

Abstract
Purpose of Review
This article summarizes the pathologic features of multiple sclerosis (MS) and other inflammatory demyelinating diseases and discusses neuropathologic studies that have yielded novel insights into potential mechanisms of demyelination.
Recent Findings
The pathologic hallmark of MS consists of focal demyelinated plaques within the CNS, with variable degrees of inflammation, gliosis, and neurodegeneration. Active MS lesions show a profound pathologic heterogeneity with four major patterns of immunopathology, suggesting that the targets of injury and mechanisms of demyelination in MS may be different in different disease subgroups. Recent pathologic studies have suggested that the subarachnoid space and cortex may be initial sites and targets of the MS disease process, that inflammatory cortical demyelination is present early in MS, and that meningeal inflammation may drive cortical and white matter injury in some MS patients.
Summary
MS is heterogeneous with respect to clinical, genetic, radiographic, and pathologic features; surrogate MRI, clinical, genetic, serologic, and/or CSF markers for each of the four immunopatterns need to be developed in order to recognize them in the general nonbiopsied MS population. Inflammatory cortical demyelination is an important early event in the pathogenesis of MS and may be driven by meningeal inflammation. These observations stress the importance of developing imaging techniques able to capture early inflammatory cortical demyelination in order to better understand the disease pathogenesis and to determine the impact of potential disease-modifying therapies on the cortex.
INTRODUCTION
The pathologic hallmark of multiple sclerosis (MS) is multiple focal areas of myelin loss within the CNS called plaques or lesions (Figure 1-1A–C).1,2 Demyelination is accompanied by variable gliosis and inflammation and by relative axonal preservation (Figure 1-1D–I). Lesions are disseminated throughout the CNS but have a predilection for optic nerves, subpial spinal cord, brainstem, cerebellum, and juxtacortical and periventricular white matter regions.1,2 Although MS has historically been considered a disease primarily affecting the CNS white matter, recent pathologic and imaging studies have established that demyelinated lesions are also commonly found in the cortical gray matter of MS patients.3–6
Figure 1-1.
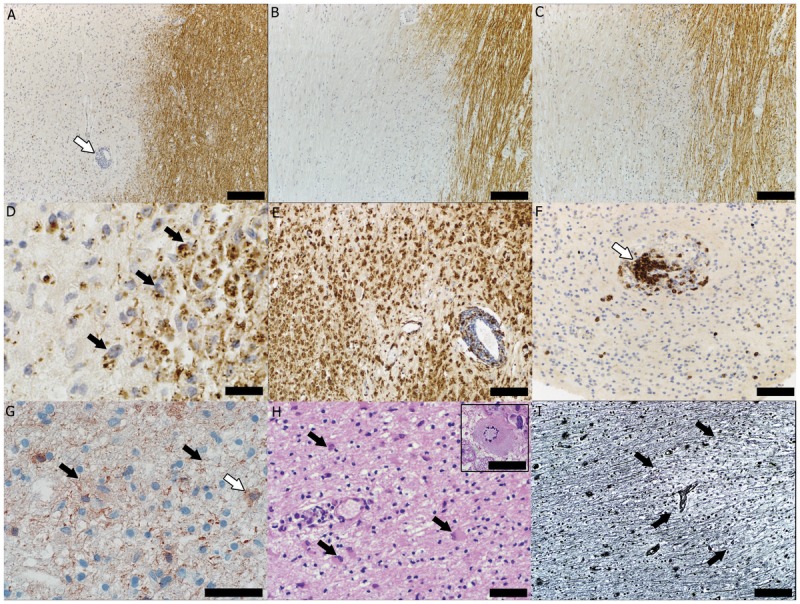
Immunopattern II multiple sclerosis lesion. A, Demyelination demonstrated as loss of immunohistochemical (IHC) staining. The myelin protein proteolipid protein (PLP) (arrow indicates a perivascular inflammatory infiltrate) (scale bar = 250 μm). B, The myelin protein myelin oligodendrocyte glycoprotein (scale bar = 250 μm). C, The myelin protein myelin-associated glycoprotein (scale bar = 250 μm). D, Active demyelination evidenced by the presence of myelin-laden macrophages (arrows) (PLP IHC, scale bar = 50 μm); E, Sea of macrophages (IHC for KiM1P, a panmacrophage marker, scale bar = 100 μm). F, T-cell perivascular inflammatory infiltrate (arrow) (IHC for cluster of differentiation 3, a marker common to all lymphocytes, scale bar = 50 μm). G, Complement activation on axons (black arrows) and phagocytosis of complement-opsonized myelin debris by macrophages (white arrow) (IHC for neoepitope on the complement component C9 of the terminal lytic complex, scale bar = 50 μm). H, Reactive astrocyte (arrows) and Creutzfeldt-Peters cells (reactive astrocytes with abundant cytoplasm and fragmented nuclear inclusions arranged in a circular pattern) (inset) (hematoxylin and eosin stain, scale bar = 100 μm, inset scale bar = 50 μm). I, Relative axonal preservation within the multiple sclerosis lesion (arrows indicate the lesion’s edge) (Bielschowsky stain, scale bar = 100 μm).
NEUROPATHOLOGY OF WHITE MATTER LESIONS
MS lesions evolve differently during early versus chronic disease phases, and within each phase, different plaque types and plaques in different stages of demyelinating activity are evident. Histologically, several basic processes drive the formation of plaques: inflammation, myelin breakdown, astrogliosis, oligodendrocyte injury, neurodegeneration and axonal loss, and remyelination. A combination of histologic and/or immunohistochemical stains can be used to visualize these processes and to neuropathologically diagnose inflammatory demyelinating lesions as consistent with MS: hematoxylin and eosin stain (demonstrates tissue and cell morphology), myelin stains (Luxol fast blue/periodic acid-Schiff, Luxol fast blue/hematoxylin/eosin, or immunohistochemistry for myelin proteins), macrophage-specific markers (immunohistochemistry for KiM1P or CD68), stains for axons (Bielschowsky silver impregnation or immunohistochemistry for neurofilament protein), stains for astrocytes (hematoxylin and eosin or immunohistochemistry for glial fibrillary acidic protein), and stains for the different lymphocyte subtypes (immunohistochemistry for CD3, CD4, CD8, CD20, and/or CD138).7
Acute Active Plaques
Acute active plaques are most frequent in acute and relapsing-remitting MS and represent the pathologic substrate of clinical attacks.1,8,9 Acute active MS lesions are hypercellular demyelinated plaques massively infiltrated by macrophages evenly distributed throughout the lesion forming the classic “sea of macrophages” (Figure 1-1E). These macrophages contain myelin debris, an indication that they have taken up and degraded the remnants of the destroyed myelin sheaths (ie, active demyelination) (Figure 1-1D). The progression of myelin fragment degradation by macrophages is reflected in different rates of their disappearance. Therefore, a stringent definition of demyelinating activity within a plaque can be obtained on the basis of the presence or absence of certain myelin degradation products within macrophages.10 Degradation of minor myelin proteins (2′,3’′-cyclic nucleotide 3’′-phosphodiesterase [CNPase], myelin oligodendrocyte glycoprotein, myelin-associated glycoprotein [MAG]) occurs rapidly, within 1 to 3 days, and the presence of minor myelin protein degradation products within macrophages denotes early active demyelination. The larger, more abundant and hydrophobic major myelin proteins (proteolipid protein, myelin basic protein) are digested more slowly and may persist in lesions for up to 10 days. Thus, the presence of major myelin protein but not minor myelin protein degradation products within macrophages indicates a late active lesion. Inactive lesions are infiltrated by macrophages that lack myelin debris, but may still contain empty vacuoles or periodic acid-Schiff–positive degradation products, the result of the macrophage’s inability to digest the myelin’s neutral lipid components that accumulate and persist in macrophages.
Perivascular and parenchymal inflammatory infiltrates are invariably present, suggesting that demyelination and axonal degeneration are inflammatory in nature (Figure 1-1A, Figure 1-1D–F).1,2,11,12 Besides activated macrophages/microglia, inflammatory infiltrates are composed of lymphocytes, the vast majority of which are CD8-positive cytotoxic T lymphocytes, and fewer CD4-positive helper T cells, B cells, and plasma cells. B cells and plasma cells tend to accumulate predominantly in the perivascular spaces.11 Gadolinium enhancement characterizes lesions with damaged blood-brain barrier (BBB), which enables infiltration of inflammatory cells into the CNS. The inflammation together with the demyelination and vasogenic edema present in early MS lesions are responsible for their pinkish-yellow, soft, and poorly demarcated appearance on fresh slices of brain and spinal cord.
Astrocytes in active lesions proliferate and become plump-shaped (gemistocytes) with homogeneous eosinophilic cytoplasm and numerous fibrillary processes (Figure 1-1H).1,2 Prominent uninucleated and multinucleated reactive astrocytes with eosinophilic hypertrophic cell bodies form a matrix in which the other cells are suspended. Mitotic astrocytes and astrocytes with fragmented nuclear inclusions (granular mitoses and Creutzfeldt-Peters cells) are also often found within early lesions (Figure 1-1H, inset). While these cells may be erroneously interpreted as a sign of malignancy, their evenly spaced pattern together with the presence of macrophages should rule out the diagnosis of astrocytic neoplasm.
