

Abstract
Cancer dormancy, a state in which cancer cells persist in a host without significant growth, is a natural forestallment of progression to manifest disease and is thus of great clinical interest. Experimental work in mice suggests that in immune-induced dormancy, the longer a cancer remains dormant in a host, the more resistant the cancer cells become to cytotoxic T-cell-mediated killing. In this work, mathematical models are used to analyse the possible causative mechanisms of cancer escape from immune-induced dormancy. Using a data-driven approach, both decaying efficacy in immune predation and immune recruitment are analysed with results suggesting that decline in recruitment is a stronger determinant of escape than increased resistance to predation. Using a mechanistic approach, the existence of an immune-resistant cancer cell subpopulation is considered, and the effects on cancer dormancy and potential immunoediting mechanisms of cancer escape are analysed and discussed. The immunoediting mechanism assumes that the immune system selectively prunes the cancer of immune-sensitive cells, which is shown to cause an initially heterogeneous population to become a more homogeneous, and more resistant, population. The fact that this selection may result in the appearance of decreasing efficacy in T-cell cytotoxic effect with time in dormancy is also demonstrated. This work suggests that through actions that temporarily delay cancer growth through the targeted removal of immune-sensitive subpopulations, the immune response may actually progress the cancer to a more aggressive state.
Keywords: cancer dormancy, immunoediting, mathematical model
1. Introduction
Cancer escape from the immune-induced dormant state involves the immunoediting process [1–3] and the adaptive immune system [4–7]. Mechanisms of escape can be classified into two broad categories: those that involve the secretion of factors, and those that involve direct cell–cell contact [8]. In general, soluble factors work to suppress anti-tumour immunity by converting the immune response (and cytokines) to a pro-tumour inflammatory reaction. Direct contact between cancer cells and cytotoxic T lymphoctyes (CTLs), on the other hand, may reduce tumour-associated antigen recognition and lymphocyte survival and increase cancer cell apoptotic resistance.
Saudemont & Quesnel [9], using a mouse model of immune-induced dormancy in acute myeloid leukaemia (AML), recently reported that the longer leukaemic cells persist in the dormant state, the more resistant they become to CTL-mediated cell lysis. This resistance was linked to cancer cell expression of B7-H1 or B7.1. The mice were vaccinated against the AML with irradiated CD154- or IL12-transduced cells, which helped to stimulate a targeted immune response against the cancer. After three weekly injections of the vaccine, the mouse was injected on the fourth week with active AML cells (DAI-3b). Mice were then sacrificed on days 35, 60, 90, 120 and 365 after challenge. Per cent lysis assays with CTLs isolated from mice spleens at day 35 and day 365 both demonstrate a decrease in lysis efficacy the longer the AML cells persist in a dormant state within the host. Additionally, production of IFN-γ and TNF-α, two known anti-tumour cytokines, was also shown to decline in cancer-CTL co-cultures with increasing time in dormancy.
The increased resistance to apoptotic signals from CTLs (as well as from imatinib mesylate) may be due to an IL-3 autocrine loop. Saudemont et al. [10] showed that IL-3 overproduction can lead to SOCS1 inactivation through DNA methylation to deregulate the JAK/STAT pathways and evoke resistance. Interestingly, natural killer cells recruited by CXCL10-transduced DAI-3b cells may provide an effective mechanism to clear dormant cancers as their predation efficacy does not decline with time in dormancy [11].
The observed decline in CTL predation strength that occurs over 1 year in dormancy [9] is considerable, but may not solely be due to overexpression of the B7-H1 or B7.1 surface proteins. For example, at the extreme effector–target ratio of 20 : 1 at day 35, the per cent of CTL-mediated cancer cell lysis is 100 per cent for wild-type (day 0) AML cells but only about 20 per cent for AML cells that persisted in the dormant state for 365 days. This loss can be recovered back up to 80 per cent using anti-B7-H1 and anti-B7.1 antibodies, but it cannot be fully recovered.
Cancer heterogeneity provides an additional mechanism for the emergence of resistance to immune predation. Experimental work by Schatton & Frank [12] and Schatton et al. [13] suggests that cancer resistance to immune cytotoxic activity may be imposed by cancer stem cells. They suggest that these tumour-initiating cells may exist as a subpopulation of the cancer cells and function in a manner that helps the cancer evade host immunity. Additionally, cancer clonal heterogeneity, acquired through branching evolutionary processes [14], can result in the emergence of clones that are more resistant to CTL attack via immunoediting [1,7].
T-cell exhaustion is another mechanism that may lead to the appearance of resistance and cancer escape from dormancy. This progressive state of cell dysfunction worsens with time and can include poor effector function and sustained activation of inhibitory pathways and immune regulators [15]. Exhaustion can lead to limiting the T-cell response during persisting infections and cancer, and strategies to avoid regulatory inhibition of the immune response such as the programmed death-1 and cytotoxic T-lymphocyte antigen-4 pathways are now being investigated as possible immunotherapy targets to improve tumour control [16–18].
Mathematical models have been used successfully to improve our understanding of cancer growth dynamics and, in particular, the dynamics of immune-induced dormancy. Some common approaches include ordinary differential equations [19–27], partial differential equations [28,29], agent-based simulations [29–31] and the kinetic theory of active particles [32,33]. Models that specifically analyse the mechanisms of tumour evasion from immune control are relatively new. A general model for the mechanisms of learning and hiding by interacting populations was proposed using the kinetic theory for active particles [32]. These mechanisms were also investigated by incorporating time-varying parameters for immune predation, growth and recruitment into an existing system of differential equations [34]. The parameters were prescribed to initially increase as the immune response learned to recognize the cancer and then to decrease as the cancer developed means of immune evasion (hiding). A constant decrease in predation efficacy was also investigated as a means of escape from tumour dormancy, and a linearly decreasing immune growth rate was investigated as an ageing mechanism leading to tumour regrowth [35]. A bifurcation analysis was performed to study the behaviour of system equilibria under the assumption of slowly varying parameters [36], but the work was theoretical with no discussion of time dynamics or connection to experimental data. Additionally, in the same paper, the role of immunoediting in cancer evasion through mutation to a resistant clone was studied analytically. Effects of both direct cell contact [37,38] and bounded noise [39] have also been investigated. Interestingly, a spatio-temporal model of emerging cancer cell resistance suggests that escape may occur through declining recognition or increasing resistance (or both) [40]. In relation to the role of B7-H1 in cancer resistance, Galante et al. [41] proposed a model to examine the interactions between CTLs and cancer cells that have low or high expression of B7-H1. While they did not consider the time-evolution of this expression, they did include both the Fas/FasL and perforin/granzyme mechanisms of CTL-induced apoptosis.
Here, we present two approaches to mathematically study the observed decline in CTL predation efficacy owing to cancer dormancy time. The first approach is more empirical and takes into account the observed decay trend for the CTL predation strength in a model of cancer–immune interactions to predict the resulting dynamics and possible escape from dormancy. Interestingly, this approach suggests that decline in predation may be accompanied by either increased recruitment without cancer escape from dormancy, or a decline in recruitment with cancer escape from dormancy. It is concluded, in this case, that predation decay must be coupled with recruitment decay (or T-cell exhaustion) in order for the model to predict escape from the dormant state, suggesting that recruitment ability may be a stronger determinant of escape potential than predation strength.
