

Abstract
Purpose: Rosuvastatin is a poorly water soluble drug and the rate of its oral absorption is often controlled by the dissolution rate in the gastrointestinal tract. Hence it is necessary to increase the solubility of the Rosuvastatin.
Methods: Several liquisolid tablets formulations containing various drug concentrations in liquid medication (ranging from 15% to 25% w/w) were prepared. The ratio of Avicel PH 102 (carrier) to Aerosil 200 (coating powder material) was kept 10, 20, 30. The prepared liquisolid systems were evaluated for their flow properties and possible drug-excipient interactions by Infrared spectra (IR) analysis, differential scanning calorimetry (DSC) and X- ray powder diffraction (XRPD).
Results: The liquisolid system showed acceptable flow properties. The IR and DSC studies demonstrated that there is no significant interaction between the drug and excipients. The XRPD analysis confirmed formation of a solid solution inside the compact matrix. The tabletting properties of the liquisolid compacts were within the acceptable limits. Liquisolid compacts demonstrated significantly higher drug release rates than those of conventional and marketed tablet due to increasing wetting properties and surface area of the drug.
Conclusion: This study shows that liquisolid technique is a promising alternative for improvement of the dissolution rate of water insoluble drug.
Keywords: Rosuvastatin calcium, Liquisolid compacts, Liquid load factor, Excipient ratio, Tablets, Dissolution rate
Introduction
Solubility is one of the important parameters to achieve desired concentration of drug in systemic circulation for achieving required pharmacological response. Poorly water soluble drugs often require high doses in order to reach therapeutic plasma concentrations after oral administration. Low aqueous solubility is the major problem with formulation development of new chemical entities. Water is the solvent of choice for liquid pharmaceutical formulations. Most of the drugs are either weakly acidic or weakly basic having poor aqueous solubility. A great number of new and possibly beneficial chemical entities do not have suitable pharmaceutical dosage form because of their poor solubility and poor dissolution rate. The oral absorption of drugs is most often controlled by dissolution in the gastrointestinal tract.1
Rosuvastatin calcium is a BCS class II drug used as a lipid lowering agent by acting as HMG CoA reductase inhibitor.2 Different methods are employed to improve the dissolution characteristics of poorly water soluble drugs like solubilization, pH adjustment, cosolvents, microemulsion, self emulsification, polymeric modification, drug complexation, particle size reduction, use of a surfactant as a solubilizing agent, the pro-drug approach and solid solutions.3 Amongst these, the most promising method for promoting dissolution is the use of the liquisolid system (LS).4-13 Liquisolid systems are acceptably flowing and compressible powdered forms of liquid medications. The term ‘liquid medication’ involves oily liquid drugs and solutions or suspensions of water insoluble solid drugs carried in suitable nonvolatile solvent systems termed liquid vehicles. Employing this liquisolid technique, a liquid medication may be converted into a dry-looking, non-adherent, free flowing and readily compressible powder by simple blending with selected powder excipients referred to as carrier and coating materials.
To attain the flowability and compressibility of liquisolid compacts, the ‘‘mathematical model for liquisolid systems” was employed as follows to calculate the appropriate quantities of excipients required to produce liquisolid systems of acceptable flowability and compressibility. Various grades of cellulose, starch and lactose may be used as the carriers, whereas very fine particle size silica powders may be used as the coating (or covering) materials.14
Due to low solubility, Rosuvastatin shows low bioavailability. Various approaches to enhance dissolution properties of Rosuvastatin are complexation with ß-cyclodextrin, solid dispersion, hydrotropy, micellar solibilisation and microemulsion.2,3
Therefore, the present work is aimed towards enhancing the solubility, dissolution and thereby the bioavailability of Rosuvastatin by using liquisolid compact technology.
Materials and Methods
Materials
The following gift samples were received: Rosuvastatin (Biocon Pvt Ltd. Benglore, India); Avicel PH 102 and Sodium starch glycolate (Maple biotech Pvt. Ltd. Pune, India); Aerosil 200( Research lab Pune India) propylene glycol (PG), polyethylene glycol 400 (PEG400), polyethylene glycol (PEG200) and Acetonitrile (Research lab, Pune, India). All reagents used were of analytical grade.
Saturation solubility studies
Solubility studies of Rosuvastatin were carried out in distilled water, propylene glycol, PEG 400 and PEG 200. Saturated solutions prepared in above vehicles were kept in an orbital shaker (Remi motors Pvt. Ltd Mumbai, India.) for 72 h at 25 °C. The solutions were filtered and their concentration was determined by UV-spectrophotometry (Shimadzu Corporation Pvt. Ltd. Nishinokyo-Kuwabara-cho, Nakagyo-ku, Kyoto 604-8511, Japan ) at 241 nm. The results were determined as the percent w/w of Rosuvastatin in its saturated solution with the solvent under investigation.
