

Abstract
The increasing availability of multi-omics cancer datasets has created a new opportunity for data integration that promises a more comprehensive understanding of cancer. The challenge is to develop mathematical methods that allow the integration and extraction of knowledge from large datasets such as The Cancer Genome Atlas (TCGA). This has led to the development of a variety of omics profiles that are highly correlated with each other; however, it remains unknown which profile is the most meaningful and how to efficiently integrate different omics profiles. We developed AMARETTO, an algorithm to identify cancer drivers by integrating a variety of omics data from cancer and normal tissue. AMARETTO first models the effects of genomic/epigenomic data on disease-specific gene expression. AMARETTO's second step involves constructing a module network to connect the cancer drivers with their downstream targets. We observed that more gene expression variation can be explained when using disease-specific gene expression data. We applied AMARETTO to the ovarian cancer TCGA data and identified several cancer driver genes of interest, including novel genes in addition to known drivers of cancer. Finally, we showed that certain modules are predictive of good versus poor outcome, and the associated drivers were related to DNA repair pathways.
Keywords: gene expression, DNA methylation, copy number, ovarian cancer, data integration
1. Introduction
The unprecedented wealth of data currently being generated for cancer patients has provided us with the challenge of its interpretation and translation to personalized medicine. Personalized medicine aims to tailor medical care to the individual through the meaningful characterization of biological heterogeneity present in cancer. Technological innovation has enabled the acquisition of multi-scale information ranging from genotypes to several phenotypic layers. For example, advances in high-throughput sequencing allow quantification of global DNA variation and RNA expression of tissue or blood samples [1–3]. These platforms produce a variety of omics profiles that, while highly correlated to each other, often raise difficulty in discerning meaningful interpretation and integration.
Previous data integration efforts in cancer have focused on integrating a subset of omics profiles. For example, Ciriello et al. [4] used a method based on mutual exclusivity to model copy number and mutation data and identified driver genes in glioblastoma. Similarly, Vandin et al. [5] developed a method to identify driver genes in cancer, but focused on finding pathways with a significant enrichment of approximately mutually exclusive genes. In addition, other groups are focusing on identifying driver genes through network analysis of copy number data to filter potential regulators in Bayesian module network analysis [6].
We selected a module network approach to integrate copy number, DNA methylation and gene expression data. We developed an algorithm called AMARETTO to unravel cancer drivers by using data integration of omics data, conditioned on differential expression between cancer and normal samples. We applied AMARETTO to the ovarian cancer data from The Cancer Genome Atlas (TCGA) project [7]. AMARETTO builds further on previous work by adding a number of new concepts [8]. AMARETTO focuses on identifying what is specifically aberrant in cancer tissue compared with normal tissue, although matched cancer and normal samples are not required for each patient; this specific aspect is integrated throughout our algorithm. In addition, AMARETTO focuses on candidate cancer drivers that are recurrent in different samples and are regarded as functional. Drivers are regarded as functional if there is a significant relationship between the genomic/epigenomic event and their resulting gene expression. AMARETTO also incorporates DNA methylation data, which to date are often not integrated because of a lack of methods to extract cancer-specific DNA methylation aberrations. To address this issue, we developed a method called MethylMix [9] that allows us to integrate DNA methylation in our workflow as a complementary driving genomic force of cancer cells, thus enabling us to expand beyond copy number aberrations.
We applied AMARETTO to identify driver genes based on multi-omic profiles from 511 ovarian cancer patients. In step 1 of AMARETTO, we identify the main cancer driver genes based on a linear model of the relationship between copy number data or DNA methylation data and disease-specific gene expression data. We observed that both copy number and DNA methylation are more explanatory of disease-specific gene expression than raw gene expression. This suggests that using normal tissue variation as a reference improves the signal and potential integration of copy number or DNA methylation data with gene expression. Next, AMARETTO's second step connects candidate cancer driver genes with their downstream targets by constructing a module network. We discuss several cancer driver genes identified by our model. We then associate the module network with the TCGA-identified molecular subtypes in ovarian cancer to identify the main cancer driver genes. Finally, we correlated the module network with overall survival and therapy response, and found some promising associations.
