

Abstract
Given the increasing number of poorly soluble and thus poorly bioavailable active pharmaceutical materials, there is a demand for innovative formulation platforms for such molecules. Thus a focus on enhancing dissolution properties of poorly soluble drugs exists. Within this study, the spin coating of acetone solutions containing 5,5-diphenyl-2,4-imidazolidinedione (phenytoin) in various concentrations is evaluated. The results reveal strong variations of the morphology of deposited phenytoin crystals at silica surfaces. Individual separated particles are obtained on low phenytoin concentrations, and closely packed particular films form when the concentration is increased. As the material is isomorphic, these various morphologies have the same crystalline structure. Dissolution experiments reveal that both the apparent maximum solubility and as the dissolution rate are strongly enhanced compared to bulk powder, suggesting that formulation based on this preparative technique will allow overcoming the low solubility problematic for a variety of drugs.
Keywords: thin film, phenytoin, solvent annealing, particle formation, spin coating
Introduction
The therapeutic action of many newly developed active pharmaceutical ingredients (APIs) or drug molecules is limited to low aqueous solubility, and they belong therefore to the Biopharmaceutics Classification System (BCS) class II or IV.1 Enhanced solubility may be achieved by various approaches; for instance, a reduction in particle size promotes dissolution and thus systemic absorption.2 However, precaution needs to be taken as API nanoparticles are developed as these are able diffusing through cell barriers causing cell malfunctions.3 In addition the crystalline structure and the morphology are important.4 While amorphous solid states are favorable in terms of solubility and dissolution rate, they often lack on long time stability, resulting in undesired crystallization on storage.5 Thus, defined crystals are preferable in solid state pharmaceutical formulations. Furthermore, APIs are able to pack in various different crystalline arrangements with each polymorph having a different long time stability6 and physical-chemical6b and therapeutic properties.6a
API loaded surfaces provide an alternative for systemic absorption through topical or buccal routes from patches7 or nanoparticle surfaces.8 Surfaces made from poly(lactic-co-glycolic) acid (PLGA)8 or cellulose9 aid in the formulation and help to transport the API to the specific side of actions (i.e., drug targeting).10 Anyway, the usage of such approaches requires deeper knowledge of API–surface interactions for optimized drug formulations. For instance, at surfaces organic molecule deposition from solutions may result in altered polymorphic structures or morphologies compared to bulk crystallization within solutions.11
In this study the formation of thin layers of a model API at a solid, flat glass substrate is investigated. Such a model system provides the possibility to study interactions with various surface sensitive methods like atomic force microscope (AFM) and gracing incidence X-ray diffraction. Further this simple model allows gaining information on important parameters useful for application relevant to other systems including patches or colloidal particles prepared in solutions or spray drying. The API used is 5,5-diphenyl-2,4-imidazolidinedione (phenytoin). While phenytoin is an anticonvulsive, antiepileptic, and antiarrhythmic drug and is typically used in solid oral dosage forms (i.e., capsules and chewable tablets) as well as in parenteral formulations, it is chosen for this study as it is also known to be isomorphic. This means that the molecules assemble in a unique crystalline phase which makes it a perfect candidate for this study. Often changes in the preparation route cause crystallization in other polymorphic structures which hardens the understanding of the film formation as other polymorphic structures results in other morphologies.11a Within this study films are prepared by a spin-casting technique12 which is a fast and reproducible preparation procedure. The variation of the API amount in the solutions and spin parameter will show differences in the phenytoin assembling at a silica surface. AFM and diffraction measurements are used to study the morphological and crystallographic properties allowing elucidating changes in the dissolution behavior.
Materials and Methods
Phenytoin was purchased from Sigma Aldrich (Sigma-Aldrich, Munich, Germany) and used without further treatment. Acetone (96%) was also purchased from Sigma Aldrich (Sigma-Aldrich, Munich, Germany) in spectroscopic grade.
Standard microscopy glass slides (Roth, Karlsruhe, Germany) were cut in 2.5 × 2.5 cm2 pieces and cleaned in ethanol, acetone, and a 0.1 M NaOH solution and dried under a nitrogen stream. This results in a hydrophilic surface with a water contact angle of 35°. Acetone and phenytoin solutions have contact angles of <20°; the low surface tension of acetone results in the liquid spreading along the surface.