Application of a mathematical model for liquisolid system
To attain the flowability and compressibility of liquisolid compacts, the ‘‘new formulation mathematical model of liquisolid systems” was employed as follows to calculate the appropriate quantities of excipients required to produce liquisolid systems of acceptable flowability and compressibility. This mathematical model was based on new fundamental powder properties (constants for each powder material with the liquid vehicle) called the flowable liquid retention potential (Φ-value) and compressible liquid retention potential (Ψ-number) of the constituent powders (carrier and coating materials) according to Spireas et al (Spireas. 2002; Spireas et al. 1999). According to the new theories, the carrier and coating powder materials can retain only certain amounts of liquid while maintaining acceptable flow and compression properties. Depending on the excipients ratio (R) or the carrier: coating ratio of the powder system used, where
R =Q/q ... (1)
As R represents the ratio between the weights of carrier (Q) and coating (q) materials present in the formulation. An acceptably flowing and compressible liquisolid system can be prepared only if a maximum liquid on the carrier material is not exceeded; such a characteristic amount of liquid is termed the liquid load factor (Lf) and defined as the ratio of the weight of liquid medication (W) over the weight of the carrier powder (Q) in the system, which should be possessed by an acceptably flowing and compressible liquisolid system. i.e.
Lf = W/Q ... (2)
Spireas et al. used the flowable liquid retention potentials (Φ -values) of powder excipients to calculate the required ingredient quantities. Hence the powder excipients ratios R and liquid load factors Lf of the formulations are related as follows:
Lf = Φ +Φ (1/R) ... (3)
So, in order to calculate the required weights of the excipients used, first, from equation (3), Φ and Φ are constants. Therefore, according to the ratio of the carrier/ coat materials (R), Lf was calculated from the linear relationship of Lf versus 1/R. Next, according to the used liquid vehicle concentration, different weights of the liquid drug solution (W) will be used. So, by knowing both Lf and W, the appropriate quantities of carrier (Q) and coating (q) powder materials required to convert a given amount of liquid medication (W) into an acceptably flowing and compressible liquisolid system, could be calculated from equations (1) and (2).15
Preparation of conventional tablet and liquisolid compacts
A conventional formulation of micronized Rosuvastatin calcium (denoted as DC) was directly compressed into cylindrical tablets, each containing 5 mg drug. In addition, each DC tablet contained the following powder excipients: 140 mg Avicel PH 102, 70 mg lactose monohydrate, 10 mg Aerosil 200, and 20 mg sodium starch glycolate. A 10 tablet batch was mixed in a mortar for 10 min. and the final admixture was compressed using a manual compression machine (Cip Machinaries.Pvt. Ltd. Ahrmadabad, Gujarat). Various liquisolid compacts containing 5 mg Rosuvastatin were prepared by dispersing in nonvolatile vehicles such as PEG 200. Then a binary mixture of carrier (Avicel PH 102) and coating material (Aerosil-200) was prepared at a ratio of 20:1,10:1 and 30:1. This binary mixture was added to the admixture of drug and vehicle. From the calculated Φ-value, the liquid load factor (Lf) was calculated.16 Depending upon the drug concentration in liquid medication, different liquid load factors were employed in liquisolid preparations. Different concentrations of Avicel and silica were used to prepare different liquisolid formulations. Finally, sodium starch glycolate as a disintegrant was added to the above powder blend and mixed. The final powder blend was subjected to compression by using manual compression machine (Cip Machinaries. Pvt. Ltd. Ahemadabad, Gujarat.). The composition of the liquisolid compacts are shown in Table 1.
Table 1. Composition of the liquisolid compacts .
| Drug in conc of PEG 200 | Batch | R value | Drug (mg) | Liquid load (Lf) | Avicel PH 102 (mg) | Aerosil (mg) | SSG (mg) | PEG 200 (mg) | Unit dose (mg) |
| F1 | 10 | 5 | 0.326 | 102.24 | 10.22 | 7.28 | 28.33 | 153.07 | |
| 15% | F2 | 20 | 5 | 0.163 | 204 | 10.20 | 12.37 | 28.33 | 259.9 |
| F3 | 30 | 5 | 0.109 | 305.77 | 10.19 | 17.46 | 28.33 | 366.75 | |
| 20% | F4 | 10 | 5 | 0.326 | 76.68 | 7.66 | 5.44 | 20 | 114.78 |
| F5 | 20 | 5 | 0.163 | 218.57 | 10.92 | 12.72 | 20 | 367.21 | |
| F6 | 30 | 5 | 0.109 | 229.35 | 7.645 | 13.09 | 20 | 275.08 | |
| 25% | F7 | 10 | 5 | 0.326 | 61.34 | 6.13 | 4.37 | 15 | 191.84 |
| F8 | 20 | 5 | 0.163 | 122.69 | 6.13 | 7.44 | 15 | 156.231 | |
| F9 | 30 | 5 | 0.109 | 183.48 | 6.11 | 10.47 | 15 | 220.06 |
Pre-compression studies of the liquisolid system
Flow properties of the liquisolid system
The flow properties of the liquisolid systems were estimated by determining the angle of repose, Carr’s index and Hausner’s ratio. The angle of repose was measured by the fixed funnel method. The bulk density and tap density were determined for the calculation of Hausner’s ratio and Carr’s index.17
Infra red spectra analysis
IR spectrum of optimized formulation was recorded by KBr method using Jasco M4100 Fourier Transform InfraRed spectrophotometer. A baseline correction was made by using dried potassium bromide and then the spectrum of powder with potassium bromide was recorded. Sample was scanned from 4000 to 400 cm-1. The compatibility of drug and other excipients in formulation was confirmed by comparing drug and formulation spectra.
