

Abstract
Understanding the origins of resistance to anti-oestrogen drugs is of critical importance to many breast cancer patients. Recent experiments show that knockdown of GRP78, a key gene in the unfolded protein response (UPR), can re-sensitize resistant cells to anti-oestrogens, and overexpression of GRP78 in sensitive cells can cause them to become resistant. These results appear to arise from the operation and interaction of three cellular systems: the UPR, autophagy and apoptosis. To determine whether our current mechanistic understanding of these systems is sufficient to explain the experimental results, we built a mathematical model of the three systems and their interactions. We show that the model is capable of reproducing previously published experimental results and some new data gathered specifically for this paper. The model provides us with a tool to better understand the interactions that bring about anti-oestrogen resistance and the effects of GRP78 on both sensitive and resistant breast cancer cells.
Keywords: anti-oestrogen resistance, breast cancer, mathematical modelling, GRP78, unfolded protein response, autophagy
1. Introduction
The majority of newly diagnosed breast cancers in women are oestrogen receptor (ERα) positive. These cancers are often treated with anti-oestrogen drugs such as tamoxifen (TAM) or ICI 182,780 (ICI; Faslodex; Fulvestrant), which block the normal functions of ERα or degrade it, resulting in cell cycle arrest or cell death [1,2]. Unfortunately, most patients receiving anti-oestrogen treatment ultimately acquire resistance during treatment [3]. While many molecular components and interactions relevant to anti-oestrogen resistance have been identified experimentally, how these components interact as a system to produce particular phenotypes is still an open question. Moreover, cross talk and feedback among these components makes reasoning about the system difficult and error prone. In this context, mathematical modelling based on experimental data provides a framework for us to analyse oestrogen signalling in a rigorous and consistent manner. The ultimate goal is an integrated model of the oestrogen receptor signalling system that explains the underlying mechanisms of endocrine resistance and enables accurate prediction of the impact of therapies on cell fate. The model may be useful for initial in silico exploration of a wide range of multi-drug therapies to combat and reverse resistance, an exploration that may be impractical to carry out experimentally. Moreover, developing such a model is an effective means for determining gaps and inconsistencies in our current knowledge and suggesting experiments for resolving these problems.
Recent experimental results point to a potentially major role for GRP78, a key player in the unfolded protein response (UPR), in anti-oestrogen resistance [4]. Consistent with previous studies [5,6] that observed increased GRP78 expression in several human breast cancer cell lines, these results showed that knockdown of GRP78 in ICI-resistant cells re-sensitized the cells to ICI, which reduced proliferation due to increased apoptosis. Furthermore, overexpression of GRP78 in an ICI-sensitive cell line conferred resistance to ICI while also upregulating autophagy. These experiments implicate the interaction of three cellular systems, UPR, autophagy and apoptosis, as critical to the role of GRP78 in anti-oestrogen resistance. The experiments also explored key cross-talk mechanisms among these systems by showing that GRP78 modulated mTOR activity (an activator of cell growth and an inhibitor of autophagy) and CHOP expression (a driver of apoptosis). (Full names of proteins, such as mTOR and CHOP, can be found in the electronic supplementary material, table S1.)
Activated by the accumulation of unfolded proteins in the endoplasmic reticulum (EnR) as a result of cellular stresses, the UPR attempts to increase the protein folding and protein degradation capacities of the EnR by upregulating synthesis of chaperones and other proteins [7,8]. A key chaperone in the EnR is GRP78, also known as BiP or HSPA5. Under unstressed conditions, GRP78 binds to EnR stress sensors IRE1, PERK and ATF6, and inhibits their activities [9]. When unfolded proteins accumulate, GRP78 binds to them and attempts to help them fold correctly. GRP78 also participates in the EnR-associated degradation (ERAD) process in which unfolded proteins are retro-translocated across the EnR membrane into the cytoplasm and degraded by proteasomes. The binding of GRP78 to unfolded proteins releases the active forms of IRE1, PERK and ATF6. Activated IRE1, PERK and ATF6 upregulate not only chaperone production but also several effectors of autophagy and apoptosis. Autophagy, a mechanism by which cells degrade unfolded or aggregated proteins and damaged organelles [10–12], reduces stress on the EnR by providing additional energy and raw materials to the cell. If homeostasis cannot be restored in response to the increased stress by means of UPR and autophagy, apoptosis may be triggered. Apoptosis, a form of programmed cell death, is an orderly process of cell death that can be initiated by either internal or external signals.
Several recent studies report on the complex interplay between UPR, autophagy and apoptosis that decides cell fate in response to various drug treatments [4,13–23]. Anti-oestrogens or other drug treatments can activate either cyto-protective or cyto-destructive UPR, and autophagy has been shown to aid in the cyto-protective role of the UPR [21–24], whereas cyto-destructive UPR leads to apoptosis [18,25].
The aim of this study is to determine whether our current mechanistic understanding of relevant cellular interactions is sufficient to explain the experimental results concerning the role of GRP78 in anti-oestrogen resistance reported in Cook et al. [4]. Our approach to modelling the interaction of these three cellular systems in response to GRP78 manipulation and ICI treatment starts with building models of the individual subsystems using data from the literature to arrive at baseline models that are not based on breast cancer cells. We then add cross-talk pathways to form an integrated model of the signalling network in breast cancer cells [26], and tune the model based on experimental data from two cell lines that reflect the clinical development of acquired endocrine resistance: LCC1, an oestrogen-independent but anti-oestrogen-sensitive line derived from the standard ERα-positive breast cancer cell line MCF-7 [27]; and LCC9, an oestrogen-independent and ICI-resistant line derived from LCC1 [28]. The mathematical model replicates the experimentally observed behaviour of the LCC1 and LCC9 cell lines when subjected to GRP78 knockdown or overexpression alone, and when combined with ICI treatment.
2. Model development
As shown in figure 1, the signalling network integrates three cellular systems: the UPR, apoptosis and autophagy. Each of these systems is modelled at a different level of detail, reflecting both differences in the degree of molecular knowledge about each system and the needs of our integrated model. For example, the molecular mechanism associated with the UPR is known in much greater detail than the mechanism of autophagy, and the molecular details of UPR interactions are necessary for understanding the influence of GRP78. By contrast, while the molecular details of the intrinsic apoptosis pathway are fairly well known, only the initial switch-like triggering of apoptosis is needed for the experimental results we are considering.
Figure 1.

Schematic of GRP78-regulated cross talk among three signalling pathways: UPR, autophagy and apoptosis. CHAP denotes the combination of GRP78 and GRP94 chaperones. ICI-triggered production of unfolded proteins relieves PERK, IRE1 and ATF6 from the inhibitory effect of chaperones and activates UPR, which results in increased GRP78 expression, increased autophagy through the IRE1–JNK path and apoptosis through the PERK–eIF2–CHOP path. GRP78 and cytosolic Ca2+ also contribute to increased autophagy by inhibiting mTORC1 activity. Solid lines denote chemical reactions (synthesis, degradation and modification) and dashed lines denote interactions (activation or inhibition).
Similarly, while there are many known pathways that produce interactions among the cellular systems in our model, we have only included those paths that appear to be the dominant ones for the experiments we consider here. In the future, as the model is used to explain additional experimental data, currently excluded pathways may become important and need to be added to the model. A complete list of the equations and parameters for our model, along with simulation code, is included in the electronic supplementary material.
2.1. Unfolded protein response module
Our representation of the UPR module is adapted from a model by Verdugo and co-workers [26]. The model captures the binding of chaperones to unfolded proteins and the resulting reduction in unfolded proteins through either correct folding or degradation (ERAD; figure 1). The binding of chaperones to unfolded proteins releases the three transmembrane sensors IRE1, PERK and ATF6 from the repressing effects of chaperones [9] and activates the three arms of the UPR. (i) Activated IRE1 converts unspliced mRNA (mXBP1u) for X-box-binding protein 1 to a spliced form (mXBP1s), which produces a transcription factor (XBP1s protein) that upregulates GRP78. (ii) Activated PERK phosphorylates eukaryotic initiation factor-2 (eIF2), which inhibits the translation of most proteins, thereby reducing the load of unfolded proteins entering the EnR. However, the translation of a few proteins, including ATF4 and CHOP, is increased by the phosphorylation of eIF2. ATF4 is a transcription factor that can increase CHOP expression, and CHOP regulates the transcription of proteins responsible for apoptosis [18]. (iii) Activated ATF6 is transported to the Golgi apparatus, where it is cleaved to produce a transcription factor that upregulates synthesis of XBP1 mRNA. ATF4, XBP1s and cleaved ATF6 also upregulate numerous EnR chaperones, which increase the protein-folding capacity of the EnR.
