive c-Kit double positive expression by week four (Figure 4 B,C), but were at week two (Figure 4 A,C). The results of long-term culturing suggest that there exists a population of cells within the Gata1s knock-in fetal liver hematopoietic progenitors that can cooperate with myrAKT signaling to promote megakaryopoiesis either through enhanced survival or proliferation. This result also suggests the mechanism is distinct from that of transcriptional enforcement of megakaryocytic potential, as administered by ERG(5, 12), in which the effects are seen within 72 hours time (Figure 1 C,D) and are persistent (Figure 4A–C). Together, these results suggest that expression of myrAKT+ERG in the Gata1s knock-in background imitates that of a dysregulated megakaryocytic progenitor.
Figure 4. Expression of c-Kit and CD41 during long-term culture of Gata1s knockin and wild-type Gata1 fetal liver derived hematopoietic progenitors display synergy between ERG and myrAKT co-expression for maintenance of pro-megakaryocyte phenotype.

(A–B) Total CD41 and c-Kit expression shown at 2 weeks (left) and 4 weeks (right) for GATA1s knock-in GFP+ mcherry+ fetal liver progenitors; average ± SEM shown. Upper left quadrant CD41 singly positive cells, middle quadrant CD41+c-Kit+ cells and bottom right quadrant singly positive c-Kit cells for both A and B. P-value ≤ 0.02 for myrAKT+ERG compared to all samples for CD41 singly positive cells in both A and B. (C) Average fold change (±SEM) at 2 and 4 weeks for the shown Gata1s knockin samples in comparison to Migr1; * p-value ≤ 0.03. (D) Average (±SEM) percentage of CD41 positive c-Kit positive population for wild-type GATA1 samples of the indicated sample type at 2 and 3 weeks of culture; * p-value ≤0.01.
The same flow cytometric analyses were performed on the samples on the wild-type GATA1 background and are shown in Figure 4D. Even though myrAKT samples experienced significant decrease in their cell survival, the cells that persist under the utilized growth conditions by week 3 of assessment are enriched for CD41 positive c-Kit double positive cells and statistically significant compared to the Migr1 control but not to either ERG or myrAKT+ERG. The results for ERG and myrAKT+ERG mirror those on the Gata1s knock-in background, with ERG’s dominance in perpetuating a megakaryocyte proliferative phenotype that is able to cooperate with constitutively active AKT regardless of GATA1 status.
Differential expression of phospho-AKT, phospho-c-Jun and FOXOs during long-term culture
As there is an active interest in identifying signaling pathways that contribute to normal and malignant hematopoiesis, especially development and maintenance of leukemia initiating cells (LIC), we monitored levels of phospho-AKT (control), phospho-c-Jun and the cellular distribution of FOXO1/FOXO3a in the various infected progenitor cells (8, 9, 14, 15). Gata1s knock-in fetal liver hematopoietic progenitors expressing Migr1, myrAKT, ERG, or myrAKT+ERG were cultured for up to 4 weeks in the presence of TPO, SCF, IL3, IL6 and serum. The distribution of total phospho-AKT signaling were analyzed by flow cytometry at 2 and 4 weeks of culture (Figure 5 A,B). As expected, samples with constitutively active AKT (i.e. myrAKT and myrAKT+ERG samples) showed consistently high levels of phosphorylation of AKT. Surprisingly, ERG alone showed increased levels of p-AKT at four weeks (Figure 5 A,B). This result suggests the dominant proliferative population of ERG expressing cells on the Gata1s knock-in background up-regulate the AKT pathway and slowly out-compete other cells in the population, suggesting why ERG is a potent oncogene in its own right and its expression is part of what has been recently defined as a leukemia stem cell signature (16).
Figure 5. Evolution of phospho-AKT and phospho-c-jun profile during long-term culturing of sorted Gata1s knock-in GFP, mcherry fetal liver derived hematopoietic samples.

(A) Bar graph of total phospho-AKT level as determined by intracellular flow cytometry at both two and four weeks of culturing under megakaryocytic conditions, N=3, average±SEM shown. (B) Representative flow cytometry histograms for phospho-AKT distribution at 2 (left) and 4 (right) weeks for indicated samples. Light blue histogram (far left) isotype control antibody.(C) Bar graph for total phospho-c-Jun as determined by intracellular flow cytometry, N=3, average±SEM shown. (D) Representative flow cytometry plot for phospho-c-jun at 2 (left) and 4 (right) weeks for indicated samples. Light blue histogram (far left) isotype control antibody. N=3 for all shown experiments.