X-ray powder diffraction
Crystallinity study was carried out by comparing XRD spectrum of drug with formulation to check peak of drug in individual state and in formulation. Study was carried out on Elementer Vario E L III XRD Sophisticated Analytical Instrument Facility, at Cochin. The data was recorded at 2θ range of 10 to 60 °C at time of 0.5 sec. the relative intensity I/I0 and inter- planar distance (d) corresponding to 2θ value were reported and compared.
Differential scanning calorimetry (DSC)
Thermograms of the rosuvastatin and its liquisolid formulations were recorded on Mettler STAR SW 9.01 instruments. The analysis was carried out by heating 2 to 3 mg of sample on an aluminum crimp pan at rate of 10 °C/min in nitrogen atmosphere.
In vitro evaluation of liquisolid compacts
Content uniformity of rosuvastatin liquisolid tablets
Ten tablets from each batch were taken randomly to examine its content uniformity. Each tablet was weighed and crushed individually. The crushed tablet powders were dissolved in acetonitrile water system. The solution was filtered using Whatman filter paper.The drug content was measured using UV spectrophotometer (Shimadzu corporation Pvt. Ltd Nishinokyo-Kuwabara-cho, Nakagyo-ku, Kyoto 604-8511, Japan ) at 241 nm.
Weight variation test
Weight variation test was performed as per USP.18
Hardness and friability
The hardness of formulated liquisolid tablets was assessed using a Monsanto hardness tester and the mean hardness of three tablets was determined. The friability of the prepared liquisolid tablets was measured in a Roche type apparatus (Electrolab Pvt. Ltd Mumbai, India.) and the percentage loss in weight was calculated and used as a measure of friability.
Disintegration test
The disintegration test was carried out using disintegration test apparatus as specified in the Indian Pharmacopoeia.
In vitro dissolution studies
The in-vitro release profiles of rosuvastatin from liquisolid compacts and directly compressed tablets were obtained using a dissolution test apparatus USP-II (Electrolab Electrolab Pvt. Ltd Mumbai, India.)The dissolution study was carried out in 900 ml phosphate buffer pH 6.8 as the dissolution medium at 37 °C ± 2 °C and 50 rpm. Then 5 ml samples were collected for up to 60 min at 5-min intervals . The dissolution medium was replaced with 5 ml fresh dissolution fluid to maintain sink conditions. The withdrawn samples were filtered and analyzed spectrophotometrically at 241 nm. The mean of three determinations was used to calculate the drug release from each of the formulations.18
Estimation of fraction of molecularly dispersed drug
The fraction (FM) of the dissolved or molecularly dispersed drug in the liquid medication is the ratio the drug’s saturation solubility (CL) in the liquid vehicle over the drug concentration (Cd) in the liquid medication.
FM = CL / Cd (1)
(Where FM = 1 when CL / Cd > 1)
The fractions of the molecularly dispersed drug in any system cannot exceed unity.
Effect of aging on tabletting properties
The study was performed under accelerated stability conditions at 40 °C ± 2 °C/75% RH ± 5% RH for three months.
Results and Discussion
UV analysis and solubility study
Rosuvastatin in acetonitrile-water solution obeyed Beer’s law and displayed linearity over the concentration range tested from 2–20 μg/ml. The solubility of the drug was studied in different solvents like propylene glycol, polyethylene glycol 200, polyethelene glycol 400, distilled water, Phosphate Buffer pH 6.8. Saturation solubility of rosuvastatin in various solvent is given in Table 2.
Table 2. Saturated solubility of rosuvastatin in various solvent.
| Solvent | Solubility (%w/w) |
| Distilled water | 0.00000043±0.037 |
| Buffer ph 6.8 | 0.0025±0.075 |
| PEG400 | 4.55±0.115 |
| Propylene glycol | 9.96 ±0.180 |
| PEG200 | 11.57 ±0.205 |
Selection of non volatile solvent
Rosuvastatin has highest solubility in PEG 200. Since the aim of this study was to enhance the dissolution rate of drug, PEG 200 was chosen as the non volatile liquid vehicle for formulation of liquisolid compacts of Rosuvastatin.
