

Abstract
Liver cancer is a leading cause of cancer death. Most patients are treated by arterial injection of chemoembolizing agents, providing a convenient avenue for local treatment by novel therapies, including gene therapy. Poly(beta-amino esters) (PBAEs) were synthesized and used to form nanoparticles for non-viral transfection of buffalo rat hepatoma (MCA-RH7777) and hepatocyte (BRL-3A) lines with eGFP and luciferase DNA. Hepatoma cells were transfected with up to 98±0.4% efficacy with no measurable cytotoxicity. Hepatocytes were transfected with as high as 73±0.4% efficacy with 10±4% non-specific cytotoxicity. In contrast, positive controls (branched polyethylenimine, Lipofectamine™ 2000, and X-tremeGENE® DNA HP) caused 30–90% toxicity in BRL-3A cells at doses required for >50% transfection. Of the 21 optimized PBAE-DNA formulations tested, 12 showed significant specificity for hepatoma cells over hepatocytes in monoculture (P<0.05 for both percentage transfected and eGFP expression intensity). Top polymers from eGFP studies also delivered luciferase DNA with 220±30-fold and 470±30-fold greater specificity for hepatoma cells than hepatocytes. Transfections of co-cultures of hepatoma and hepatocytes with eGFP DNA also showed high specificity (1.9±0.1- to 5.8±1.4-fold more transfected hepatoma cells than hepatocytes, measured by percentage transfected and flow cytometry). By eGFP intensity, up to 530±60-fold higher average expression per cell was measured in hepatoma cells. One top formulation caused 95±0.2% transfection in hepatoma cells and 27±0.2% in hepatocytes (96±9% relative hepatocyte viability). PBAE-based nanoparticles are a viable strategy for liver cancer treatment, delivering genes to nearly 100% of cancer cells while maintaining high biomaterial-mediated specificity to prevent toxic side-effects on healthy hepatocytes.
Introduction
Nearly 750,000 people worldwide are diagnosed with liver cancer each year, with hepatocellular carcinoma (HCC) the second leading cancer death in men and the sixth in women, accounting for nearly 700,000 deaths each year.1 Unfortunately, for most HCC patients, current chemotherapeutic and local therapy options provide only modest benefit. The median survival is less than 1 year for patients with unresectable disease, and only 30% of patients are able to undergo potentially curative treatment (i.e. surgical resection).3 For patients not qualifying for surgical intervention, transarterial chemoembolization (TACE) provides a non-invasive therapeutic alternative, albeit with only modest improvement of morbidity and mortality.4 TACE employs emulsified oil-drug droplets that are injected into the hepatic artery under real-time fluoroscopic image guidance in an interventional radiology suite5; importantly, tumor perfusion is almost exclusively from the hepatic artery while the majority of normal liver perfusion is provided by the portal vein. By injecting an emulsion of a chemotherapeutic and aqueous contrast agent, this allows both local delivery of a drug and real-time, qualitative imaging of the tumor uptake. Other groups have previously explored the possibility of using the TACE procedure for local delivery of DNA encoding either a therapeutic protein or a luminescent protein for imaging, in particular the VIPER system (vector of iopamidol, protamine, and ethiodized oil reagents).6 However, taken together, these strategies been hampered by low in vitro delivery efficacy and high toxicity of the delivery agent(s) used to enhance specificity for cancer cells.
While gene therapy has great promise as a therapeutic in diseases refractory to systemic chemoradiation treatments, such as hepatocellular carcinoma, the translation of such technologies has been limited by a number of difficulties in DNA delivery. Viruses tend to have high efficiency but at the cost of safety concerns, including excessive immune response and high rate of mutagenesis.7 In recent years, progress has been made in the field of synthetic gene delivery agents, such as cationic lipids and polymers, which can be tailored to the cells and application of interest and can be modified to decrease toxicity.8,9
Here, the class of polycations used as DNA delivery agents are synthetic poly(beta-amino esters) (PBAEs),10,11 which we have previously shown to be effective for in vitro DNA delivery to a number of hard-to-transfect cell types, including primary human cells or tissue,12–14 and for in vivo delivery in various animal disease models.15,16 In contrast to the previously studied VIPER system, our PBAE-DNA nanoparticles are hydrolytically degradable and have been shown to be biocompatible, and many of them suggest intrinsic biomaterial-mediated cell specificity.17,18 By using a set of polymers optimized from an initial high-throughput screening of a combinatorial library,10 we report here the identification of PBAE-based non-viral gene delivery nanoparticles with (1) high in vitro efficacy for gene delivery to hepatoma cells; (2) low non-specific cytotoxicity; and (3) intriguing cell-specificity, enabling the targeting of hepatoma over hepatocytes in co-culture.