In contrast to the increased levels of phospho-AKT seen in the myrAKT expressing cells, Migr1 and ERG alone expressing Gata1 mutant cells displayed increased phosphorylated c-Jun compared to both myrAKT expressing samples at 2 weeks (Figure 5 C,D). This difference suggests this pathway is activated early to provide critical survival signals. Both Migr1 and ERG expressing Gata1s cells continued to maintain their phospho-c-Jun activity over the time course. In contrast, the myrAKT expressing samples (myrAKT and myrAKT+ERG) up-regulated their phospho-c-Jun activity over time to match or exceed the other two samples: this is most easily seen by comparing the distribution of signaling and phospho-c-jun histogram overlays at 2 versus 4 weeks (Figure 5 C, D). Together, these results suggest that the dominant proliferative Gata1s population favors and selects for this survival mechanism, regardless of expression of myrAKT and/or ERG.
Loss of PTEN and loss of FOXO1/3/4 were recently shown to enhance megakaryopoiesis, particularly in the presence of Notch signaling (8). Therefore, we evaluated changes in FOXO1 and FOXO3a sub-cellular localization in GATA1s progenitors expressing combinations of myrAKT and ERG, as only un-phosphorylated FOXOs are present in the nucleus. In the Migr1 and ERG alone samples, we detected FOXO1 and FOXO3a predominantly in the cytoplasm at 4 weeks (Figure 6A, B), suggesting that the FOXOs are being rapidly phosphorylated and shuttled to the cytoplasm by the cytokine mediated signaling. In contrast, FOXO1 and FOXO3a expression was markedly reduced in the myrAKT and myrAKT+ERG expressing cells at 4 weeks (Figure 6A, B) suggesting that the persistent AKT activity leads not only to exclusion from the nucleus but to degradation of both FOXO proteins (17). Additionally, we observed obvious changes in FOXO3a phosphorylation in DS-AMKL cell lines upon treatment with the AKT inhibitor, MK2206, with loss of phosphorylation upon AKT inhibition (Figure 7).
Figure 6. Decreased FOXO1/3a expression mediated by myrAKT in cultured Gata1s knock-in GFP, mcherry fetal liver derived hematopoietic samples.

(A) Comparison of all samples at four weeks of culturing for FOXO1 (pink) expression as demonstrated by immunofluorescence; Blue nuclear stain (Hoechst 33342). Comparison of all samples (B) at four weeks of culturing for FOXO3a (pink) expression as demonstrated by immunofluorescence; Blue nuclear stain (Hoechst 33342); N=3 for all experiments. Depicted bar 20 micron for scale reference.
Figure 7. AMKL cell lines are susceptible to inhibition of AKT signaling by the allosteric AKT inhibitor MK2206.

(A) IC50 (green dotted line) determination for the CMK, CMY, CMS and CHRF cell lines via assessment of cell proliferation after 48 hour incubation with increasing doses of MK2206. The average value of the percentages of proliferating cells calculated compared to the DMSO control are shown; N=3. Individual IC50 values for the cell lines to right of graph in (A). (B) Flow cytometric analysis of BrdU incorporation for increasing doses of MK2206 in CMK (left DS-AMKL cell line) and CHRF (right non-DS-AMKL cell line) cell lines. N=3 with average and SEM shown. P-value ≤ 0.04 for changes in S phase, G1, and G2/M for all three doses of MK2206 in comparison to DMSO control. (C) Western blots for CMY (DS-AMKL cell line) and CMS (non-DS AMKL cell line) treated with DMSO (control) or 10 µM MK2206 and assessed for the indicated proteins.(D–E) Western blots for CMK (top) and CHRF (bottom) treated with increasing doses of MK2206 with DMSO shown for comparison for the same proteins as in D. (F) Phospho-flow analysis demonstrates a progressive decrease in phospho-S6 ribosomal protein after treatment with increasing doses of MK2206 in comparison to the DMSO control after 48 hours of treatment for CMK (left) and CHRF(right) N=3, representative flow plot shown.