Application of a mathematical model for liquisolid system
To determine the quantities of the ingredients of the compacts,, the flowable liquid-retention potentials (Φ-values) and liquid load factor(Lf value) and the carrier to coating ratio (R value).
Determination of flowable liquid-retention potential (Φ-values)
‘‘Angle of slide” measurement was used to evaluate the flow property of powder excipients (Avicel PH102 and Aerosil20 with PEG200). Several uniform liquid vehicle/powder admixtures which contain 10 g of the carrier or coating materials with increasing amounts of liquid vehicle (PEG200) were prepared. To measure the angle of slide, the prepared liquid/powder admixtures were placed on polished metal plates, the plate was then tilted gradually until the liquid/powder admixture was about to slide. The angle formed between the plate and the horizontal surface was defined as the angle of slide (h). The flow properties of excipients will be change due to adsorption of the liquid vehicle. The flowable liquid-retention potential (Φ values) of each liquid/powder admixture was calculated using the following equation.
Φ value=weight of liquid/weight of solid
The Φ -values for Avicel PH102 and Aerosil with PEG 200 were 0.007 and 3.26 respectively.
Determination of liquid load factor
Using the Φ –values the liquid load factor, Lf, was calculated according to equation
Lf = Φ +Φ (1/R)
The Lf values were used to calculate the required quantities of excipients.
Pre-compression studies of the liquisolid system
Flow properties of the Rosuvastatin liquisolid system
The flow properties of the liquisolid powder system are influenced by physical, mechanical as well as environmental factors. Therefore, different flow parameters were employed and results are depicted in Table 3. Batch F3 showed good flow properties with a θ value of 30.02 and was considered as the liquisolid system with acceptable flowability. Carr’s index up to 16 was considered acceptable as a flow property. Hausner’s ratio was related to the inter particle friction; powders with a low interparticle friction had a ratio of approximately 1.25 indicating a good flow. Batch F3 with a Carr’s index of 12.10 and a Hausner’s ratio of 1.17 was considered for further study for preparing batches of rosuvastatin liquisolid compacts.
Table 3. Flow properties of rosuvastatin liquisolid system .
| Batch | Tap density (gm/cm3) | Bulk density (gm/cm3) | Angle Of repose (θ) | Cars index | Hausner’s ratio |
| F1 | 0.502±0.04 | 0.421±0.04 | 28.14±1.46 | 16.14±2.78 | 1.18±0.03 |
| F2 | 0.472±0.01 | 0.410±0.009 | 29.14±1.03 | 14.12±1.59 | 1.16±0.02 |
| F3 | 0.503±0.17 | 0.442±0.01 | 30.02±1.88 | 12.10±0.30 | 1.13±0.005 |
| F4 | 0.456±0.02 | 0.390±0.042 | 30.10±2.30 | 16.25±0.791 | 1.16±0.05 |
| F5 | 0.497±0.009 | 0.437±0.007 | 29.66±1.15 | 11.98±0.18 | 1.13±0.00 |
| F6 | 0.417±0.009 | 0.379±0.007 | 30.94±0.554 | 11.29±1.55 | 1.12±0.023 |
| F7 | 0.465±0.01 | 0.386±0.01 | 31.77±1.74 | 16.33±2.20 | 1.2±0.04 |
| F8 | 0.620±0.03 | 0.516±0.02 | 32.85±0.39 | 16.33±0.62 | 1.19±0.01 |
| F9 | 0.497±0.01 | 0.406±0.01 | 25.49±1.53 | 18.33±0.433 | 1.22±0.001 |
IR spectra analysis
The IR spectrum showing percentage transmission (T%) versus wave number of Rosuvastatin is shown in Figure 1 with characteristic peaks of aromatic N-H stretching and C=O stretching at 3316 cm-1 and 1600 cm-1, respectively. From the figure it was observed that functional group of rosuvastatin was retained in liquisolid compact, suggesting absence of chemical interaction with any of the excipients used in the preparation of liquisolid compacts.10
Figure 1 .