We assume that the association of UPR sensors with GRP78 is not the sole regulatory mechanism of IRE1, PERK and ATF6 activity, since these sensors can still upregulate the EnR stress response in cells manipulated by either GRP78 knockdown or disrupted binding of GRP78 with the sensors [4,29]. These results suggest that, in the absence of GRP78, alternate inhibitory mechanisms (e.g. other chaperones) release the sensors for further activation upon additional EnR stress (reviewed in Ma & Hendershot [30]). While numerous chaperones are likely to play a role in sensor inhibition, such as BI-1 [31,32], AIP1 [33] and RACK1 [34] for IRE1, and WFS1 [35] for ATF6, for simplicity of the model, we lump the effects of these other chaperones into the effect of a single chaperone, GRP94. While not redundant to GRP78, GRP94 plays an important role in EnR stress control [36–38]. Furthermore, GRP78 knockdown strongly induces GRP94 expression [4,39], indicating that cells can use an alternative mechanism to increase protein-folding capacity. To account for the effect of other chaperones, we modify Verdugo's UPR model by including GRP94 as an alternative stress regulator. While other chaperones play an essential role in EnR homeostasis, their modes of action on UPR sensors are still obscure relative to the well-characterized role of GRP78. Hence, we assume that GRP94 controls the UPR arms in the same way as GRP78, and modify the equations accordingly. We assume a constant level of GRP94 protein, ignoring its upregulation by EnR stress, since its enhanced expression under GRP78 knockdown does not relieve the stress, as discussed in Cook et al. [4], and so its upregulation may not be significant.
2.2. Autophagy module
Autophagy is principally an adaptive response to protect cells from stress, but, in certain settings, it can lead to cell death [40,41]. The process is controlled by a set of evolutionarily conserved autophagy-related proteins (ATG proteins). ATG6/Beclin-1 (BECN1), ATG8 (also known as microtubule-associated protein 1 light chain 3, LC3), ATG5, ATG12, ATG9 and ATG13 are essential for autophagosome formation in mammalian cells [42–44]. We model only the initial step of autophagy, namely the initiation of an isolation membrane, which is subsequently elongated to form the autophagosome, a vesicle enclosing the cellular material to be recycled. Autophagosomes then fuse with lysosomes, and their contents are degraded. BECN1 and ATG13 are indispensable for the formation of the isolation membrane. Under unstressed conditions, BCL2EnR (BCL2 in the EnR) binds to BECN1 and inhibits its activity [20,44,45]. Phosphorylation of BCL2EnR by activated JNK releases BECN1 from BCL2EnR, which then leads to an induction of autophagy. The active mammalian target of rapamycin complex (mTORC1*) negatively regulates autophagy by suppressing the activity of the ATG1 : ATG13 : ATG17 complex [44,46,47]. mTORC1 is regulated by a variety of upstream components such as AMPK, TSC1/2, AKT and intracellular Ca2+, depending upon the type of stress [4,15,48–50]. The transport of Ca2+ from the EnR to the cytoplasm, which plays a key role in the model, is regulated by the inositol-1,4,5-triphosphate receptor (IP3R), a Ca2+ channel in the EnR membrane [15].
Our model of autophagy initiation, based on a model by Tavassoly and co-workers [26], comprises the following steps (figure 1): (i) phosphorylation of BCL2EnR by phosphorylated JNK, and subsequent release of BECN1 and IP3R from inhibition by BCL2EnR, (ii) deactivation of mTORC1 by cytoplasmic Ca2+ and GRP78 [4], relieving its repressive effect on ATG13 and inducing expression of ATG9, and (iii) formation of autophagosomes by BECN1, ATG13 and ATG9 interaction. We have added ATG9 to Tavassoly's model because experiments show increased expression of ATG9 and increased autophagy upon GRP78 overexpression. Without ATG9, overexpression of GRP78 in the model leads to repression of IRE1 activity, inhibition of JNK phosphorylation, decreased binding of BECN1 to ATG13 and ultimately to downregulation of autophagy. Including ATG9 provides an alternative pathway that compensates for this downregulation.
2.3. Apoptosis module
We consider the intrinsic apoptosis pathway in which mitochondrial outer membrane permeabilization (MOMP) represents the key decision process and point of no return [51]. Ignoring downstream details, we model only the switch-like MOMP event by considering three classes of proteins: BCL2mito (anti-apoptotic BCL2 family members in mitochondria), BH3 (BH3-only BCL2 family members) and BAX (pro-apoptotic BCL2 family members directly involved in MOMP). Under non-apoptotic conditions, BCL2mito binds and sequesters BAX in an inactive form. The intrinsic apoptosis pathway responds to elevated intracellular stresses by activating BH3 proteins, which release BAX from the inhibitory effect of BCL2mito. Released BAX self-oligomerizes and forms pores in the mitochondrial outer membrane, releasing the mitochondrial intermembrane protein cytochrome c (Cyt-C) into the cytosol [52,53]. Cyt-C activates pro-caspases that subsequently activate effector caspases [51]. The model includes interactions between BCL2mito, BH3 and BAX as shown in figure 1 [26,54]. In the model, initiation of apoptosis is signalled by the rapid increase in free BAX, whereas, in the experiments, it was quantified in terms of the levels of both cleaved CASPASE-7 (a downstream effector caspase) and annexin V staining.
2.4. Integration of unfolded protein response, autophagy and apoptosis modules
EnR stress is a potent trigger of autophagy [16,55]. Autophagy is usually considered a pro-survival response, protecting cells against starvation, infectious agents and treatment with anti-cancer drugs. However, under certain circumstances, autophagy can promote death [40,41]. When EnR stress is extensive or sustained, and the UPR fails to restore normal functioning, apoptosis is triggered [19]. Several studies have shown that apoptosis and autophagy may be interconnected in some settings [56–60].
IRE1 and increased cytoplasmic Ca2+ have been implicated as inducers of autophagy linked to EnR stress in mammalian cells [15,16]. Activation of the IRE1 branch of the UPR can lead to phosphorylation and activation of JNK [16,61,62], and activated JNK phosphorylating BCL2EnR leads to the dissociation of BCL2EnR from BECN1 and activation of autophagy [20]. EnR stress can also lead to release of Ca2+ from the EnR into the cytosol through the IP3R channel, which in turn can activate autophagy by inhibiting mTORC1 activity [15,63]. BCL2EnR has been proposed as a regulator of IP3R function, and thus the cytoplasmic Ca2+ level [15,45]. In particular, phosphorylation of BCL2EnR by phosphorylated JNK renders the IP3R active and increases the cytoplasmic Ca2+ concentration [64]. These pathways are shown in figure 1.
In addition, Cook et al. [4] proposed that GRP78 inhibits mTOR phosphorylation and decreases mTORC1 activity by activating AMPK and phosphorylating TSC2 (also reviewed in Cook & Clarke [65]). For simplicity, the details of mTOR phosphorylation and mTORC1 formation are modelled as a simple reaction that activates or deactivates the mTORC1 complex. Thus, the effect of GRP78 on mTORC1 has been modelled as a direct effect, excluding the unknown intermediate reactions between GRP78, AMPK and TSC2. To account for the pro-survival effect of autophagy, we add an autophagy-dependent negative feedback into the equation for cellular stress.
The UPR is usually linked to apoptosis through its PERK signalling branch, which downregulates the expression of anti-apoptotic BCL2mito and upregulates pro-apoptotic BH3 proteins [66,67]. In the model, this regulation is accomplished via increased expression of CHOP, which has been shown to promote apoptosis in several contexts [18,25,66,67]. The UPR can also signal to apoptosis via an IRE1–JNK pathway. We have not explicitly included this pathway in our initial modelling, lumping all UPR signalling to apoptosis into the CHOP pathway.
In addition, increased cytoplasmic Ca2+ can stimulate the production of reactive oxygen species (ROS) in mitochondria, impair electron transport and oxidative phosphorylation and increase expression of pro-apoptotic BH3 proteins. In the model, we lump these effects of cytoplasmic Ca2+ on apoptosis into its action on BH3 protein expression. This role for cytoplasmic Ca2+ in triggering apoptosis via the UPR is consistent with a study by Ouyang et al. [68], who observed that increasing GRP78 protects astrocytes from ischaemic injury by decreasing the net flux of Ca2+ from the EnR to the mitochondria.
2.5. ICI treatment
ICI treatment increases ERα aggregation in the cytoplasm, which generates a weak signal that activates the UPR [69]. Furthermore, Cook et al. [4] demonstrated that ICI treatment induces a low level of autophagy in breast cancer cells, consistent with its modest effect on the UPR. Therefore, in the model, we consider a weak activation of UPR by ICI that comes through a parameter ‘stress’, which represents the rate of production of unfolded proteins.
In response to ICI treatment of anti-oestrogen-sensitive breast cancer cells, the major apoptotic stimulus is through ERα degradation not UPR activation. Expression of the anti-apoptotic protein BCL2mito is promoted by ERα [70], and since ICI degrades ERα, ICI therapy results in a decreased level of BCL2mito, favouring apoptosis [71]. In the model, the BCL2mito level is controlled by ERα and NFκB. We do not model details of ERα signalling but merely allow for its synthesis and degradation, including an ICI-dependent degradation term.