Some MS neuropathologic studies suggest that oligodendrocytes are preferentially destroyed in early lesions.13 However, oligodendroglial injury is variable, with numerous oligodendrocytes present in some lesions, often displaying signs of concurrent early remyelination.10
On the basis of specific myelin protein loss, plaque extent and topography, oligodendrocyte destruction, presence or absence of remyelination, immunoglobulin deposition, and complement activation, early active white matter MS lesions show a profound pathologic heterogeneity, and Lucchinetti and colleagues have demonstrated that these lesions can be classified into four immunopatterns, suggesting that the targets of injury and mechanisms of demyelination in MS may differ between patients.9
Pattern I lesions, found in 15% of MS patients who have been biopsied, are sharply demarcated perivascular lesions characterized by active demyelination with equal loss of all myelin components, lack of immunoglobulin deposition, and lack of complement activation on a T lymphocyte and activated macrophage/microglia inflammatory background. The destruction of myelin in pattern I MS may therefore be mediated by toxic factors produced by activated macrophages. Loss of oligodendrocytes is variable at the active lesional border, but numerous oligodendrocytes reappear in the inactive plaque center, and there is a high incidence of remyelinated plaques.
Pattern II lesions, found in about 58% of MS biopsies, are sharply demarcated lesions characterized by active demyelination (Figure 1-1D) with equal loss of all myelin components (Figure 1-1A–C) associated with immunoglobulin and complement deposition on myelin, as well as phagocytosis of complement-opsonized myelin debris by macrophages (Figure 1-1G), on an inflammatory background (Figure 1-1E–F). Pattern II lesions also exhibit a variable loss of oligodendrocytes at the active border, with their reappearance in the inactive plaque center and a high incidence of remyelinated shadow plaques. These findings suggest that demyelination and tissue injury in pattern II MS lesions may be induced by antibody-mediated and complement-mediated mechanisms. Elevated immunoglobulins and activated complement are present in the CSF of MS patients, and serum and CSF from MS patients can cause demyelination in CNS organotypic cultures in vitro. However, the specific target of these complement-activating antibodies has not yet been identified.14 Myelin oligodendrocyte glycoprotein has been the prime candidate target antigen because it is localized on the extracellular surface of the myelin sheath and is accessible to humoral immune reactions. However, autoantibodies that recognize myelin oligodendrocyte glycoprotein in the process of demyelination have been detected only in a small subset of MS patients, consisting of children with atypical disease course.15 Moreover, by analogy with neuromyelitis optica (NMO),16 it is possible that antibodies in MS patients may recognize other nonmyelin targets,17 causing secondary demyelination.
Pattern III lesions, found in 26% of biopsied MS patients, are ill-defined lesions that show active demyelination with oligodendrocyte apoptosis and preferential loss of the periaxonal myelin components (MAG and CNPase) on an inflammatory background (Figure 1-2). Loss of oligodendrocytes is pronounced at the active plaque border and extends into the apparently normal periplaque white matter. The inactive center is devoid of oligodendrocytes, and remyelinated shadow plaques are absent. No evidence of immunoglobulin deposition and complement activation has been reported. The early alterations affecting the most distal site from the oligodendrocyte cell body suggest a dying-back oligodendrogliopathy. These changes have been observed in inflammatory demyelination induced by viruses, damage of oligodendrocytes induced by toxicity (ie, cuprizone), and white matter injury caused by acute ischemia and/or hypoxia; they may be driven by reactive oxygen and/or reactive nitrogen species, mitochondrial dysfunction, or increased expression of heme oxygenase 1, which contributes to mitochondrial iron accumulation.9,18
Figure 1-2.
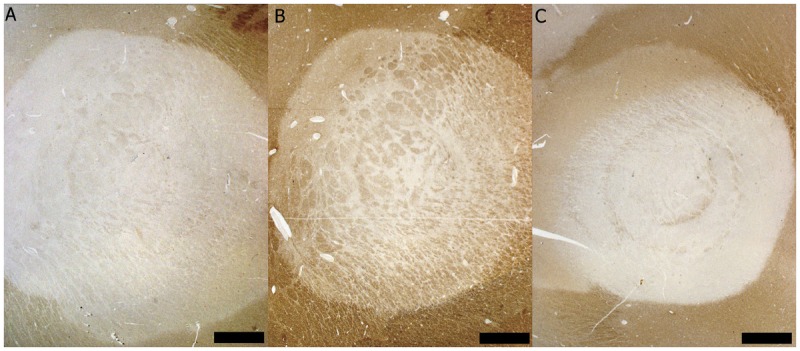
Immunopattern III multiple sclerosis/Baló concentric sclerosis. Demyelination is characterized by A, a preferential loss of the myelin protein myelin-associated glycoprotein (MAG) seen on MAG immunohistochemical (IHC) staining (scale bar = 1.5 mm), while other myelin proteins such as B, proteolipid protein (PLP) (PLP IHC, scale bar = 1.5 mm), and C, myelin oligodendrocyte glycoprotein (MOG) (MOG IHC, scale bar = 1.5 mm), are partially preserved.
Pattern IV lesions, found in only 1% of MS biopsies, are extremely rare and show a profound nonapoptotic death of oligodendrocytes in the periplaque white matter, suggesting a potential primary metabolic oligodendrocyte disturbance that renders them particularly vulnerable to the toxic action of inflammatory mediators.
A small neuropathologic series by Barnett and Prineas of patients with MS dying during or shortly after a relapse challenged the concept of pathologic heterogeneity.19 In these patients, the earliest pathologic changes described were prephagocytic lesions and consisted of widespread oligodendrocyte apoptosis on a background of microglial activation and myelin preservation. Such regions had few lymphocytes and phagocytes. These changes were followed by the disappearance of oligodendrocytes and the formation of intramyelinic edema. Finally, macrophages in the presence of inflammatory T cells phagocytosed the fragmented myelin. These macrophages were positive for markers of complement. The authors of this study interpreted this overlap of complement-containing macrophages and oligodendrocyte apoptosis within the same case as overlap between MS patterns II and III, suggesting that immunopathologic heterogeneity was dependent on the age of the lesions and not on the patient.19 However, recent neuropathologic findings have shown that pathologic features resembling MS pattern II and pattern III (ie, macrophages containing antibody or complement markers, and oligodendrocyte apoptosis with preferential loss of MAG, respectively) can be simultaneously found in a subset of active demyelinating NMO supraspinal lesions. These features strongly resemble the characteristics of the stage-dependent heterogeneity that Barnett and Prineas described in the prephagocytic/active demyelinating lesions and raise the possibility that the index case described by Barnett and Prineas was NMO rather than MS.20 This is further supported by the aggressive clinical course in their pediatric patient with relapse-related disability, including an episode of myelitis as well as severe brainstem dysfunction, leading to death within 1 year of onset.19
Demyelination with relative axonal sparing (Figure 1-1I) is important for the diagnosis of demyelinating lesions, whereas an infarct is more likely if axons and myelin in lesions are depleted to the same extent.1,2,7 Despite relative axonal sparing, axonal injury does occur, evidenced by the presence of axonal swellings (irregularly swollen axons with a beaded appearance), accumulation of amyloid-β precursor protein (a marker of focal accumulations of proteins that are typically moved along axons by fast axonal transport), and mild axonal loss.21 Axonal injury is most pronounced during active inflammatory demyelination. Acute axonal injury occurring in early MS lesions likely contributes to relapse-related disability observed predominantly during the inflammatory disease phases.1,8 The extent of axonal damage in active lesions correlates significantly with the number of lymphocytes and activated microglia. This damage is caused by the release of toxic inflammatory and nonspecific immune mediators in the lesion, such as proteases, cytokines, excitotoxins, and free radicals from the inflammatory cells seen in close apposition to injured axons, which in turn causes mitochondrial damage, increased oxidative stress, and energy deficiency.22,23
Chronic Plaques
Chronic plaques are more frequently seen than active plaques in patients with progressive MS. Chronic active plaques are sharply demarcated demyelinated lesions where numerous myelin-laden macrophages are concentrated at the centrifugally expanding edges of the plaque and diminish toward its hypocellular inactive center, while smoldering plaques may contribute to progression1,8,24 and are characterized by a slowly expanding rim of activated microglia (few of which contain myelin degradation products) surrounding their inactive center. Chronic inactive plaques are completely demyelinated, sharply circumscribed hypocellular lesions characterized by substantial loss of axons and oligodendrocytes, astrogliosis, and minor infiltration by macrophages/microglia and lymphocytes.1,2
Inflammation is invariably present in almost all lesion types and disease stages of MS. However, its severity decreases with the age of patients and disease duration, and it may even decline to levels seen in age-matched controls, but only in chronic inactive lesions of aged patients at the very late stage of the disease.11 Perivascular inflammatory infiltrates are often encountered in chronic lesions, but the BBB remains intact or its damage is too limited to be visible by gadolinium enhancement on MRI.11 Lymphoid follicular structures are formed in large perivascular spaces, and plasma cells accumulate in later disease stage and persist within the CNS even when T-cell and B-cell inflammation is cleared.11 This dissociation between inflammation and BBB disturbance may be explained by inflammation in chronic progressive MS becoming trapped within the CNS behind a closed or repaired BBB.
As the plaque progresses from acute active to chronic inactive, its edema resolves, inflammation decreases, and macrophages and microglia gradually disappear. Astrocytes produce glial fibers, and ultimately a glial scar fills the demyelinated plaque (astrocytic fibrillary gliosis). These characteristics prompted Charcot to name the disease “sclerose en plaque” and are also responsible for the distinctly demarcated, firm, retracted, and brownish discoloration of the long-standing chronic MS plaques on gross pathology.