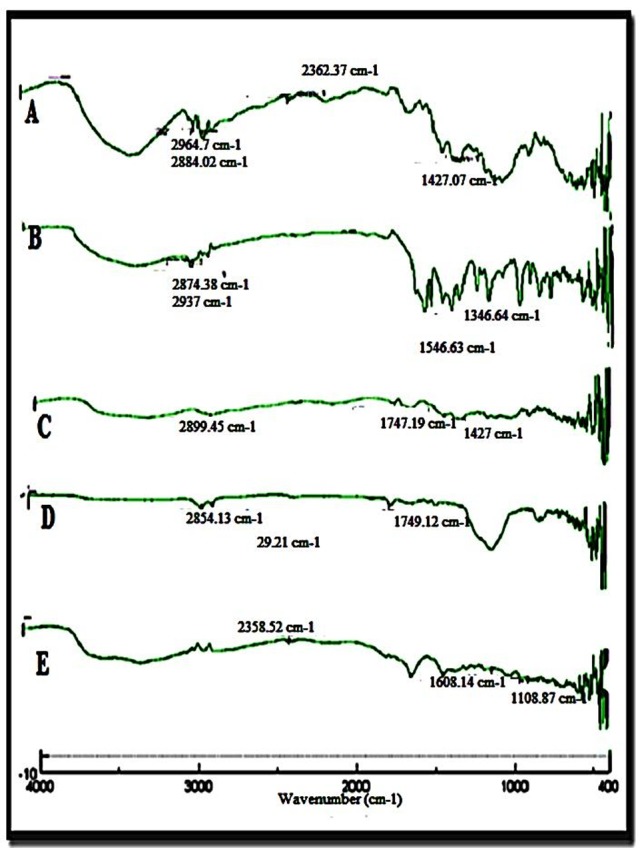
FTIR spectra A) Liquisolid formulation, F3 B) Rosuvastatin C) Avicel D) Aerosil E) Sodium starch glycolate.
X-ray powder diffraction (XRPD)
Powder X-RD is used to determine the crystallinity of compounds. Polymorphic changes of drug are important factor which might affect the dissolution rate and in turn bioavailability. Crystallinity of the drug and the liquisolid compacts samples was determined. The X-Ray Diffraction pattern (Figure 2) of pure drug (Rosuvastatin) showed sharp diffraction peaks at 2θ values of 16.04, 22.45, and 34.3 while liquisolid powder showed sharp peak at 2θ values of 19.87. The absence of characteristics peak in formulation indicated that drug had probably converted from crystalline to amorphous form.10
Figure 2 .
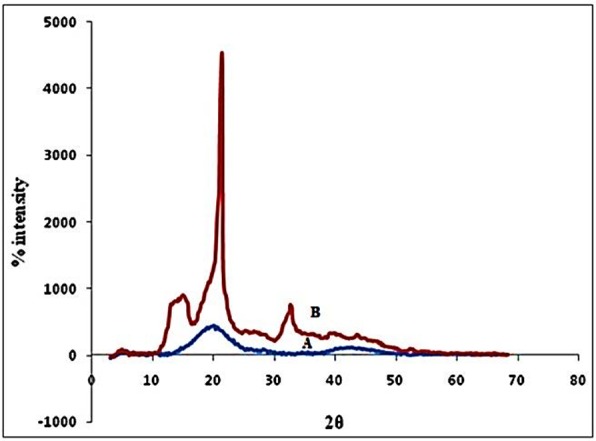
X-ray diffractograms of A) Liquisolid powder system (F3) and B) Pure drug, Rosuvastatin.
Differential scanning calorimetry (DSC)
The possible interactions between a drug entity and excipients in liquisolid compacts were determined by DSC. Figure 3 shows the thermal behavior of the pure components as well as of the final liquisolid system prepared. The rosuvastatin peaks appeared clear, demonstrating a sharp characteristic endothermic peak at 125.38 °C (Figure 3A) corresponding to its melting temperature (Tm); such a sharp endothermic peak shows that the rosuvastatin used was in a pure crystalline state. On the other hand, the liquisolid system (Figure 3B) showed that the characteristic peaks of rosuvastatin had disappeared; this agrees with the formation of a solid solution in the liquisolid powdered system, i.e., the drug was molecularly dispersed within the liquisolid matrix. That was accompanied by the formation of a new endothermic peak at 93.5.°C indicating the melting and decomposition of whole liquisolid system. This disappearance of drug peaks upon formulation into a liquisolid system was in agreement with McCauley and Brittain who declared that the complete suppression of all drug thermal features undoubtedly indicates the formation of an amorphous solid solution. In addition, Mura et al. found that the total disappearance of the drug melting peak indicates that drug amorphization had taken place.10
Figure 3 .
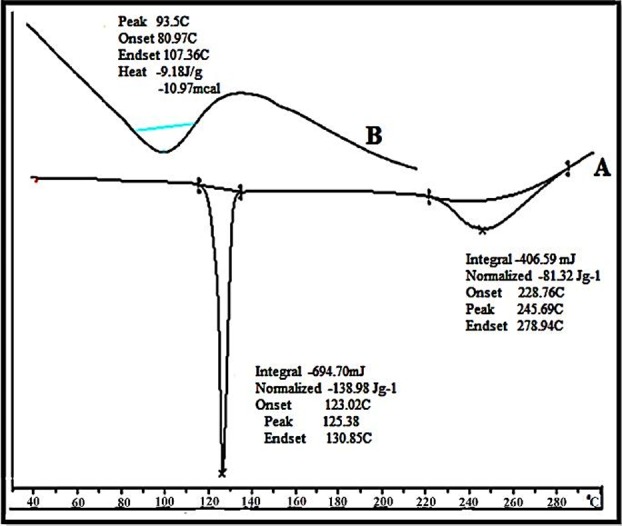
DSC thermogram of (A) Pure drug Rosuvastatin, (B) Liquisolid powder system (F3).