The addition of ICI to anti-oestrogen-resistant cells that have had GRP78 knocked down increases mTORC1 activity and decreases autophagy [4]. The exact mechanism of mTORC1 activation is unknown. However, it is known that ICI can induce AKT or ERK1/2 phosphorylation in ERα-negative breast cancer cells [72] or in ERα-positive and HER2-abundant breast cancer cells [73], which can result in mTOR phosphorylation and enhanced mTORC1 activity. This ICI-mediated activation of AKT/ERK depends on ERα36, a membrane-bound ERα [72] that is not degraded by ICI. However, given the complex bidirectional cross talk between EnR stress and mTOR signalling [74], we cannot rule out other mechanisms involving downstream effectors of UPR. In the model, we add a phenomenological term to account for the effect of ICI on mTORC1.
2.6. Proliferation
The deterministic model we have developed captures the average behaviour of one cell in a population of N cells. To model the expansion or contraction of the population, we use a simple birth–death equation of the form
| 2.1 |
where the specific birth and death rates of the population, kB and kD, are functions of the signalling network that determines cellular responses to stress. In particular, phosphorylated (active) mTOR, through the active protein complex mTORC1*, regulates global rates of protein synthesis. Since the rate of cell division must match the rate of cell growth, we write the specific birth rate kB as a function of activated mTORC1 in the form
| 2.2 |
where T0 is the intrinsic doubling time of the cell culture (28 h). With our choice of parameter values (w1 = 1.9, wmTORC1 = 0.07), kB increases smoothly from 0.0215 h−1 at [mTORC1*] = 0 to 0.0247 h−1 at [mTORC1*] 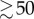 (see the electronic supplementary material, figure S1(a)).
(see the electronic supplementary material, figure S1(a)).
The specific death rate in experiments and in the model is determined by the rate of apoptosis. In our model, the rate of apoptosis is determined by the relative levels of BCL2mito and BH3 proteins. Individual cells in a population will have varying levels of these proteins owing to stochastic effects within the cells. Cells with higher levels of BCL2mito or lower levels of BH3 will be more immune to apoptotic stimuli, whereas cells with less BCL2mito or more BH3 will be more likely to die in response to such stimuli. For this reason, we write the specific death rate kD as a function of BCL2mito and BH3 in the form
| 2.3 |
where 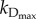 is the specific death rate of apoptotic cells (when [BH3]
is the specific death rate of apoptotic cells (when [BH3] 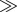 [BCL2mito]). With our choice of parameter values (w2 = 0.1, wBCL2 = −0.07, wBH3 = 0.05),
[BCL2mito]). With our choice of parameter values (w2 = 0.1, wBCL2 = −0.07, wBH3 = 0.05), 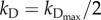 when [BH3] = 1.4 × [BCL2mito] + 12. For smaller values of [BH3], kD drops rapidly to approximately 10−3 h−1 (see the electronic supplementary material, figure S1(b)).
when [BH3] = 1.4 × [BCL2mito] + 12. For smaller values of [BH3], kD drops rapidly to approximately 10−3 h−1 (see the electronic supplementary material, figure S1(b)).
To compute the expansion of a cell population over a specified time period, 0 ≤ t ≤ Tend, we first compute the steady-state values of active mTORC1, BCL2 and BH3 from the model under specific experimental conditions. Then, we compute kB and kD according to equations (2.2) and (2.3). Finally, equation (2.1) implies that N(Tend) = N(0) · exp[(kB − kD) · Tend].
2.7. Matching experimental data
Our baseline model refers to LCC1 cells. To model LCC9 cells, we make a small number of parameter changes to account for the biological differences observed between these two cell lines. In anti-oestrogen-resistant LCC9 cells, growth factor signalling replaces ERα signalling as the driver of proliferation [75–77]. Also, compared with LCC1 cells, LCC9 cells have higher NFκB activity [78] and lower levels of expression of ERα [79]. Thus, for modelling LCC9 cells, the synthesis rate of the ERα is decreased and the amount of active NFκB is increased relative to the LCC1 model. In addition, to account for increased GRP78 levels in LCC9 cells, we assume a higher basal level of stress and hence of unfolded protein production than in LCC1 cells. This assumption is consistent with the fact that LCC9 cells have higher basal autophagy than LCC1 cells [4].
In the experimental protocol, transient transfection was performed 24 h before ICI treatment, and ICI treatment was continued for 6 days before final measurements were made [4]. To reproduce this protocol in our simulations, the initial conditions (at t = −24 h) for the simulation were chosen to correspond to a steady state of the model, knockdowns or overexpressions were initiated at −24 h and ICI was added at t = 0. The model was then simulated for an additional 144 h (6 days) to allow direct comparison between the model endpoint and the experimental measurements.
3. Results
The subsystem models and cross-talk mechanisms discussed above were merged into an overall model and the parameters were adjusted to reproduce the experimental measurements at 144 h reported by Cook et al. [4]. In addition, some time-course data were gathered specifically for this study, to ensure that the model captures key transient characteristics of the system in addition to capturing its end-to-end performance. We compare model simulations with observations on four cell types: LCC1, LCC1/GRP78+, LCC9 and LCC9/GRP78−. After showing that the model is consistent with observed cell proliferation for all cases, we consider each case individually and show that the model captures key internal states of the system related to apoptosis, growth (mTORC1* level) and autophagy.
When LCC1 cells are treated with increasing doses of ICI, they exhibit a marked decrease in relative proliferation at 6 days (figure 2a). By contrast, LCC9 cells show no change in proliferation as a function of ICI dose (figure 2b). Figure 2 also shows that the sensitivity of LCC1 cells and the resistance of LCC9 cells can be reversed by GRP78 overexpression and GRP78 knockdown, respectively. The end-to-end performance of our model for these four cases shows good agreement with the experimental results in figure 2.
Figure 2.

GRP78 level affects cell proliferation in response to ICI. (a) The proliferation of ICI-sensitive LCC1 cells (left) decreases with increasing dose of ICI, whereas GRP78-overexpressing LCC1 cells (right) are insensitive to ICI treatment. Experiments (with error bars) and model simulations (without error bars) show good agreement. (b) Proliferation of ICI-resistant LCC9 cells is not sensitive to ICI, but knockdown of GRP78 in these cells restores their sensitivity to ICI. Knockdown of autophagy, via BECN1 knockdown, has little effect on ICI sensitivity, but joint knockdown of BECN1 and GRP78 has a synergistic effect. Experimental results in (a) and (b) adapted from Cook et al. [4].
3.1. LCC1
For the case of LCC1 cells treated with ICI, the reduction in proliferation appears mainly to be the result of increased levels of apoptosis, as illustrated by the marked increase in the number of cells positive for annexin V staining in response to 100 nM ICI (figure 3a). A simulation of BAXmT (the active form of BAX in our model) in response to treatment with 100 nM ICI (figure 3b) indicates that an ‘average’ cell would commit to apoptosis near the 24 h time point. The apoptotic response is due mainly to the degradation of ERα by ICI and the resulting decrease of anti-apoptotic BCL2mito. In our deterministic model, we simulate a single cell, and for specified initial conditions, the cell either commits to apoptosis or it does not. In a population of cells, however, a specified treatment will cause some cells to commit to apoptosis and others to live, owing (presumably) to the fact that protein levels in individual cells are distributed over a range of values [80,81]. Thus, cells with lower levels of BCL2mito are more likely to commit apoptosis, whereas those with higher levels are less likely to do so. The model captures this effect in the formula for the specific death rate, kD, in the proliferation equation.
Figure 3.

GRP78 level affects the onset of apoptosis in response to ICI treatment. (a) In LCC1 cells, ICI induces apoptosis (as measured by annexin V staining), and GRP78 overexpression reverses this effect. (b) In deterministic simulations of LCC1 cells, ICI induces apoptosis (as evidenced by BAX activation), and a higher dose of ICI moves the activation point earlier in time, as does knockdown of autophagy via ATG9. GRP78 overexpression inhibits apoptosis in response to ICI, but this effect is reversed if ATG9 is also knocked down. (c) In ICI-resistant LCC9 cells, GRP78 knockdown, with or without ICI, induces apoptosis. (d) Deterministic simulations of LCC9 cells show activation of apoptosis in response to GRP78 knockdown, but not in response to ICI alone. The combination of GRP78 knockdown and ICI moves the commitment to apoptosis earlier in time. Experimental results in (a) and (c) adapted from Cook et al. [4].