Materials and Methods
Materials
Monomers used for synthesizing polymers (Figure 1) were purchased as follows: 1,3-propanediol diacrylate (B3; Monomer-Polymer and Dajac Labs, Trevose, PA); 1,4-butanediol diacrylate (B4; Alfa Aesar, Ward Hill, MA); 1,5-pentanediol diacrylate (B5, Monomer-Polymer and Dajac Labs); 1,6-hexanediol diacrylate (B6, Alfa Aesar); 3-amino-1-propanol (S3, Alfa Aesar); 4-amino-1-butanol (S4, Alfa Aesar); 5-amino-1-pentanol (S5, Alfa Aesar); 6-amino-1-hexanol (S6, Sigma Aldrich, St. Louis, MO); 1,3-diaminopentane (E3; TCI America, Portland, OR); 2-(3-aminopropylamino)ethanol (E6, Sigma Aldrich); and 1-(3-aminopropyl)-4-methylpiperazine (E7, Alfa Aesar. Lipofectamine™ 2000 and Opti-MEM I from Invitrogen (Carlsbad, CA) and X-tremeGENE HP from Roche (Indianapolis, IN) were optimized according to manufacturer instructions. DNA plasmids pEGFP-N1 (eGFP) and pCMV-Luc (luciferase) were amplified and purchased from Aldevron (Fargo, ND) and Elim Biopharmaceuticals (Hayward, CA), respectively. Piggybac transposase and nuclear H2B-cherry Piggybac transposon plasmids were kindly provided by Dr. Karl Wahlin of Dr. Don Zack’s lab at Johns Hopkins. 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) was purchased from Sigma (Saint Louis, MO). All materials were reagent grade and used as received.
Figure 1.
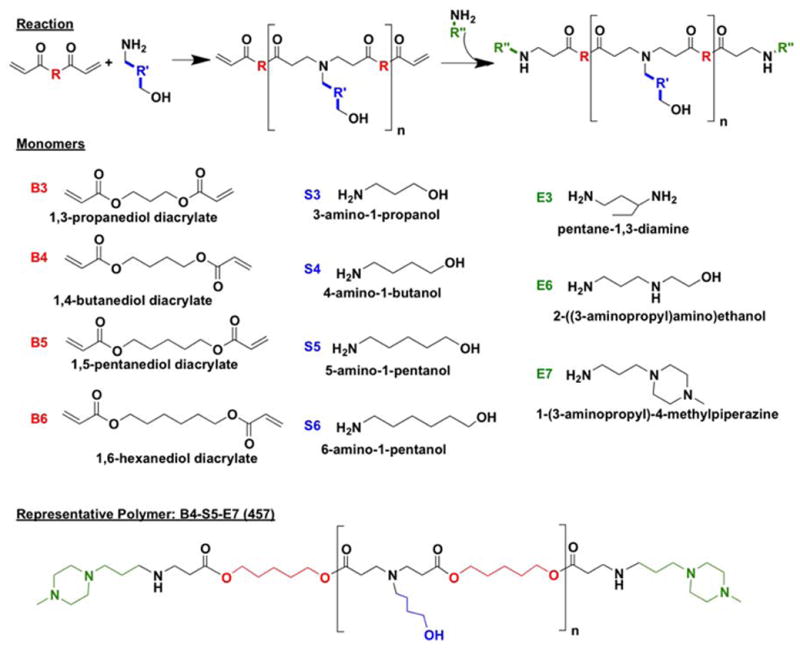
For PBAE synthesis, one monomer from each of B, S, and E react to form an amine-terminated polymer.
Polymer synthesis
For initial screening, polymers were synthesized as previously reported (Figure 1).19 Briefly, one acrylate-terminated backbone (“B”) monomer was mixed with one amine-terminated side-chain (“S”) monomer and stirred at 90°C for 24 hr at 1.05:1, 1.1:1, or 1.2:1 molar ratio of B:S. The resulting acrylate-terminated base polymer (B-S) was dissolved in anhydrous DMSO, and 10-fold molar excess of one end-cap (“E”) monomer in DMSO was added. The mixture was vortexed for 20 sec at room temperature, incubated at room temperature for 1 hr, and stored at 4°C until use at 100 mg/mL (measured by base polymer concentration) in DMSO. Polymers are referred to henceforth by their components BSE and their B:S molar ratio. For example, B4 polymerized with S5 at 1.1:1 ratio B:S and then end-capped with E7 is abbreviated “457, 1.1:1.”
After initial screenings, top polymer candidates were re-synthesized in a purified form. After base polymer synthesis, the polymer was dissolved in anhydrous THF and mixed with 10-fold molar excess of the end-cap, stirred for 1 hr at room temperature, and precipitated into anhydrous diethyl ether. The resulting mixture was centrifuged to isolate the polymer precipitate, and solvent and precipitant were decanted by pouring. The polymer was washed once more with ether and kept under vacuum with desiccant for 48 hr to remove final traces of ether. The purified polymer was then dissolved in DMSO at 100 mg/mL (measured by total polymer) and stored at −20°C with desiccant until use. Ether-purified polymers have “e” included in their names (for example, “457e, 1.1:1”).
Cell culture
Buffalo rat hepatoma cells (MCA-RH7777) and hepatocytes (BRL-3A) were purchased from ATCC (Manassas, VA). MCA-RH7777 cells were cultured in complete hepatoma medium [high glucose DMEM (Invitrogen) with 10% heat-inactivated fetal bovine serum (FBS) and 100 U/mL penicillin and 10 μg/mL streptomycin (pen/strep)]. BRL-3A cells were cultured in complete hepatocyte medium [Minimal Essential Medium with GlutaMAX™-I, 10% FBS, pen/strep, 2 mM L-glutamine, 1 mM sodium pyruvate, and 100 μM non-essential amino acids].
Transfections
MCA-R7777 or BRL-3A cells were seeded separately into 96-well plates in complete medium at 1.5*104 cells/well and incubated at 37°C and 5% CO2 overnight. For luciferase studies, opaque white plates with clear bottoms were used. Immediately before the transfection, all media were changed to fresh complete hepatocyte medium to eliminate media components as variables for direct comparison of hepatoma cells and hepatocyes.