Films were prepared via spin coating. The spin coating experiments were performed by dropping 200 μL of various concentrated acetone solutions onto the glass slides and subsequently rotated at 25 rounds per second (rps) for 30 s around its surface normal. This results in homogeneous structures forming over the entire glass surface. All samples were stored under a vacuum of 10–6 mbar for 2 h at ambient temperatures prior the experiments to reduce residuals of the solvent.
AFM images were taken with a Nanosurf Easyscan 2 machine in tapping mode. Tap190-Al cantilevers (Budgetsensors, Sofia, Bulgaria) were used for all experiments. Data evaluation and image processing was performed by the software package Gwyddeon.13
Specular X-ray diffraction experiments were performed with a Siemens 500D diffractometer in Bragg–Brentano configuration. The copper radiation (wavelength λ = 0.154 nm) was narrowed with primary and secondary side slit systems. The diffracted intensity was guided toward a graphite monochromator and collected by a scintillation counter. The angular measurement (2Θ) is represented in the scattering vector (qz) notation which calculates as qz = 4π sin(Θ)/λ.14
Grazing incidence X-ray diffraction (GIXD) experiments were performed with a modified lab based Bruker D8 Discover.15 The copper radiation is parallelized via a Goebell mirror and defined by parallel plate collimators. The diffracted beam is led trough a collimator system and is collected by a Vantech 1D detector. The GIXD data were evaluated using the software package PyGID.16
The apparent solubility (Cs) of bulk phenytoin and from spin-coated samples was evaluated, and the content uniformity of prepared spin-coated samples was determined using standard routines according to European Pharmacopoeia V 7.0. Dissolution experiments, that is, the amount of API dissolved over time of both samples, were conducted on a horizontal shaker (IKA yellow line RS 10 control, IKA-Werke GmbH & Co. KG, Germany) at 200 rpm and a temperature of 25 °C. The dissolution experiments were setup by adding a phenytoin coated glass slide (i.e., 94.02 ± 8.16 μg of phenytoin) or 4.7 mg of bulk material into dissolution vessels, and 20 mL (spin-coated samples) or 1000 mL (bulk material), respectively, of Milli-Q-water was added. An aliquot of 1 mL was withdrawn after defined times over 6 h. The concentration was determined by UV–vis adsorption measurement at a wavelength of 220 nm using a nanophotometer (Implen, Munich, Germany) and standard quartz cuvettes (Hellma, Müllheim, Germany). For statistics, four samples of each kind were investigated, and the standard deviation was elucidated.
Results and Discussion
The variation of phenytoin concentration within acetone solution results in the formation of various morphologies at a glass surface as the solutions are spin coated at a spin speed of 25 rps. In Figure 1 AFM height images of films deposited from 0.05 to 1.5 wt % acetonic solutions are shown. At the lowest concentration of 0.05 wt % separated islands of phenytoin are present. The islands (bright areas in the image) consist of small spot like structures which are connected by flat areas. A mean thickness of about 30 nm is observed for the islands. In between adjacent islands (dark areas) no material could be identified; thus a layer that fully covers the substrate is absent. At a slightly increased concentration of 0.1 wt % again separated structures are observed (see Figure 1b). Anyway the morphology has drastically changed. Large drop-like particles are distributed over the entire film. The particles have a height of about 70 nm and are a result of the solution favoring dewetting from the silica surface. Besides these particles elongated needle like structures are visible which correspond to the typical structure observed on slow solvent removal (data will be shown elsewhere). These needles have an extension of up to 6 μm. A height of 40 nm shows that these structures are significantly lower compared to the drop like particles, suggesting that this morphology stronger interacts with the surface; thus a rod-like growth along the surface is present.
Figure 1.