In terms of deterministic simulations, movement of apoptosis initiation earlier in time indicates that even more BCL2mito would be required to avoid apoptosis for this case; thus, we would expect a larger fraction of cells in a population to die than in a simulation in which apoptosis was initiated later. Figure 3b shows that the initiation of apoptosis in the model does indeed move earlier in response to increasing ICI dose. Although ICI decreases the proliferation rate mainly by inducing apoptosis, the experimentally observed decrease in phospho-mTOR after ICI treatment (figure 4a) may also contribute to the reduced proliferation. The model captures the effect of ICI on mTOR through the reduction of active mTORC1 (figure 4b), and the effect on growth through the dependence of the specific birth rate, kB, on mTORC1*. We assume that mTORC1 activity, which we model directly, reflects the active (phosphorylated) mTOR level. For the case of treatment with 100 nM ICI, our model predicts a decrease in proliferation of 42 per cent (figure 2a), with 37 percentage points of the decrease coming from increased apoptosis and 5 percentage points from decreased growth, showing that apoptosis is the dominant mechanism.
Figure 4.
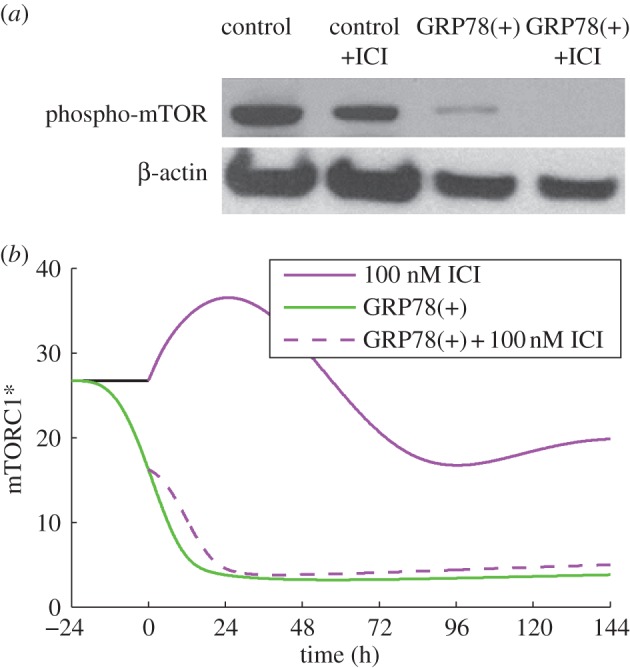
GRP78 regulates mTOR activity. (a) ICI treatment of LCC1 cells decreases mTOR phosphorylation slightly, while GRP78 overexpression, with or without ICI, markedly reduces it. Adapted from Cook et al. [4] with permission. (b) Model simulations are consistent with the experimental results at the 6 day time point. The model variable, mTORC1*, is related to the experimental variable phospho-mTOR.
ICI treatment modestly increases autophagy in LCC1 cells in experiments (figure 5a) and model simulations (figure 5b). Experiments blocking autophagy via ATG5 knockdown enhanced the killing effect of ICI [4], showing that the ICI-induced autophagic response is pro-survival. We simulate these experiments by knocking down ATG9 in our model, which causes apoptosis to initiate earlier in time (figure 3b), indicating that a larger fraction of cells will die. The reduction in autophagy upon ATG9 knockdown decreases the negative feedback to stress and thereby increases the ICI-induced stress level. The increased stress intensifies UPR activation resulting in increased CHOP expression, which drives apoptosis earlier.
Figure 5.

GRP78 regulates autophagy. (a) GRP78 overexpression in LCC1 cells results in a marked increase in autophagy as indicated by LC3-II. (b) Model simulations reproduce this effect in terms of autophagosome level. (c) GRP78 knockdown in LCC9 cells does not change the autophagy level at the 6 day time point, but knockdown plus ICI significantly reduces the level. (d) Model simulations are consistent with the experimental results at the 6 day time point. Experimental results in (a) and (c) adapted from Cook et al. [4].
3.2. LCC1/GRP78(+)
For the case of LCC1 cells overexpressing GRP78, ICI treatment does not induce apoptosis, as shown in figure 3a. To produce the corresponding simulation (figure 3b), GRP78 was overexpressed by 3.5-fold, in accord with the experimentally observed level in fig. 2d of Cook et al. [4]. We see no triggering of apoptosis and thus expect no substantial decrease in proliferation due to cell death. While ICI treatment decreases ERα levels and thereby BCL2mito, apoptosis in this simulation is averted by GRP78 overexpression through three mechanisms: (i) a decrease in pro-apoptotic signalling from PERK to CHOP, (ii) an increase in autophagy via GRP78-mediated mTORC1 inactivation and consequent reduction in UPR activity, and (iii) a decrease in pro-apoptotic signalling from IRE1 to cytoplasmic Ca2+ via phospho-JNK. To determine the relative strengths of these mechanisms in the model, we can hold the levels of CHOP, autophagy or Ca2+ equal to their levels in ICI-treated LCC1 cells at 144 h and examine how this changes proliferation for the LCC1/GRP78+ case. For treatment with 100 nM ICI, the proliferation of LCC1/GRP78+ cells decreases by 51, 28 or 25 per cent when CHOP, autophagy or Ca2+, respectively, are held at LCC1 levels, indicating that a decrease in pro-apoptotic signalling from PERK to CHOP is the dominant mechanism.
Figure 4a shows that GRP78 overexpression dramatically suppresses mTOR activity in experiments. ICI treatment, either alone or in combination with GRP78 overexpression, has no additional effect. Model simulations (figure 4b) exhibit the same qualitative response as experiments. In the model, the reduction in proliferation owing to GRP78 overexpression (observed in figure 2a) is due to a slowdown in cell growth captured by the dependence of the birth rate on active mTORC1. Biologically, GRP78-induced dephosphorylation of mTOR, and hence decreased mTORC1 activity, causes the phosphorylation and inactivation of initiation factors eIF4E and eIF2E, which reduces the rate of protein synthesis in cells [82].
Because mTORC1* significantly inhibits autophagy, both experimental and simulation results show that autophagy is markedly upregulated by GRP78 overexpression (figure 5a,b). Experiments further show that ATG5 knockdown restores sensitivity of LCC1/GRP78+ cells to ICI, suggesting a significant role for autophagy in the resistance of LCC1/GRP78+ (results not shown). Similarly, BAXmT simulations (figure 3b) show that overexpression of GRP78 eliminates apoptosis, whereas knocking down autophagy through ATG9 restores the initiation of apoptosis in response to ICI treatment.
Experimental fold-changes at the 6 day time point due to GRP78 overexpression were reported for mTORC1* (down 6.9-fold), CHOP (down 3.0-fold), ATG9 (up 3.0-fold) and total BCL2 (up 2.0-fold) in Cook et al. [4]. Model simulation results are in good agreement: mTORC1* (down 7.0-fold), CHOP (down 3.1-fold), ATG9 (up 2.6-fold) and total BCL2mito (up 2.1-fold). In the model, total BCL2mito was taken as the sum of BCL2mito, BCL2 : BH3 and BAXm : BCL2 levels.
3.3. LCC9
Normal LCC9 cells are insensitive to ICI treatment (figure 2b), as seen by the lack of apoptosis as measured by annexin V staining (figure 3c). Simulation of an LCC9 cell with ICI treatment shows no activation of BAX, and hence no apoptosis, over 6 days (figure 3d). LCC9 cells behave this way in our simulation because their BCL2 level is elevated by their increased NFκB activity. (NFκB directly binds the promoters and induces the expression of several anti-apoptotic proteins, including BCL-2 family members [83].) LCC9 cells are also less sensitive to degradation of ERα by ICI since they have less ERα to begin with. Figure 6d shows that the mTORC1* level in our model of LCC9 cells does not change significantly in response to ICI, so cell growth rate is not expected to change significantly. The combination of no apoptosis and unchanged growth rate renders our simulated LCC9 cells insensitive to ICI. ICI treatment produces a small increase in autophagy in both experiments and simulations (figure 5c,d). As might be expected, knocking down autophagy via BECN1 has little effect on the insensitivity of LCC9 cells to ICI in the experiments or simulations (figure 2b), although high doses of ICI do reduce proliferation somewhat.
Figure 6.

Time courses of GRP78total (a), CHOP (b), p-JNK (c) and mTORC1* (d) measured for LCC9 cells subjected to GRP78 knockdown with and without added ICI. Simulation results (lines) are compared with experiments (circles and squares with error bars). Experimental results are new to this work (see §5).
3.4. LCC9/GRP78(−)
When GRP78 is knocked down in LCC9 cells, proliferation decreases significantly (approx. 25%) and treatment with ICI causes an intensifying decrease with increasing dose (figure 2b). These effects are due to increasing levels of apoptosis, as indicated by the experimentally observed increase in annexin V staining (figure 3c). Model simulations of these experiments (figure 3d) show that BAX is activated when GRP78 is knocked down and adding ICI moves the activation earlier in time; thus, a larger fraction of cells in a population will die. The increases in apoptosis come from four sources: (i) knocking down GRP78 in the model increases activation of the UPR, which in turn provides pro-apoptotic signalling via increased CHOP expression and increased cytoplasmic Ca2+ levels; (ii) ICI dramatically increases mTORC1 activation (figure 6d), inhibiting autophagy (figure 5d) and causing increased UPR activation and pro-apoptotic signalling; (iii) ICI reduces the level of ERα, via enhanced degradation, leading to a decrease in the level of BCL2mito that increases the likelihood of apoptosis; and (iv) ICI increases EnR stress and causes a further activation of apoptotic signalling.