Nanoparticles were formed by diluting DNA and polymer separately in 25 mM sodium acetate buffer (pH 5) and mixing at varying polymer-to-DNA weight/weight ratios (w/w). After 10 min incubation at room temperature for complexation to occur, nanoparticles were added directly to the cell culture medium (1:5 v/v ratio of particles in sodium acetate to medium). Unless otherwise stated, the dosage was 600 ng DNA per well (5 μg/mL final concentration in medium). Positive controls Lipofectamine™ 2000 and X-tremeGENE DNA HP were prepared according to manufacturers’ instructions in Opti-MEM I, and PEI was prepared in 150 mM NaCl. A replicate set of wells for each condition was used for viability assays (see below). The cells were incubated with PBAE-DNA nanoparticles for 4 hr at 37°C. The media and particles were then aspirated and replaced with fresh complete culture medium.
Forty-eight hours after transfections, cells were analyzed for expression levels of delivered genes. For eGFP DNA delivery, cells were trypsinized, centrifuged, and resuspended in PBS with 2% FBS and 5 μg/mL propidium iodide (PI) for flow cytometry. Cells were scanned using an Accuri C6 flow cytometer with an Intellicyt high-throughput 96-well plate sampler. PI+ cells (FL3, emission >670 nm) were excluded from analysis as dead or dying. The percent of cells eGFP+ (FL1, emission 533±15 nm) and the intensity of eGFP expression (geometric mean of total population of cells) were used to measure transfection efficacy. For luciferase DNA delivery, BrightGlo assay (Promega) was used according to manufacturer’s instructions. Luminscence was measured with a Synergy 2 multiplate reader with Gen5 software (Biotek, Winooski, VT) and reported as fold luciferase expression over untreated cells. Polymers with statistically significant specificity for hepatoma cells over hepatocytes, measured by both higher percent of cells transfected and also higher average expression intensity for at least two of three polymer:DNA w/w ratios, were considered top candidates for further studies.
Viability assessments
24 hr after transfections, viability was assessed by metabolic activity and by cell count. For the former, MTS assay was used (CellTiter 96® Aqueous One, Promega, Madison, WI) according to manufacturer’s instructions. Absorbance at 490 nm was measured with a Synergy 2 multiplate reader with Gen5 software (Biotek, Winooski, VT). For cell counts, cells were fixed with 10% buffered formalin (pH 7.4) for 20 min at room temperature. The formalin was removed and the cells washed twice with 1xPBS. Cells were then incubated with 750 nM DAPI in PBS for 5 min at room temperature, shielded from light. The solution was removed, and the stained cells were washed once with 1xPBS. Cells were stored at 4°C covered from light until use. A Zeiss Axio Observer with Axiovision 5 software was used to take one fluorescent image from the center of each well (n=4 for all groups). ImageJ was used to count number of nuclei per well. For both metabolic activity and cell count methods, reported viability is relative to untreated cells. Polymers showing more than 10% cytotoxicity in BRL-3A cells (non-cancer) were removed from consideration as leading materials for further studies.
Hepatocyte and hepatoma co-culture
For co-culture experiments, MCA-RH7777 cells were labeled with nuclear H2B-Cherry in order to be distinguishable from BRL-3A cells. Using a top PBAE from initial MCA-RH7777 transfection experiments (457, 1.1:1; see below for results), 10 μg H2B-Cherry plasmid and 2 μg Piggybac transposase plasmid were delivered to cells in 6-well plates at 50 w/w ratio (457:total DNA) in complete culture medium and incubated for 4 hr at 37°C. The media and particles were aspirated and replaced with fresh medium. The cells were monitored by fluorescence microscopy for H2B-Cherry expression over time and were passaged every 4–7 days as necessary. After 4 wk in culture, the cells were sorted by FACS with 98% purity (Supplemental Figure 1) and were expanded under the same conditions as unlabeled MCA-RH7777 cells as described above.
BRL-3A cells and labeled MCA-RH7777 cells were expanded separately in their respective complete media. Before a transfection, cells were seeded at a 1:1 ratio in 96-well plates (1.5*104 total cells/well) in complete hepatocyte medium and allowed to adhere overnight. Top polymers from initial screenings on eGFP and luciferase transfections on individual cell populations were selected based on expression and viability data. Using these polymers, transfections were carried out as described above. Efficacy was measured by fluorescence microscopy and flow cytometry. For the latter, labeled MCA-RH7777 cells and unlabeled BRL-3A cells were gated using the red fluorescence signal from hepatoma cells on the FL3 channel to minimize overlap with the FL1 (eGFP channel) and utilizing the automated FlowJo 7 gating tool (Treestar, Inc., Ashland, OR), which is based on cell density contours.
Statistics
Results are reported as mean±standard error of the mean (SEM). To compare results of a set of experimental groups to a control, a one-way ANOVA with a post-hoc Dunnett test is utilized. For paired comparisons, two-tailed Student’s t-test were used with a Bonferroni correction for multiple comparisons.
Results
Initial screening transfections on hepatoma cells
A wide variety of PBAE structures was initially tested to find the most promising groups of polymers for transfection of MCA-RH7777 hepatoma cells. Among the variables tested were the base polymer structure (B-S), end-cap monomer (E), polymer:DNA w/w ratio, and polymer chain length (Figure 2).