The second approach is more mechanistic in that it considers the immune system as a process that selectively prunes a heterogeneous population of cancer cells to produce a more homogeneous population that may correspond to the overexpressing B7-H1/B7.1, highly resistant, population of cells described by Saudemont & Quesnel [9], or to a cancer stem cell population as described by Schatton et al. [13]. This mechanistic approach uses the immunoediting hypothesis to transform a mostly sensitive population into a mostly resistant population, while still maintaining the dormant state. In silico per cent lysis assays predict a decline in CTL efficacy with time in dormancy owing to the evolving heterogeneity of the population. The predicted decline in predation strength agrees with the experimentally observed decline while providing an intuitive and mechanistic explanation for its cause. Repetition of the described transition from more sensitive to more resistant populations will eventually lead to cancer escape from the dormant state.
2. Material and methods
2.1. Experimental data
Saudemont & Quesnel [9] use a mouse model of AML (DA1-3b) to investigate the immune-induced dormant state. Mice were immunized with three injections of irradiated and IL12- or CD154-transduced DA1-3b cells, challenged with wild-type cells and randomly sacrificed over a 1 year period. CTL activity was measured with effector cells from vaccinated mice, and the specific lysis assays were conducted after various times in dormancy (0, 35, 60, 90, 120 and 365 days). The data suggest that the longer the cancer cells were kept in an immune-induced dormant state, the more resistant they became to CTL predation. For the sake of simplicity, we interpret this as a loss of T-cell predation efficacy and immunological function. Plots of the experimental data are shown in figure 1a,b, demonstrating the decay in efficacy as either a function of effector–target (immune–cancer) cell ratio or as a function of time in dormancy (for constant effector–target ratios).
Figure 1.

Immune cytotoxicity assay data and model fits for predation decay after 0, 35, 60, 90, 120 or 365 days in dormancy. (a) Experimental specific lysis assay data published by Saudemont & Quesnel [9] (figure 2e) show predation efficacy after various days in dormancy as a function of the effector–target (immune–cancer) cell ratio, and (b) another view of these data that shows the decay of each effector–target ratio with time in dormancy. (c) The predation efficacy parameter a0 was estimated using our MCMC algorithm and the experimental data at each time point, resulting in the values indicated by the red dots. Three fits for the decay of predation efficacy were then estimated by least squares fitting. Fit 1 is an exponential decay, fit 2 is an exponential decay but the initial value (day 0) is matched to the day 0 data estimate a0d = a0, and fit 3 is a modified exponential decay. (d–f) The simulated cytotoxic assays assuming the predation decays according to fit 1, 2 or 3.
In addition to the marked decline in cytotoxic efficacy, production of IFN-γ and TNF-α was found to decrease in CTL-cancer cell co-cultures the longer the DA1-3b cells were maintained in the dormant state. Because both these cytokines are signalling proteins associated with activating and maintaining anti-tumour immunity [42], we interpret this decline as a reduction in recruitment potential. In our model, we consider only the presence of activated CTLs, and thus the primary action of these cytokines within the framework is to stimulate the presence of active CTLs through chemoattraction, growth and differentiation. This is, by necessity, a simplified view of these multifaceted cytokines.
2.2. Markov chain Monte Carlo parameter estimation
A Markov chain Monte Carlo (MCMC) algorithm [43,44] is used to estimate the predation strength parameter, at with t = 0, 35, 60, 90, 120 and 365, where the estimated values are anticipated to decrease with time in dormancy. The parameter estimation algorithm works as follows: from an initial guess of at, an MC of permitted parameter values is created by randomly perturbing the previous value and accepting this perturbed value with a probability determined by a measure of the goodness of fit. Here, we use the sum of squared deviations between simulated and experimentally measured percentage of cancer cell lysis at each effector–target ratio. The algorithm is repeated 10 times with each run having 20 000 iterations and varying initial guesses. The 10 runs produced consistent results indicating a mathematically consistent system. The simulated lysis assay time was taken to be 2 h. The resulting estimated values of predation strength at each time point (time held in dormancy) are denoted {a0, a35, a60, a90, a120, a365} and indicated by the red dots in figure 1c.
To model the decay trend observed in the experimental data, we prescribe three functional forms based on an exponential decay and fit their parameters to the data. Fit 1 is a typical exponential decay function, fit 2 is an exponential decay function with the initial value forced to match the day 0 estimate, that is 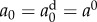 , and fit 3 is a modified exponential decay with a square-root to slow the effect of progressing time. That is,
, and fit 3 is a modified exponential decay with a square-root to slow the effect of progressing time. That is,
| 2.1 |
| 2.2 |
| 2.3 |
For fits 1 and 3, parameters a0 and τ are fit, in the least squares sense, to the values of {a0, a35, a60, a90, a120, a365} previously estimated by fitting the model to the per cent lysis data at each time point with an MCMC algorithm. For fit 2, only τ is fit in the least squares sense, as the initial value is forced to match the data estimate, or 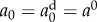 . Note that in the MCMC fitting algorithm, the time of the simulated lysis assay is 2 h.
. Note that in the MCMC fitting algorithm, the time of the simulated lysis assay is 2 h.
Figure 1c shows these three fits and the estimated predation strength values that they are fitted to. Figure 1d–f shows the simulated cytotoxic assays assuming the three fits. From these plots, it appears that the best fit to these data is fit 3, because the error of the fit is smallest, i.e. the spread between the per cent lysis curves is captured most accurately by fit 3.
2.3. Mathematical modelling and numerical simulations
We consider homogeneous populations of cells that can vary in time but have no spatial dimensions. Thus, the mathematical formulation outlined below uses ordinary differential equations to describe the rate of change of each population whose size is measured by the number of cells. We consider two populations of cells: cancer cells, C(t), and immune cells, I(t). The cancer cells are assumed to grow according to a logistic growth function and to be inhibited by immune cells (mostly comprised of CD8+ T-cell effectors) according to the law of mass action. The equation governing tumour growth is thus,
| 2.4 |
Here α is the proliferation rate and K is the carrying capacity, or maximum size of the cancer population.
We note that the assumption of mass action to govern the predation term may be viewed as overly simple, but we argue that it is sufficient for our purposes. For example, de Pillis et al. [45] claim that a ratio-dependent Hill-formulation predation term better fits the shape of experimental per cent lysis curves. The ratio-dependent form presented, however, better captured the saturation effect observed in the experimental data they were fitting [46,47]. The saturation effect describes the appearance of a limit on per cent lysis, or the lack of increase in predation strength with increasing effector–target ratio. In the experimental data of Saudemont & Quesnel [9], however, there is no indication of saturation in the per cent lysis curves, and thus we feel that the assumed mass-action form adequately captures the trends observed in these data.
In the mass-action formulation, the strength of CTL predation is controlled by parameter a0. This parameter controls the lysis activity and immunological function of the activated CTLs interacting with the cancer cells. Initially, we will consider the predation effectiveness to be constant such that the lysis rate is proportional to the interactions of the two populations, CI. Next, we will allow the effectiveness to decrease in time, simulating the observed decay in predation strength with time in dormancy. Exponential or modified exponential decay functions will be assumed, which will represent the accumulation of immune-resistance mutations within the cancer population. Saudemont & Quesnel [9] correlate this increase in resistance to increased cancer cell expression of B7-H1 and B7.1. Physically speaking, the decay in predation strength and immune function (or increase in cancer cell resistance) should occur as a result of the interactions of the two populations, but we simplify these interactions here to their time dynamics, which are captured by the exponential decay.