The experimental results in figure 6d indicate no dramatic changes in mTORC1 activity when GRP78 is knocked down, so we expect that the changes in proliferation observed in figure 2b are due to changing levels of apoptosis. The experimental results also show that adding ICI to LCC9/GRP78(−) cells significantly intensifies the effect of GRP78 knockdown on mTORC1 activation. Simulations of the model in figure 6d are in good agreement with these results. Simulations of normal LCC9 cells with ICI treatment (figure 6d) show little change in mTORC1 activity because the initial increase in mTORC1 activity by ICI is offset by ICI activation of UPR, resulting in an increased GRP78 expression and cytosolic Ca2+ level that both inhibit mTORC1 activity. The dramatic increase when ICI is combined with GRP78 knockdown is a result of the direct action of ICI on mTORC1 activation in the model combined with the saturation of the mTORC1 deactivation reaction.
Experimentally, we also find that GRP78 knockdown has little effect on autophagy after 6 days (figure 5c), whereas the model shows a small decrease from the baseline value (figure 5d). The addition of ICI in the presence of a GRP78 knockdown in LCC9 cells significantly reduces the extent of autophagy in both experiments and simulations (figure 5c,d). This reduction in the model is a consequence of ICI's role in promoting mTORC1 activation. Furthermore, knocking down both autophagy (via BECN1) and GRP78 decreases proliferation in experiments and simulations (figure 2b).
We have collected time-course data for four key proteins, GRP78, phospho-JNK, CHOP and mTORC1*, for LCC9 cells treated with GRP78 siRNA alone and in combination with ICI (symbols, figure 6). The data show the fold change of each protein, compared with its unstressed level, at several time points. Our simulations capture the experimental data quite well (lines, figure 6). The experimental data in figure 6a show that GRP78 siRNA successfully decreases GRP78 protein expression by 80 per cent within 72 h. This decrease activates the UPR and ultimately causes JNK phosphorylation and increases CHOP expression (figure 6b,c). Owing to the inverse relationship between GRP78 expression and mTORC1 activity, GRP78 knockdown stimulates mTORC1 activation (figure 6d). ICI treatment of GRP78(−) cells does not have a major effect on GRP78 expression (figure 6a), but it increases phospho-JNK, CHOP expression and mTORC1 activity significantly compared with GRP78 knockdown alone (figure 6b–d).
4. Discussion
Our model provides a clear, consistent and quantitatively accurate account of how we think apoptosis, autophagy and UPR interact to produce both sensitivity and resistance to anti-oestrogen therapy under various conditions. The model allows us to characterize the relative importance of the multiple pathways that create the observed phenotypes and demonstrates that the mechanisms we have chosen to model are sufficient to explain the experimental results under consideration. However, this success does not prove that other pathways are unimportant to the current experimental results or will not need to be considered as we move forward with modelling other cellular responses to anti-oestrogen therapy.
An interesting aspect of the model, and the experiments, is that, whether GRP78 is overexpressed (in LCC1 cells) or knocked down (in LCC9 cells), proliferation in both cases decreases when there is no ICI treatment. This is due to the nonlinear nature of the model. If GRP78 is sufficiently overexpressed, proliferation ultimately decreases owing to inactivation of mTOR. Similarly, if GRP78 is sufficiently knocked down, proliferation ultimately decreases owing to increasing apoptosis. Somewhere between these two extremes, there is an optimal level of GRP78 at which proliferation is a maximum. For cells not at this maximum level, if a small decrease in GRP78 causes proliferation to increase, then a small increase in GRP78 will cause proliferation to decrease. This is the case for LCC9 cells. But for large changes in GRP78, proliferation ultimately decreases regardless of whether GRP78 is increased or decreased.
In the model, GRP78 overexpression creates resistance by suppressing pro-apoptotic signalling, using the three mechanisms pointed out in §3. However, contributions to resistance from other mechanisms under the direct control of GRP78 cannot be excluded. For example, GRP78 can suppress EnR stress-induced apoptosis by sequestering caspase-7 and inhibiting its activation [84,85]. GRP78 can also inhibit pro-apoptotic protein BIK [86]. There may also be a contribution from growth factor receptor (GFR) signalling to GRP78 overexpression-induced resistance, since membrane-bound GRP78 can activate RAS–MAPK and PI3K–AKT pathways in other cells [87,88]. The effect of growth factor signalling may not be significant in our cells, as Cook et al. [4] observed no change in anti-oestrogen resistance when cell surface signalling was blocked in LCC9 cells. Measurements of AKT and ERK activity in LCC1/GRP78(+) cells may clarify whether GFR signalling is important or not.
To model LCC9 cells, we assumed a higher basal rate of unfolded protein production than in LCC1 cells. Persistent ROS stress or persistent production of unfolded proteins may induce adaptive stress responses, enabling cells to maintain viability [89]. Since LCC9 cells were obtained from LCC1 cells by a stepwise selection using ICI, we speculate that LCC9 cells are adapted to survive higher stress levels than their parental LCC1 cells. Furthermore, LCC9 cells have a higher basal level of autophagy than LCC1 cells, indicating higher basal stress. The increased GRP78 mRNA and protein levels in LCC9 cells may be a consequence of mechanisms other than just increased basal stress, however. Dai et al. [90] found that PI3K/AKT promotes increased accumulation of GRP78 in HEK293 cells by increasing protein stability and not by increasing the GRP78 mRNA level. This may be the case in LCC9 cells as well, since they have high PI3K/AKT activity [91]. Other UPR-independent mechanisms that can result in increased GRP78 mRNA and protein levels include IGF-1R/PI3K/AKT/mTORC1 signalling [92] and Raf1–MEK–ERK signalling [93], observed in mouse embryonic fibroblast cells and in human gastric tumour cells, respectively. It would be interesting to know whether the increased level of GRP78 is a consequence of epigenetic changes or is actively maintained to deal with higher rates of unfolded proteins.
In the model, GRP78 knockdown restores anti-oestrogen sensitivity in LCC9 cells by increased apoptotic signalling through CHOP and Ca2+. However, other mechanisms may be involved. As mentioned above, GRP78 can sequester caspases and pro-apoptotic proteins, so GRP78 knockdown may release these proteins and enhance apoptotic signalling. In addition, EnR stress-induced signalling to apoptosis via CASPASE-4/12 [94,95] or the IRE1–JNK pathway may be important. GRP78 knockdown is also known to induce apoptosis by blocking AKT activation in colon, PTEN-null leukaemia and prostate cancer cell lines [88,96,97]. Hence, it will be important to measure the effect of knocking down GRP78 on AKT signalling in LCC9 cells.
By providing a temporal interaction mechanism capable of explaining key aspects of anti-oestrogen resistance, this model is a step towards a better understanding of an important clinical problem affecting a large number of breast cancer patients. Accurate prediction, however, will require a model that is based on a much larger set of experimental results. Future work will improve the model by considering data involving knockdowns and overexpression of other key resistance players, such as XBP1, NFκB and ERα. In addition, we plan to extend the model to include relevant pathways from growth factor signalling.
5. Experimental material and methods
5.1. Materials
The following materials were obtained as indicated: ICI 182,780 (Tocris Bioscience, Ellisville, MO); improved minimal essential medium (IMEM; Gibco Invitrogen BRL, Carlsbad, CA); bovine calf charcoal stripped serum (Equitech-Bio Inc., Kerrville, TX); Lipofectamine RNAiMax reagent (Invitrogen); GRP78 siRNA (On-Target plus SMART pool (consisting of three different siRNA for the same target), ThermoScientific Dharmacon, Lafayette, CO); antibodies were obtained from the following sources: GRP78, CHOP, JNK, phospho-JNK and mTORC1 (Cell Signalling Technology); β-actin and polyclonal and horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA).
5.2. Cell culture
MCF7/LCC9 (LCC9) breast carcinoma cells were grown in phenol-red-free IMEM medium containing 5 per cent charcoal-treated calf serum. Cells were grown at 37°C under a humidified 5 per cent CO2/95 per cent air atmosphere. Cells were plated in a six-well plate and transfected with control or GRP78 siRNA. The next day, the medium was replaced and cells were treated with vehicle or 100 nM ICI for 0, 24 or 48 h before protein was harvested.
5.3. Western blot hybridization
Treated cell monolayers were solubilized in radioimmunoprecipitation assay lysis buffer. Protein concentrations of cell lysates were measured using a standard bicinchoninic acid assay. Proteins were size fractionated by polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane. Non-specific binding was blocked by incubation with Blotto (Tris-buffered saline containing 5% powdered milk and 1% Triton X-100). Membranes were incubated overnight at 4°C with primary antibodies. The next day, membranes were washed and then incubated with polyclonal HRP-conjugated secondary antibodies (1 : 2000) for 1 h at room temperature. Immunoreactive products were visualized and quantified by densitometry using the ImageJ digital densitometry software (http://rsbweb.nih.gov/ij/). Protein loading control was determined by incubation of stripped membranes with a monoclonal antibody to β-actin (1 : 1000).