Figure 2.

Initial screenings of a wide range of different PBAE structures (shown on x-axis). Polymers for further study were chosen based on (A) high transfection efficacy on MCA-RH7777 cells and by (B) low cytotoxicity.
Base polymers found to be most effective in transfection while maintaining low toxicity were 44, 45, and 53. End-cap E7 was the most effective, followed by E6 and E3, respectively. E3 tended to cause the most cytotoxicity and was excluded from further studies except in the case of 453, 1.2:1, which achieved up to 88±3% transfection efficacy with low cytotoxicity. Although 90 w/w was generally more effective than 60 w/w, this was not the case for some polymers, particularly more cytotoxic ones like 54-based polymers; therefore, 50 w/w and 75 w/w were the focus of further studies. Based on results seen in early hepatocyte transfections (see below), 25 w/w was also included.
Lipofectamine™ 2000, X-tremeGENE DNA HP, and PEI were optimized for use as positive controls for transfection (Supplemental Figure 2). While PEI had both low efficacy and high cytotoxicity, the other two lipid-based reagents achieved up to 89±4% and 76±1% transfection, respectively, accompanied by 24±6% and 24±1% toxicity in hepatoma cells.
Transfection of hepatocytes and hepatoma cells with purified polymers
Purified polymers generally had higher efficacy than their unpurified counterparts (Figure 3), with 457e, 1.1:1 transfecting up to 98±0.4% MCA-RH7777 cells and 68±4% BRL-3A cells. Significant specificity was seen between the two cell types for most polymers. The highest transfection efficacy seen in BRL-3A cells was 77±1% (537e, 1.05:1, 75 w/w), while 10 of the 21 PBAE formulations evaluated transfected >90% of the MCA-RH7777 cells. One exception is the relatively high transfection of BRL-3A cells with low w/w ratios (25 w/w) compared to the low MCA-RH7777 transfection, particularly 457e, 1.1:1 and 1.2:1, with this discrepancy statistically significant in the former.
Figure 3.
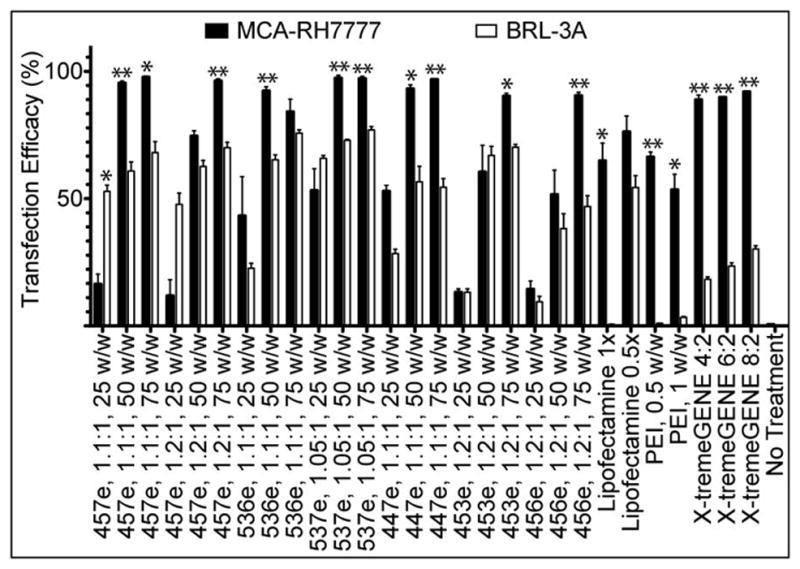
When cultured and transfected separately, most of the PBAEs and controls tested showed statistically significantly higher rate of transfection in hepatoma cells (MCA-RH7777) than in non-cancer hepatocytes (BRL-3A) (*α=0.05, **α=0.01).
Much more striking than the difference in percent transfection is the difference in the amount of protein produced, measured as eGFP fluorescence intensity (Figure 4). Fluorescence micrographs show that much higher exposure was needed to visualize eGFP+ BRL-3A hepatocytes compared with the very intensely bright eGFP+ MCA-RH7777 hepatoma cells. Furthermore, the geometric mean fluorescence of hepatoma cells as measured by flow cytometry was significantly higher in many of the PBAE formulations tested. For polymers 457e, 1.1:1; 537e, 1.05:1; 447e, 1.1:1; 453e, 1.2:1; and 456e, 1.2:1, at least 10-fold higher eGFP expression was seen in hepatoma cells when used at both 50 and 75 w/w, with some conditions showing up to 330±96-fold higher expression.
Figure 4.

Hepatoma cells show much higher transgene expression compared with hepatocytes as measured by (A) flow cytometry and by (B) fluorescence microscopy. All micrographs were taken with the same exposure time (200 ms) on the green fluorescence channel.
As can be seen in Figure 4B, certain groups caused some cell death. Viability was tested in two ways to ensure that the polymers used for later studies would not cause non-specific cytotoxicity, particularly in non-cancer BRL-3A cells. While the CellTiter 96® MTS assay was useful as a quick assessment in the initial large screens, several groups, especially in the MCA-RH7777 cells, showed greater metabolic activity than untreated wells (Supplemental Figure 3). Although cell counts also show a slight (but not statistically significant) increase in many groups, the increased MTS signal was higher still. Therefore, cell counts were used as the method of choice for determining polymer-mediated non-specific cytotoxicity (Figure 5).