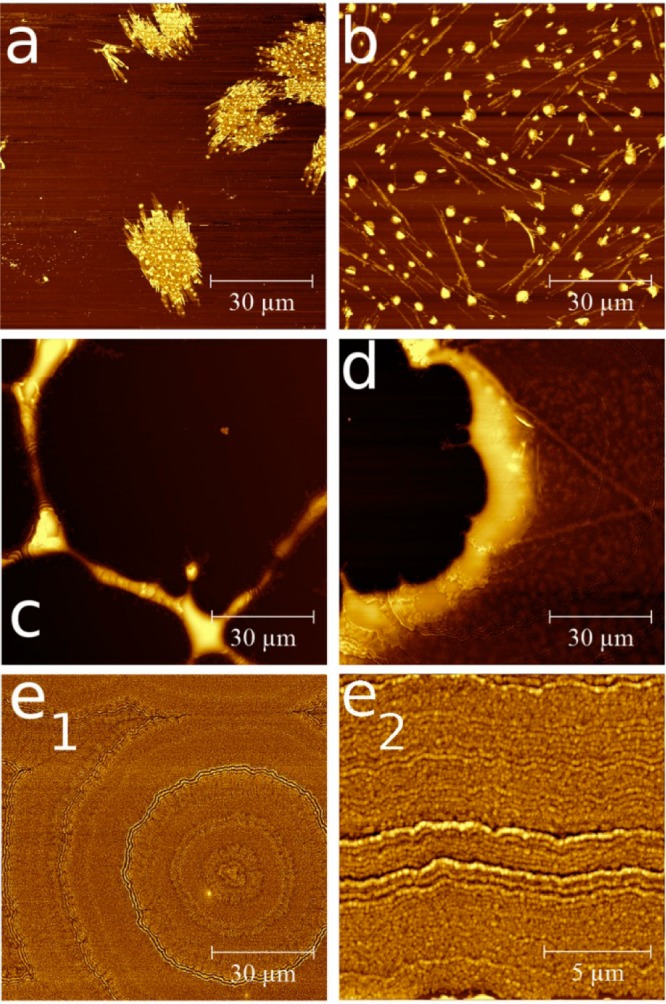
AFM height images of phenytoin films spin coated from 0.05 wt % (a), 0.1 wt % (b), 0.5 wt % (c), 1 wt % (d), and 1.5 wt % (e1). e2 is a higher resolution image of e1. The color scale is chosen individually for clarity.
Increasing the concentration further results in a phenytoin “network” being present on the silica surface. Elongated structures run over several tens of micrometers along the surface. A width of 3.3 μm and height of 240 nm can be deduced from the AFM image (Figure 1c). At the connection points of these structures larger islands form. Such structures are typical for materials trying to dewet the surface, but as sufficient material is present interconnections develop during drying.
At a concentration of 1 wt % the gaps in between the elongated structures start to fill resulting in a 90 nm thick film with nearly full coverage but with remaining holes (see Figure 1d). However, increasing the concentration to 1.5 wt % results in a film that entirely covers the glass substrate (see Figure 1e1). Surprisingly, the AFM height image reveals a tree-ring like structure with concentric rings forming around a common center. A high resolution image reveals that this tree-like structures are a result of small particles packing together (Figure 1e2); thus the tree-like rings form on account of particle packing during solvent removal. Furthermore the results suggest that various phenytoin nuclei rapidly form during the spinning process, which then assemble in this observed tree-like structure.
All samples shown in Figure 1 were prepared at the same spin speed of 25 rps. At low concentrations dewetting structures are observed. Typically, such structures are a result of the molecules at the API–glass interface disfavoring their contact;17 that is, the adhesive forces are smaller than the cohesive forces which results from the difference in their polar and apolar interaction sides. Phenytoin is a highly hydrophobic molecule, while glass is hydrophilic which results in a disjoining force and thus in dewetting.17 Within very thin films the interface is the dominating factor. Anyway, as the film thickness increases, the balance of short-range (e.g., double layer forces) and long-range forces (e.g., van der Waals forces) is changed. As a result, the film stabilizes, and a homogeneous layer without holes results.18 In the case of phenytoin deposited from a 1.5 wt % acetone solution at 25 rps, a fully closed layer with a thickness of 121 nm forms.
The variation of the solution concentration has a drastic effect on the film morphology. Anyway, the variation of the spin speed also strongly affects the morphology. In Figure 2 the AFM images of phenytoin thin films prepared by various spin speeds from the same 1.5 wt % aceton solution are shown. At a spin speed of 10 rps the resulting film covers the entire glass slide. The morphology shows again various tree-ring like structures with centers around the middle and in the right up corner of the image (see Figure 2a). Compared to the structure observed at 25 rps the number of rings and the packing density are increased (compare Figure 1e and Figure 2a).The corresponding high resolution image reveals that the structure results from particles packing closely together but with the particle size being increased compared to samples spin-coated at 25 rps. The situation remains similar for samples which are prepared at 50 rps (see Figure 2b), but the particle size is reduced compared to the other two samples.