2. Material and methods
2.1. AMARETTO
We developed a method called AMARETTO to identify cancer drivers by using data integration of omics data that considers the differential expression between cancer and normal samples. AMARETTO accomplishes this using a multi-step algorithm that integrates copy number, DNA methylation and gene expression data to identify cancer driver genes and subsequently associates them with their downstream targets through module network analysis. Figure 1 gives an overview of how the steps are linked to each other and which algorithms are used for each step.
— Step 1 attempts to identify cancer driver genes by modelling the relationship between genomic and transcriptomic data on an individual gene basis. In addition, we add two important filters to this model. First, we take gene expression variation in normal tissue into account and focus on disease-specific expression. Second, we focus only on genomic events such as copy number alterations or aberrant DNA methylation that are recurrent in the population of cancer samples.
— Step 2 uses the cancer driver genes identified from step 1 and now takes a global approach by dissecting global gene expression data into modules of co-expressed genes. Each module also has an associated gene regulatory program that connects the cancer driver genes from step 1 with their downstream targets. This gene regulatory program is modelled using linear regression with elastic net regularization.
Figure 1.

Workflow of AMARETTO. Step 1 involves generating a list of candidate cancer driver genes by using a linear model capturing the relationship between genomic and transcriptomic data for each gene separately. First, we create disease-specific gene expression (DSE) profiles by taking normal gene expression profiles into account. Second, only genes identified by GISTIC and MethylMix are modelled. Step 2 connects the cancer drivers from step 1 with their downstream targets by reconstructing a module network. This module network associates a set of co-expressed genes with cancer driver genes through learning a regulatory program. The regulatory program is modelled using linear regression with elastic net regularization.
2.1.1. Step 1: generating a list of candidate cancer driver genes
To generate a list of candidate cancer driver genes, we used a linear regression model to estimate the effect of copy number and DNA methylation on gene expression. This step focuses on modelling the cis-regulatory effects of genomic data on gene expression. This linear regression model was built for each gene independently. Next, we evaluated whether copy number data had a significant positive effect on gene expression and whether DNA methylation had a significant effect on gene expression. These effects were quantified using the R2 statistic. Significance of each genomic event was selected using 10-fold cross validation. We modelled disease-specific gene expression and only focused on genes that have either copy number or DNA methylation alterations. We describe the algorithms used for these filters in the following paragraphs.
Disease-specific gene expression analysis: we were specifically interested in modelling disease-specific gene expression. Our rationale is that genes which exhibit high variability in normal tissue are less likely to be candidate cancer drivers. Therefore, we integrated disease-specific gene expression data into our models using disease-specific genomic analysis (DSGA; [10]). DSGA is a mathematical method that first models the normal gene expression data and subsequently extracts the disease-specific component as a deviation from normal [10]. The normal gene expression data are modelled by dimensionality reduction using modified principal component analysis. Next, each cancer gene expression profile is then fitted to this normal model and the residual is defined as the disease-specific component [10].
Filtering for recurrent genomic events using GISTIC and MethylMix: next, we focused on identifying recurrent genomic alterations by using several model-based approaches. Copy number data have been extensively studied in this context, and several methods are available to identify recurrent amplifications and deletions [11,12]. We selected the GISTIC method and extracted the amplified and deleted genes from the GISTIC output. GISTIC focuses on identifying focal copy number alterations by separately modelling arm-level and focal alterations. This method identifies candidate driver genes and reduces the confounding effects of broad-level alterations. Few methods have been developed that statistically model DNA methylation aberrations in cancer. We recently developed a method called MethylMix to identify methylation states by differentiating normal DNA methylation from aberrations found in cancer [9]; a summary of MethylMix has been provided as electronic supplementary material associated with this paper. MethylMix uses a mixture model to identify the major methylation states, compares each state with normal DNA methylation and identifies which genes are hyper- and hypo-methylated. We combined both GISTIC and MethylMix to identify candidate cancer driver genes and only these genes were used as input for downstream analysis.