In vitro evaluation of liquisolid compacts
Rosuvastatin liquisolid compact content uniformity
A fundamental quality attribute for all pharmaceutical preparations is the requirement for a constant dose of drug between individual tablets. The percent drug content as shown in Table 4 varied from 90 to 97% w/w.
Table 4. Post compression evaluation of Rosuvastatin liquisolid tablets .
| Batch | Thickness (mm) | Hardness (kg/cm2) | Weight variation (mg) | Friability | Disintegration time (sec) | Drug content (%) | FM |
| F1 | 3.02±0.01 | 4.20±0.24 | 151.45±0.95 | 0.095 | 175.00±0.63 | 96.66±1.31 | 0.771 |
| F2 | 3.53±0.01 | 4.08±0.25 | 256.51±0.95 | 0.126 | 210.00±0.89 | 92.83±1.32 | 0.771 |
| F3 | 3.45±0.01 | 4.00±0.15 | 363.64±0.85 | 0.274 | 143.33±0.81 | 96.66±1.31 | 0.771 |
| F4 | 2.06±0.01 | 3.33±0.51 | 110.96±1.10 | 0.089 | 253.5±1.37 | 92.85±1.28 | 0.578 |
| F5 | 3.42±0.01 | 4.00±0.44 | 363.62±0.91 | 0.072 | 307.0±0.89 | 96.66±1.31 | 0.578 |
| F6 | 3.63±0.00 | 3.66±0.51 | 272.27±0.95 | 0.099 | 226.83±1.16 | 90.58±1.36 | 0.578 |
| F7 | 3.00±0.02 | 3.5±0.44 | 187.25±1.19 | 0.094 | 354.66±0.81 | 90.58±1.36 | 0.462 |
| F8 | 2.84±0.00 | 3.58±0.58 | 151.8±0.92 | 0.199 | 182.83±0.75 | 92.11±1.28 | 0.462 |
| F9 | 2.65±0.01 | 4.00±0.31 | 216.20±0.44 | 0.168 | 254.66±1.03 | 95.13±1.32 | 0.462 |
| *All readings are average ± SD (n=3) | |||||||
Hardness, Friability, Weight variation, Disintegration test
The results of thickness, hardness, weight variation, friability, disintegration test and fraction of molecularly dispersed drug (FM )of liquisolid tablet are mentioned in Table 4. All the selected Rosuvastatin tablets had acceptable friability as none of the tested formulae had percentage loss in tablet weights that exceed 0.5%. Also none of the tablets was cracked, split or broken. The liquisolid tablet disintegrated in less than 5 minutes which is as per specifications given for the uncoated tablets in the IP. Uniform drug content was observed for all the formulation (90 to 100).
In vitro dissolution studies
The drug dissolution profile of the liquisolid compact, the directly compressed compacts and marketed tablet of rosuvastatin at different dissolution volume and medium was studied. Phosphate buffer pH 6.8, 900 ml, was used as the dissolution medium. As seen in Figure 4, liquisolid compacts (F3) showed better in-vitro release than those of the directly compressed tablet and marketed tablet. The liquisolid compacts contain a solution of the drug in PEG 200 which facilitates the wetting of drug particle by decreasing the interfacial tension between tablet and dissolution medium. The drug surface available for dissolution is tremendously increased. After disintegration, the liquisolid primary particles suspended in the dissolving medium contain the drug in a molecularly dispersed state, whereas the directly compressed compacts are merely exposed micronized drug particles. Therefore, in the case of liquisolid compacts, the surface area of drug available for dissolution is much greater than that of the directly compressed compacts. According to Noyes and Whitney, the drug dissolution rate (DR) is directly proportional to the concentration gradient (Cs-C) of the drug in the stagnant diffusion layer and its surface area (S) available for dissolution. The significantly increased surface area of the molecularly dispersed rosuvastatin in the liquisolid compacts may be principally responsible for their observed higher dissolution rates. The consistent and higher dissolution rate displayed by liquisolid compacts will improve the absorption of drug from the GI tract.
Figure 4 .
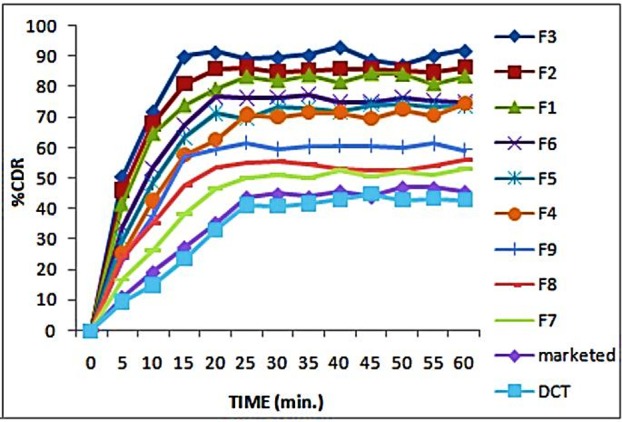
Dissolution profile of liquisolid, marketed and directly compressed tablet.