Figure 2.

AFM height images of phenytoin thin films spin-cast at rotation speeds of 10 rps (a), 50 rps (b), and 100 rps (c). The right hand row shows the same samples at a higher resolution.
Spin-casting of the film at a rotation speed of 100 rps results in a strong deviation of the morphology (see Figure 2c). Other than the previous films, clear dewetting structures with large areas remaining uncovered are present (e.g., left top corner). In addition small holes in the film are noticeable as dark spots within Figure 2c and c1. The morphology of the particles is slightly more elongated compared to the sample prepared at a spin speed of 50 rps.
For a quantitative evaluation of the surface properties on the variation of the rotation speed, the layer thickness (d), the root-mean-square roughness (σrms), and the power spectral density (PSD) were evaluated. The data are summarized in Table 1. For the film prepared at a spin speed of 10 rps d = 159 nm and σrms = 17.5 nm are obtained. As the spin speed is increased to 25 rps, d reduces to 121 nm as well as the σrms reduces to 16.2 nm. For the 50 and 100 rps samples d = 54 and 45 nm, respectively, are obtained. The corresponding σrms reduces to 12.0 and 8.6 nm. Thus the increase of the rps results in a decreasing thickness and roughness.
Table 1. Summary of the Extracted Parameters of the Spin-Cast Phenytoin Film at Various Speedsa.
| sp-sp (rps) | d (nm) | σrms (nm) | C (nm) |
|---|---|---|---|
| 10 | 159 | 17.5 | 2200/571 |
| 25 | 121 | 16.2 | 398 |
| 50 | 54 | 12.0 | 363 |
| 100 | 45 | 8.6 | 311 |
sp-sp, spin speed; d, layer thickness; σrms, root-mean-square roughness; C, correlation length.
The power spectral density of the various samples, calculated from Figure 2a1–2c1, are summarized in Figure 3. Within such an evaluation common distances on a surface are extractable.19 For the sample prepared at 10 rps a high spectral density is observed for small wave vectors (k). As k is increased, the density drops from the plateau and reaches the background level above 0.2 nm–1. For the evaluation of common distances, linear regression fits in the plateau, and the linear decreasing regions are performed (lines in Figure 3). The intersection point of the lines typically describes a correlation length (C) (or common distance), which calculates as C = 2π/k. The 10 rps sample reveals a real space correlation of 570 nm which agrees well with the mean particle sizes observed in Figure 2a1. The same evaluation of the 25 rps and 50 rps samples reveals a very similar behavior but with the correlation “drop off” shifting to larger wave vectors; a real space correlation length of 398 and 363 nm is obtained for the 25 and 50 rps, respectively. Thus the increase in spin speed does not only decrease the layer thickness but also decreases the particle size. The PSD spectrum of the 100 rps is distinct from the previous samples. The curve reveals the presence of two correlation lengths. The first is at low k = 0.0028 nm–1 (corresponding to a real space distance of 2.2 μm), and a second is present at a higher wavevector of 0.021 nm–1 (real space distance of 311 nm). Inspecting the AFM image shows that the large correlation length is due to the holes within the sample; that is, the holes have a mean separation of 2.2 μm. The 311 nm correlation is due to common particle size which are smaller compared to the previous samples.
Figure 3.

Power spectral density spectra for samples prepared from the 1.5 wt % solution deposited at various spin speeds. Symbols denote the experimental data and lines linear fits to the regions. Curves are shifted for clarity.
The determined particle sizes for all samples have lateral extensions which are larger than the film height. From this follows that the films consist of small cylinder like particles with an aspect ratio (lateral size/film thickness) of 3.5 for the thickest film and increases to 6.6 for the thinnest film. This means that particles forming during the spin coating process become more “flake like” as the spin speed is increased.