The immune population is assumed to grow logistically, with a proliferation rate of γ. The carrying capacity is non-constant, and may increase due to recruitment by cancer–immune interactions, Ie + μCI. Note that the initial (i.e. post-immunization) level of immune presence in the host is Ie. The immune population may also be directly recruited by the cancer population, γρC, but this recruitment is ultimately limited by the logistic growth term. Putting all these together gives
To simplify the analysis, however, we shall assume that direct recruitment by the cancer population is negligible, that is ρ = 0, so that the only recruitment occurs through cancer-initiated increases in the carrying capacity. Thus, the immune population is assumed to grow according to
| 2.5 |
Model equations are numerically simulated in Maple (www.maplesoft.com) with the parameter values listed in table 1. Parameter values were chosen to simulate an immune-induced dormant tumour state. Note that these parameter values were not estimated from experimental data, but that they are biologically relevant values, as they are intended to allow the model to demonstrate, quantitatively, the phenomenon of immune-induced tumour dormancy and the emergence of immune-resistance as observed in AML [9].
Table 1.
Parameter values used in the simulations.
| parameter | definition |
|---|---|
| α = 0.2 | cancer growth rate |
| K = 1010 | cancer carrying capacity |
| a0 = 10−4 | cancer sensitivity to immune predation (predation strength) |
| γ = 0.2 | immune growth rate |
| Ie = 100 | immune homeostatic state |
| μ = 10−3 | immune recruitment potential |
| C0 = 104 | cancer initial condition for homogeneous population per cent lysis simulations |
| C0 = 1 | cancer initial condition for homogeneous population growth simulations with decays |
| C0 = 9 × 103 | sensitive subpopulation initial condition for heterogeneous population simulations |
| R0 = 103 | resistant subpopulation initial condition for heterogeneous population simulations |
3. Decaying immune predation and recruitment efficacy
With this mathematical system, we can investigate the effect on cancer growth dynamics of decay in immune predation or recruitment owing to dormancy. The increasing ability of cancer cells to resist CTL-mediated apoptotic signals will be interpreted mathematically as a decrease in the immune predation strength with time in dormancy. Similarly, the decreased production of IFN-γ and TNF-α by CTLs in culture with dormant cancer cells will be interpreted as a decrease in immune recruitment potential. Thus, we focus here on decay in two of the model parameters: immune predation strength a0, and immune recruitment potential μ. Analytical and numerical results will be discussed in relation to cancer dormancy and escape from the dormant state.
3.1. Analytical results
We first establish the baseline behaviour of the mathematical system defined by equations (2.4) and (2.5) using a bifurcation analysis on the predation strength parameter. Equilibrium points of the system, found by solving (dI/dt = dC/dt = 0), are (I, C) = (0, 0), (0, K), (Ie, 0) and (Id, Cd), where
and
As (0, 0) and (Ie, 0) correspond to zero cancer presence, and (0, K) corresponds to zero immune presence (and a presumably lethal cancer size), the focus of our analysis will be on the dormant state given by (Id, Cd), where both the cancer and immune populations are non-growing.
Assuming positive growth rates (α > 0, γ > 0) implies that the equilibrium point (0, 0) is unstable and that the point (0, K) is an unstable saddle. If the immune predation parameter lies in the physical region 0 < a0 < α/Ie, then the equilibrium point (Ie, 0) is also an unstable saddle, leaving the point (Id, Cd) as the only stable attractor in the system. This implies that any non-trivial cancer, given sufficient time, will be controlled to the dormant state by the immune response. Note that this accurately models the experimental set-up of Saudemont & Quesnel [9] where mice were immunized before cancer cell injection to guarantee the formation of an immune-induced dormant state. Furthermore, note that if a0 > α/Ie, then the dormant cancer state is negative and thus unphysical. In this situation, the equilibrium point (Ie, 0) becomes stable, and thus all tumours will be eliminated, given sufficient time, because the predation strength with homeostatic immune surveillance is strong enough to overcome the tumour growth rate.
The behaviour of the dormant state depends not only on the predation strength parameter, a0 but also on the immune recruitment parameter, μ. Figure 2 shows bifurcation diagrams for the cancer dormant population, Cd, the immune dormant population, Id and the relationship between the two. Two regions are displayed in each graph, one region contains solutions where μ > 1/K (shaded blue) and the other where μ < 1/K (shaded green).
Figure 2.
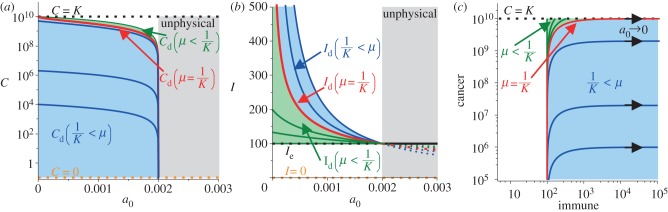
Bifurcation diagrams and bifurcation phase portrait for the cancer–immune dormant state. The bifurcation parameter, predation strength, has a physical range of a0 ∈ [0, α/Ie], as larger values correspond to negative (and unphysical) dormant cancer populations. The immune recruitment parameter, μ, controls the behaviour of the dormant state. In (a), the dormant cancer population increases as predation strength decreases, but it is ultimately limited in size if the recruitment is strong enough. In (b), the dormant immune population increases without bound as predation strength decreases, but will be ultimately limited in size if the recruitment is too weak. The relation between the immune and cancer dormant sizes as predation strength weakens is shown in (c). Each curve is the progression of (Id, Cd) as predation strength weakens. Small dormant cancer populations will be maintained even though predation weakens provided that recruitment is strong enough to grow an infinitely large immune response.
The cancer dormant state (Cd) tends to zero as immune predation approaches the maximal size a0 = α/Ie, but behaviour differs based on three cases of the recruitment potential as predation strength weakens. That is, when 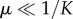 then
then 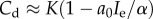 , which tends to Cd = K as predation weakens. Also, when μ = 1/K then
, which tends to Cd = K as predation weakens. Also, when μ = 1/K then  , which lies below the previously described case, but also tends to Cd = K as a0 → 0. Interestingly, however, when
, which lies below the previously described case, but also tends to Cd = K as a0 → 0. Interestingly, however, when 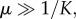 then
then 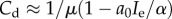 , which tends to
, which tends to 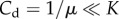 as predation weakens. This suggests that when recruitment is strong, the dormant cancer state may be achieved and maintained below the maximum possible size, even as predation strength weakens.
as predation weakens. This suggests that when recruitment is strong, the dormant cancer state may be achieved and maintained below the maximum possible size, even as predation strength weakens.
For the dormant immune population, Id, when recruitment is very strong, that is, in the limit as μ → ∞ (μ > 1/K), then Id → α/a0, which tends to infinity as a0 → 0. Similarly, if μ = 1/K, then 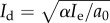 , which is also unbounded as predation weakens. In the range μ < 1/K, if
, which is also unbounded as predation weakens. In the range μ < 1/K, if 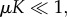 then
then 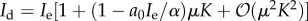 , which shows that for
, which shows that for 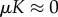 ,
,  and for
and for 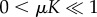 ,
, 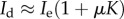 as a0 → 0, indicating that in these cases, the immune dormant population is bounded when predation weakens owing to negligible recruitment potential. The cases for strong recruitment suggest that as predation weakens the immune response will grow infinitely large to compensate for the loss in efficacy.
as a0 → 0, indicating that in these cases, the immune dormant population is bounded when predation weakens owing to negligible recruitment potential. The cases for strong recruitment suggest that as predation weakens the immune response will grow infinitely large to compensate for the loss in efficacy.