References
- 1.Dauvois S, Danielian PS, White R, Parker MG. 1992. Antiestrogen ICI 164,384 reduces cellular estrogen receptor content by increasing its turnover. Proc. Natl Acad. Sci. USA 89, 4037–4041 10.1073/pnas.89.9.4037 (doi:10.1073/pnas.89.9.4037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.O'Regan RM, et al. 1998. Effects of the antiestrogens tamoxifen, toremifene, and ICI 182,780 on endometrial cancer growth. J. Natl Cancer Inst. 90, 1552–1558 10.1093/jnci/90.20.1552 (doi:10.1093/jnci/90.20.1552) [DOI] [PubMed] [Google Scholar]
- 3.Jordan VC, O'Malley BW. 2007. Selective estrogen-receptor modulators and antihormonal resistance in breast cancer. J. Clin. Oncol. 25, 5815–5824 10.1200/JCO.2007.11.3886 (doi:10.1200/JCO.2007.11.3886) [DOI] [PubMed] [Google Scholar]
- 4.Cook KL, Shajahan AN, Warri A, Jin L, Hilakivi-Clarke LA, Clarke R. 2012. Glucose-regulated protein 78 controls cross-talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer Res. 72, 3337–3349 10.1158/0008-5472.CAN-12-0269 (doi:10.1158/0008-5472.CAN-12-0269) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez PM, Tabbara SO, Jacobs LK, Manning FCR, Tsangaris TN, Schwartz AM, Kennedy KA, Patierno SR. 2000. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res. Treat. 59, 15–26 10.1023/A:1006332011207 (doi:10.1023/A:1006332011207) [DOI] [PubMed] [Google Scholar]
- 6.Gazit G, Lu J, Lee AS. 1999. De-regulation of GRP stress protein expression in human breast cancer cell lines. Breast Cancer Res. Treat. 54, 135–146 10.1023/A:1006102411439 (doi:10.1023/A:1006102411439) [DOI] [PubMed] [Google Scholar]
- 7.Ron D, Walter P. 2007. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 10.1038/nrm2199 (doi:10.1038/nrm2199) [DOI] [PubMed] [Google Scholar]
- 8.Wu J, Kaufman RJ. 2006. From acute ER stress to physiological roles of the unfolded protein response. Cell Death Differ. 13, 374–384 10.1038/sj.cdd.4401840 (doi:10.1038/sj.cdd.4401840) [DOI] [PubMed] [Google Scholar]
- 9.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. 2000. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2, 326–332 10.1038/35014014 (doi:10.1038/35014014) [DOI] [PubMed] [Google Scholar]
- 10.Codogno P, Meijer AJ. 2005. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 12, 1509–1518 10.1038/sj.cdd.4401751 (doi:10.1038/sj.cdd.4401751) [DOI] [PubMed] [Google Scholar]
- 11.Kroemer G, Jaattela M. 2005. Lysosomes and autophagy in cell death control. Nat. Rev. Cancer 5, 886–897 10.1038/nrc1738 (doi:10.1038/nrc1738) [DOI] [PubMed] [Google Scholar]
- 12.Levine B, Klionsky DJ. 2004. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6, 463–477 10.1016/S1534-5807(04)00099-1 (doi:10.1016/S1534-5807(04)00099-1) [DOI] [PubMed] [Google Scholar]
- 13.Bernales S, McDonald KL, Walter P. 2006. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 4, e423. 10.1371/journal.pbio.0040423 (doi:10.1371/journal.pbio.0040423) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boya P, Cohen I, Zamzami N, Vieira HLA, Kroemer G. 2002. Endoplasmic reticulum stress-induced cell death requires mitochondrial membrane permeabilization. Cell Death Differ. 9, 465–467 10.1038/sj/cdd/4401006 (doi:10.1038/sj/cdd/4401006) [DOI] [PubMed] [Google Scholar]
- 15.Hoyer-Hansen M, et al. 2007. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol. Cell 25, 193–205 10.1016/j.molcel.2006.12.009 (doi:10.1016/j.molcel.2006.12.009) [DOI] [PubMed] [Google Scholar]
- 16.Imaizumi K. 2007. Autophagy is activated for cell survival after ER stress. J. Pharmacol. Sci. 103, 45 [Google Scholar]
- 17.Kouroku Y, et al. 2007. ER stress (PERK/eIF2 α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 14, 230–239 10.1038/sj.cdd.4401984 (doi:10.1038/sj.cdd.4401984) [DOI] [PubMed] [Google Scholar]
- 18.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. 2004. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077 10.1101/gad.1250704 (doi:10.1101/gad.1250704) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szegezdi E, Logue SE, Gorman AM, Samali A. 2006. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880–885 10.1038/sj.embor.7400779 (doi:10.1038/sj.embor.7400779) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. 2008. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30, 678–688 10.1016/j.molcel.2008.06.001 (doi:10.1016/j.molcel.2008.06.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clarke R, et al. 2009. Gene network signaling in hormone responsiveness modifies apoptosis and autophagy in breast cancer cells. J. Steroid Biochem. Mol. Biol. 114, 8–20 10.1016/j.jsbmb.2008.12.023 (doi:10.1016/j.jsbmb.2008.12.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cook KL, Shajahan AN, Clarke R. 2011. Autophagy and endocrine resistance in breast cancer. Expert Rev. Anticancer Ther. 11, 1283–1294 10.1586/Era.11.111 (doi:10.1586/Era.11.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samaddar JS, et al. 2008. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol. Cancer Ther. 7, 2977–2987 10.1158/1535-7163.Mct-08-0447 (doi:10.1158/1535-7163.Mct-08-0447) [DOI] [PubMed] [Google Scholar]
- 24.Schoenlein PV, Periyasamy-Thandavan S, Samaddar JS, Jackson WH, Barrett JT. 2009. Autophagy facilitates the progression of ER α-positive breast cancer cells to antiestrogen resistance. Autophagy 5, 400–403 10.4161/auto.5.3.7784 (doi:10.4161/auto.5.3.7784) [DOI] [PubMed] [Google Scholar]
- 25.Zinszner H, Kuroda M, Wang XZ, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. 1998. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 12, 982–995 10.1101/gad.12.7.982 (doi:10.1101/gad.12.7.982) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tyson JJ, Baumann WT, Chen C, Verdugo A, Tavassoly I, Wang Y, Weiner LM, Clarke R. 2011. Dynamic modelling of oestrogen signalling and cell fate in breast cancer cells. Nat. Rev. Cancer 11, 523–532 10.1038/nrc3081 (doi:10.1038/nrc3081) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brunner N, Boulay V, Fojo A, Freter CE, Lippman ME, Clarke R. 1993. Acquisition of hormone-independent growth in MCF-7 cells is accompanied by increased expression of estrogen-regulated genes but without detectable DNA amplifications. Cancer Res. 53, 283–290 [PubMed] [Google Scholar]
- 28.Brunner N, et al. 1997. MCF7/LCC9: an antiestrogen-resistant MCF-7 variant in which acquired resistance to the steroidal antiestrogen ICI 182,780 confers an early cross-resistance to the nonsteroidal antiestrogen tamoxifen. Cancer Res. 57, 3486–3493 [PubMed] [Google Scholar]
- 29.Oikawa D, Kimata Y, Kohno K, Iwawaki T. 2009. Activation of mammalian IRE1 alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp. Cell Res. 315, 2496–2504 10.1016/j.yexcr.2009.06.009 (doi:10.1016/j.yexcr.2009.06.009) [DOI] [PubMed] [Google Scholar]
- 30.Ma YJ, Hendershot LM. 2004. ER chaperone functions during normal and stress conditions. J. Chem. Neuroanat. 28, 51–65 10.1016/j.jchemneu.2003.08.007 (doi:10.1016/j.jchemneu.2003.08.007) [DOI] [PubMed] [Google Scholar]