Figure 5.
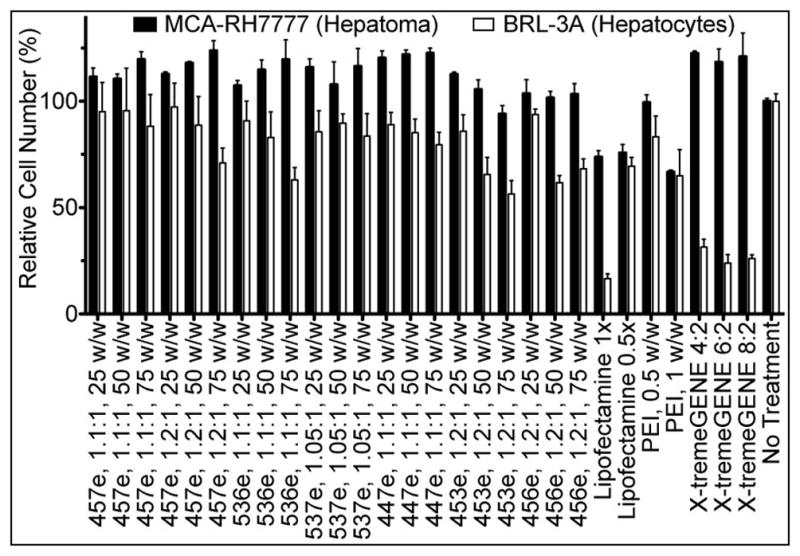
Viability of MCA-RH7777 cells and BRL-3A cells was assessed by automated cell counting 24 hr after transfection with eGFP DNA.
In agreement with what was observed with unpurified polymers, the ether-purified E3-terminated polymer (453e, 1.2:1) caused the highest cytotoxicity of the polymers tested in both MCA-RH7777 cells and BRL-3A cells. While hepatoma cells and hepatocytes followed similar trends, with the hepatoma cells generally somewhat more robust than the hepatocytes, there were certain compounds for which this was not the case. In particular, the two lipid-based positive controls, Lipofectamine™ 2000 and X-tremeGENE® DNA HP, were highly effective for DNA delivery to hepatoma cells but were also very cytotoxic to non-cancer hepatocytes. Because the cytotoxicity of transfection reagents to healthy cells was considered a primary safety concern, only polymers that caused <10% cytotoxicity to BRL-3A cells continued to be used in the following studies.
Luciferase DNA Transfection
To ensure that the high rate of transfection in MCA-RH7777 cells and specificity over BRL-3A cells was not affected by any elements of the plasmid used or the eGFP marker protein, transfection experiments were repeated with luciferase DNA with a different vector backbone using top polymers from eGFP studies (Figure 6). Interestingly, the cell specificity became even more clear using the luminescence assay, with many polymers causing at least 200-fold higher luminescence in MCA-RH7777 cells compared with BRL-3A cells when used at 50 and 75 w/w. 537e, 1.05:1, 75 w/w showed the highest expression as well as the most cell-type specificity, with up to 470 ±25-fold more luminescence in hepatoma cells over hepatocytes.
Figure 6.
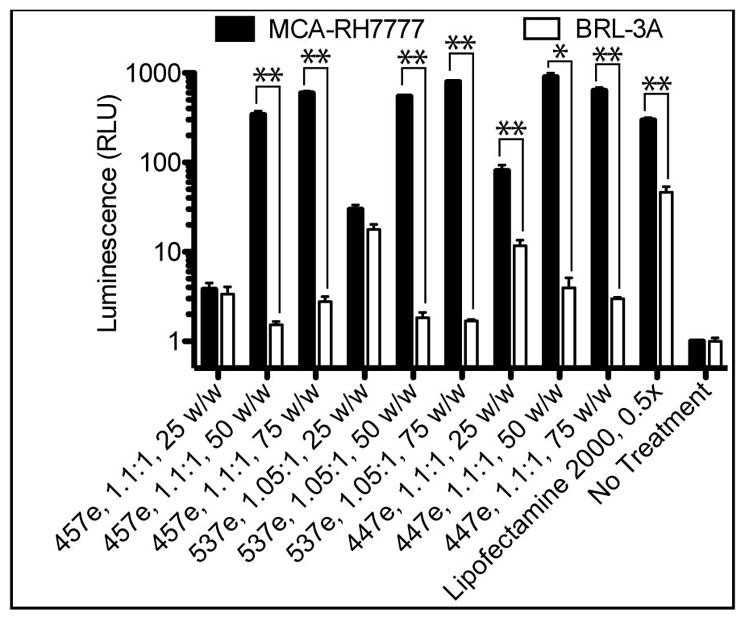
MCA-RH7777 cells and BRL-3A cells cultured and transfected separately with luciferase DNA showed high specificity for hepatoma cells over hepatocytes, measured by luminescence per well.
Co-culture
The cell-type specificity observed in monoculture is also strongly observed in co-culture experiments. In addition to the Cherry dye, labeled MCA-RH7777 cells could be distinguished from BRL-3A cells by morphology of the individual cells as well as the multicellular structures formed by each cell type. Even though both cell types spread on a 2D surface when grown separately, forming relatively even monolayers, the hepatoma cells formed dense clusters when in co-culture with hepatocytes, which themselves remained in monolayer (Figure 7).