This analysis suggests that strong recruitment potential is essential for the maintenance of the dormant state given that predation strength decays in time. It also suggests that to compensate for the decreasing efficacy, the immune population must grow infinitely large, which may result in either of two outcomes. First, the host may not be able to survive such a large immune onslaught, and second, the immune response may become exhausted at some point and be incapable of producing any more immune cells.
3.2. Numerical simulations and results
While the earlier-mentioned analytical results are very informative, they do not illuminate the time dynamics of the induction of dormancy or the escape from dormancy—both of which are critical to host survival. Numerical simulations will now be presented to examine the dynamics of the system over time.
Note. The oscillations present in these simulations may be a modelling artefact, or they may be an observable biological phenomenon. In this model, the immune response necessarily lags behind the predation level required to exactly control the tumour, because the immune response grows only in response to cancer presence. This lag produces the oscillations observed in the simulations. However, both clinical and experimental observations of oscillatory behaviour in immune cells [48,49] and cancer burden [50–53] have been reported.
3.2.1. Decay in predation efficacy
Figure 3 shows the simulated cancer and immune growth curves assuming that the predation efficacy decays according to these three fits. As predicted, with strong recruitment, the immune population grows large to compensate for the diminishing predation efficacy, while still maintaining the cancer in the dormant state. Both the growth curves and phase portraits demonstrate the slower progression captured by fit 3 compared with fits 1 and 2. Because of the unlimited immune recruitment, the dormant cancer state will always (theoretically) be maintained, and thus the cancer escape time for these fits is infinite. Note that we define the escape time as the time it takes the cancer population to reach the size of 109 cells, a presumably symptomatic and near-lethal size.
Figure 3.

Simulations of immune-mediated tumour dormancy assuming the three fits to predation decay. Predation strength is assumed to decay according to fit 1 in (a), fit 2 in (b) and fit 3 in (c). Cancer–immune phase portraits for each fit are shown in (d–f). Note that the recruitment is strong enough to increase the immune population sufficiently to compensate for the diminishing predation strength, thus maintaining the dormant state. Theoretically, in long time, this will require the immune population to grow infinitely large, which is unphysical.
3.2.2. Limited immune potential
Theoretically, the immune response will grow unbounded to compensate for the decay in predation strength with our assumption of strong recruitment potential. However, physically, this should be impossible as either the host will succumb to symptoms associated with such a large immune presence, or, the host will limit the immune response to some maximal tolerable size through T-cell exhaustion. Here, we consider the latter case, where the immune response is limited to 109 cells. Owing to the oscillations in population size, we limit the immune size once the mean value over one oscillation is greater than or equal to the theoretical limit. After this point in time, the immune response is large and constant, and thus cannot compensate for any further decay in predation strength. As a result, the cancer population will escape dormancy with further decay in predation efficacy.
T-cell exhaustion, which includes the inability to proliferate or recruit new T cells in addition to the decline in predation strength, provides a possible mechanism for cancer escape from dormancy. Figure 4 shows simulations of the cancer growth curves with limited immune responses assuming the three fits in predation decay. Escape times for the cancer population are 8.5 years for fit 1, 6.5 years for fit 2 and 47.9 years for fit 3. The much slower decay captured by fit 3 results in a much larger prediction of escape time. In these simulations, the limit imposed on immune presence provides the mechanism for cancer escape following the decline in predation strength, as without this limit, escape would theoretically never occur.
Figure 4.
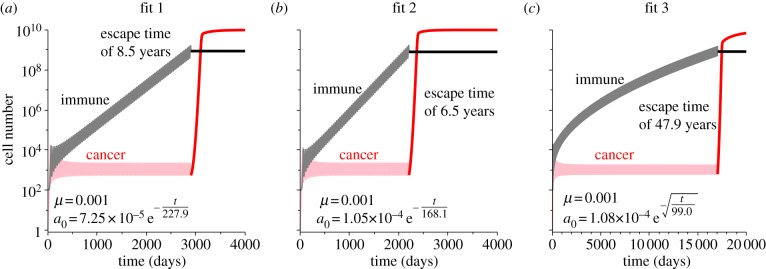
Population growth curves assuming the three fits in predation decay and that the immune response is limited. In all simulations, once the mean value over one oscillation of the immune response is greater than 109 cells, our theoretical limit, it is forced to be constant. After this occurs, the cancer can escape the dormant state owing to the continuing decay in predation efficacy. Escape times for each fit are indicated in the plots. Fit 1 in (a), fit 2 in (b) and fit 3 in (c).
3.2.3. Decay in recruitment potential
The decreased production of anti-tumour-associated cytokines by cancer–immune co-cultures after increasing time in dormancy suggests that immune recruitment potential, in addition to predation efficacy, may decay. For the sake of simplicity, we assume that the same three fits of decay, equations (2.1)–(2.3), apply for the recruitment potential with the same timescale τ as determined by the fitting procedure (table 2). The initial value for μ is as listed in table 1.
Table 2.
Time for the cancer population to reach the size of 109 cells with parameter values as listed in table 1 if not specified. Three fits to the predation decay are assumed and applied to decaying predation, decaying recruitment and decaying predation and recruitment.
| fit 1 | fit 2 | fit 3 | |
|---|---|---|---|
|
a0exp(−t/τ) a0 = 7.25 × 10−5 τ = 227.9 |
a0exp(−t/τ) a0 = a0 = 1.054 × 10−4 τ = 168.1 |
a0exp(−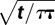 ) )a0 = 1.08 × 10−4 τ = 99.0 |
|
| predation decay | n.a. | n.a. | n.a. |
| predation decay with limited immune response | 3100 days or 8.5 years (200 days after the immune limit is reached) | 2367 days or 6.5 years (159 days after the immune limit is reached) | 17 486 days or 47.9 years (438 days after the immune limit is reached) |
| recruitment decay | 3159 days or 8.6 years | 2334 days or 6.4 years | 19 077 days or 52.2 years |
| predation and recruitment decay | 3135 days or 8.6 years | 2312 days or 6.3 years | 18 901 days or 51.7 years |
With decay in recruitment potential, the immune response becomes unable to grow in response to the cancer presence and thus allows the cancer population to slowly escape from the dormant state. Figure 5 shows the effect on tumour growth of decay in recruitment potential, assuming all three fits. The escape time, when the cancer population reaches 109 cells, is 8.6 years for fit 1, 6.4 years for fit 2 and 52.2 years for fit 3, respectively.
Figure 5.

Simulations of immune-mediated tumour dormancy assuming the three fits for decay are applied to the recruitment potential while the predation strength is constant. Recruitment is assumed to decay according to fit 1 in (a), fit 2 in (b) and fit 3 in (c) with the initial value of μ = 10−3, and the timescale of the decay determined by the fit. Cancer–immune phase portraits for each fit are shown in (d–f). Note that as recruitment decays, even though predation strength is strong, the immune population cannot maintain the dormant state. The escape time for each fit is indicated in the corresponding plot.
3.2.4. Decay in predation and recruitment
Finally, we assume that both immune predation strength and recruitment potential decay with time in dormancy according to the fits described in table 2. Figure 6 shows the simulations corresponding to this dual decay. As predation strength and recruitment both decay, the cancer population is allowed to grow larger, and as a result, the dormant state is escaped. Interestingly, these two factors combined do not significantly alter the escape time compared with recruitment decay alone. In these dual-decay simulations, the cancer population will reach 109 cells in 8.6 years for fit 1, 6.3 years for fit 2, and 51.7 years for fit 3—only slightly faster than was predicted by decay in recruitment alone.