- 31.Bailly-Maitre B, et al. 2010. Hepatic Bax inhibitor-1 inhibits IRE1alpha and protects from obesity-associated insulin resistance and glucose intolerance. J. Biol. Chem. 285, 6198–6207 10.1074/jbc.M109.056648 (doi:10.1074/jbc.M109.056648) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Castillo K, et al. 2011. BAX inhibitor-1 regulates autophagy by controlling the IRE1α branch of the unfolded protein response. EMBO J. 30, 4465–4478 10.1038/emboj.2011.318 (doi:10.1038/emboj.2011.318) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo DH, He Y, Zhang HF, Yu LY, Chen H, Xu Z, Tang SB, Urano F, Min W. 2008. AIP1 is critical in transducing IRE1-mediated endoplasmic reticulum stress response. J. Biol. Chem. 283, 11 905–11 912 10.1074/jbc.M710557200 (doi:10.1074/jbc.M710557200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiu YF, et al. 2010. A crucial role for RACK1 in the regulation of glucose-stimulated IRE1 α activation in pancreatic β cells. Sci. Signal 3, 7. 10.1126/scisignal.2000514 (doi:10.1126/scisignal.2000514) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fonseca SG, et al. 2010. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J. Clin. Invest. 120, 744–755 10.1172/Jci39678 (doi:10.1172/Jci39678) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marzec M, Eletto D, Argon Y. 2012. GRP94: an HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Res. 1823, 774–787 10.1016/j.bbamcr.2011.10.013 (doi:10.1016/j.bbamcr.2011.10.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eletto D, Dersh D, Argon Y. 2010. GRP94 in ER quality control and stress responses. Semin. Cell Dev. Biol. 21, 479–485 10.1016/j.semcdb.2010.03.004 (doi:10.1016/j.semcdb.2010.03.004) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mao CH, Wang MA, Luo BQ, Wey SA, Dong DZ, Wesselschmidt R, Rawlings S, Lee AS. 2010. Targeted mutation of the mouse Grp94 gene disrupts development and perturbs endoplasmic reticulum stress signaling. PLoS ONE 5 10.1371/journal.pone.0010852 (doi:10.1371/journal.pone.0010852) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. 2008. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ. 15, 1460–1471 10.1038/Cdd.2008.81 (doi:10.1038/Cdd.2008.81) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Debnath J, Baehrecke EH, Kroemer G. 2005. Does autophagy contribute to cell death? Autophagy 1, 66–74 10.4161/auto.1.2.1738 (doi:10.4161/auto.1.2.1738) [DOI] [PubMed] [Google Scholar]
- 41.Kroemer G, Levine B. 2008. Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell Biol. 9, 1004–1010 10.1038/Nrm2529 (doi:10.1038/Nrm2529) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 10.1093/emboj/19.21.5720 (doi:10.1093/emboj/19.21.5720) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. 2004. The role of autophagy during the early neonatal starvation period. Nature 432, 1032–1036 10.1038/nature03029 (doi:10.1038/nature03029) [DOI] [PubMed] [Google Scholar]
- 44.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. 1999. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402, 672–676 10.1038/45257 (doi:10.1038/45257) [DOI] [PubMed] [Google Scholar]
- 45.Pattingre S, Tassa A, Qu XP, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. 2005. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939 10.1016/j.cell.2005.07.002 (doi:10.1016/j.cell.2005.07.002) [DOI] [PubMed] [Google Scholar]
- 46.Chang YY, Neufeld TP. 2009. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol. Biol. Cell 20, 2004–2014 10.1091/mbc.E08-12-1250 (doi:10.1091/mbc.E08-12-1250) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. 2009. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20, 1992–2003 10.1091/mbc.E08-12-1249 (doi:10.1091/mbc.E08-12-1249) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guertin DA, Sabatini DM. 2007. Defining the role of mTOR in cancer. Cancer Cell 12, 9–22 10.1016/j.ccr.2007.05.008 (doi:10.1016/j.ccr.2007.05.008) [DOI] [PubMed] [Google Scholar]
- 49.Hardie DG. 2008. AMPK: a key regulator of energy balance in the single cell and the whole organism. Int. J. Obes. (Lond) 32(Suppl. 4), S7–S12 10.1038/ijo.2008.116 (doi:10.1038/ijo.2008.116) [DOI] [PubMed] [Google Scholar]
- 50.Huang JX, Manning BD. 2009. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 37, 217–222 10.1042/Bst0370217 (doi:10.1042/Bst0370217) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Green DR, Kroemer G. 2004. The pathophysiology of mitochondrial cell death. Science 305, 626–629 10.1126/science.1099320 (doi:10.1126/science.1099320) [DOI] [PubMed] [Google Scholar]
- 52.Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC. 1999. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 144, 891–901 10.1083/jcb.144.5.891 (doi:10.1083/jcb.144.5.891) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. 2002. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111, 331–342 10.1016/S0092-8674(02)01036-X (doi:10.1016/S0092-8674(02)01036-X) [DOI] [PubMed] [Google Scholar]
- 54.Zhang T, Brazhnik P, Tyson JJ. 2009. Computational analysis of dynamical responses to the intrinsic pathway of programmed cell death. Biophys. J. 97, 415–434 10.1016/j.bpj.2009.04.053 (doi:10.1016/j.bpj.2009.04.053) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yorimitsu T, Nair U, Yang ZF, Klionsky DJ. 2006. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 281, 30 299–30 304 10.1074/jbc.M607007200 (doi:10.1074/jbc.M607007200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bauvy C, Gane P, Arico S, Codogno P, Ogier-Denis E. 2001. Autophagy delays sulindac sulfide-induced apoptosis in the human intestinal colon cancer cell line HT-29. Exp. Cell Res. 268, 139–149 10.1006/excr.2001.5285 (doi:10.1006/excr.2001.5285) [DOI] [PubMed] [Google Scholar]
- 57.Lockshin RA, Zakeri Z. 2004. Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol. 36, 2405–2419 10.1016/j.biocel.2004.04.011 (doi:10.1016/j.biocel.2004.04.011) [DOI] [PubMed] [Google Scholar]
- 58.Xue LZ, Fletcher GC, Tolkovsky AM. 1999. Autophagy is activated by apoptotic signalling in sympathetic neurons: an alternative mechanism of death execution. Mol. Cell. Neurosci. 14, 180–198 10.1006/mcne.1999.0780 (doi:10.1006/mcne.1999.0780) [DOI] [PubMed] [Google Scholar]
- 59.Lemasters JJ, et al. 1998. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta 1366, 177–196 10.1016/S0005-2728(98)00112-1 (doi:10.1016/S0005-2728(98)00112-1) [DOI] [PubMed] [Google Scholar]
- 60.Cui Q, Tashiro S, Onodera S, Minami M, Ikejima T. 2007. Autophagy preceded apoptosis in oridonin-treated human breast cancer MCF-7 cells. Biol. Pharm. Bull. 30, 859–864 10.1248/bpb.30.859 (doi:10.1248/bpb.30.859) [DOI] [PubMed] [Google Scholar]
- 61.Urano F, Bertolotti A, Ron D. 2000. IRE1 and efferent signaling from the endoplasmic reticulum. J. Cell Sci. 113, 3697–3702 [DOI] [PubMed] [Google Scholar]
- 62.Urano F, Wang XZ, Bertolotti A, Zhang YH, Chung P, Harding HP, Ron D. 2000. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 10.1126/science.287.5453.664 (doi:10.1126/science.287.5453.664) [DOI] [PubMed] [Google Scholar]
- 63.Demarchi F, Bertoli C, Copetti T, Tanida I, Brancolini C, Eskelinen EL, Schneider C. 2006. Calpain is required for macroautophagy in mammalian cells. J. Cell Biol. 175, 595–605 10.1083/jcb.200601024 (doi:10.1083/jcb.200601024) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ. 2004. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 23, 1207–1216 10.1038/sj.emboj.7600104 (doi:10.1038/sj.emboj.7600104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cook KL, Clarke R. 2012. Heat shock 70 kDa protein 5/glucose-regulated protein 78 ‘AMP'ing up autophagy. Autophagy 8, 1827–1829 10.4161/auto.21765 (doi:10.4161/auto.21765) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. 2001. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bc12 and perturbing the cellular redox state. Mol. Cell. Biol. 21, 1249–1259 10.1128/MCB.21.4.1249-1259.2001 (doi:10.1128/MCB.21.4.1249-1259.2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Puthalakath H, et al. 2007. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell 129, 1337–1349 10.1016/j.cell.2007.04.027 (doi:10.1016/j.cell.2007.04.027) [DOI] [PubMed] [Google Scholar]