2.1.2. Step 2: associating candidate drivers with their downstream targets
The second step of AMARETTO involves connecting the cancer drivers of step 1 with their downstream targets by reconstructing a module network using an approach built upon previous work [8,13]. This step focuses on modelling the trans-effects of cancer drivers identified in step 1. A major change in this version of AMARETTO is that in step 2 we model disease-specific expression. The algorithm is initiated by clustering the disease-specific gene expression data into gene modules of co-expressed genes and then assigns a regulatory program to each module. This regulatory program is defined as a sparse linear combination of cancer driver genes selected in step 1 that predict the module's mean expression. The sparseness of the regulatory program is induced using elastic net regularization. After initial clustering of the data, the module network algorithm is run iteratively by learning the regulatory program and reassigning genes to modules based on the updated regulatory program. Genes are reassigned to the module to which they are closest, based on Pearson correlation. We used k-means clustering with 100 clusters as the initial clustering algorithm. Next, our algorithm is run until convergence corresponding to less than 1 per cent of the genes being assigned to new modules is achieved.
2.2. Data preprocessing
We used gene expression, copy number and DNA methylation data from TCGA ovarian cancer [7]. The gene expression data were produced using Agilent G4502A microarrays. Preprocessing was done by log-transformation and quantile normalization of the arrays. Next, we used DNA methylation data generated using the Illumina Infinium Human Methylation 27 Bead Chip. We used the level 3 methylation data containing methylation data on 27 578 CpG sites in 14 473 genes. DNA methylation is quantified using β-values ranging from 0 to 1, with values close to 0 versus 1 indicating low versus high levels of DNA methylation, respectively. We removed CpG sites with more than 10 per cent of missing values in all samples, reducing the number of usable CpG sites to 23 030 for ovarian cancer. We used the 15-K nearest neighbour algorithm to estimate the remaining missing values in the dataset [14]. Next, we used copy number data produced by the Agilent Sure Print G3 Human CGH Microarray Kit 1 M × 1 M platform. This platform has high redundancy at the gene level, but we observed high correlation between probes matching the same gene. Therefore, probes matching the same gene were merged by taking the average. For all data sources, gene annotation was translated to official gene symbols based on the HUGO Gene Nomenclature Committee (version August 2012). Owing to the size of the TCGA project, the TCGA samples are analysed in batches and a significant batch effect was observed based on a one-way analysis of variance in most data modes. We applied Combat to adjust for these effects [15]. In total, we used 511 primary tumour and eight normal fallopian tube samples for ovarian cancer [7]. These normal samples were profiled using the same TCGA pipeline and platform. In addition, fallopian tube tissue has been shown as a cell or origin for serous ovarian cancer [16,17]. All TCGA data are accessible at the TCGA data portal (https://tcga-data.nci.nih.gov/tcga/).
2.3. Gene set enrichment analysis
To evaluate the enrichment of modules with gene sets, we used several databases, namely: MSigDB v. 3 [18], GeneSetDB v. 4 [19], CHEA for CHIP-X gene sets v. 2 [20] and manually curated gene sets related to stem cells and immune gene sets. We used a hyper-geometric test to check for enrichment of gene sets in the lists of hyper- and hypo-methylated genes. We corrected for multiple hypothesis testing using the false discovery rate (FDR; [21]).
2.4. Survival analysis
We used Cox proportional hazards modelling to investigate univariate relationships between modules and overall survival (survival R package v. 2.36–10). Hazard ratios were used to report the direction of the survival effect, and the Wald test was used to determine the significance of Cox models. We applied the FDR to correct for multiple hypothesis testing [21].
2.5. Supervised modelling
We used linear regression with lasso regularization to build supervised models predicting clinical and prognostic markers [22], and investigated whether modules can accurately predict these outcomes by applying 10-fold cross validation to estimate the performance.
3. Results
3.1. Disease-specific candidate cancer driver genes
Using gene expression, copy number and DNA methylation data for 511 patients with ovarian cancer from TCGA, AMARETTO identified both known and novel ovarian cancer driver genes. We first looked at how disease-specific copy number alterations and DNA methylation influence disease-specific gene expression on a genome-wide scale in the ovarian cancer TCGA data. We used a mathematical method to estimate the disease-specific gene expression [10] and compared our results with the raw gene expression data, which were unadjusted for the variation in normal samples. We used a linear regression model and estimated for each gene whether copy number, DNA methylation or both are significantly predictive of gene expression.