Figure 6.

Simulations of immune-mediated tumour dormancy assuming the three fits for decay are applied to both the predation strength and the recruitment potential. Predation and recruitment are assumed to decay according to fit 1 in (a), fit 2 in (b) and fit 3 in (c) with initial values of μ = 10−3 and the initial predation strengths and timescales of the decays determined by the fit. Cancer–immune phase portraits for each fit are shown in (d–f). Note that as predation strength and recruitment decay together, the immune population cannot maintain the dormant state, but the two populations grow large together. After long time, however, the immune response returns to the homeostatic level since recruitment has become negligible. The escape time for each fit is indicated in the corresponding plot.
The escape time results for these four cases assuming the three fits are summarized in table 2. The fastest escape times are predicted by fit 2, and the slowest times by fit 3. Predation decay alone cannot explain escape from dormancy so long as the immune response can grow to compensate for the lost efficacy. If the immune response is somehow limited, however, then escape will occur after 2367 days (or 6.5 years) of growth assuming fit 2. This is 159 days after the immune response was limited. In comparison, if recruitment decays alone, cancer escape will occur in 2334 days (or 6.4 years), and if both recruitment and predation decay, then escape will occur in 2312 days (or 6.3 years). This suggests that decay in recruitment potential is the stronger mechanism of cancer escape than decay in predation strength, especially because their combined actions do not significantly alter the escape time of recruitment decay alone.
4. Emergence of a resistant cancer subpopulation
We now investigate one possible mechanism for the observed decay in predation strength with time in dormancy. The immunoediting hypothesis [1] proposes that during dormancy, immune selection processes can eliminate sensitive subpopulations of the cancer, leaving more resistant (and less immunogenic) subpopulations to grow in their place. Experiments in mice have demonstrated this fact, by clearly showing how the immunoediting process can drive cancer progression by selecting cancer subpopulations with decreased sensitivity to immune attack and decreased immunogenicity [5,6]. Presumably, this process occurs iteratively as new mutations lead to the production of more or less sensitive subpopulations over time. Here, we consider a simple analogy of the immunoediting process with two subpopulations: sensitive cancer cells, C(t), and resistant cancer cells, R(t). Subject to the predation imposed by immune cells, I(t), we demonstrate how a population that is initially composed of mostly sensitive cells, can, over the course of 1 year in dormancy, transform into a population composed mostly of resistant cells, and thus cause the appearance of decreased immune predation efficacy, as observed by Saudemont & Quesnel [9].
4.1. Modification of the mathematical model
We now extend our mathematical model to allow for two subpopulations within the cancer: C(t), an immunogenic and immuno-sensitive subpopulation, and R(t), a less-immunogenic and immuno-resistant subpopulation. The sensitive subpopulation is assumed to grow according to
| 4.1 |
and the resistant subpopulation is assumed to grow according to
| 4.2 |
Note that both subpopulations contribute to the total carrying capacity of the cancer, which introduces a competition mechanism for space and nutrients between the two subpopulations.
The immune population is assumed to be strongly recruited by the sensitive subpopulation (with recruitment potential μ) and weakly recruited by the resistant subpopulation (with 0.1% of the recruitment potential μ). Thus, immune population growth is governed by
| 4.3 |
4.2. Analytical results
In the logistic growth terms of (4.1) and (4.2), the sum of the two subpopulations contribute to the total cancer carrying capacity, which allows each subpopulation to inhibit the other through the biological mechanisms of competition for space and nutrients. For example, if C is much larger than R, then C will continue to grow while the growth of R will be inhibited by the large presence of C. Because of this competition, if one subpopulation proliferates slower than the other, there is an increased chance that the inhibition will smother the slower proliferating subpopulation. Additionally, there is no evidence to suggest that the immunoediting process selects for cancer subpopulations with shortened proliferation times. Thus, we assume that both cancer subpopulations in our model have equal proliferation rates and set α = β.
Non-trivial dormant solutions to the system of equations given by (4.1)–(4.3) are found by setting dC/dt = dR/dt = dI/dt = 0. Solving this system indicates that dormancy can exist with either one of the cancer subpopulations eliminated, (C, R, I) = (Cd, 0, Id) or (C, R, I) = (0, Rd, Id), or where both subpopulations exist (C, R, I) = (Cd, Rd, Id). This last case, where both subpopulations are non-trivial, requires the following two criteria:
| 4.4 |
The first criterion requires that the immune response has satisfied the recruitment signals emitted by the cancer population, and the second requires that the ratio of the predation effectiveness parameter to the proliferation rate is the same for the two cancer subpopulations. Under our assumption that α = β, this second criterion requires b0 = a0, so that the two populations have the same sensitivity to immune predation—contradicting our assumption of one sensitive and one resistant subpopulation.
Therefore, if both cancer subpopulations exist simultaneously in the dormant state, with the same proliferation rates, they must both be equally sensitive to immune predation. If, however, one subpopulation is more resistant to predation than the other, that population will be maintained in dormancy while the sensitive population will be eliminated.
4.3. Numerical simulations and results
Simulations of this model system are shown in figure 7. Parameter values are listed in table 1 with the addition of β = α and the value of b0 indicated in each subfigure. In figure 7a, both the growth rates and the predation sensitivities are equal between the two cancer subpopulations as required by the second criterion mentioned earlier. Using initial conditions C0 = 9 × 103 and R0 = 103 (so that resistant cells are 10% of the population), dormancy is induced by the immune response with decaying oscillations. In figure 7b, this induction of dormancy is indicated by the black diagonal line that connects the initial composition to the dormant point in the phase portrait for resistant-to-sensitive cancer subpopulations. Also demonstrated here is the effect of increasing or decreasing the predation strength for the resistant subpopulation. Note that if b0 < a0, then the resistant subpopulation is less sensitive to immune predation and thus increases to a larger dormant size while simultaneously eliminating the sensitive subpopulation through contact inhibition. Figure 7c demonstrates the time-course of this process. In comparison, if the resistant subpopulation is more sensitive to immune predation (contrary to its definition), then the resistant subpopulation will be eliminated by the immune response, leaving a homogeneous sensitive population in the dormant state.
Figure 7.

The transition of a heterogeneous cancer population to a mostly homogeneous but resistant population can explain the observed decline in predation efficacy. Induction of the dormant state for the heterogeneous population (90% sensitive and 10% resistant cancer cells) is shown in (a) and cancer heterogeneity is shown via the phase portrait in (b). The dormant state from (a) is represented by the black diagonal line in (b) indicated by b0 = a0. As time increases, the composition is represented by a point on the line that oscillates up and down, starting at the initial composition point, (R, C) = (103, 9 × 103), and tending towards the dormant state value of (R, C) = (106, 950). From this curve, if b0 is increased, so that b0 > a0 contrary to the ‘resistant’ definition, then the resistant population is eliminated leaving the sensitive population in immune-induced dormancy (blue curve). But, if b0 is decreased, so that b0 < a0, then the resistant population is less sensitive to immune predation and so grows to a larger but still dormant size. This larger population causes the sensitive population to be eliminated through contact inhibition (red curve). The time-evolution of this process for b0 = 0.9a0 is shown in (c). To match the experimental data, we also simulate this process with b0 = 0.5a0 (d) and show the transition of the population composition in (e). Taking the composition at the corresponding time points and simulating a lysis assay with this composition gives the specific lysis curves shown in (f) overlaid with the experimental data from Saudemont & Quesnel [9].