Axonal damage and loss are also apparent in chronic MS, where axonal density is reduced up to 80% within the plaque.11,12 Neurodegeneration in all demyelinated lesions is invariably associated with inflammation, and in chronic inactive lesions from aged patients with long-standing progressive MS where the inflammatory process has died out, the neurodegeneration is also reduced to levels seen in non-MS control patients.11 Several mechanisms have been proposed to account for chronic axonal damage and neurodegeneration in MS: (1) repeated demyelination within previously remyelinated lesions; (2) axonal degeneration due to the lack of trophic support from myelin and oligodendrocytes; (3) chronic mitochondrial failure in the setting of increased energy demands; (4) oxidative burst by activated microglia; (5) alterations in the expression or activity of axonal ion channels; (6) oxidative stress caused by mitochondrial dysfunction, inflammation, or dysregulation of iron homeostasis; and (7) Wallerian degeneration.22,25,26
Remyelinated Plaques
Remyelinated plaques are characterized by the presence of thinly myelinated axons with short internodal distances. Extensive remyelination, indicated by the presence of newly formed myelin sheaths and oligodendrocyte precursor cells, is frequently encountered within active plaques of early MS and may represent an early phase of remyelination.1,2,10 Remyelinated lesions are present within active immunopattern I and II MS plaques: oligodendrocytes are lost in the expanding actively demyelinating regions, but oligodendrocyte precursor cells are recruited and reappear in the inactive plaque center. Immunopattern III and IV MS lesions show massive loss of oligodendrocytes, typically without signs of oligodendrocyte precursor cell recruitment and remyelination. The presence and extent of remyelination therefore depends in part on the availability of oligodendrocyte precursor cells and the pro- or anti-inflammatory balance and varies among patients, possibly because of pathologic heterogeneity of oligodendrocyte damage.9
Complete remyelinated lesions, so-called shadow plaques, are sharply demarcated areas with reduced myelin density and disproportionately thin myelin sheaths, and reflect a late phase of remyelination. Older remyelinated plaques show an almost normal thickness of myelin and are therefore difficult to distinguish from normal white matter. Shadow plaques are extensive in progressive MS cases as well as in patients with relapsing MS, as evidence for remyelination is seen in almost half of chronic MS lesions,27 and in some patients with MS almost all plaques are shadow plaques.28 However, remyelinated plaques are more susceptible to a second-hit inflammatory demyelinating attack than the normal-appearing white matter.29 The mechanisms of remyelination may progressively fail in patients with MS, and the failure to remyelinate axons may be due to an age-dependent loss of trophic support from microglia, oligodendrocyte precursor cell exhaustion by repeated demyelinating insults, inappropriate interactions between axons and oligodendrocytes, or the dense glial scar that may function as a barrier preventing the migration of oligodendrocyte precursor cells into lesions.
NEUROPATHOLOGY OF CORTICAL LESIONS
The cortex in MS may be involved either as neuronal loss and atrophy or as classical cortical demyelinated lesions. The former is the result of the anterograde or retrograde degeneration from lesions located in the white matter, deep gray nuclei, or other areas of the cortex.30,31 However, classical cortical demyelination occurs spatially and is anatomically independent of white matter or deep gray pathology.12 Although the existence of cortical lesions has been acknowledged for a long time, their study has been largely disregarded because of their poor visualization when using classical myelin histochemical staining methods.5 Immunohistochemistry for myelin proteins is superior to Luxol fast blue for detecting cortical demyelinating lesions.5 Three cortical lesion types have been described based on their location within the cortex: subpial lesions extend from the pial surface to cortical layer three or four, or to the entire width of the cortex, and may involve several gyri; intracortical lesions are small, perivascular demyelinated lesions confined within the cortex with the sparing of both superficial cortex and adjacent white matter; leukocortical lesions involve both gray and white matter at the gray matter–white matter junction with sparing of the superficial cortical layers. Conventional MRI techniques are relatively insensitive in detecting intracortical and subpial lesions. While recent imaging protocols using double inversion recovery and high-field MRI have substantially improved their in vivo detection, most cortical lesions are still not visualized by any MRI technique.4,6,8,32 Patients with early cortical involvement may have a worse prognosis, and the accumulation of cortical involvement is also linked with disease progression and disability. Because extensive cortical involvement may be associated with more rapid progression, there is a pressing need to develop imaging techniques able to reliably capture this pathology, both to better understand the disease pathogenesis and to determine the potential impact of disease-modifying therapies on the cortex.
Cortical Demyelinated Lesions in Early Multiple Sclerosis
Cortical demyelinated lesions in early MS are common and may represent the pathologic substrate of cognitive impairment and epilepsy in relapsing-remitting MS. Minimal cortical pathology has been associated with a benign MS course, and the cortical lesion load correlates positively with clinical disability, white matter T2 lesion load, and brain atrophy.3,4,33
The majority of all cortical lesion types in early MS show evidence of active demyelination as illustrated by the presence of myelin-laden macrophages (Figure 1-3A, B).34,35 Perivascular and parenchymal inflammatory infiltrates are invariably present in early cortical lesions and are composed mainly of macrophages, T cells, and fewer B cells and plasma cells (Figure 1-3C, D).34,35 Breakdown of BBB is also present (Figure 1-4A).34,35 Inflammatory cells can be observed in close apposition to neurons and neurites in early cortical lesions34,35; the observed oligodendrocyte, axonal, and neuronal injury may be the result of the acute inflammatory damage (Figure 1-3E, F). Thus, histopathologic evidence of inflammatory cortical demyelination in early MS suggests that neuronal and axonal injury in early cortical demyelination typically occurs on a background of inflammation.
Figure 1-3.
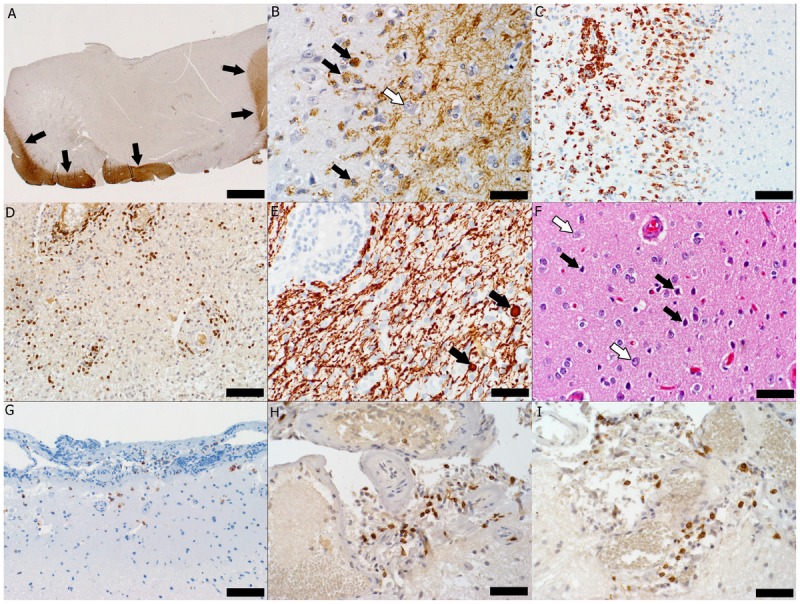
Pathology of multiple sclerosis cortical onset. A, Subpial cortical lesion visualized as the absence of proteolipid protein (PLP) immunohistochemical (IHC) staining; arrows indicate the border between the cortical lesion and the normally myelinated gray matter and white matter (PLP IHC, scale bar = 500 μm). B, Active demyelination (black arrows indicate myelin-laden macrophages) in the cortical lesion (white arrow indicates a neuron) (PLP IHC, scale bar = 50 μm). C, Macrophage infiltration (IHC for cluster of differentiation [CD] 68, a macrophage marker, scale bar = 100 μm). D, Perivascular and parenchymal infiltration with T cells visible as CD3-positive brown-stained cells (CD3 IHC, scale bar = 100 μm). E, Axonal injury evidenced by the presence of axonal swellings (arrows) in a lesional area with relative axonal preservation (IHC for neurofilament protein, scale bar = 50 μm). F, Injured neurons (black arrows) scattered among normal-appearing neurons (white arrows) (hematoxylin and eosin, scale bar = 50 μm). G, Macrophages/microglia (brown-stained cells) present in the upper cortex and meninges (CD68 IHC, scale bar = 100 μm). H, Meningeal inflammation with T lymphocytes (brown-stained cells) (CD3 IHC, scale bar = 50 μm). I, Most T cells are cytotoxic T cells as evidenced by their brown CD8 immunohistoreactivity (IHC for CD8, a marker of cytotoxic lymphocytes, scale bar = 50 μm).
Figure 1-4.
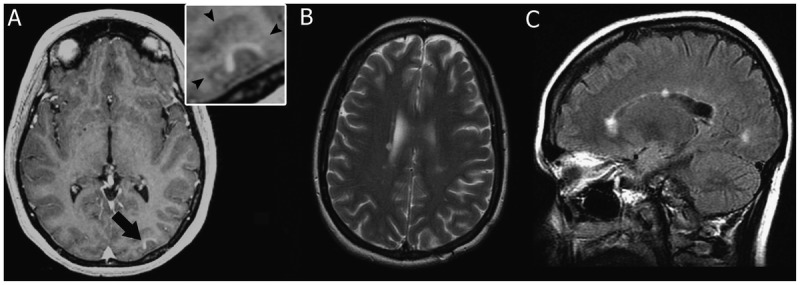
MRI of multiple sclerosis cortical onset. A, Prebiopsy axial T1-weighted image with contrast showing enhancement of the cortical lesion (arrow). Inset shows that contrast enhancement is within the cortical gray matter (arrowheads indicate the gray/white matter junction). B, Axial T2-weighted image 69 months after biopsy showing the appearance of new periventricular white matter lesions. C, Sagittal fluid-attenuated inversion recovery (FLAIR) image 69 months after biopsy showing the appearance of multiple new white matter lesions, with two of the lesions involving the corpus callosum.