- 68.Ouyang YB, Xu LJ, Emery JF, Lee AS, Giffard RG. 2011. Overexpressing GRP78 influences Ca(2+) handling and function of mitochondria in astrocytes after ischemia-like stress. Mitochondrion 11, 279–286 10.1016/j.mito.2010.10.007 (doi:10.1016/j.mito.2010.10.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ishii Y, Papa L, Bahadur U, Yue ZY, Aguirre-Ghiso J, Shioda T, Waxman S, Germain D. 2011. Bortezomib enhances the efficacy of fulvestrant by amplifying the aggregation of the estrogen receptor, which leads to a proapoptotic unfolded protein response. Clin. Cancer Res. 17, 2292–2300 10.1158/1078-0432.Ccr-10-1745 (doi:10.1158/1078-0432.Ccr-10-1745) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Perillo B, Sasso A, Abbondanza C, Palumbo G. 2000. 17 β-estradiol inhibits apoptosis in MCF-7 cells, inducing bcl-2 expression via two estrogen-responsive elements present in the coding sequence. Mol. Cell. Biol. 20, 2890–2901 10.1128/MCB.20.8.2890-2901.2000 (doi:10.1128/MCB.20.8.2890-2901.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Diel P, Smolnikar K, Michna H. 1999. The pure antiestrogen ICI 182780 is more effective in the induction of apoptosis and down regulation of BCL-2 than tamoxifen in MCF-7 cells. Breast Cancer Res. Treat. 58, 87–97 10.1023/A:1006338123126 (doi:10.1023/A:1006338123126) [DOI] [PubMed] [Google Scholar]
- 72.Zhang X, Ding L, Kang L, Wang ZY. 2012. Estrogen receptor-α 36 mediates mitogenic antiestrogen signaling in ER-negative breast cancer cells. PLoS ONE 7, e30174. 10.1371/journal.pone.0030174 (doi:10.1371/journal.pone.0030174) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Emde A, Mahlknecht G, Maslak K, Ribba B, Sela M, Possinger K, Yarden Y. 2011. Simultaneous inhibition of estrogen receptor and the HER2 pathway in breast cancer: effects of HER2 abundance. Transl. Oncol. 4, 293–300 10.1593/Tlo.11127 (doi:10.1593/Tlo.11127) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Appenzeller-Herzog C, Hall MN. 2012. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends Cell Biol. 4, 274–282 10.1016/j.tcb.2012.02.006 (doi:10.1016/j.tcb.2012.02.006) [DOI] [PubMed] [Google Scholar]
- 75.Knowlden JM, Hutcheson IR, Jones HE, Madden T, Gee JMW, Harper ME, Barrow D, Wakeling AE, Nicholson RI. 2003. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology 144, 1032–1044 10.1210/en.2002-220620 (doi:10.1210/en.2002-220620) [DOI] [PubMed] [Google Scholar]
- 76.Nicholson RI, Hutcheson IR, Harper ME, Knowlden JM, Barrow D, McClelland RA, Jones HE, Wakeling AE, Gee JMW. 2001. Modulation of epidermal growth factor receptor in endocrine-resistant, oestrogen receptor-positive breast cancer. Endocr. Relat. Cancer 8, 175–182 10.1677/erc.0.0080175 (doi:10.1677/erc.0.0080175) [DOI] [PubMed] [Google Scholar]
- 77.Nicholson RI, Hutcheson IR, Harper ME, Knowlden JM, Barrow D, McClelland RA, Jones HE, Wakeling AE, Gee JMW. 2002. Modulation of epidermal growth factor receptor in endocrine-resistant, estrogen-receptor-positive breast cancer. Ann. N. Y. Acad. Sci. 963, 104–115 10.1111/j.1749-6632.2002.tb04101.x (doi:10.1111/j.1749-6632.2002.tb04101.x) [DOI] [PubMed] [Google Scholar]
- 78.Gu Z, et al. 2002. Association of interferon regulatory factor-1, nucleophosmin, nuclear factor-κB, and cyclic AMP response element binding with acquired resistance to Faslodex (ICI 182,780). Cancer Res. 62, 3428–3437 [PubMed] [Google Scholar]
- 79.Kuske B, Naughton C, Moore K, MacLeod KG, Miller WR, Clarke R, Langdon SP, Cameron DA. 2006. Endocrine therapy resistance can be associated with high estrogen receptor α (ER α) expression and reduced ER α phosphorylation in breast cancer models. Endocr. Relat. Cancer 13, 1121–1133 10.1677/Erc.1.01257 (doi:10.1677/Erc.1.01257) [DOI] [PubMed] [Google Scholar]
- 80.Skommer J, Brittain T, Raychaudhuri S. 2010. Bcl-2 inhibits apoptosis by increasing the time-to-death and intrinsic cell-to-cell variations in the mitochondrial pathway of cell death. Apoptosis 15, 1223–1233 10.1007/s10495-010-0515-7 (doi:10.1007/s10495-010-0515-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK. 2009. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 459, 428–432 10.1038/Nature08012 (doi:10.1038/Nature08012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ma XM, Blenis J. 2009. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 10, 307–318 10.1038/nrm2672 (doi:10.1038/nrm2672) [DOI] [PubMed] [Google Scholar]
- 83.Karin M, Lin A. 2002. NF-κB at the crossroads of life and death. Nature Immunol. 3, 221–227 10.1038/ni0302-221 (doi:10.1038/ni0302-221) [DOI] [PubMed] [Google Scholar]
- 84.Li C, Harada A, Oh Y. 2012. IGFBP-3 sensitizes antiestrogen-resistant breast cancer cells through interaction with GRP78. Cancer Lett. 325, 200–206 10.1016/j.canlet.2012.07.004 (doi:10.1016/j.canlet.2012.07.004) [DOI] [PubMed] [Google Scholar]
- 85.Reddy RK, Mao C, Baumeister P, Austin RC, Kaufman RJ, Lee AS. 2003. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J. Biol. Chem. 278, 20 915–20 924 10.1074/jbc.M212328200 (doi:10.1074/jbc.M212328200) [DOI] [PubMed] [Google Scholar]
- 86.Fu Y, Li J, Lee AS. 2007. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 67, 3734–3740 10.1158/0008-5472.CAN-06-4594 (doi:10.1158/0008-5472.CAN-06-4594) [DOI] [PubMed] [Google Scholar]
- 87.Zhang LH, Yang XL, Zhang XA, Cheng JX, Zhang W. 2011. Association of elevated GRP78 expression with increased astrocytoma malignancy via Akt and ERK pathways. Brain Res. 1371, 23–31 10.1016/j.brainres.2010.11.063 (doi:10.1016/j.brainres.2010.11.063) [DOI] [PubMed] [Google Scholar]
- 88.Wey S, Luo BQ, Tseng CC, Ni M, Zhou H, Fu Y, Bhojwani D, Carroll WL, Lee AS. 2012. Inducible knockout of GRP78/BiP in the hematopoietic system suppresses PTEN-null leukemogenesis and AKT oncogenic signaling. Blood 119, 817–825 10.1182/blood-2011-06-357384 (doi:10.1182/blood-2011-06-357384) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gibellini L, Pinti M, Nasi M, De Biasi S, Roat E, Bertoncelli L, Cossarizza A. 2010. Interfering with ROS metabolism in cancer cells: the potential role of quercetin. Cancers 2, 1288–1311 10.3390/cancers2021288 (doi:10.3390/cancers2021288) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dai RY, Chen SK, Yan DM, Chen R, Lui YP, Duan CY, Li J, He T, Li H. 2010. PI3K/Akt promotes GRP78 accumulation and inhibits endoplasmic reticulum stress-induced apoptosis in HEK293 cells. Folia Biol. (Praha) 56, 37–46 [PubMed] [Google Scholar]
- 91.Cerqueira V. 2009. Role of intracellular signalling pathways in conferring resistance to endocrine therapies in breast cancer. Thesis, ch. 6, pp. 108–131, University of Edinburgh, Edinburgh, UK [Google Scholar]
- 92.Pfaffenbach KT, Pong M, Morgan TE, Wang H, Ott K, Zhou B, Longo VD, Lee AS. 2012. GRP78/BiP is a novel downstream target of IGF-1 receptor mediated signaling. J. Cell Physiol. 227, 3803–3811 10.1002/jcp.24090 (doi:10.1002/jcp.24090) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Song MS, Park YK, Lee JH, Park K. 2001. Induction of glucose-regulated protein 78 by chronic hypoxia in human gastric tumor cells through a protein kinase C-epsilon/ERK/AP-1 signaling cascade. Cancer Res. 61, 8322–8330 [PubMed] [Google Scholar]
- 94.Hitomi J, et al. 2004. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and a β-induced cell death. J. Cell Biol. 165, 347–356 10.1083/jcb.200310015 (doi:10.1083/jcb.200310015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim SJ, Zhang ZJ, Hitomi E, Lee YC, Mukherjee AB. 2006. Endoplasmic reticulum stress-induced caspase-4 activation mediates apoptosis and neurodegeneration in INCL. Hum. Mol. Genet. 15, 1826–1834 10.1093/Hmg/Ddl105 (doi:10.1093/Hmg/Ddl105) [DOI] [PubMed] [Google Scholar]
- 96.Chang YJ, Huang YP, Li ZL, Chen CH. 2012. GRP78 knockdown enhances apoptosis via the down-regulation of oxidative stress and AKT pathway after epirubicin treatment in colon cancer DLD-1 cells. PLoS ONE 7, e35123. 10.1371/journal.pone.0035123 (doi:10.1371/journal.pone.0035123) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fu Y, Wey S, Wang M, Ye R, Liao CP, Roy-Burman P, Lee AS. 2008. PTEN null prostate tumorigenesis and AKT activation are blocked by targeted knockout of ER chaperone GRP78/BiP in prostate epithelium. Proc. Natl Acad. Sci. USA 105, 19 444–19 449 10.1073/pnas.0807691105 (doi:10.1073/pnas.0807691105). [DOI] [PMC free article] [PubMed] [Google Scholar]