To eliminate the noise introduced by the oscillations, we now choose initial conditions close to the dormant state induced when b0 = a0 to demonstrate the effect of immunoediting on observed predation efficacy when b0 < a0. This simulates the acquisition though mutation of a small resistant subpopulation within a dormant mostly homogeneous and sensitive cancer population. The new initial conditions are C0 = 950, R0 = 106 and I = 2000. Figure 7d shows the time dynamics of the transition from one dormant state to the other when b0 = 0.5a0. Figure 7e shows how the per cent composition of the tumour changes over this transition, with the days where experimental measurements were obtained indicated. And finally, figure 7f shows the simulated per cent lysis curves that would be obtained at each time point in dormancy assuming the heterogeneous composition indicated for that time by figure 7e.
The observed decay in immune cell predation efficacy [9] is captured by the simulated per cent lysis curves, and the simulated curves fit the experimental data well. In this example, the process of immunoediting converted a heterogeneous population of about 90 per cent sensitive cells and 10 per cent resistant cells to about 100 per cent resistant cells over the course of 1 year. In doing so, the dormant cancer burden was increased from about 104 cells to about 106 cells, which still represents a small cancer burden to the host.
5. Discussion
5.1. Decline in recruitment a strong mechanism to escape dormancy
Assuming strong immune recruitment potential, our work suggests that decay in immune predation may not be sufficient to explain cancer escape from dormancy. Theoretically, the immune response may be able to sustain the dormant state by increasing immune presence to compensate for the loss in predation strength. In long time, however, this may require an almost infinite immune presence, which is unphysical. Therefore, we consider that the immune response may be ultimately limited through an immune-exhaustion mechanism wherein the CTLs become unable to proliferate or recruit more immune cells in addition to the decline in predation efficacy. With this additional mechanism of limiting the immune response, our simulations predict cancer escape in as little as 6.5 years (fit 2).
Interestingly, decay in immune recruitment potential alone can explain cancer escape from dormancy. Our simulations show that even with strong predation strength, declining recruitment will lead to cancer escape. The escape times predicted in this case range from 6.4 (fit 2) to 52.2 years (fit 3). When both predation strength and recruitment potential are allowed to decay the escape times are not significantly shortened when compared with the previous case. The smallest escape time predicted is 6.3 years (fit 2).
This work suggests that recruitment decay is a stronger determinant of cancer escape than decay in predation efficacy because cancer escape is only predicted when recruitment is declining or limited. Additionally, escape times are not significantly shortened when predation decay is added to recruitment decay. Theoretically, if recruitment is strong enough, it can maintain a dormant cancer state until the required immune presence overwhelms the host.
Incidentally, the escape times predicted by our model with fits 1 and 2 seem to correspond to recurrence rates for immune-induced cancer dormancy. In clinical trials reported by Davis et al. [54] individuals with B-cell lymphomas were treated with anti-idiotype antibodies to stimulate an immunological reaction to their cancer. In five of the reported patients, residual disease (evidence of cancer dormancy) was found after 3–8 years, and these five continued recurrence-free for up to 3 years after the study. This suggests that the escape time for these immune-induced dormant cancers was at least 6 years.
In the least squares fitting procedure, fit 3 provided the best fit to the experimental per cent lysis curves from [9]. Fits 1 and 2, however, seem to predict cancer escape times that are more biologically relevant. This observation may be a consequence of over-fitting the data—or using a more complex model to capture an aspect of the data that is not as significant as the general trend. Another explanation may be that this data-driven modelling approach may misrepresent the actual causative mechanism of cancer escape from dormancy, which may be better captured by the mechanistic (immunoediting) approach described in §4.
5.2. Immunoediting a mechanism for declining cytotoxic T lymphoctye efficacy
Immunoediting has been proposed as one mechanism behind the escape of cancers from dormancy [1,5,6], and here we have demonstrated that this mechanism can also explain the observed decline in predation efficacy. Increase in cancer cell resistance to CTL-induced apoptotic signals has been linked to an increase in B7-H1 and B7.1 surface markers and a decrease in the production of anti-tumour immunity cytokines IFN-γ and TNF-α [9]. These observations were incorporated into our simulation of the immunoediting process, wherein a largely sensitive and immunogenic population was transformed over the course of 1 year into a population consisting of cancer cells that were less sensitive to immune attack and less immunogenic (lower recruitment potential). This transformation recapitulates the decay observed in CTL per cent lysis curves with time in dormancy through a mechanistic explanation.
Over time, mutations may arise in the cancer population creating more subpopulations of increasingly resistant cancer cells with low immunogenicity or increasingly sensitive cancer cells with high immunogenicity. The immunoediting process will quickly clear the mutations leading to more sensitive cancer cells, whereas the mutations leading to more resistant cancer cells will transform the population as demonstrated here. Through repetition of this transformation process, the cancer will eventually escape the dormant state, leaving behind an evolutionary history of increasingly resistant, decreasingly immunogenic, subpopulations.
6. Conclusions
This work is a first attempt to mathematically investigate the dynamics and mechanisms of immune-induced dormancy and immunoescape by incorporating relevant timescales of decay from experimental data. Other theoretical investigations have been made [32,34–37,40], but these lacked a connection to experimental cancer dormancy data. We have made many simplifying assumptions in this work to distil the process of cancer escape down to the fundamental players: cancer cells and CTLs. By necessity, we have thus ignored other cell types that may contribute to immune suppression (such as T regulatory cells and myeloid derived suppressor cells). We have also ignored other confounding factors such as the tumour microenvironment, tumour vasculature and other stromal cells. These additional players, as well as the incorporation of patient data, would enhance this model and lead to a more biologically complete predictive framework for the study of cancer dormancy and immunoevasion.
The goal of this work is to investigate the escape mechanisms from the immune-induced dormant state. To do this, we impose the dormant state, as is done in the AML mouse model [9], through the assumption of high immune recruitment and predation parameter values. Conversely, in patients, minimal residual disease may be imposed following therapies, and the immune response may play a role in maintaining this dormant state. All models, whether mouse or mathematical, are simplifications of the complex real-world system, but are nonetheless useful tools to investigate causative mechanisms. Here, we design the mathematical model to ensure the dormant state is reached (mimicking the mouse model and not patient outcome) so that the mechanisms of escape might be analysed.
The concept of treating cancers and minimizing recurrence through long-term maintenance of the dormant state [55] requires a complete understanding of all mechanisms of escape. Mathematical models and analyses provide an alternate modality to investigate these biological mechanisms, and continued interdisciplinary work can only lead to a more rapid attainment of this goal.
Acknowledgements
This project emerged from discussions during the First Annual Center of Cancer Systems Biology Workshop, ‘Systems Biology of Tumour Dormancy’, hosted at Steward Saint Elizabeth's Medical Center, Tufts University School of Medicine [56]. The authors wish to thank the participants of the immune working group where this problem was discussed. This work was supported by the National Cancer Institute under award no. U54CA149233 (to L. Hlatky) and by the Office of Science (BER), US Department of Energy under award no. DE-SC0001434 (to P.H.).