The AFM images reveal the formation of particular films on the spin coating process. Depending on the spin speed, the particles and the surface morphology change. Typically, spinning at a low speed means that the system has more time to assemble until the solvent is evaporated. During the solvent evaporation, supersaturation takes place, resulting in phenytoin nucleation, and these initial nuclei grow into larger crystallites until the entire acetone is vanished from the sample. At larger rotation speeds the initial solution gets spin off faster from the surface. In addition the evaporation of the acetone is enhanced. As a result the layer thickness is reduced and is composed of an increased number but smaller crystallites compared to “slower prepared” samples. At the fastest speed investigated, that is, 100 rps, the particles are not able to entirely wet the surface. Often liquids form dewetting structures at solid surfaces. These structures may assist in the movement of already formed particles as well as the dissolved molecules along certain channels within the surface; thus the observed dewetting structures form.
For the characterization of the crystalline structure of the phenytoin films, X-ray diffraction experiments are performed, and the results for the 1.5 wt % at 25 rps spin coated samples are shown in Figure 4, for example. To our knowledge there exists only one polymorphic modification of phenytoin in literature;20 the phenytoin molecules pack in a tetragonal unit cell with a = 0.62 nm, b = 1.36 nm, and c = 1.55 nm which reveal a calculated powder pattern demonstrated in Figure 4. Within the experimental scan of the thin film, only one peak at 8.09 nm–1 is present belonging to the 002 Bragg reflection of the crystalline phase. (The broad amorphous hump results from the underlying glass substrate). Within a specular scan, only net planes which are parallel to the substrate surface are detectable; thus it follows that the sample shows a preferred alignment (texture) with respect of the surface.21 Anyway, GIXD measurements result in a slightly different conclusion (see Figure 5a,b). In such a scan net planes which are parallel (or close to) to the surface normal are investigated.14 The GIXD pattern reveals high intensity spots (highlighted by white X’s in Figure 3b) and are a result of Bragg reflection of certain Miller’s indices (hkl’s). Using the information of the unit cell shows that these hkl’s are a result of phenytoin assembling in a 001 texture in accordance with the specular scan. However, ring-like structures are also noted. Such rings are a result of random orientations of the crystallites or powder like behavior; that is, within an ideal powder, no preferred orientation exists. From this follows that fractions of the crystallites adapt a random conformation with respect of the surface. As the film is thin (only 121 nm), the amount of random particles is probably very low, which causes an absence of Bragg peaks in the specular scan. Within a powder sample all Bragg reflections which have sufficient intensity should be visible, as observed for the as-delivered powder sample investigated in Figure 4.
Figure 4.

Specular X-ray diffraction scan of the phenytoin sample prepared from 1.5 wt % actone solution at a spin speed of 25 rps (top), experimental powder diffraction measurement of the as-delivered powder (middle), and the calculation of an ideal powder (bottom).20 Curves are shifted for clarity.
Figure 5.

GIXD pattern of the phenytoin sample prepared from 1.5 wt % solution and spin coated at 25 rps (a). Dashed box indicates the area over which the pattern is integrated to generate a line scan (b). Part c shows the molecular packing within the unit cell.
GIXD is a very surface sensitive technique allowing order22 and disorder23 within films as thin as monolayers to be determined. As there is no indication of an amorphous hump within the GIXD pattern, this means that the film consists solely of crystalline phenytoin.
In Figure 5c the packing of the molecules viewed along the crystal a-axis is shown. In such a representation the 001 net-planes are parallel to the crystal b-axis, or in other words the a- and b-axis are in contact with the glass surface. The phenytoin molecules consist of two phenyl rings and an imidazolidin ring. All rings are twisted with respect to each other, whereby the imidazolidin ring runs along the crystal a-axis. Furthermore the imidazolidin ring adapts a position at which the terminal oxygen is in contact with the glass surface within a 001 textured growth. As glass surfaces typically have highly polar and/or hydrogen bonding sites, this contact is favored, and as a result the 001 orientation is preferentially obtained. Anyway, the additional powder characteristic is most likely a consequence of fast removal of acetone during the spin-casting process; the API concentration rapidly increases resulting in the formation of various nuclei simultaneously. For nuclei close to the surface interaction are sufficient to achieve a preferred orientational growth. For nuclei further off the surface an adaption with respect of the surface is not present; thus the “bulk” crystalline particles adapt a random orientation.