Figure 2a shows the number of genes that have a significant effect of copy number, DNA methylation or both. We compared this with the uncorrected gene expression data and found that more gene expression variation, captured using the R2 statistic, can be explained when using the disease-specific gene expression data. This was also found when calculating the difference in explained gene expression values in both cases. We found that this differential R2 distribution was skewed towards an increased R2 when using the disease-specific gene expression data (p-value 3.7261 × 10−20, Wilcoxon rank-sum test; figure 2b). In addition, when focusing on cancer-specific genes from the cancer gene census [23], we observed a tendency towards higher R2 values for cancer-specific genes after correction for normal gene expression data (p-value 0.0652, Wilcoxon rank-sum test).
Figure 2.
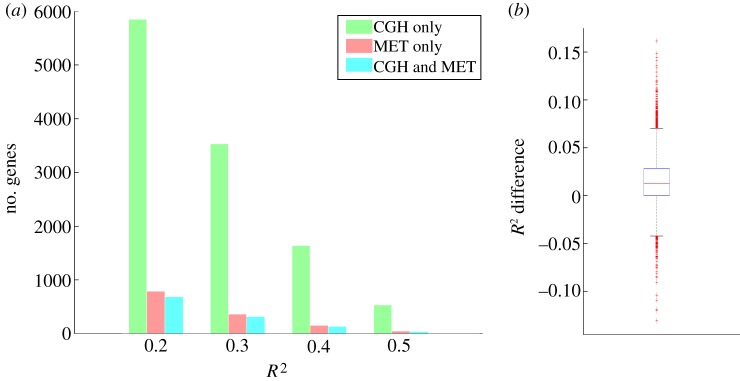
(a) Number of genes whose disease-specific expression is significantly explained by only copy number, only DNA methylation or both. The number of genes is shown at different thresholds for the R2 value. CGH, copy number data; MET, DNA methylation. (b) Box plot of the difference in the R2 value of the disease-specific gene expression data versus the uncorrected gene expression data, showing a bias towards a higher R2 for the disease-corrected gene expression data.
Regarding STAT3, for example, we observed an increase of 14 per cent in the R2 value when explaining its disease-specific gene expression (36% versus 22% for the disease-specific model versus the model not adjusted for normal variation), solely based on its copy number. Similarly, TNFRSF1A shows an increase of 11 per cent for the R2 in the disease-specific model versus the model not adjusted for normal. Both genes have functions related to cell growth and apoptosis, which represent major processes that are deregulated in cancer. Next, we looked at examples of a decreasing R2 in the disease-specific gene expression. This corresponds to genes where adjusting for normal variation reduces the correlation between genomic markers and gene expression. We found, for example, that LY86 shows a decrease of 13 per cent, indicating that normal gene expression variation is confounding the correlation between genomic data and gene expression data, and, after correcting for normal gene expression variation, the genomic data explain less of the disease-specific gene expression.
3.2. Recurrent genomic or epigenomic events
In addition to focusing on disease-specific gene expression, we select genes that are recurrently altered genomically or epigenomically. In the case of copy number data, this entails that we look for genes that are recurrently amplified or deleted. Copy number data have been extensively studied and several methods are available to identify recurrent amplifications and deletions [11,12]. We used GISTIC to identify recurrent amplifications and deletions across 481 TCGA ovarian cancer samples. GISTIC identified 670 and 2353 genes that are in recurrently amplified and deleted regions, respectively, consistent with earlier reports that ovarian cancer has a large number of copy number alterations.
In the case of DNA methylation, we focused on genes that are significantly and differentially methylated in a subset of patients. To accomplish this, we developed an algorithm called MethylMix that uses a mixture model to identify subgroups of patients with similar DNA methylation for a specific gene [9]. This allows us to define hyper- and hypo-methylation states when compared with DNA methylation in normal tissue. MethylMix identified 205 and 251 significantly and differentially hyper- and hypo-methylated genes, respectively, in the TCGA ovarian cancer dataset. This includes well-known hyper-methylated ovarian cancer genes such as BRCA1, RAB25 and ATM.