Figure 7.
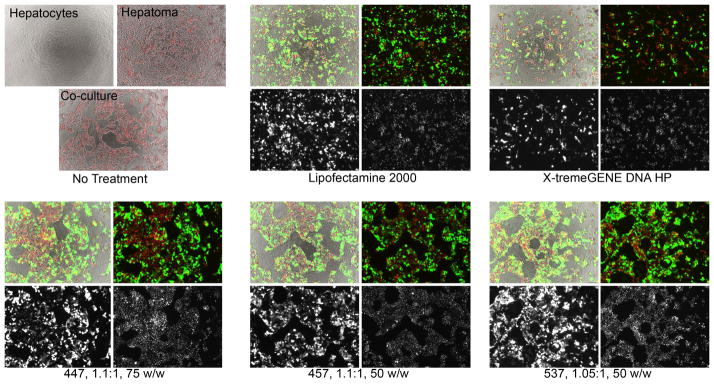
Labeled MCA-RH7777 cells and unlabeled BRL-3A cells grown and transfected in co-culture showed high transgene expression in hepatoma cells with much lower levels of transfection in hepatocytes. Brightfield images are uncolored, eGFP signal is green, and H2B-cherry signal is red. Each set of four images shows brightfield merged with eGFP and H2B-cherry (top left), eGFP and H2B-cherry (top right), eGFP only (bottom left), and H2B-cherry only (bottom right).
In co-cultures, hepatoma cells were preferentially transfected over hepatocytes with very high specificity. Fluorescence microscopy showed that green eGFP signal overlapped almost exclusively with the clusters of red H2B-Cherry signal, indicating that transfection occurred largely in hepatoma cells and infrequently in hepatocytes. Close inspection of the images does reveal some BRL-3A cells that are eGFP+; however, the expression intensity is once again very low, and relatively few cells were transfected compared to hepatoma cells. This co-culture experiment also supported the previous findings that Lipofectamine and X-tremeGENE, while relatively effective at transfecting hepatoma cells, were also quite toxic to hepatocytes.
Flow cytometry quantitatively showed similar results (Figure 8; see Supplemental Figure 4 for detailed gating information). In the case of all PBAEs tested, transfection efficacy was statistically significantly higher for hepatoma cells compared with hepatocytes (α=0.01) measured both by percent of cells transfected and by eGFP fluorescence intensity. This is true even for 25 w/w formulations, which did not usually show statistically higher specificity for hepatoma cells in when the two cell types were cultured in monoculture separately. In the least effective group, hepatoma cells were transfected at 83±1% and 160±24-fold higher fluorescence intensity over hepatocytes (457e, 1.1:1, 25 w/w). The best conditions had up to 98±0.3% transfection (537e, 1.05:1, 50 w/w) and up to 528±57-fold higher intensity (457e, 1.1:1, 50 w/w). While the highest increase in fluorescence over hepatocytes was 330-fold when cells were cultured separately, both 457e and 447e, 1.1:1, at 50 and 75 w/w showed higher specificity in co-culture.
Figure 8.

(A) H2B-cherry signal of hepatoma cells allowed separate gating of the two populations with flow cytometry. The top population (tinted pink) shows H2B-cherry+ hepatoma cells; the bottom (tinted yellow) is unlabeled hepatocytes. Transfection efficacy measured by (B) percent of cells transfected and (C, D) geometric mean fluorescence intensity showed high specificity for MCA-RH7777 cells over BRL-3A cells in co-culture studies.
Discussion
In this study, PBAEs were evaluated for their potential as specific gene delivery agents to MCA-RH7777 hepatoma cells, minimizing the off-target effect on healthy BRL-3A hepatocytes. At the dosages and formulations tested, low cytotoxicity was seen in hepatoma cells for most of the polymers tested. Interestingly, the two most effective commercially-available controls, Lipofectamine and X-tremeGENE, are very cytotoxic to hepatocytes. This may suggest that, while malignant hepatocytes can survive when treated with lipid-based materials at the doses tested and may even have high transfection, healthy hepatocytes are more susceptible to cell death in such conditions. This does not contradict previous results that showed increased luminescence from MCA-RH7777 cells transfected in the presence with Lipiodol® but no change or decrease in luminescence from BRL-3A cells,6 and it further emphasizes the importance of delivery vectors that can be modified or tailored for a specific application or organ system.
The ability of PBAEs to target certain disease cell types more than healthy cell types has been shown. Although the mechanism of this specificity is not fully understood, increased uptake by hepatoma cells of certain compounds has been attributed at least in part to differences in cell surface molecules in different cell types,20 with previous work on HCC-specific drug delivery focusing primarily on the use of targeting ligands for receptors overexpressed on hepatoma cells.21–23 Many current cancer therapeutics achieve some degree of specificity because of the shorter division cycle of cancer cells; this is not the case in the current study, as measurement of MCA-RH7777 and BRL-3A cells showed doubling times of 24 hr and 20 hr, respectively (Supplemental Figure 5). Other factors may play a role; for example, we showed here that MCA-RH7777 cells have increased metabolic activity after transfection with certain polymers that cannot be fully explained by a slight increase in proliferation (Supplemental Figure 3). Many obstacles must normally be overcome to achieve successful gene delivery,24 each of which could vary from one cell type to another to cause the preferential transfection of hepatoma cells seen here. Regardless of which steps in the delivery or gene expression pathway are the major bottlenecks, the specificity with which hepatoma cells are transfected is promising for further mechanistic study or for in vivo experiments.