References
- 1.Dunn GP, Old LJ, Schreiber RD. 2004. The three Es of cancer immunoediting. Annu. Rev. Immunol. 22, 329–360 10.1146/annurev.immunol.22.012703.104803 (doi:10.1146/annurev.immunol.22.012703.104803) [DOI] [PubMed] [Google Scholar]
- 2.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. 2002. Cancer immunoediting: from immunosurveillance to tumor escape. Nat. Immunol. 3, 991–998 10.1038/ni1102-991 (doi:10.1038/ni1102-991) [DOI] [PubMed] [Google Scholar]
- 3.Teng MWL, Swann JB, Koebel CM, Schreiber RD, Smyth MJ. 2008. Immune-mediated dormancy: an equilibrium with cancer. J. Leukoc. Biol. 84, 988–993 10.1189/jlb.1107774 (doi:10.1189/jlb.1107774) [DOI] [PubMed] [Google Scholar]
- 4.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD. 2007. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903–907 10.1038/nature06309 (doi:10.1038/nature06309) [DOI] [PubMed] [Google Scholar]
- 5.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. 2012. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482, 405–409 10.1038/nature10803 (doi:10.1038/nature10803) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsushita H, et al. 2012. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482, 400–404 10.1038/nature10755 (doi:10.1038/nature10755) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quesnel B. 2013. Tumor dormancy: long-term survival in a hostile environment. Systems biology of tumor dormancy, pp. 181–200 New York, NY: Springer; [DOI] [PubMed] [Google Scholar]
- 8.Stewart TJ, Abrams SI. 2008. How tumours escape mass destruction. Oncogene 27, 5894–5903 10.1038/onc.2008.268 (doi:10.1038/onc.2008.268) [DOI] [PubMed] [Google Scholar]
- 9.Saudemont A, Quesnel B. 2004. In a model of tumor dormancy, long-term persistent leukemic cells have increased B7-H1 and B7.1 expression and resist CTL-mediated lysis. Blood 104, 2124–2133 10.1182/blood-2004-01-0064 (doi:10.1182/blood-2004-01-0064) [DOI] [PubMed] [Google Scholar]
- 10.Saudemont A, Hamrouni A, Marchetti P, Liu J, Jouy N, Hetuin D, Colucci F, Quesnel B. 2007. Dormant tumor cells develop cross-resistance to apoptosis induced by CTLs or imatinib mesylate via methylation of suppressor of cytokine signaling 1. Cancer Res. 67, 4491–4498 10.1158/0008-5472.CAN-06-1627 (doi:10.1158/0008-5472.CAN-06-1627) [DOI] [PubMed] [Google Scholar]
- 11.Saudemont A, Jouy N, Hetuin D, Quesnel B. 2005. NK cells that are activated by CXCL10 can kill dormant tumor cells that resist CTL-mediated lysis and can express B7-H1 that stimulates T cells. Blood 105, 2428–2435 10.1182/blood-2004-09-3458 (doi:10.1182/blood-2004-09-3458) [DOI] [PubMed] [Google Scholar]
- 12.Schatton T, Frank MH. 2009. Antitumor immunity and cancer stem cells. Ann. NY Acad. Sci. 1176, 154–169 10.1111/j.1749-6632.2009.04568.x (doi:10.1111/j.1749-6632.2009.04568.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schatton T, et al. 2010. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res. 70, 697–708 10.1158/0008-5472.CAN-09-1592 (doi:10.1158/0008-5472.CAN-09-1592) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerlinger M, et al. 2012. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883–892 10.1056/NEJMoa1113205 (doi:10.1056/NEJMoa1113205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wherry EJ. 2011. T cell exhaustion. Nat. Immunol. 131, 492–499 10.1038/ni.2035 (doi:10.1038/ni.2035) [DOI] [PubMed] [Google Scholar]
- 16.Peggs KS, Quezada SA, Korman AJ, Allison JP. 2006. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr. Opin. Immunol. 18, 206–213 10.1016/j.coi.2006.01.011 (doi:10.1016/j.coi.2006.01.011) [DOI] [PubMed] [Google Scholar]
- 17.Topalian SL, et al. 2012. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 10.1056/NEJMoa1200690 (doi:10.1056/NEJMoa1200690) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Curran MA, Montalvo W, Yagita H, Allison JP. 2010. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc. Natl Acad. Sci. USA 107, 4275–4280 10.1073/pnas.0915174107 (doi:10.1073/pnas.0915174107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilkie KP. 2013. A review of mathematical models of cancer–immune interactions in the context of tumor dormancy. Systems Biology of Tumor Dormancy, pp. 201–234 New York, NY: Springer; [DOI] [PubMed] [Google Scholar]
- 20.Kuznetsov VA. 1988. Mathematical modeling of the development of dormant tumors and immune stimulation of their growth. Cybern. Syst. Anal. 23, 556–564 10.1007/BF01078916 (doi:10.1007/BF01078916) [DOI] [Google Scholar]
- 21.Page KM, Uhr JW. 2005. Mathematical models of cancer dormancy. Leuk. Lymphoma. 46, 313–327 10.1080/10428190400011625 (doi:10.1080/10428190400011625) [DOI] [PubMed] [Google Scholar]
- 22.Kuznetsov VA, Makalkin IA, Taylor M, Perelson AS. 1994. Nonlinear dynamics of immunogenic tumors: parameter estimation and global bifurcation analysis. Bull. Math. Biol. 56, 295–321 [DOI] [PubMed] [Google Scholar]
- 23.Eftimie R, Bramson JL, Earn DJ. 2010. Interactions between the immune system and cancer: a brief review of non-spatial mathematical models. Bull. Math. Biol. 73, 2–32 10.1007/s11538-010-9526-3 (doi:10.1007/s11538-010-9526-3) [DOI] [PubMed] [Google Scholar]
- 24.d'Onofrio A. 2005. A general framework for modeling tumor-immune system competition and immunotherapy: Mathematical analysis and biomedical inferences. Physica D 208, 220–235 10.1016/j.physd.2005.06.032 (doi:10.1016/j.physd.2005.06.032) [DOI] [Google Scholar]
- 25.de Pillis LG, Radunskaya AE. 2001. A mathematical tumor model with immune resistance and drug therapy: an optimal control approach. J. Theor. Med. 24, 79–100 10.1080/10273660108833067 (doi:10.1080/10273660108833067) [DOI] [Google Scholar]
- 26.Kirschner DE, Panetta JC. 1998. Modeling immunotherapy of the tumor-immune interaction. J. Math. Biol. 37, 235–252 10.1007/s002850050127 (doi:10.1007/s002850050127) [DOI] [PubMed] [Google Scholar]
- 27.DeLisi C, Rescigno A. 1977. Immune surveillance and neoplasia—I A minimal mathematical model. Bull. Math. Biol. 39, 201–221 10.1007/BF02462859 (doi:10.1007/BF02462859) [DOI] [PubMed] [Google Scholar]
- 28.Matzavinos A, Chaplain MAJ, Kuznetsov VA. 2004. Mathematical modelling of the spatio-temporal response of cytotoxic T-lymphocytes to a solid tumour. Math. Med. Biol. 21, 1–34 10.1093/imammb/21.1.1 (doi:10.1093/imammb/21.1.1) [DOI] [PubMed] [Google Scholar]