We refined the list of potential driver genes from 8464 to 1572 by applying the GISTIC and MethylMix filters. These resultant genes have significant relationships between genomic data and disease-specific gene expression data, and have recurrent genomic or epigenomic alterations, and constitute likely cancer driver genes.
3.3. Cancer driver genes associated with their downstream targets
The candidate driver genes still constitute a large set, even after selecting for significant relationships between genomic and transcriptomic data, and recurrent events. The second step of AMARETTO associates candidate driver genes with their downstream targets. This provides insight into the processes that candidate driver genes are regulating, and also serves to focus only on driver genes that are predictive of downstream gene expression.
We selected the top half of the genes that exhibited the highest variance to build the module network, producing 8907 genes on 560 samples. This variance-based filter was also applied to the candidate driver genes, further refining the list to 865 candidate driver genes. We built a network consisting of 100 modules with corresponding regulatory programs (see figure 3 and the electronic supplementary material, figure S1) that are functionally enriched for key processes in cancer such as cell cycle, immune response, RNA regulation and extracellular matrix signalling. Electronic supplementary material, table S1, contains the modules and regulatory programs of the complete ovarian cancer module network. This network contains 339 selected cancer driver genes including 213 that are copy number driven and 144 that are DNA methylation driven. Interestingly, a higher proportion of genes selected are DNA methylation driven compared with the initial number of DNA methylation candidate driver genes. The selected cancer driver genes include well-known genes such as CCNE1, CDKN2A, KRAS, PTEN and RB1 but also genes with unknown functions in cancer such as EVI2A, C1orf114 and LCP2.
Figure 3.

Visualization of the module network. Grey nodes are modules (co-expressed gene sets); blue nodes are cancer driver genes. Blue links represent membership of a cancer driver to a module, and green lines represent membership to a regulatory program of a module. We have annotated key gene set enrichments on the network. ECM, extracellular matrix. A high-resolution version of this network is available as electronic supplementary material, figure S1.
A number of biological hypotheses can be deduced from this network. For example, the network suggests that CCL5 is a master immune system response regulator. This gene is significantly hypo-methylated in a subset of samples. CCL5 is one of the top cancer drivers in the network and is part of five regulatory programs. Each of the corresponding modules is highly enriched with gene sets related to immune response or defence response. For example, module 37 is highly enriched with immune response genes and is also significantly increased in the immunoreactive molecular subtype (figure 4; p-value 1.2009 × 10−29, Wilcoxon rank-sum test; [7]). CCL5 is a chemokine that facilitates disease progression by recruiting and modulating the activity of inflammatory cells, which subsequently remodel the tumour microenvironment. Moreover, CCL5 has been shown to promote metastasis in basal breast cancer cells [24].
Figure 4.

Visualization of (a) module 37 and (b) module 32 regulatory programs and expression profiles. (A) Visualization of the regulatory programs. (B) Comparison of the accuracy of the estimated mean expression of the module based on the regulatory program with the average module expression. (C) Expression of the genes in the module. (D) Indication of membership of samples to the immunoreactive (panel (a)) and mesenchymal (panel (b)) molecular subtype. (E) Box-plot comparison of the gene expression in the immunoreactive (panel (a)) and mesenchymal (panel (b)) versus the remaining samples.
Next, we analysed how frequently certain cancer drivers co-occur in a regulatory program. This analysis showed that NUAK1 and PCOLCE always co-occur and are part of four regulatory programs. NUAK1 is known to directly phosphorylate TP53 and regulate cell proliferation. PCOLCE is a pro-collagen and is active in the extracellular matrix. All four NUAK and PCOLCE modules are highly correlated with the mesenchymal molecular subtype, suggesting that both genes are major drivers of this ovarian cancer subtype. For example, module 32 is regulated by NUAK1 and PCOLCE and is significantly upregulated in the mesenchymal subtype (figure 4; Wilcoxon rank-sum test, p-value 1.3125 × 10−51). It is interesting to note that both NUAK1 and PCOLCE are aberrantly methylated genes.