Comparing the GIXD line scans pattern of the thin film sample with the powder pattern of the as-delivered material reveals an excellent agreement of the peak positions (compare Figure 4 middle and Figure 5b). Variations in the peak heights are a result from varying texture being present within both samples resulting in enhancing or decreasing Bragg peak intensities. Additionally, these results are in excellent agreement with a calculated powder pattern (Figure 4 bottom).20 From this follows that the preparation route via spin coating does not influence the crystallographic properties of phenytoin, that is, an incorporation of acetone within the thin film is very unlikely, and the same polymorph forms.
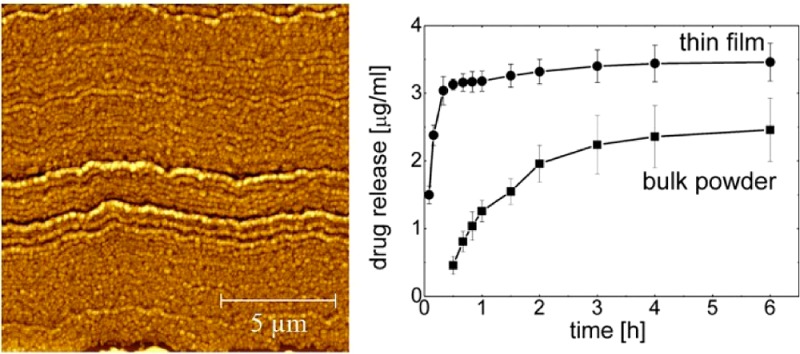
For the evaluation of the solubility properties of phenytoin deposited on the glass surface dissolution profiles are determined, i.e. the increase in solute concentration over time (see Figure 6). Milli-Q water was chosen as dissolution media as it minimizes the impact of surface affine species within other dissolution media (e.g., salts) onto the particle removal from the surface. At a short period of time (5 min) a concentration of 1.5 ± 0.1 μg/mL is already obtained. As time progresses the concentration increases linearly and reaches a value of 3.1 ± 0.1 μg/mL after 30 min. At this point the increase in solution concentration with time is slowed down. It took another five and a half hours to reach a value of 3.5 ± 0.3 μg/mL.
Figure 6.

Phenytoin concentration as a function of time for thin films samples and the as-purchased powder.
Additionally, dissolution studies of bulk phenytoin were conducted for sake of classification, that is, if the spinning process results in improved dissolution properties. The dissolution studies were performed under same conditions (i.e., same amount of API relative to dissolution-media volume, temperature, same apparatus, same parameters) to make results comparable. As shown in Figure 6 (bottom curve), API release of bulk phenytoin is noticeable after 30 min at which a concentration of 0.5 ± 0.1 μg/mL is determined. (API concentrations prior 30 min were under the detection limit of the experimental setup.) From this point, a linear increase in drug concentration is observed, and at 1 h a concentration of 1.3 ± 0.2 μg/mL is reached. After 1 h the increase in API concentration is slowed down. At 6 h a solution concentration of 2.5 ± 0.5 μg/mL is measured.
Within the dissolution experiments the amount of phenytoin dissolved from the thin spin-cast sample is significantly higher at all times compared to the bulk powder sample. Furthermore, the spin-cast sample reveals a rapid dissolution at the very beginning with most of the phenytoin being dissolved. In contrast the dissolution of the bulk material after this time was just initiated. Basically, thin films of phenytoin crystals exhibit an improved solubility behavior with higher and faster dissolution rate due to higher specific surface area of the smaller particles being available for dissolution media. The bulk phenytoin shows a particle size distribution of d0.9 = 25.0 μm, d0.5 = 14.1 μm, and a d0.1 = 7.7 μm whereby the AFM height images reveal crystal sizes around 400 nm. The smaller particle size also affects the maximum apparent solubility of both. It was observed that twice as much phenytoin could be dissolved when prepared as thin film (i.e., 23.6 ± 0.6 μg/mL) compared to untreated bulk phenytoin (i.e.,11.5 ± 0.5 μg/mL).