Though it is unclear what role the cytoplasmic FOXOs are playing under these conditions in the Migr1 and ERG expressing cells, the simultaneous disappearance of both FOXOs correlate with the pro-megakaryocyte phenotype of myrAKT+ERG expressing cells at four weeks of culture, which are either CD41+c-Kit double positive or CD41 singly positive cells of a more mature phenotype and suggest that loss of FOXOs and enhanced AKT activity does not affect the function of ERG or GATA1s to enforce a proliferative megakaryocyte progenitor and may facilitate their ability to do so.
Inhibition of AKT signaling leads to increased cell death and decreased proliferation in both DS and non-DS AMKL cell lines
The CMK and CMY megakaryoblastic cell lines that were derived from patients with DS-AMKL harbor mutations in GATA1 and JAK3 (JAK3A572V) and are dependent upon IGF/IGFR signaling for survival (1, 2, 18). Since a previous study indicated that CMK and CMY cells are sensitive to rapamycin, we investigated whether the allosteric AKT inhibitor MK2206 would affect their survival or proliferation. We also assayed MK2206 activity against two non-DS AMKL cell lines, CMS and CHRF, as non-DS AMKL also harbors mutations in surface receptors and tyrosine kinases (e.g. JAK kinases, MPL, FLT3, KIT) that activate the PI3K/AKT/mTOR pathway (1, 2, 19). We discovered that MK2206 leads to a decrease in cell survival, mediated by both apoptotic and necrotic cell death in all four cell lines in a dose responsive manner as monitored at both 24 and 48 hour time points (Figure 7A and Supplementary Figure 3A). Live cell counts via trypan blue exclusion were dramatically and significantly reduced by 48 hours for all four lines with increasing concentrations of MK2206 and used for IC50 determinations of all four cell lines (Figure 7A). BrdU incorporation assays with both CMK (DS-AMKL cell line) and CHRF (non-DS AMKL cell line) in the presence of increasing concentrations of MK2206 at the 48 hour time point revealed a dose dependent statistically significant increase of cells in G1 and concomitant decreases in S and G2/M phase cells in comparison to the DMSO control (Figure 7B). By western blot and phospho-flow cytometric analysis in the multiple AMKL cell lines treated with MK2206, we confirmed that MK2206 induced a decrease in the degree of phosphorylated AKT (serine473) and decreased phosphorylation of multiple downstream targets/effectors, including p-FOXO3a, p-4EBP1, phospho-S6 ribosomal protein (Figure 7C–F). In contrast, expression of ERG, which is expressed at higher levels in both types of AMKL in comparison to AML in general (20), remains relatively unchanged and phosphorylation of c-Jun, a target of the JNK family of kinases and a component of AP-1, a known co-factor of Gata1, was also unchanged or reduced, respectively. Finally, synergy of MK2206 with cytarabine (Ara-C), a common therapeutic for AML, was also demonstrated with both the CMK and CHRF cell lines (Supplementary Figure 3B, C). These experiments demonstrate that inhibition of AKT signaling by MK2206 leads to defective proliferation and survival of both DS- and non-DS AMKL cell lines and suggest that PI3K/AKT inhibition may be a feasible strategy for treatment of both DS-AMKL and non-DS AMKL.
Discussion
Activating mutations in Flt3, Jak2, Jak3 and Mpl are associated with DS-AMKL(2, 18, 21). Other signaling networks, including the IGF pathway, have also been implicated in the pathogenic growth of megakaryocytes in DS-AMKL(1, 21). All of these signaling pathways are associated with activation of PI3K/AKT, ERK1/2 and STAT cascades. Here, we demonstrate that activated AKT collaborates with both ERG and GATA1s to promote expansion of fetal liver derived megakaryoblasts and that direct inhibition of AKT blocks the growth of DS-AMKL cells.
It has been suggested that there exists a specific population of hematopoietic progenitors that are uniquely susceptible to acquired GATA1 mutations in the background of trisomy 21 (1, 10, 22–24). We used in vitro fetal liver megakaryopoiesis assays to uncover an interaction between AKT and the chromosome 21 gene ERG. ERG expression enforces a megakaryocytic phenotype, which is exacerbated by the presence of both GATA1s and myrAKT while the combination of myAKT+ERG led to an increase in CD41 and significant decrease in CD42 expression. This suggests that there is immediate cooperation between the two factors and excessive AKT signaling to promote expansion of a megakaryocytic progenitor with poor differentiation. Furthermore, our results with the Gata1s knock-in mice indicate there is a small population of cells that can expand and perturb megakaryocytic differentiation when combined with myrAKT. This observation reaffirms the leukemogenic potential of GATA1s and confirms that the combination of ERG and altered signaling can alter megakaryopoiesis during fetal mediated megakaryopoiesis of Gata1 mutant cells.