Similarly, CHEK1 and FBXO5 co-regulate two modules: module 55 and module 16. These modules are the top modules enriched for cell proliferation and cell cycle. This suggests that both genes are disease-specific drivers of cell proliferation in ovarian cancer. CHEK1 is a well-known cell cycle gene required for checkpoint-mediated cell cycle arrest in response to DNA damage. FBXO5 is a well-known regulator of the mitotic cell cycle. Both CHECK1 and FBXO5 are recurrently deleted genes.
Finally, EVI2A is among the top regulators in the network. EVI2A is a membrane protein that has unknown function. The predicted targets of EVI2A are highly enriched in the KEGG pathways, ECM receptor interaction and focal adhesion, indicating that this gene may have a role in interactions between cancer cells and the extracellular matrix. EVI2A is a methylation-driven gene expressed in neutrophils and leads us to speculate that we may be measuring the infiltration of neutrophils in ovarian cancer cells, which may reflect an immune-mediated driver of oncogenesis.
3.4. Modules that accurately predict molecular subtypes
Next, we focused on how modules can be combined to predict the ovarian cancer molecular subtypes [7]. This allows further identification of the cancer drivers of each of the molecular subtypes. We used linear regression with lasso regularization to build a model for each of the four molecular subtypes based on module expression. The performance was estimated using the area under the receiver operating characteristic (ROC) curve (AUC) in a 10-fold cross-validation loop (figure 5).
Figure 5.

Receiver operating characteristic (ROC) curves of supervised classification of the ovarian cancer molecular subtypes using module expression for each of the four subtypes. AUC, area under the ROC curve; s.e., standard error. (Online version in colour.)
All four subtypes can be accurately predicted with AUCs ranging from 0.857 to 0.975. The mesenchymal subtype is most accurately predicted. This model is based on only one module: module 48. Module 48 is one of the four modules regulated by NUAK1 and PCOLCE. Functionally, this module is highly enriched in extracellular matrix genes and focal adhesion (see figure 3 and electronic supplementary material, figure S1). Module 48 is also significantly enriched in genes upregulated in ovarian cancer metastasis versus primary tumours [25]. This is consistent with the mesenchymal subtype. Similarly, the proliferative subtype is also predicted based on only one module: module 34. This module has limited functional enrichment in genes related to protein kinase activity but is otherwise not well characterized. The other two subtypes need an average of 2.9 modules for the differentiated subtype and 2.3 modules for the immunoreactive subtype.
3.5. Correlation of modules with ovarian cancer outcome data
We investigated whether the modules can be used to predict therapy response and survival. We used Cox proportional hazards modelling to correlate the module expression with overall survival. We found that eight modules were significantly correlated with overall survival (table 1; Wald test < 0.05 at FDR < 15%).
Table 1.
Correlation with survival of the top 20 modules. HR, hazard ratio; CI, 95% confidence interval; FDR, false discovery rate.