Conclusion
By varying the spin coating process the film appearance as well as the local morphology (shape and size) of phenytoin could be manipulated. The results show that the API crystallizes fast on the removal of the solvent and particles form whereby the size can be manipulated by the spin speed. Furthermore the variation of solution concentration reveals morphologies that varied from separated islands to extended dewetting structures and finally fully closed layers. The dissolution properties of films prepared by spin coating technique are significantly improved compared to those of the bulk powder. This gives confidence that the other system including multilayer application forms may be improved as well by the usage of this approach.
Acknowledgments
The work was funded by the Austrian Science Fund (FWF): [P25541-N19]. E.R. would like to thank the NAWI-Graz, and the authors would like to thank Y. Cooper for helpful discussions.
The authors declare no competing financial interest.
References
- Cook J.; Addicks W.; Wu Y. H. Application of the Biopharmaceutical Classification System in Clinical Drug Development—An Industrial View. AAPS J. 2008, 10, 306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mosharraf M.; Nyström C. The effect of particle size and shape on the surface specific dissolution rate of microsized practically insoluble drugs. Int. J. Pharmaceutics 1995, 1221–235–47. [Google Scholar]; b Kesisoglou F.; Panmai S.; Wu Y. Nanosizing--oral formulation development and biopharmaceutical evaluation. Adv. Drug Delivery Rev. 2007, 59, 631–644. [DOI] [PubMed] [Google Scholar]
- a Roblegg E.; Fröhlich E.; Meindl C.; Teubl B.; Zaversky M.; Zimmer A. Evaluation of a physiological in-vitro system to study the transport of nanomaterials through the buccal mucosa. Nanotoxicology 2012, 6, 399–413. [DOI] [PubMed] [Google Scholar]; b Fröhlich E.; Meindl C.; Roblegg E.; Griesbacher A.; Pieber T. R. Cytotoxity of nanoparticles is influenced by size, proliferation and embryonic origin of the cells used for testing. Nanotoxicology 2012, 6, 424–439. [DOI] [PubMed] [Google Scholar]
- Newman A. W.; Byrn S. R. Solid-state analysis of the active pharmaceutical ingredient in drug products. Drug Discovery Today 2003, 819898–905. [DOI] [PubMed] [Google Scholar]
- Ng Y. C.; Yang Z.; McAuley W. J.; Qi S. Stabilisation of amorphous drugs under high humidity using pharmaceutical thin films. Eur. J. Pharm. Biopharm. 2013, 84, 555–565. [DOI] [PubMed] [Google Scholar]
- a Singhal D.; Curatolo W. Drug polymorphism and dosage form design: a practical perspective. Adv. Drug Delivery Rev. 2004, 56, 335–347. [DOI] [PubMed] [Google Scholar]; b Miller J. M.; Collman B. M.; Greene L. R.; Grant D. J.; Blackburn A. C. Identifying the stable polymorph early in the drug discovery-development process. Pharm. Dev. Technol. 2005, 10, 291–297. [DOI] [PubMed] [Google Scholar]
- Paudel K. S.; Milewski M.; Swadley C. L.; Brogden N. K.; Ghosh P.; Stinchomb A. L. Challenges and opportunities in dermal/transdermal delivery. Ther. Delivery 2010, 1, 109–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govender T.; Stolnik S.; Ganett M. C.; Illum L.; Davis S. S. PLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water soluble drug. J. Controlled Release 1999, 57, 171–185. [DOI] [PubMed] [Google Scholar]
- Shukla A. J.; Price J. C. Effect of Drug Loading and Molecular Weight of Cellulose Acetate Propionate on the Release Characteristics of Theophylline Microspheres. Pharm. Res. 1991, 8, 1396–1400. [DOI] [PubMed] [Google Scholar]