Studies of MPL signaling have demonstrated that signaling proceeds through PI3K/AKT/mTOR (25–27) and that activation can vary between adult versus fetal megakaryopoiesis. It has also been recently published that loss of PTEN and dys-regulation of the PI3K/AKT pathway is developmentally regulated (28). We used fetal liver progenitors to study the sensitivity of this developmental stage to changes in megakaryopoiesis when constitutively active AKT is combined with the oncogene ERG. We were surprised to see that Gata1s mutant cells tolerated excessive AKT signaling. Multiple groups have reported that there is a specific population of GATA1s expressing cells that mediates a pro-megakaryocytic progenitor that is the target population for dysregulated pathogenic signaling and susceptible to oncogenic cooperation mediated through trisomy 21 constituents (1, 10, 22–24). Our results with the Gata1s knock-in background indicate there is a small population of cells present in the lineage depleted fetal liver population that, when combined with myrAKT, can expand and allow perpetuation of a megakaryocytic progenitor. This reaffirms the leukemogenic potential of GATA1s and provides further insights into why DS-AMKL is susceptible to pathogenic signaling pathways within in the fetal compartment, but not the bone marrow(1, 10).
The downstream effectors of PI3K/AKT/mTOR, such as FOXO proteins, have been implicated in normal myeloid and AML biology (8, 9, 17). Our study demonstrates that long-term cultured Gata1s knock-in myrAKT and myrAKT+ERG cells dramatically decrease their expression of FOXO1 and FOXO3a compared to both Migr1 and ERG controls. This is not surprising as part of the normal function of activated AKT is to inhibit FOXO family members (including FOXO1 and FOXO3a) by direct phosphorylation, which facilitates their export from the nucleus and ubiquitination/proteosome mediated degradation in the cytoplasm (17, 25). Nuclear FOXO localization has been seen with leukemia initiating cells harboring specific oncogenes (MLL fusions, BCR-ABL), while in contrast, myeloid progenitors (GMPs, cycling CD34- LSKs, CD34+ LSKs) display predominantly cytoplasmic localization of FOXOs (9, 14, 29). Here, we observed that the Gata1s mutant ERG and Migr1 cultures resemble GMP myeloid progenitors/CD34+LSKs/ cycling CD34-LSKs, whereas myrAKT and myrAKT+ERG cells exhibit a more extreme case, most similar to cycling/freshly isolated CD34+ LSKs, with cytoplasmic localization and degradation of FOXO3a (9, 14, 29, 30). In our study of long-term cultured Gata1s cells with myrAKT and myrAKT+ERG, the dominant population of propagating cells reinforced the degradation of FOXO1/FOXO3a, demonstrating specific clonal advantage for cells utilizing this pathway under conditions of our assay.
In our study the combination of GATA1s, ERG and myrAKT was insufficient to produce a self-renewing megakaryocytic progenitor, suggesting additional chromosome 21 constituents or pathogenic signaling pathways are needed to fully transform these cells. Recent work on the loss of PTEN revealed resistance to leukemia in neonatal hematopoietic cells underscoring the unique and specific temporal controls on signaling mechanisms between the adult and developing hematopoietic system of children, as over-expression of myrAKT in bone marrow transplanted from 6–8 week old mice leads to AML, T-ALL and myeloproliferative disease (7, 28, 31). Taken together, our results highlight the critical role of AKT in aberrant megakaryopoiesis and suggest that AKT inhibition may be a useful strategy for treatment of DS-AMKL.
Supplementary Material
Acknowledgements
The authors are grateful to Thomas Mercher for plasmids and Merck for supplying MK2206 and thank the Robert H. Lurie Comprehensive Cancer Center supported Flow Cytometry Core and Cell Imaging Cores at Northwestern University. Additional thanks to Benjamin Goldenson for assistance with CalcuSyn software calculations. This work was supported in part by the Samuel Waxman Cancer Research Foundation and a grant from the NCI (R01CA101774).