Some patients with MS may present with cortical lesions as their earliest pathologic event. Therefore, inflammatory demyelinating disease should be considered in the differential diagnosis of patients presenting on MRI with a solitary cortical enhancing lesion.35,36 Case 1-1 is an example of inflammatory cortical-onset MS.35
Pathologic studies have established that meningeal inflammation is prominent in early MS (Figure 1-3G–I).34 Both focal perivascular meningeal inflammation and diffuse meningeal inflammation are topographically and strongly associated with cortical lesions in early MS.34 The presence of diffuse meningeal inflammation increases the odds of cortical demyelination 45-fold, whereas perivascular meningeal inflammation increases the odds of cortical demyelination 15-fold.34 Experimental autoimmune encephalomyelitis models have emphasized the importance of pathogenic T-lymphocyte trafficking into the CSF in the subarachnoid space, where they are restimulated by meningeal antigen-presenting cells, undergo clonal expansion, and produce cytokines.37 These events promote a second wave of T-cell infiltration across pia vessels and an upregulation of vascular adhesion molecules in the deeper brain vasculature, followed by parenchymal invasion and disease onset.38 These observations support the hypothesis that production and release of inflammatory cytokines in the subarachnoid space in early MS may drive cortical demyelination and promote subsequent inflammation and demyelination of the deeper subcortical white matter.34
Case 1-1
A 33-year-old white woman presented with sudden-onset headache associated with right periorbital pain. Her medical history was notable only for a history of migraine, a vaginal delivery 4 months ago, and an upper respiratory tract infection 1 week ago. The neurologic examination was normal. Brain MRI revealed a high signal intensity lesion on T2-weighted imaging involving the left occipital cortex (visual association cortex) that enhanced with gadolinium (Figure 1-4A). No other cortical or white matter lesions were noted.
An open brain biopsy performed 1 month after symptom onset to exclude neoplasm revealed a subpial cortical demyelinated lesion (Figure 1-3A) heavily infiltrated by phagocytic macrophages involved in active demyelination (Figure 1-3B). Parenchymal and perivascular cortical inflammatory infiltrates were present and composed of macrophages, microglia, CD3-positive and CD8-positive T cells, B lymphocytes, and plasma cells (Figure 1-3C, D). Acute axonal injury was evidenced by axonal swellings (Figure 1-3E), and neuronal injury was evidenced by the presence of scattered pyknotic neurons (Figure 1-3F). Macrophages, microglia, and T lymphocytes were found in close apposition to neurons as well as concentrated subpially in the cortical molecular layer and in the subarachnoid space (Figure 1-3G). Mild meningeal inflammation consisting of CD3-positive T lymphocytes, CD8-positive T and B lymphocytes, and plasma cells was present (Figure 1-3H, I).
Because of the patient’s persistent headaches, a CSF analysis was done 4 months after biopsy and demonstrated an elevated CSF IgG synthesis rate with normal protein, glucose, and cell count and one oligoclonal band. Lyme serology was negative. Six months after biopsy, the patient complained of increasing left-sided chest numbness, and spine MRI showed two subtle T2-weighted lesions at C4-5 and C6 levels. Eleven months after biopsy, she developed burning dysesthesias in the lower extremities. Brain MRI revealed new periventricular T2-weighted white matter lesions. She fulfilled diagnostic criteria for multiple sclerosis (MS) with a relapsing-remitting disease course. At the time of last follow-up (69 months after biopsy), brain MRI demonstrated an interval increase in the number of white matter lesions typical of MS (Figure 1-4B, C), and she was started on glatiramer acetate after referral to the authors’ clinic.
Comment. The first lesion detected in this case was a cortical lesion with pathologic features typical for acute inflammatory demyelinating lesions of MS.35 Subpial demyelinated lesions, as demonstrated in this case, may occur early in MS.34 Gadolinium enhancement of the newly forming cortical lesion is an uncommon observation; however, it was observed in this case, which highlights that acute cortical lesion formation is characterized by similar, albeit more transient MRI features compared to white matter lesions in MS. Possible reasons for the paucity of this phenomenon include the lack of symptoms associated with lesion formation in small focal subpial cortical areas (therefore no MRI is acquired during this stage), limited blood-brain barrier breakdown, and more rapid resolution of inflammation in the cortex compared to the white matter.
Cortical Demyelinated Lesions in Chronic Multiple Sclerosis
Cortical demyelinated lesions in chronic MS may be the pathologic substrate of irreversible disability, progression, and cognitive deficits,39 since they are prominent and extensive in patients with progressive MS and cognitive impairment. In progressive MS, extensive cortical demyelination has been detected in the frontal, temporal, insular, and cerebellar cortices in addition to the cingulate gyrus and hippocampus.39
Pathologically, in chronic progressive MS, cortical lesions lack breakdown of the BBB, inflammatory infiltrates, and complement deposition.5 Most phagocytes in cortical lesions are microglia located in close apposition to neurons and neurites,5 with atrophy and apoptosis of neurons; oligodendrocytes are also damaged.5,40
Profound meningeal inflammatory infiltrates have been described and extensively characterized in late-stage progressive MS. They are composed of T cells, B cells, and macrophages; are topographically associated with subpial lesions; and are found in patients with both primary and secondary progressive MS.41–43 The extent of meningeal inflammation correlates with microglia activation and the extent of demyelination and neurodegeneration in the underlying cortex. Patients with extensive meningeal inflammation show a more severe clinical course, shorter disease duration, and younger age at death. Meningeal inflammatory infiltrates resembling ectopic B-cell follicular structures have been described in secondary progressive MS. They are located in the deep sulci of the temporal, cingulate, insular, and frontal cortices of secondary progressive MS patients with accelerated clinical course and topographically associated with subpial lesions.43 Such lymphoid follicle-like structures have not been identified in primary progressive MS.42 Therefore, meningeal inflammatory aggregates may drive cortical injury, and soluble factors produced by meningeal activated lymphocytes may diffuse into the cortical tissue, causing demyelination and neurodegeneration either directly or indirectly through microglia activation.
NEUROPATHOLOGY OF OTHER INFLAMMATORY DEMYELINATING DISEASES
Baló Concentric Sclerosis
Baló concentric sclerosis is an MS variant with large concentric lesions characterized by circumferential rings of myelin loss alternating with rings of myelin preservation (Figure 1-2).1,2,44 Due to this spectacular concentric arrangement, Baló MS lesions can be occasionally recognized on MRI and even on fresh pathologic specimens. Concentric Baló-like alternating rims of demyelinated and myelinated tissue are found at the periphery of pattern III MS lesions. Active Baló lesions are associated with preferential loss of MAG9,45 and oligodendrocyte apoptosis, suggesting that the oligodendrocyte apoptosis in the demyelinated rings is the main pathogenetic insult.45,46 Baló concentric sclerosis is now believed to be a variant of pattern III MS, although other mechanisms of demyelination have been hypothesized.1,2
Neuromyelitis Optica
NMO is an autoimmune astrocytopathy that causes secondary demyelination (Figure 1-5B–D). The water channel aquaporin-4 (AQP4), expressed on astrocytes and highly concentrated in the astrocytic foot processes that abut capillaries and pia in the CNS, has been identified as the target antigen in NMO. AQP4 exists as two different heterotetramers (M-1 and M-23 AQP4), that further assemble in various ratios into higher ordered structures named orthogonal array of particles. Clinical, serologic, CSF, neuroimaging, and neuropathologic criteria that distinguish NMO from MS are now available.16,47–52 Case 1-2 is an example of NMO. The neuropathologic characteristic of all NMO lesions, irrespective of the stage of demyelinating activity, is the striking loss of AQP4 that extends beyond the demyelinated area (Figure 1-5E, J). This characteristic is unlike MS, in which AQP4 loss is stage dependent: MS active lesions show increased AQP4 expression on astrocytes within the lesions (Figure 1-5L), while AQP4 is lost in MS lesions that are no longer active.50 Other important neuropathologic features of NMO lesions include a vasculocentric pattern of immunoglobulin deposition and complement activation in active NMO lesions; decrease or complete loss of glial fibrillary acidic protein (GFAP) immunoreactivity in parallel to the loss of AQP4 (Figure 1-5F) in destructive NMO lesions; the presence of perivascular and parenchymal inflammatory infiltrates that contain eosinophils and neutrophils in demyelinated and nondemyelinated areas; axonal loss, necrosis, and cavitation in destructive regions (Figure 1-5A); blood vessels with thickened and hyalinized walls (Figure 1-5I); and intramyelinic edema.48,50,56 The affinity of NMO-IgG binding to AQP4 is influenced by the ratio M1:M23.53,57,58 Furthermore, the binding of NMO-IgG to AQP4 has isoform-specific outcomes: M1 is completely internalized, whereas M23 resists internalization and is aggregated into larger-order orthogonal arrays of particles that activate complement more effectively than M1 when bound by NMO-IgG.56 Therefore, differences in the nature and anatomical distribution of NMO lesions and in the clinical and imaging manifestations of NMO may be influenced by regional and maturational differences in the ratio of the two AQP4 isoforms (M1 and M23) in the astrocytic membranes. A subset of active demyelinating supraspinal NMO brain lesions simultaneously show pathologic features resembling MS pattern II (antibody or complement deposition in macrophages) and pattern III (preferential MAG loss and apoptosis of oligodendrocytes).20 Misclassification of these cases as MS rather than NMO may erroneously support the concept of intraindividual heterogeneity in MS.20
Figure 1-5.