The high specificity for delivery to liver cancer while avoiding healthy liver cells in co-culture is intriguing. This experiment more closely approximates the in vivo situation, in which tumors and non-cancerous liver tissue would both be present and exposed simultaneously to a therapeutic. The difference in expression between hepatoma and hepatocytes is larger when the two cells are transfected together in the same well.
The potential of this technology for clinical translation is supported by a number of factors. First, there is low or insignificant cytotoxicity caused by the leading polymer formulations identified in this study. Gene delivery is also inherently versatile in the types of therapeutic proteins that can be delivered, because, as we have previously shown, PBAEs form the same nanoparticles regardless of the sequence of the DNA plasmid or its size within a range of <2 kb to >25 kb, allowing virtually any gene to be delivered.19 Additional layers of specificity can also be built into the DNA construct being delivered, such as promoter control to allow expression only in cancer cells,25–27 with some promoters causing several-fold to nearly an order of magnitude higher expression in cancer cells over healthy controls. Using cytomegalovirus (CMV) promoter for our plasmids, with no transcriptional targeting, we were able to achieve approximately 500-fold higher expression in hepatoma cells using two different plasmids expressing different marker proteins. Significantly, well over 90% of hepatoma cells are using our top PBAE polymers. The ability of a cancer therapeutic to kill all of the cells in a tumor is imperative to its efficacy, as tumor recurrence is an important factor in many cases of hepatoma.28 Even without additional engineering of a therapeutic plasmid, our system could provide a safe, effective, and innately specific method to deliver DNA. We also show here that PBAEs are effective for delivery to hepatocytes in isolation from hepatoma cells, with over 70% transfection in some cases, though with low levels of expression; these PBAE-based nanoparticles could be an attractive method of treating other hepatic diseases as well.
Conclusion
We have synthesized and identified PBAEs that are highly effective for DNA delivery to hepatoma cells that normally demonstrate low transfection efficiency as well as low cytotoxicity in healthy hepatocytes. A high degree of specificity is observed when transfecting with either of the two DNA plasmids tested as well as in a co-culture setting with both hepatoma cells and hepatocytes exposed to the PBAE nanoparticles at the same time. This work represents nanobiotechnology that has high potential for eventual translation to in vivo or clinical applications in the field of cancer gene delivery.
Supplementary Material
Acknowledgments
This work was supported in part by the NIH (R21CA152473) and CCNE (U54 CA151838). Labeled MCA-RH7777 cells were sorted by flow cytometry by Drs. Hao Zhang and Joseph Margolick at the Johns Hopkins Bloomberg School of Public Health using MoFlo (Beckman Coulter). SYT thanks the National Science Foundation for fellowship support.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.El-Serag HB. Hepatocellular carcinoma: Recent trends in the United States. Gastroenterology. 2004;127(5):S27–S34. doi: 10.1053/j.gastro.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 3.Llovet JM, Di Bisceglie AM, Bruix J, Kramer BS, Lencioni R, Zhu AX, Sherman M, Schwartz M, Lotze M, Talwalkar J, et al. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst. 2008;100(10):698–711. doi: 10.1093/jnci/djn134. [DOI] [PubMed] [Google Scholar]
- 4.Marelli L, Stigliano R, Triantos C, Senzolo M, Cholongitas E, Davies N, Tibballs J, Meyer T, Patch DW, Burroughs AK. Transarterial therapy for hepatocellular carcinoma: which technique is more effective? A systematic review of cohort and randomized studies. Cardiovasc Intervent Radiol. 2007;30(1):6–25. doi: 10.1007/s00270-006-0062-3. [DOI] [PubMed] [Google Scholar]
- 5.Huppert P. Current concepts in transarterial chemoembolization of hepatocellular carcinoma. Abdominal Imaging. 2011;36(6):677–83. doi: 10.1007/s00261-011-9755-4. [DOI] [PubMed] [Google Scholar]
- 6.Higgins LJ, Hwang GL, Rosenberg J, Katzenberg RH, Kothary N, Sze DY, Hofmann LV. In vitro design and characterization of the nonviral gene delivery vector iopamidol, protamine, ethiodized oil reagent. J Vasc Interv Radiol. 2011;22(10):1457–1463. e2. doi: 10.1016/j.jvir.2011.06.025. [DOI] [PubMed] [Google Scholar]
- 7.Putnam D. Polymers for gene delivery across length scales. Nature Materials. 2006;5(6):439–51. doi: 10.1038/nmat1645. [DOI] [PubMed] [Google Scholar]
- 8.Viola JR, El-Andaloussi S, Oprea II, Smith CIE. Non-viral nanovectors for gene delivery: factors that govern successful therapeutics. Expert Opinion on Drug Delivery. 2010;7(6):721–735. doi: 10.1517/17425241003716810. [DOI] [PubMed] [Google Scholar]