Footnotes
Conflict of Interest Statement The authors declare that they have no relevant conflicts of interest.
References
- 1.Klusmann JH, Godinho FJ, Heitmann K, Maroz A, Koch ML, Reinhardt D, et al. Developmental stage-specific interplay of GATA1 and IGF signaling in fetal megakaryopoiesis and leukemogenesis. Genes Dev. 2010;24(15):1659–1672. doi: 10.1101/gad.1903410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malinge S, Ragu C, Della-Valle V, Pisani D, Constantinescu SN, Perez C, et al. Activating mutations in human acute megakaryoblastic leukemia. Blood. 2008;112(10):4220–4226. doi: 10.1182/blood-2008-01-136366. [DOI] [PubMed] [Google Scholar]
- 3.Malinge S, Izraeli S, Crispino JD. Insights into the manifestations, outcomes, and mechanisms of leukemogenesis in Down syndrome. Blood. 2009;113(12):2619–2628. doi: 10.1182/blood-2008-11-163501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roy A, Roberts I, Norton A, Vyas P. Acute megakaryoblastic leukaemia (AMKL) and transient myeloproliferative disorder (TMD) in Down syndrome: a multi-step model of myeloid leukaemogenesis. Br J Haematol. 2009;147(1):3–12. doi: 10.1111/j.1365-2141.2009.07789.x. [DOI] [PubMed] [Google Scholar]
- 5.Stankiewicz MJ, Crispino JD. ETS2 and ERG promote megakaryopoiesis and synergize with alterations in GATA-1 to immortalize hematopoietic progenitor cells. Blood. 2009;113(14):3337–3347. doi: 10.1182/blood-2008-08-174813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muntean AG, Crispino JD. Differential requirements for the activation domain and FOG-interaction surface of GATA-1 in megakaryocyte gene expression and development. Blood. 2005;106(4):1223–1231. doi: 10.1182/blood-2005-02-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kharas MG, Okabe R, Ganis JJ, Gozo M, Khandan T, Paktinat M, et al. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood. 2010;115(7):1406–1415. doi: 10.1182/blood-2009-06-229443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cornejo MG, Mabialah V, Sykes SM, Khandan T, Lo Celso C, Lopez CK, et al. Crosstalk between NOTCH and AKT signaling during murine megakaryocyte lineage specification. Blood. 2011;118(5):1264–1273. doi: 10.1182/blood-2011-01-328567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sykes SM, Lane SW, Bullinger L, Kalaitzidis D, Yusuf R, Saez B, et al. AKT/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Cell. 2011;146(5):697–708. doi: 10.1016/j.cell.2011.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Z, Godinho FJ, Klusmann JH, Garriga-Canut M, Yu C, Orkin SH. Developmental stage-selective effect of somatically mutated leukemogenic transcription factor GATA1. Nat Genet. 2005;37(6):613–619. doi: 10.1038/ng1566. [DOI] [PubMed] [Google Scholar]
- 11.Ng AP, Hyland CD, Metcalf D, Carmichael CL, Loughran SJ, Di Rago L, et al. Trisomy of Erg is required for myeloproliferation in a mouse model of Down syndrome. Blood. 2010;115(19):3966–3969. doi: 10.1182/blood-2009-09-242107. [DOI] [PubMed] [Google Scholar]
- 12.Salek-Ardakani S, Smooha G, de Boer J, Sebire NJ, Morrow M, Rainis L, et al. ERG is a megakaryocytic oncogene. Cancer Res. 2009;69(11):4665–4673. doi: 10.1158/0008-5472.CAN-09-0075. [DOI] [PubMed] [Google Scholar]
- 13.Tsuzuki S, Taguchi O, Seto M. Promotion and maintenance of leukemia by ERG. Blood. 2011;117(14):3858–3868. doi: 10.1182/blood-2010-11-320515. [DOI] [PubMed] [Google Scholar]
- 14.Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, et al. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. 2010;463(7281):676–680. doi: 10.1038/nature08734. [DOI] [PubMed] [Google Scholar]
- 15.Chapuis N, Park S, Leotoing L, Tamburini J, Verdier F, Bardet V, et al. IkappaB kinase overcomes PI3K/Akt and ERK/MAPK to control FOXO3a activity in acute myeloid leukemia. Blood. 2010;116(20):4240–4250. doi: 10.1182/blood-2009-12-260711. [DOI] [PubMed] [Google Scholar]