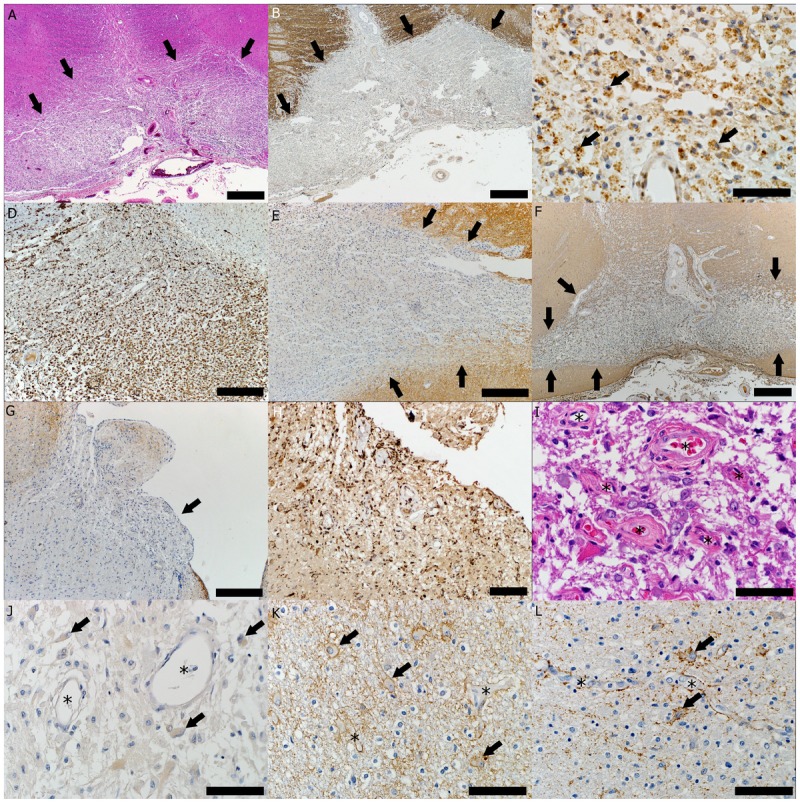
Pathology of a neuromyelitis optica (NMO) lesion located in the ventral medulla. A, Destructive hypercellular lesion; arrows indicate the border between the lesion that shows atypical hematoxylin and eosin (HE) staining (area below arrows) and normal-appearing white matter with preserved HE staining (area above arrows) (HE, scale bar = 500 μm). B, Demyelinated lesion; arrows indicate the border between lesion that shows loss of proteolipid protein (PLP) immunohistoreactivity (area below arrows) and normal-appearing white matter with preserved PLP immunohistoreactivity (area above arrows) (PLP immunohistochemical [IHC] staining, scale bar = 500 μm). C, Active demyelination (arrows indicate myelin-laden macrophages) (PLP, scale bar = 50 μm). D, The lesion shows macrophage infiltration (KiM1P IHC, scale bar = 250 μm). E, Loss of aquaporin-4 (AQP4) is characteristic for active NMO lesions; arrows indicate the border between lesion that shows loss of AQP4 immunohistoreactivity (area below arrows) and increased AQP4 immunohistoreactivity seen at the lesion’s edge (area above arrows) (AQP4 IHC, scale bar = 250 μm). F, Loss of glial fibrillary acidic protein (GFAP) is also frequently encountered in NMO-active demyelinating lesions; arrows indicate the border between the lesion that shows loss of GFAP immunohistoreactivity (area below arrows) and normal GFAP immunohistoreactivity of the normal-appearing white matter surrounding the lesion (area above arrows) (GFAP IHC, scale bar = 500 μm). G, Loss of aquaporin in the area postrema (arrow) (AQP4 IHC, scale bar = 250 μm). H, Macrophage/microglia infiltration in the area postrema (KiM1P IHC, scale bar = 100 μm). I, Area postrema blood vessels (asterisks) with thickened and hyalinized walls (HE; scale bar = 50 μm). J, Active NMO lesions with loss of AQP4 immunoreactivity seen as the disappearance of the perivascular rim or rosette AQP4 pattern (asterisks) and loss of AQP4 outlining of the astrocytic surface (arrows) (AQP4 IHC; scale bar = 50 μm). K, Increased AQP4 immunoreactivity outlining the cytoplasmic surface of reactive astrocytes (arrows) with preservation of the typical perivascular distribution (asterisks) at the border of the NMO lesion (AQP4 IHC; scale bar = 50 μm). L, Increased AQP4 immunoreactivity outlining the cytoplasmic surface of reactive astrocytes (arrows) with preservation of the typical perivascular distribution (asterisks) within an active multiple sclerosis lesion (AQP4 IHC; scale bar = 50 μm).
Inflammatory but nondestructive and nondemyelinating lesions have also been described in NMO. These lesions have been observed in the area postrema, which lacks a BBB, suggesting that this and other circumventricular organs could be sites of initial NMO-IgG access to the CNS (Figure 1-5G–I). Area postrema lesions in NMO are believed to be the pathologic substrate for the intractable but reversible nausea and vomiting that sometimes precede episodes of optic neuritis and transverse myelitis; they may even represent the initial heralding NMO symptom.53,54
While the normal human cortex is rich in AQP4, in NMO, the cortical distribution of AQP4 is preserved with no pathologic or MRI evidence of cortical demyelinated lesions.59–61 The absence of cortical demyelination, which is another neuropathologic feature distinguishing NMO from MS, may explain the typical absence of a secondary progressive clinical course in NMO.59,62 Regional differences in the permeability of the BBB or differential expression of the two AQP4 isoforms may be responsible for the absence of cortical lesions in NMO.
Case 1-2
A 39-year-old woman of African, Hispanic, and Asian ancestry developed epigastric pain and right foot numbness, followed by similar symptoms in the left foot a week later. Over the following 2 weeks she developed lower extremity upper motor neuron weakness and frequent tonic flexor spasms. She spontaneously improved from this event.
Her second relapse 4 months later included left-sided weakness, diplopia, and spasticity. She had a left Babinski sign and bilateral lower extremity hyperreflexia with loss of pinprick sensation up to the right hip and left knee. She also experienced constipation and recurrent tonic flexor spasms. Carbamazepine and 5 days of IV methylprednisolone were administered without improvement. MRIs showed a longitudinally extensive T2 lesion in the cervical cord and medulla and no hemispheric abnormalities. CSF examination revealed 10 nucleated cells per μL.
Her third relapse–less than a month after the second–included left upper extremity pain and dystonic movements in the context of high fever, as well as urinary incontinence, leading to urinary retention and quadriplegia. Repeat brain MRI showed periventricular and corpus callosum lesions, along with brainstem lesions (Figure 1-6A, B). Spinal cord MRI showed lesions throughout the cervical and thoracic cord. The patient failed to respond to repeat IV methylprednisolone treatment. Worsening quadriplegia and diplopia with new-onset dysphagia was documented 2 years after her initial presentation in the context of urosepsis. Neuromyelitis optica (NMO)–IgG screening was positive. MRI revealed brain and spinal cord atrophy with gadolinium enhancement in the medulla (Figure 1-6C, D).
Figure 1-6.
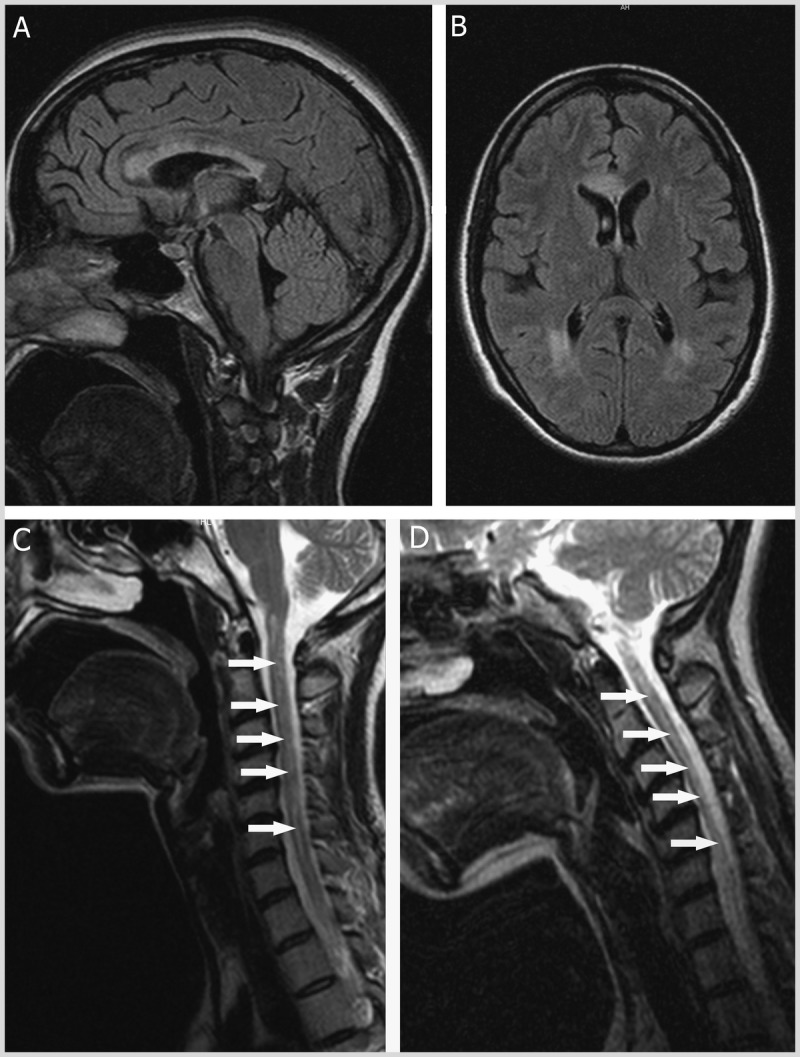
MRI of neuromyelitis optica. A, Sagittal, and B, axial fluid-attenuated inversion recovery (FLAIR) images demonstrating heterogeneous T2-hyperintense corpus callosum and periventricular lesions with poorly defined margins. C, 2 months later, and, D, 17 months later, sagittal T2-weighted images of the cervical spinal cord and posterior fossa. Note the longitudinally extensive central T2-hyperintense lesion of the spinal cord on both images (arrows). Spinal cord atrophy is appreciated as a more narrow appearance of the cord in panel D.