- 29.Roose T, Chapman SJ, Maini PK. 2007. Mathematical models of avascular tumor growth. SIAM Rev. 49, 179–208 10.1137/S0036144504446291 (doi:10.1137/S0036144504446291) [DOI] [Google Scholar]
- 30.Takayanagi T, Kawamura H, Ohuchi A. 2006. Cellular automaton model of a tumor tissue consisting of tumor cells, cytotoxic T lymphocytes (CTLs), and cytokine produced by CTLs. IPSJ Digital Courier 2, 138–144 10.2197/ipsjdc.2.138 (doi:10.2197/ipsjdc.2.138) [DOI] [Google Scholar]
- 31.Enderling H, Hlatky LR, Hahnfeldt P. 2012. Immunoediting: evidence of the multifaceted role of the immune system in self-metastatic tumor growth. Theor. Biol. Med. Model. 9, 31. 10.1186/1742-4682-9-31 (doi:10.1186/1742-4682-9-31) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bellomo N. 2010. Modeling the hiding–learning dynamics in large living systems. Appl. Math. Lett. 23, 907–911 10.1016/j.aml.2010.04.008 (doi:10.1016/j.aml.2010.04.008) [DOI] [Google Scholar]
- 33.Bellomo N, Delitala M. 2008. From the mathematical kinetic, and stochastic game theory to modelling mutations, onset, progression and immune competition of cancer cells. Phys. Life Rev. 5, 183–206 10.1016/j.plrev.2008.07.001 (doi:10.1016/j.plrev.2008.07.001) [DOI] [Google Scholar]
- 34.Cattani C, Ciancio A, d'Onofrio A. 2010. Metamodeling the learning–hiding competition between tumours and the immune system: a kinematic approach. Math. Comp. Model. 52, 62–69 10.1016/j.mcm.2010.01.012 (doi:10.1016/j.mcm.2010.01.012) [DOI] [Google Scholar]
- 35.Kuznetsov VA, Knott GD. 2001. Modeling tumor regrowth and immunotherapy. Math. Comp. Model. 33, 1275–1287 10.1016/S0895-7177(00)00314-9 (doi:10.1016/S0895-7177(00)00314-9) [DOI] [Google Scholar]
- 36.d'Onofrio A. 2007. Tumor evasion from immune control: strategies of a MISS to become a MASS. Chaos Soliton Fract. 31, 261–268 10.1016/j.chaos.2005.10.006 (doi:10.1016/j.chaos.2005.10.006) [DOI] [Google Scholar]
- 37.d'Onofrio A, Ciancio A. 2011. Simple biophysical model of tumor evasion from immune system control. Phys. Rev. E 84, 031910. 10.1103/PhysRevE.84.031910 (doi:10.1103/PhysRevE.84.031910) [DOI] [PubMed] [Google Scholar]
- 38.d'Onofrio A. 2013. Multifaceted kinetics of immuno-evasion from tumor dormancy. Systems biology of tumor dormancy, pp. 111–143 New York, NY: Springer; [DOI] [PubMed] [Google Scholar]
- 39.d'Onofrio A. 2010. Bounded-noise-induced transitions in a tumor-immune system interplay. Phys. Rev. E 81, 021923. 10.1103/PhysRevE.81.021923 (doi:10.1103/PhysRevE.81.021923) [DOI] [PubMed] [Google Scholar]
- 40.Al-Tameemi MM, Chaplain MA, d'Onofrio A. 2012. Evasion of tumours from the control of the immune system: consequences of brief encounters. Biol. Direct. 7, 31. 10.1186/1745-6150-7-31 (doi:10.1186/1745-6150-7-31) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galante A, Tamada K, Levy D. 2012. B7-h1 and a mathematical model for cytotoxic T cell and tumor cell interaction. Bull. Math. Biol. 74, 91–102 10.1007/s11538-011-9665-1 (doi:10.1007/s11538-011-9665-1) [DOI] [PubMed] [Google Scholar]
- 42.DeNardo DG, Andreu P, Coussens LM. 2010. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metast. Rev. 29, 309–316 10.1007/s10555-010-9223-6 (doi:10.1007/s10555-010-9223-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robert CP, Casella G. 2010. Metropolis–Hastings algorithms. introducing Monte Carlo methods with R, pp. 167–97 New York, NY: Springer [Google Scholar]
- 44.Cirit M, Haugh JM. 2012. Data-driven modelling of receptor tyrosine kinase signalling networks quantifies receptor-specific potencies of PI3K- and Ras-dependent ERK activation. Biochem. J. 441, 77–85 10.1042/BJ20110833 (doi:10.1042/BJ20110833) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Pillis LG, Radunskaya AE, Wiseman CL. 2005. A validated mathematical model of cell-mediated immune response to tumor growth. Cancer Res. 65, 7950–7958 10.1158/0008-5472 (doi:10.1158/0008-5472) [DOI] [PubMed] [Google Scholar]
- 46.Dudley ME, et al. 2002. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298, 850–854 10.1126/science.1076514 (doi:10.1126/science.1076514) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Diefenbach A, Jensen ER, Jamieson AM, Raulet DH. 2001. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature 413, 165–171 10.1038/35093109 (doi:10.1038/35093109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chikkappa G, Chanana AD, Chandra P, Cronkite EP, Thompson KH. 1980. Cyclic oscillation of blood neutrophils in a patient with multiple myeloma. Blood 55, 61–66 [PubMed] [Google Scholar]
- 49.Rodriguez AR, Lutcher CL. 1976. Marked cyclic leukocytosis–leukopenia in chronic myelogenous leukemia. Am. J. Med. 60, 1041–1047 10.1016/0002-9343(76 (doi:10.1016/0002-9343(76)90578-7) [DOI] [PubMed] [Google Scholar]
- 50.Krikorian JG, Portlock CS, Cooney P, Rosenberg SA. 1980. Spontaneous regression of non-Hodgkin's lymphoma: a report of nine cases. Cancer 46, 2093–2099 (doi:10.1002/1097-0142(19801101)46:9<2093::AID-CNCR2820460931>3.0.CO;2-4) [DOI] [PubMed] [Google Scholar]
- 51.Mehta BC, Agarwal MB. 1980. Cyclic oscillations in leukocyte count in chronic myeloid leukemia. Acta Haematol. 63, 68–70 10.1159/000207373 (doi:10.1159/000207373) [DOI] [PubMed] [Google Scholar]
- 52.Posadas EM, Criley SR, Coffey DS. 1996. Chaotic oscillations in cultured cells: rat prostate cancer. Cancer Res. 56, 3682–3688 [PubMed] [Google Scholar]
- 53.Uhr JW, Tucker T, May RD, Siu H, Vitetta ES. 1991. Cancer dormancy: studies of the murine BCL1 lymphoma. Cancer Res. 51(Suppl. 18), 5045s–5053s [PubMed] [Google Scholar]
- 54.Davis TA, Maloney DG, Czerwinski DK, Liles T-M, Levy R. 1998. Anti-idiotype antibodies can induce long-term complete remissions in non-Hodgkin's lymphoma without eradicating the malignant clone. Blood 92, 1184–1190 [PubMed] [Google Scholar]
- 55.Hensel JA, Flaig TW, Theodorescu D. 2012. Clinical opportunities and challenges in targeting tumour dormancy. Nat. Rev. Clin. Oncol. 10, 41–51 10.1038/nrclinonc.2012.207 (doi:10.1038/nrclinonc.2012.207) [DOI] [PubMed] [Google Scholar]
- 56.Enderling H, Hahnfeldt P, Hlatky LR, Almog N. 2012. Systems Biology of tumor dormancy: linking biology and mathematics on multiple scales to improve cancer therapy. Cancer Res. 72, 2172–2175 10.1158/0008-5472.CAN-11-3269 (doi:10.1158/0008-5472.CAN-11-3269) [DOI] [PMC free article] [PubMed] [Google Scholar]