- 16.Eppert K, Takenaka K, Lechman ER, Waldron L, Nilsson B, van Galen P, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17(9):1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 17.Tothova Z, Gilliland DG. FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell. 2007;1(2):140–152. doi: 10.1016/j.stem.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 18.Walters DK, Mercher T, Gu TL, O'Hare T, Tyner JW, Loriaux M, et al. Activating alleles of JAK3 in acute megakaryoblastic leukemia. Cancer Cell. 2006;10(1):65–75. doi: 10.1016/j.ccr.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 19.Gruber TA, Larson Gedman A, Zhang J, Koss CS, Marada S, Ta HQ, et al. An Inv(16)(p13.3q24.3)-Encoded CBFA2T3-GLIS2 Fusion Protein Defines an Aggressive Subtype of Pediatric Acute Megakaryoblastic Leukemia. Cancer Cell. 2012;22(5):683–697. doi: 10.1016/j.ccr.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, et al. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J Clin Invest. 2012;122(3):948–962. doi: 10.1172/JCI60455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan I, Malinge S, Crispino J. Myeloid leukemia in Down syndrome. Crit Rev Oncog. 2011;16(1–2):25–36. doi: 10.1615/critrevoncog.v16.i1-2.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou ST, Opalinska JB, Yao Y, Fernandes MA, Kalota A, Brooks JS, et al. Trisomy 21 enhances human fetal erythro-megakaryocytic development. Blood. 2008 doi: 10.1182/blood-2008-05-157859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izraeli S. Trisomy 21 tilts the balance. Blood. 2008;112(12):4361–4362. doi: 10.1182/blood-2008-09-176719. [DOI] [PubMed] [Google Scholar]
- 24.Mundschau G, Crispino J. GATA1s goes germline. Nat Genet. 2006;38(7):741–742. doi: 10.1038/ng0706-741. [DOI] [PubMed] [Google Scholar]
- 25.Martelli AM, Chiarini F, Evangelisti C, Grimaldi C, Ognibene A, Manzoli L, et al. The phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin signaling network and the control of normal myelopoiesis. Histol Histopathol. 2010;25(5):669–680. doi: 10.14670/HH-25.669. [DOI] [PubMed] [Google Scholar]
- 26.Raslova H, Baccini V, Loussaief L, Comba B, Larghero J, Debili N, et al. Mammalian target of rapamycin (mTOR) regulates both proliferation of megakaryocyte progenitors and late stages of megakaryocyte differentiation. Blood. 2006;107(6):2303–2310. doi: 10.1182/blood-2005-07-3005. [DOI] [PubMed] [Google Scholar]
- 27.Liu ZJ, Italiano J, Jr, Ferrer-Marin F, Gutti R, Bailey M, Poterjoy B, et al. Developmental differences in megakaryocytopoiesis are associated with up-regulated TPO signaling through mTOR and elevated GATA-1 levels in neonatal megakaryocytes. Blood. 2011;117(15):4106–4117. doi: 10.1182/blood-2010-07-293092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magee JA, Ikenoue T, Nakada D, Lee JY, Guan KL, Morrison SJ. Temporal Changes in PTEN and mTORC2 Regulation of Hematopoietic Stem Cell Self-Renewal and Leukemia Suppression. Cell Stem Cell. 2012;11(3):415–428. doi: 10.1016/j.stem.2012.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamazaki S, Iwama A, Takayanagi S, Morita Y, Eto K, Ema H, et al. Cytokine signals modulated via lipid rafts mimic niche signals and induce hibernation in hematopoietic stem cells. Embo J. 2006;25(15):3515–3523. doi: 10.1038/sj.emboj.7601236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miyamoto K, Araki KY, Naka K, Arai F, Takubo K, Yamazaki S, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1(1):101–112. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 31.Kentsis A, Look AT. Distinct and dynamic requirements for mTOR signaling in hematopoiesis and leukemogenesis. Cell Stem Cell. 2012;11(3):281–282. doi: 10.1016/j.stem.2012.08.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.