- 9.Sunshine JC, Bishop CJ, Green JJ. Advances in polymeric and inorganic vectors for nonviral nucleic acid delivery. Therapeutic Delivery. 2011;2(4):493–521. doi: 10.4155/tde.11.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Green JJ. 2011 Rita Schaffer Lecture: Nanoparticles for Intracellular Nucleic Acid Delivery. Annals of Biomedical Engineering. 2012;40(7):1408–18. doi: 10.1007/s10439-012-0550-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynn DM, Langer R. Degradable poly(beta-amino esters): Synthesis, characterization, and self-assembly with plasmid DNA. J Am Chem Soc. 2000;122:10761–10768. [Google Scholar]
- 12.Tzeng SY, Guerrero-Cazares H, Martinez EE, Sunshine JC, Quinones-Hinojosa A, Green JJ. Non-viral gene delivery nanoparticles based on Poly(beta-amino esters) for treatment of glioblastoma. Biomaterials. 2011;32(23):5402–10. doi: 10.1016/j.biomaterials.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhise NS, Gray RS, Sunshine JC, Htet S, Ewald AJ, Green JJ. The relationship between terminal functionalization and molecular weight of a gene delivery polymer and transfection efficacy in mammary epithelial 2-D cultures and 3-D organotypic cultures. Biomaterials. 2010;31(31):8088–96. doi: 10.1016/j.biomaterials.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tzeng SY, Hung BP, Grayson WL, Green JJ. Cystamine-terminated poly(beta-amino ester)s for siRNA delivery to human mesenchymal stem cells and enhancement of osteogenic differentiation. Biomaterials. 2012;33(32):8142–51. doi: 10.1016/j.biomaterials.2012.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sunshine JC, Sunshine SB, Bhutto I, Handa JT, Green JJ. Poly(beta-Amino Ester)-Nanoparticle Mediated Transfection of Retinal Pigment Epithelial Cells In Vitro and In Vivo. Plos One. 2012;7(5) doi: 10.1371/journal.pone.0037543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green JJ, Zugates GT, Tedford NC, Huang Y, Griffith LG, Lauffenburger DA, Sawicki JA, Langer R, Anderson DG. Combinatorial modification of degradable polymers enables transfection of human cells comparable to adenovirus. Advanced Materials. 2007;19(19):2836–2842. [Google Scholar]
- 17.Shmueli RB, Sunshine JC, Xu Z, Duh EJ, Green JJ. Gene delivery nanoparticles specific for human microvasculature and macrovasculature. Nanomedicine. 2012 doi: 10.1016/j.nano.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sunshine J, Green JJ, Mahon K, Yang F, Eltoukhy A, Nguyen DN, Langer R, Anderson DG. Small molecule end group of linear polymer determine cell-type gene delivery efficacy. Advanced Materials. 2009;21(48):4947–4951. doi: 10.1002/adma.200901718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tzeng SY, Green JJ. Subtle changes to polymer structure and degradation mechanism enable highly effective nanoparticles for siRNA and DNA delivery to human brain cancer. Advanced Healthcare Materials. 2012 doi: 10.1002/adhm.201200257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chou FI, Fang KC, Chung C, Lui WY, Chi CW, Liu RS, Chan WK. Lipiodol uptake and retention by human hepatoma cells. Nuclear Medicine and Biology. 1995;22(3):379–86. doi: 10.1016/0969-8051(94)00112-w. [DOI] [PubMed] [Google Scholar]
- 21.Mohr L, Yeung A, Aloman C, Wittrup D, Wands JR. Antibody-directed therapy for human hepatocellular carcinoma. Gastroenterology. 2004;127(5 Suppl 1):S225–31. doi: 10.1053/j.gastro.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 22.Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, Hanna TN, Liu J, Phillips B, Carter MB, et al. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nature Materials. 2011;10(5):389–97. doi: 10.1038/nmat2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolschek MF, Thallinger C, Kursa M, Rossler V, Allen M, Lichtenberger C, Kircheis R, Lucas T, Willheim M, Reinisch W, et al. Specific systemic nonviral gene delivery to human hepatocellular carcinoma xenografts in SCID mice. Hepatology. 2002;36(5):1106–14. doi: 10.1053/jhep.2002.36372. [DOI] [PubMed] [Google Scholar]
- 24.Green JJ, Langer R, Anderson DG. A combinatorial polymer library approach yields insight into nonviral gene delivery. Accounts of Chemical Research. 2008;41(6):749–759. doi: 10.1021/ar7002336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim YI, Ahn BC, Ronald JA, Katzenberg R, Singh A, Paulmurugan R, Ray S, Gambhir SS, Hofmann LV. Intratumoral versus intravenous gene therapy using a transcriptionally targeted viral vector in an orthotopic hepatocellular carcinoma rat model. J Vasc Interv Radiol. 2012;23(5):704–11. doi: 10.1016/j.jvir.2012.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhang HE, Gabrielson KL, Laterra J, Fisher PB, Pomper MG. Tumor-specific imaging through progression elevated gene-3 promoter-driven gene expression. Natural Medicines. 2011;17(1):123–9. doi: 10.1038/nm.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su ZZ, Sarkar D, Emdad L, Duigou GJ, Young CS, Ware J, Randolph A, Valerie K, Fisher PB. Targeting gene expression selectively in cancer cells by using the progression-elevated gene-3 promoter. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(4):1059–64. doi: 10.1073/pnas.0409141102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanazaki K, Kajikawa S, Shimozawa N, Mihara M, Shimada K, Hiraguri M, Koide N, Adachi W, Amano J. Survival and recurrence after hepatic resection of 386 consecutive patients with hepatocellular carcinoma. J Am Coll Surg. 2000;191(4):381–8. doi: 10.1016/s1072-7515(00)00700-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.