Six months later she experienced progressive worsening of all neurologic symptoms, new-onset intractable hiccupping, and an active urinary tract infection. Within the following year she required a feeding tube. She had recurrent nausea and vomiting episodes. Progressively worsening dyspnea resulted in her death 1 month later.
Autopsy revealed the presence of hypercellular demyelinating lesions (Figure 1-5A, B) infiltrated by macrophages/microglia involved in active demyelination (Figure 1-5C, D), as well as lymphocytes and plasma cells. Lesional blood vessels had thickened and hyalinized walls (Figure 1-5I). These active lesions exhibited aquaporin-4 (AQP4) and glial fibrillary acidic protein (GFAP) loss consistent neuropathologically with NMO (Figure 1-5E, F, J). Inflammatory, nondestructive nondemyelinating lesions with loss of AQP4 were noted in the area postrema (Figure 1-5G–I).
Comment. The pathologic diagnosis is strongly consistent with NMO in this case, highlighting several key features of the disease, including the presence of nondemyelinated as well as destructive, demyelinating lesions, associated with AQP4 and GFAP loss.48–50,53 The original spinal cord MRI with a longitudinally extensive T2-hyperintense lesion and a “swollen cord” appearance is typical for NMO.52 Intractable hiccups, nausea, and vomiting are common features of NMO, likely related to area postrema involvement.53–55 While a progressive course as demonstrated in this case is uncommon in NMO, and frequent severe attacks are more typical, the presence of a progressive stage does not exclude this diagnosis.52 NMO-IgG is a highly specific biomarker of NMO; however, in up to 30% of cases this serum test may be negative.16,47 Respiratory involvement leading to death is more common in fulminant NMO cases compared to MS.
This patient had not been treated at the authors’ institution, and her treatment predated current discoveries related to NMO. Aggressive treatment of acute relapses and robust maintenance therapies are now available and should be utilized in severe NMO cases.
Acute Disseminated Encephalomyelitis
Acute disseminated encephalomyelitis (ADEM) lesions are often bilateral, affecting the cerebral white matter, brainstem, cerebellum, and spinal cord. ADEM may also involve the peripheral ganglia and roots of the spinal and cranial nerves. Case 1-3 is an example of ADEM. Pathologically, ADEM consists of “sleeves” of demyelination centered on small, engorged venules. Significant inflammatory infiltrates are present and consist of myelin-laden macrophages, variable T and B lymphocytes, and occasional plasma cells and granulocytes (Figure 1-7A–D).2,7,63,64 Despite relative axonal preservation, axonal injury may be present. Many perivenous demyelinating lesions may coalescence to form larger areas of demyelination, but the MS-characteristic joint areas of demyelination, macrophage infiltration, and reactive astrocytes are not typically seen in ADEM, although transitional cases have been described.
Figure 1-7.
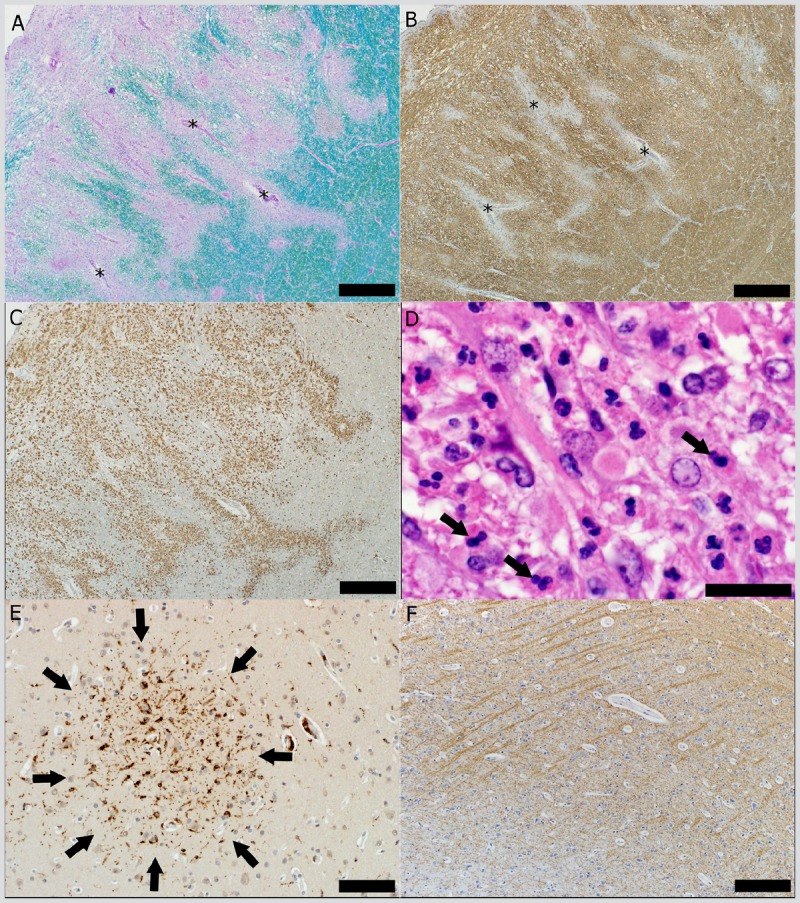
Pathology of acute disseminated encephalomyelitis. A, Sleeves of perivascular demyelination (Luxol fast blue/periodic acid–Schiff staining) seen as the disappearance of the normal blue staining of the myelin conferred by the Luxol fast blue (several blood vessels with perivascular demyelination are marked with asterisks) (scale bar = 500 μm). B, Sleeves of perivascular demyelination (asterisks mark several blood vessels with perivascular loss of proteolipid protein [PLP]) (PLP, scale bar = 500 μm). C, Infiltration with macrophages (KiM1P, scale bar = 500 μm). D, Perivascular inflammatory infiltrate with neutrophils (arrows) (hematoxylin and eosin, scale bar = 50 μm); E, Cortical microglial aggregate (arrows) (KiM1P, scale bar = 100 μm). F, The cortex in the region of the microglial aggregate does not show cortical demyelination (PLP, scale bar = 500 μm).
Cortical pathology evidenced by the presence of subpial and intracortical demyelinated lesions with mild lymphocytic meningitis is frequent in ADEM.63 Cortical microglial aggregates that are not associated with cortical demyelinated lesions may be found dispersed throughout the cortex (Figure 1-7E, F).63 This pattern-of multifocal cortical microglial aggregates scattered throughout the cortex, not associated with cortical demyelination-is unique to ADEM and may reflect the pathologic substrate of the depressed level of consciousness seen in this disorder.63
Case 1-3
A 24-year-old white woman with a 1-year history of IV drug abuse developed intermittent headache with occasional blurry vision. One month later she experienced right fronto-occipital headache with right eye blurred vision and right arm and left leg paresthesias in the context of an upper respiratory infection with right ear pain, sore throat, cough, fever, and chills. Three days later she noted severe photophobia with worsening neurologic symptoms, leading to hospitalization. Examination was notable for Kernig sign, right Babinski sign, meningismus, right visual field cut, an unsteady gait, and a left pupillary dilation. Brain CT was normal. Lumbar puncture revealed a protein level of 86 mg/dL, glucose level of 54 mg/dL, and 37 nucleated cells per μL, with 97% lymphocytes. Bacterial, viral, and fungal CSF cultures were negative, as were HIV, monospot, Venereal Disease Research Laboratory (VDRL), and purified protein derivative (PPD) tests. Ophthalmologic evaluation, cerebral angiogram, and nerve conduction studies were unrevealing. EEG showed diffuse generalized slowing. Serology studies revealed IgM seropositivity for Legionella and Mycoplasma.
Over the course of 3 days, the patient became quadriparetic with multiple brainstem/cranial nerve signs and worsening mental status, eventually becoming completely unresponsive and requiring mechanical ventilation. MRI showed extensive signal abnormalities in the brainstem and medulla. The patient remained comatose and continued to decline despite high-dose IV methylprednisolone treatment and supportive care in the intensive care unit. She died after support was withdrawn.