- a Active pharmaceutical ingredient adsorbed on solid support. EP Patent 2,238,979: 2010;; b Arya A.; Chandra A.; Sharma V.; Pathak K. Fast dissolving oral films: an innovative drug delivery system and dosage form. Int. J. Chem. Technol. Res. 2010, 21576–583. [Google Scholar]
- a Wedl B.; Resel R.; Leising G.; Kunert B.; Salzmann I.; Oehzelt M.; Koch N.; Vollmer A.; Duhm S.; Werzer O.; Gbabode G.; Sferrazza M.; Geerts Y. H. Crystallisation kinetics in thin films of dihexyl-terthiophene: the appearance of polymorphic phases. RSC Adv. 2012, 2, 4404–4414. [Google Scholar]; b Werzer O.; Boucher N.; de Silva J. P.; Gbabode G.; Geerts Y. H.; Konovalov O.; Moser A.; Novak J.; Resel R.; Sferrazza M. Interface Induced Crystal Structures of Dioctyl-Terthiophene Thin Films. Langmuir 2012, 28, 8530–8536. [DOI] [PubMed] [Google Scholar]
- a Temple-Boyer P.; Mazenq L.; Doucet J. B.; Conedera V.; Torbiero B.; Launay J. Theoretical studies of the spin coating process for the deposition of polymer-based Maxwellian liquids. Microelectron. Eng. 2010, 872163–166. [Google Scholar]; b Schwartz L. W.; Roy R. V. Theoretical and numerical results for spin coating of viscous liquids. Phys. Fluids 2004, 163569–584. [Google Scholar]; c Chang Y.; Wu W. C.; Chen W. C. Theoretical analysis on spin coating of polyimide precursor solutions. J. Electrochem. Soc. 2001, 1484F77–F81. [Google Scholar]
- Necas D.; Klapetek P. Gwyddion: an open-source software for SPM data analysis. Cent. Eur. J. Phys. 2012, 101181–188. [Google Scholar]
- Pietsch U.; Holy V.; Baumbach T.. High-Resolution X-Ray Scattering: From Thin Films to Lateral Nanostructures; Springer-Verlag: New York, 2004. [Google Scholar]
- Neuschitzer M.; Moser A.; Neuhold A.; Kraxner J.; Stadlober B.; Oehzelt M.; Salzmann I.; Resel R.; Novak J. Grazing-incidence in-plane X-ray diffraction on ultra-thin organic films using standard laboratory equipment. J. Appl. Crystallogr. 2012, 45, 367–370. [Google Scholar]
- Moser A.; Werzer O.; Flesch H.-G.; Koini M.; Smilgies D.-M.; Nabok D.; Puschnig P.; Ambrosch-Draxl C.; Schiek M.; Rubahn H.-G.; Resel R. Crystal structure determination from two-dimensional powders: A combined experimental and theoretical approach. Eur. Phys. J. Spec. Top. 2009, 167, 59–65. [Google Scholar]
- Yadavali S.; Krishna H.; Kalyanaraman R. Morphology transitions in bilayer spinodal dewetting systems. Phys. Rev. B 2012, 85, 235446(1–8). [Google Scholar]
- Xie R.; Karim A.; Douglas J. F.; Han C. C.; Weiss R. A. Spinodal Dewetting of Thin Polymer Films. Phys. Rev. Let. 1998, 81, 1251–1254. [Google Scholar]
- Itoh T.; Yamauchi N. Surface morphology characterization of pentacene thin film and its substrate with under-layers by power spectral density using fast Fourier transform algorithms. Appl. Surf. Sci. 2007, 253146196–6202. [Google Scholar]
- Camerman A.; Camerman N. The stereochemical basis of anticonvulsant drug action. I. The crystal and molecular structure of diphenylhydantoin, a noncentrosymmetric structure solved by centric symbolic addition. Acta Crystallogr., Sect. B 1971, 27, 2205–2211. [Google Scholar]
- Birkholz M.Thin Film Analysis by X-ray Scattering; Wiley-VCH: Weinheim, 2006. [Google Scholar]
- Smits E. C. P.; Mathijssen S. G. J.; van Hal P. A.; Setayesh S.; Geuns T. C. T.; Mutsaers K. A. H. A.; Cantatore E.; Wondergem H. J.; Werzer O.; Resel R.; Kemerink M.; Kirchmeyer S.; Muzafarov A. M.; Ponomarenko S. A.; de Boer B.; Blom P. W. M.; de Leeuw D. M. Bottom-up organic integrated circuits. Nature 2008, 4557215956–959. [Google Scholar]
- Dohr M.; Werzer O.; Shen Q.; Salzmann I.; Teichert C.; Ruzie C.; Schweicher G.; Geerts Y. H.; Sferrazza M.; Resel R. Dynamics of Monolayer-Island Transitions in 2,7-Dioctyl-benzothienobenzthiophene Thin Films. ChemPhysChem 2013, 14112554–2559. [DOI] [PubMed] [Google Scholar]