Effect of R Value on drug release from Rosuvastatin Liquisolid compacts: Spireas et al in their patent have stated that R value ranging from 10 to 30 gives optimal result. In the present study different drug concentrations in liquid medication were used between 15-25%. Further for each drug to non volatile solvent R value was varied from 10 to 30 and its in vitro drug release patterns were studied. The drug release followed the following pattern: R30 > R20 > R10
Liquisolid compacts with lower R-values contain relatively smaller amounts of carrier powder and larger quantities of fine drug loaded silica particles. Also, the ratios of the amounts of their liquid medication per powder substrate are relatively higher. On the other hand, liquisolid compacts with higher R-values contain low liquid/powder ratios, high presence of cellulose and low presence of silica. This could be directly associated with enhanced wicking, disintegration and deaggregation properties. Therefore, the liquisolid tablets with low R-values showed relatively poor dissolution.
Effect of different drug concentrations in liquid medication on drug release: PEG 200 was selected as nonvolatile vehicle as the drug rosuvastatin showed maximum solubility in it. Concentrations of drug in liquid medication were varied from 15 to 25%. The drug release when compared suggested that 15% showed highest dissolution profile followed by 20 and there on . The drug release was high as higher fraction of drug was in solubilized or molecular state as compared to other concentrations.
Thus, formulations with smaller drug concentration (15%w/w) have a higher dissolution than higher drug concentration (25%w/w) in liquisolid tablets formulated with PEG 200. This can be explained by the dissolved drug in the liquid medication. The drug release follows the following pattern: F3 > F6 > F9
FM = Cl/ Cd ---------------------- (4)
Where FM is the fraction of molecularly dispersed or dissolved drug in liquid medication of the prepared liquisolid formulation, Cl is the saturation solubility of Rosuvastatin in the liquid vehicle and Cd is the concentration of the liquid. According to Spireas et al. FM value cannot exceed unity. The saturation solubility of rosuvastatin in PEG 200 is 11.57 % w/w. By applying Equation 4, it was again seen that FM value is not greater than 1 .
Effect of aging on tabletting properties
The optimized formulation was subjected to stability studies to evaluate any change in the formulation. Stability study of optimized formulation was performed under accelerated stability conditions at 40 °C ± 2 °C/75% RH ± 5% RH for three months. The hardness, drug content and dissolution rate were measured for the aged tablets. The results showed that there was no significant different between hardness of fresh (4Kg/Cm2) and aged (4Kg/Cm2) of the liquisolid tablets. Figure 5. too shows similar dissolution profiles between the fresh and aged formulation. This means that aging had no significant effect on drug release profile of rosuvastatin liquisolid tablets. The study concluded that the tested formulations are found to be stable.
Figure 5 .
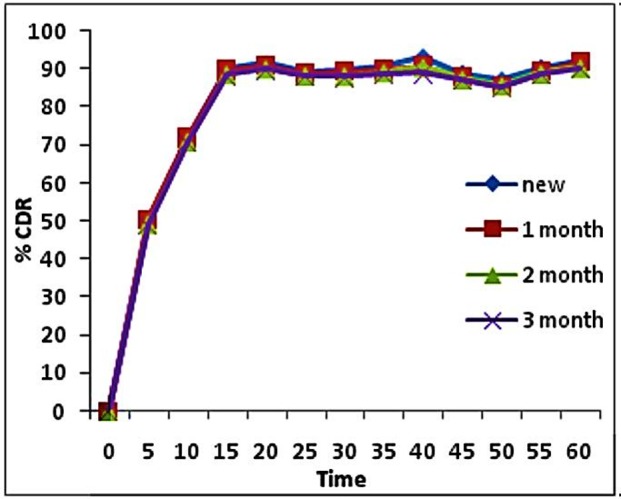
Disolution profile of F3 during Stability studies.
Conclusion
Rosuvastatin exhibits high permeability through biological membranes, but its absorption after oral administration is limited by its low dissolution rate due to its very low aqueous solubility. Hence, the use of the liquisolid technique was chosen to enhance the dissolution properties of rosuvastatin. The rosuvastatin liquisolid compacts were prepared using Avicel PH 102 and Aerosil 200 as the carrier and coating material, respectively. The P-XRD studies showed complete inhibition of crystallinity in the rosuvastatin liquisolid compacts. The DSC study confirmed the absence of any interaction between the drug and excipients used in the preparation of Rosuvastatin liquisolid compacts. The hardness, friability, weight variation and disintegration tests were within acceptable limits. The in vitro dissolution study confirmed enhanced drug release from liquisolid compacts compared with directly compressed tablet and marketed tablet. It was observed that aging had no significant effect on the hardness, disintegration time and dissolution profile of the liquisolid compacts.