Autopsy revealed the presence of “sleeves” of demyelination surrounding small, engorged venules (Figure 1-7A, B), associated with significant inflammatory infiltrates dominated by macrophages, T and B lymphocytes, and granulocytes (Figure 1-7C, D). Findings were consistent with the neuropathologic diagnosis of acute disseminated encephalomyelitis (ADEM). Multifocal cortical microglial aggregates, not associated with cortical demyelination, were scattered throughout the cortex (Figure 1-7E, F).
Comment. A monophasic course over several weeks with signs and symptoms of systemic infections are characteristic for fulminant ADEM, as highlighted in this case.2,65 Headache, meningismus, and encephalopathy are uncommon in multiple sclerosis (MS) but may accompany ADEM, and overall a polysymptomatic onset is also more typical of ADEM.2,65 While ADEM is more common among pediatric populations, adult cases do occur.2,65 When tissue is available, ADEM can be diagnosed neuropathologically; its most typical features include perivenular sheets of demyelination with variable inflammatory infiltrates consisting of macrophages, lymphocytes, and granulocytes.2,7,64 This is in contrast with MS, where confluent demyelination with sheets of macrophage infiltration admixed with reactive astrocytes are more typical. In addition, a distinct pattern of cortical microglial activation and aggregation without demyelination may be observed in ADEM, especially in cases with depressed levels of consciousness.63
Acute Hemorrhagic Leukoencephalitis
Acute hemorrhagic leukoencephalitis, or Hurst disease, is considered a hyperacute variant of ADEM. It consists histologically of hemorrhagic demyelinating lesions around blood vessels that are small and often necrotic, with prominent edema and axonal injury.64 Mononuclear cells and neutrophils comprise perivascular and meningeal inflammatory infiltrates. The presence of perivascular hemorrhage and the extent of microvascular damage serve to distinguish acute hemorrhagic leukoencephalitis from ADEM lesions.
CONCLUSIONS
Recent neuropathologic studies have provided new fundamental insights into the pathogenesis of MS and have helped to distinguish MS from other inflammatory demyelinating diseases. MS is heterogeneous, and each immunopattern needs surrogate MRI, clinical, genetic, serologic, or CSF markers to be developed that would allow their differentiation in the nonbiopsied MS population, and a specifically tailored therapeutic strategy to each. Cortical lesions are common in early MS, where they may even represent the earliest pathologic event in some MS patients and are inflammatory and topographically associated with meningeal inflammation. These new observations set the stage for a new “outside-in” view of MS pathogenesis: the extension of the inflammatory process from the meninges and the subarachnoid space, where the CNS immune surveillance takes place, to the cortex, and then further to the deep white matter with subsequent demyelination and neurodegeneration. These new concepts may help identify novel inflammatory targets for the treatment of MS.
KEY POINTS
The pathologic hallmark of multiple sclerosis is multiple focal areas of myelin loss within the CNS called plaques or lesions, accompanied by variable gliosis and inflammation and by relative axonal preservation.
Active multiple sclerosis lesions are infiltrated by macrophages containing myelin debris.
Lymphocytic inflammatory infiltrates in multiple sclerosis are composed mainly of CD8-positive cytotoxic T lymphocytes, and fewer CD4-positive helper T cells, B cells, and plasma cells, unlike experimental autoimmune encephalomyelitis models in which inflammation starts with profound infiltration of the tissue by CD4-positive T cells.
Gadolinium enhancement characterizes lesions with a damaged blood-brain barrier, which enables the infiltration of inflammatory cells into the CNS.
Active multiple sclerosis lesions show a profound pathologic heterogeneity and can be classified into four immunopatterns, suggesting that the targets of injury and mechanisms of demyelination in multiple sclerosis are different in different disease subgroups.
Axonal injury in multiple sclerosis is most pronounced during active inflammatory demyelination, and acute axonal injury occurring in early multiple sclerosis lesions likely contributes to the relapse-related disability observed predominantly during the inflammatory disease phases.
Inflammation is invariably present in all lesion types and disease stages of multiple sclerosis, but its severity decreases with patient age and disease duration.
The neurodegeneration in all demyelinated lesions is invariably associated with inflammation, and in chronic inactive lesions from aged patients with long-standing progressive multiple sclerosis where the inflammatory process has died out, the neurodegeneration is also reduced to levels seen in control patients.
Extensive remyelination, illustrated by the presence of newly formed myelin sheaths and oligodendrocyte precursor cells, is frequently encountered within active plaques of early multiple sclerosis.
Cortical demyelinated lesions are present and common in early multiple sclerosis, are highly inflammatory, and may represent the pathologic substrate of cognitive impairment and epilepsy in relapsing-remitting multiple sclerosis.
The presence of inflammatory cortical demyelination in early multiple sclerosis argues against a primary neurodegenerative process at this stage of disease and suggests that neuronal and axonal injury in early cortical demyelination occur on a background of inflammation.
Meningeal inflammation is present in early multiple sclerosis and topographically associated with cortical lesions; it may drive the cortical demyelination but also set the stage for subsequent subcortical white matter inflammation and demyelination.
Profound meningeal inflammatory infiltrates are composed of T cells, B cells, and macrophages; topographically associate with subpial lesions; and may drive the cortical demyelination and neurodegeneration in patients with both primary and secondary progressive multiple sclerosis.
Baló concentric sclerosis is now believed to be a variant of pattern III multiple sclerosis.
Clinical, neuroimaging, and neuropathologic criteria-as well as serologic and CSF markers that distinguish neuromyelitis optica from multiple sclerosis-are available.
In acute disseminated encephalomyelitis, multifocal cortical microglial aggregates not associated with cortical demyelination are scattered throughout the cortex or are concentrated adjacent to neurons in the external pyramidal layer and may represent the pathologic substrate of the depressed level of consciousness in these patients.
ACKNOWLEDGMENTS
The authors have been supported by grants from the Saskatchewan Health Research Foundation (2633, to Dr Popescu), the Canada Research Chairs program (to Dr Popescu), the National Multiple Sclerosis Society (NMSS RG3185-B-3, to Dr Lucchinetti), and the NIH (NIH 1R01NS058698, to Dr Pirko; and NIH 1R01NS049577, to Dr Lucchinetti).
REFERENCES
- 1. Popescu BF, Lucchinetti CF. Pathology of demyelinating diseases. Ann Rev Pathol 2012; 7: 185– 217 [DOI] [PubMed] [Google Scholar]
- 2. Sobel RA, Moore GRW. Demyelinating diseases. In: Love S, Louis DN, Ellison DW, editors. Greenfield’s neuropathology, 8th ed London, England: Oxford University Press, 2008: 1513– 1608 [Google Scholar]
- 3. Calabrese M, Filippi M, Gallo P. Cortical lesions in multiple sclerosis. Nat Rev Neurol 2010; 6 (8): 438– 444 [DOI] [PubMed] [Google Scholar]
- 4. Geurts JJ, Barkhof F. Grey matter pathology in multiple sclerosis. Lancet Neurol 2008; 7 (9): 841– 851 [DOI] [PubMed] [Google Scholar]
- 5. Peterson JW, Bo L, Mork S, et al. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol 2001; 50 (3): 389– 400 [DOI] [PubMed] [Google Scholar]
- 6. Pirko I, Lucchinetti CF, Sriram S, Bakshi R. Gray matter involvement in multiple sclerosis. Neurology 2007; 68 (9): 634– 642 [DOI] [PubMed] [Google Scholar]
- 7. Kuhlmann T, Lassmann H, Bruck W. Diagnosis of inflammatory demyelination in biopsy specimens: a practical approach. Acta Neuropathol 2008; 115 (3): 275– 287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Filippi M, Rocca MA, Barkhof F, et al. Association between pathological and MRI findings in multiple sclerosis. Lancet Neurol 2012; 11 (4): 349– 360 [DOI] [PubMed] [Google Scholar]
- 9. Lucchinetti C, Bruck W, Parisi J, et al. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000; 47 (6): 707– 717 [DOI] [PubMed] [Google Scholar]
- 10. Bruck W, Porada P, Poser S, et al. Monocyte/macrophage differentiation in early multiple sclerosis lesions. Ann Neurol 1995; 38 (5): 788– 796 [DOI] [PubMed] [Google Scholar]
- 11. Frischer JM, Bramow S, Dal-Bianco A, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009; 132 (pt 5): 1175– 1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kutzelnigg A, Lucchinetti CF, Stadelmann C, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005; 128 (pt 11): 2705– 2712 [DOI] [PubMed] [Google Scholar]
- 13. Prineas JW, McDonald WI. Demyelinating diseases. In: Graham DI, Lantos PL, editors. Greenfield’s neuropathology, 6th ed London, England: Arnold, 1997: 813– 896 [Google Scholar]
- 14. Burgoon MP, Gilden DH, Owens GP. B cells in multiple sclerosis. Front Biosci 2004; 9: 786– 796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brilot F, Dale RC, Selter RC, et al. Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann Neurol 2009; 66 (6): 833– 842 [DOI] [PubMed] [Google Scholar]
- 16. Lennon VA, Kryzer TJ, Pittock SJ, et al. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005; 202 (4): 473– 477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Srivastava R, Aslam M, Kalluri SR, et al. Potassium channel KIR4.1 as an immune target in multiple sclerosis. N Engl J Med 2012; 367 (2): 115– 123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Aboul-Enein F, Lassmann H. Mitochondrial damage and histotoxic hypoxia: a pathway of tissue injury in inflammatory brain disease? Acta Neuropathol 2005; 109 (1): 49– 55 [DOI] [PubMed] [Google Scholar]
- 19. Barnett MH, Prineas JW. Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann Neurol 2004; 55 (4): 458– 468 [DOI] [PubMed] [Google Scholar]
- 20. Bruck W, Popescu B, Lucchinetti CF, et al. Neuromyelitis optica lesions may inform multiple sclerosis heterogeneity debate. Ann Neurol 2012; 72 (3): 385– 394 [DOI] [PubMed] [Google Scholar]
- 21. Bjartmar C, Wujek JR, Trapp BD. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J Neurol Sci 2003; 206 (2): 165– 171 [DOI] [PubMed] [Google Scholar]
- 22. Dutta R, Trapp BD. Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog Neurobiol 2011; 93 (1): 1– 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fischer MT, Sharma R, Lim JL, et al. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 2012; 135 (pt 3): 886– 899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Prineas JW, Kwon EE, Cho ES, et al. Immunopathology of secondary-progressive multiple sclerosis. Ann Neurol 2001; 50 (5): 646– 657 [DOI] [PubMed] [Google Scholar]
- 25. Coleman M. Axon degeneration mechanisms: commonality amid diversity. Nat Rev Neurosci 2005; 6 (11): 889– 898 [DOI] [PubMed] [Google Scholar]
- 26. Raff MC, Whitmore AV, Finn JT. Axonal self-destruction and neurodegeneration. Science 2002; 296 (5569): 868– 871 [DOI] [PubMed] [Google Scholar]
- 27. Barkhof F, Bruck W, De Groot CJ, et al. Remyelinated lesions in multiple sclerosis: magnetic resonance image appearance. Arch Neurol 2003; 60 (8): 1073– 1081 [DOI] [PubMed] [Google Scholar]
- 28. Patrikios P, Stadelmann C, Kutzelnigg A, et al. Remyelination is extensive in a subset of multiple sclerosis patients. Brain 2006; 129 (pt 12): 3165– 3172 [DOI] [PubMed] [Google Scholar]
- 29. Bramow S, Frischer JM, Lassmann H, et al. Demyelination versus remyelination in progressive multiple sclerosis. Brain 2010; 133 (10): 2983– 2998 [DOI] [PubMed] [Google Scholar]
- 30. Cifelli A, Arridge M, Jezzard P, et al. Thalamic neurodegeneration in multiple sclerosis. Ann Neurol 2002; 52 (5): 650– 653 [DOI] [PubMed] [Google Scholar]
- 31. Kolasinski J, Stagg CJ, Chance SA, et al. A combined post-mortem magnetic resonance imaging and quantitative histological study of multiple sclerosis pathology. Brain 2012; 135 (pt 10): 2938– 2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seewann A, Kooi EJ, Roosendaal SD, et al. Postmortem verification of MS cortical lesion detection with 3D DIR. Neurology 2012; 78 (5): 302– 308 [DOI] [PubMed] [Google Scholar]
- 33. Popescu BF, Lucchinetti CF. Meningeal and cortical grey matter pathology in multiple sclerosis. BMC Neurol 2012; 12: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lucchinetti CF, Popescu BFG, Bunyan RF, et al. Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med 2011; 365 (23): 2188– 2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Popescu BF, Bunyan RF, Parisi JE, et al. A case of multiple sclerosis presenting with inflammatory cortical demyelination. Neurology 2011; 76 (20): 1705– 1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Calabrese M, Gallo P. Magnetic resonance evidence of cortical onset of multiple sclerosis. Mult Scler 2009; 15 (8): 933– 941 [DOI] [PubMed] [Google Scholar]
- 37. Reboldi A, Coisne C, Baumjohann D, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol 2009; 10 (5): 514– 523 [DOI] [PubMed] [Google Scholar]
- 38. Bartholomaus I, Kawakami N, Odoardi F, et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature 2009; 462 (7269): 94– 98 [DOI] [PubMed] [Google Scholar]
- 39. Kutzelnigg A, Lassmann H. Cortical demyelination in multiple sclerosis: a substrate for cognitive deficits? J Neurol Sci 2006; 245 (1–2): 123– 126 [DOI] [PubMed] [Google Scholar]
- 40. Wegner C, Esiri MM, Chance SA, et al. Neocortical neuronal, synaptic, and glial loss in multiple sclerosis. Neurology 2006; 67 (6): 960– 967 [DOI] [PubMed] [Google Scholar]
- 41. Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 2007; 130 (pt 4): 1089– 1104 [DOI] [PubMed] [Google Scholar]
- 42. Choi SR, Howell OW, Carassiti D, et al. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 2012; 135 (pt 10): 2925– 2937 [DOI] [PubMed] [Google Scholar]
- 43. Howell OW, Reeves CA, Nicholas R, et al. Meningeal inflammation is widespread and linked to cortical pathology in multiple sclerosis. Brain 2011; 134 (pt 9): 2755– 2771 [DOI] [PubMed] [Google Scholar]
- 44. Moore GR, Neumann PE, Suzuki K, et al. Balo’s concentric sclerosis: new observations on lesion development. Ann Neurol 1985; 17 (6): 604– 611 [DOI] [PubMed] [Google Scholar]
- 45. Stadelmann C, Ludwin S, Tabira T, et al. Tissue preconditioning may explain concentric lesions in Balo’s type of multiple sclerosis. Brain 2005; 128 (pt 5): 979– 987 [DOI] [PubMed] [Google Scholar]
- 46. Yao DL, Webster HD, Hudson LD, et al. Concentric sclerosis (Balo): morphometric and in situ hybridization study of lesions in six patients. Ann Neurol 1994; 35 (1): 18– 30 [DOI] [PubMed] [Google Scholar]
- 47. Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004; 364 (9451): 2106– 2112 [DOI] [PubMed] [Google Scholar]
- 48. Lucchinetti CF, Mandler RN, McGavern D, et al. A role for humoral mechanisms in the pathogenesis of Devic’s neuromyelitis optica. Brain 2002; 125 (pt 7): 1450– 1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Misu T, Fujihara K, Kakita A, et al. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain 2007; 130 (pt 5): 1224– 1234 [DOI] [PubMed] [Google Scholar]
- 50. Roemer SF, Parisi JE, Lennon VA, et al. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 2007; 130 (pt 5): 1194– 1205 [DOI] [PubMed] [Google Scholar]
- 51. Takano R, Misu T, Takahashi T, et al. Astrocytic damage is far more severe than demyelination in NMO: a clinical CSF biomarker study. Neurology 2010; 75 (3): 200– 201 [DOI] [PubMed] [Google Scholar]
- 52. Wingerchuk DM, Lennon VA, Pittock SJ, et al. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006; 66 (10): 1485– 1489 [DOI] [PubMed] [Google Scholar]
- 53. Popescu BF, Lennon VA, Parisi JE, et al. Neuromyelitis optica unique area postrema lesions: nausea, vomiting and pathogenic implications. Neurology 2011; 76 (14): 1229– 1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Apiwattanakul M, Popescu BF, Matiello M, et al. Intractable vomiting as the initial presentation of neuromyelitis optica. Ann Neurol 2010; 68 (5): 757– 761 [DOI] [PubMed] [Google Scholar]
- 55. Rodriguez M, Scheithauer B. Ultrastructure of multiple sclerosis. Ultrastruct Pathol 1994; 18 (1–2): 3– 13 [DOI] [PubMed] [Google Scholar]
- 56. Hinson SR, Romero MF, Popescu BF, et al. Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc Natl Acad Sci U S A 2012; 109 (4): 1245– 1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fenton RA, Moeller HB, Zelenina M, et al. Differential water permeability and regulation of three aquaporin 4 isoforms. Cell Mol Life Sci 2010; 67 (5): 829– 840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Crane JM, Lam C, Rossi A, et al. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin-4 M1/M23 isoforms and orthogonal arrays. J Biol Chem 2011; 286 (18): 16516– 16524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Popescu BF, Parisi JE, Cabrera-Gomez JA, et al. Absence of cortical demyelination in neuromyelitis optica. Neurology 2010; 75 (23): 2103– 2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sinnecker T, Dorr J, Pfueller CF, et al. Distinct lesion morphology at 7-T MRI differentiates neuromyelitis optica from multiple sclerosis. Neurology 2012; 79 (7): 708– 714 [DOI] [PubMed] [Google Scholar]
- 61. Calabrese M, Oh MS, Favaretto A, et al. No MRI evidence of cortical lesions in neuromyelitis optica. Neurology 2012; 79 (16): 1671– 1676 [DOI] [PubMed] [Google Scholar]
- 62. Wingerchuk DM, Pittock SJ, Lucchinetti CF, et al. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology 2007; 68 (8): 603– 605 [DOI] [PubMed] [Google Scholar]
- 63. Young NP, Weinshenker BG, Parisi JE, et al. Perivenous demyelination: association with clinically defined acute disseminated encephalomyelitis and comparison with pathologically confirmed multiple sclerosis. Brain 2010; 133 (pt 2): 333– 348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hart MN, Earle KM. Haemorrhagic and perivenous encephalitis: a clinical-pathological review of 38 cases. J Neurol Neurosurg Psychiatry 1975; 38 (6): 585– 591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wingerchuk DM. The clinical course of acute disseminated encephalomyelitis. Neurol Res 2006; 28 (3): 341– 347 [DOI] [PubMed] [Google Scholar]