Acknowledgements
The authors are thankful to Maple Biotech Pvt. Ltd. Bhosari Pune for providing gift samples of the polymers and Biocon Pharma Bangalore for providing gift sample of the rosuvastatin.
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Vemula VR, Lagishetty V, Lingala S. Solubility enhancement techniques. Int J Pharm Sci Rev Res . 2010;5(1):41–51. [Google Scholar]
- 2.Akbari BV, Valaki BP, Mardiya VH, Akbari AK, Vidyasagar G. Enhancement of solubility and dissolution rate of rosuvastatin calcium by complexation with β cyclodextrin. Int J Pharm Biol Arch . 2011;2(1):511–20. [Google Scholar]
- 3.Nainwal P, Sinha P, Singh A, Nanda D, Jain DA, Bhoomi D. A comparative solubility enhancement study of rosuvastatin using solubilization technique. Int J Appl Biol Pharm Technol . 2011;2(4):14–8. [Google Scholar]
- 4.Fahmy RH, Kassem MA. Enhancement of Loratidine dissolution rate through liquisolid tablets formulation: in vitro and in vivo evaluation. Eur J Pharm Biopharm . 2008;69(3):993–1003. doi: 10.1016/j.ejpb.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 5.Spireas S, Sadu S. Enhancement of prednisolone dissolution properties using liquisolid compacts. Int J Pharm . 1998;166(2):177–88. [Google Scholar]
- 6.Spireas S, Wang T, Grover R. Effect of powder substrate on the dissolution properties of methyclothiazide liquisolid compacts. Drug Dev Ind Pharm . 1999;25(2):163–8. doi: 10.1081/ddc-100102156. [DOI] [PubMed] [Google Scholar]
- 7.Karmarkar AB, Gonjari ID, Hosmani AH, Dhabale PN, Bhise SB. Dissolution rate enhancement of fenofibrate using liquisolid tablet technique.Part II: Evaluation of in vitro dissolution profile comparison methods. Lat Am J Pharm. 2009;28(4):538–43. [Google Scholar]
- 8.Nokhodchi A, Javadzadeh Y, Siahi-Shadbad MR, Barzegar-Jalali M. The effect of type and concentration of vehicles on the dissolution rate of a poorly soluble drug (indomethacin) from liquisolid compacts. J Pharm Pharm Sci . 2005;8(1):18–25. [PubMed] [Google Scholar]
- 9.Khaled KA. Formulation and evaluation of hydrochlorothiazide liquisolid tablets. Saudi Pharm J . 1998;6(1):39–46. doi: 10.1016/s0378-5173(01)00633-0. [DOI] [PubMed] [Google Scholar]
- 10.Gubbi SR, Jarag R. Formulation and characterization of atorvastatin calcium liquisolid compacts. Asian J Pharm Sci . 2010;5(2):50–60. [Google Scholar]
- 11.Burra S, Kudikala S, Reddy GJ. Formulation and evaluation of Simvastatin liquisolid tablets. Der Pharmacia Lettre . 2011;3(2):419–26. [Google Scholar]
- 12.Javadzadeh Y, Jafari-Navimipour B, Nokhodchi A. Liquisolid technique for dissolution rate enhancement of a high dose water-insoluble drug (carbamazepine) Int J Pharm. 2007;341(1-2):26–34. doi: 10.1016/j.ijpharm.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 13.Singh SK, Srinivasan KK, Gowthamarajan K, Prakash D, Gaikwad NB, Singare DS. Influence of formulation parameters on dissolution rate enhancement of glyburide using liquisolid technique. Drug Dev Ind Pharm . 2012;38(8):961–70. doi: 10.3109/03639045.2011.634810. [DOI] [PubMed] [Google Scholar]
- 14.Gavali SM, Pacharane SS, Sankpal SV, Jadhav KR, Kadam VJ. Liquisolid compact: A new technique for enhancement of drug dissolution. Int J Res Pharm Chem . 2011;1(3):705–13. [Google Scholar]
- 15.Vaskula S, Vemula SK, Bontha VK, Garrepally P. Liquisolid Compacts: An Approach to Enhance the Dissolution Rate of Nimesulide. J Appl Pharm Sci . 2012;2(5):115–21. [Google Scholar]
- 16.Tiong N, Elkordy AA. Effects of liquisolid formulations on dissolution of naproxen. Eur J Pharm Biopharm . 2009;73(3):373–84. doi: 10.1016/j.ejpb.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 17.Ansel HC, Allen LV, Popovich NG. Pharmaceutical dosage forms and drug delivery systems. Philadelphia: Lippincott williams and wilkins; 1999. [Google Scholar]
- 18.United States Pharmacopeia and National Formulary. 29th ed. Rockville, MD, USA: United States Pharmacopeial Convention; 2006.