| module number | Wald test | HR | HR lower CI | HR upper CI | FDR |
|---|---|---|---|---|---|
| module_41 | 0.0013115 | 0.60921 | 0.45029 | 0.82422 | 0.13115 |
| module_17 | 0.0063686 | 1.6636 | 1.1541 | 2.398 | 0.14577 |
| module_18 | 0.0067449 | 0.6607 | 0.48953 | 0.89171 | 0.14577 |
| module_76 | 0.0094106 | 0.66192 | 0.48479 | 0.90377 | 0.14577 |
| module_39 | 0.0098352 | 0.64297 | 0.45979 | 0.89911 | 0.14577 |
| module_69 | 0.010812 | 1.5165 | 1.101 | 2.0889 | 0.14577 |
| module_81 | 0.011528 | 0.63858 | 0.45091 | 0.90435 | 0.14577 |
| module_44 | 0.011662 | 1.3305 | 1.0657 | 1.6609 | 0.14577 |
| module_52 | 0.021869 | 0.70067 | 0.51695 | 0.94969 | 0.20173 |
| module_37 | 0.022771 | 1.3389 | 1.0415 | 1.7211 | 0.20173 |
| module_65 | 0.023899 | 1.2892 | 1.0342 | 1.607 | 0.20173 |
| module_73 | 0.026206 | 0.69138 | 0.49935 | 0.95725 | 0.20173 |
| module_62 | 0.026224 | 0.72201 | 0.54177 | 0.96221 | 0.20173 |
| module_45 | 0.030562 | 1.2241 | 1.0191 | 1.4702 | 0.2183 |
| module_47 | 0.033869 | 0.68353 | 0.48095 | 0.97142 | 0.2223 |
| module_7 | 0.037937 | 0.72948 | 0.54157 | 0.98259 | 0.2223 |
| module_57 | 0.03836 | 0.69547 | 0.49319 | 0.98072 | 0.2223 |
| module_59 | 0.040014 | 1.2658 | 1.0108 | 1.5852 | 0.2223 |
| module_15 | 0.043767 | 0.70292 | 0.49898 | 0.9902 | 0.23035 |
| module_13 | 0.050691 | 1.3951 | 0.999 | 1.9482 | 0.25157 |
We compared patients with good survival with those with poor survival to investigate whether analysis of the extreme cases results in more significant results. More specifically, we compared patients who had no recurrence or progression for at least 1000 days versus patients who have treatment-refractory disease. We found that four modules are significantly correlated with this outcome (Wilcoxon rank sum test < 0.01, FDR < 15%). Module 16 in particular was significantly correlated with this outcome (p-value 8.2757 × 10−06, FDR 8.2757 × 10−06). Module 16 is functionally enriched for cell cycle and DNA repair, and by the co-regulators CHEK1 and FBXO5 (figure 6). Expression of module 16 is highly upregulated for good versus poor survival. This is consistent with the copy number data of CHEK1 and FBXO5, both recurrently deleted, corresponding to a tumour suppressor function. Thus, patients with intact CHEK1 and FBXO5 expression have a better prognosis, consistent with these genes’ function in DNA repair or cell cycle control. In addition, supervised modelling using 10-fold cross validation of this outcome resulted in an accurate model using only module 16 expression (AUC 0.83; figure 6).
Figure 6.

Visualization of module 16, significantly correlated with very good versus poor outcome in ovarian cancer. (a) Regulatory program of module 16. (b) Estimated expression of module 16 using the regulatory program versus actual mean expression. (c) Module 16 gene expression. (d) Patients with very good outcome are indicated in black versus poor outcome patients in white. (e) Box-plot of the module 16 expression in very good versus poor outcome patients. (f) ROC curve of the supervised model based on module 16 to predict the very good versus poor outcome.
Finally, we investigated whether any of the modules are correlated with drug response. More specifically, we looked at sensitivity (time to failure greater than 365 days) or resistance (time to failure less than 365 days) to platinum-based chemotherapy. We found that three modules were significantly correlated with this outcome (p-value < 0.01, FDR < 15%). Module 86 was highly correlated with platinum sensitivity (p-value 0.00046015, FDR 4.6%). Module 86 is functionally enriched with the Notch signalling pathway, which has been tied to platinum-based chemotherapy response [26].
4. Discussion
AMARETTO is an analytical approach that aims to address the challenges associated with integrating and interpretating multi-omics cancer datasets, for example through the TCGA project. TCGA now has over 20 cancers that are being studied extensively with multiple omics technologies. This clearly creates a need for methods such as AMARETTO to extract knowledge that leverages all the data.
AMARETTO identifies cancer driver genes by considering that genes which are recurrently altered at the genome or epigenome level with functional consequences, as measured by their gene expression, are the most likely candidates. In addition, AMARETTO takes into account only disease-specific expression variations. This eliminates genes that are naturally expressed in normal tissue and are most likely not to be cancer drivers. Finally, AMARETTO only focuses on cancer drivers that explain downstream gene expression in the form of modules.
AMARETTO is being continuously developed and improved. Future plans involve integrating microRNA and DNA sequencing data into our models. We investigated the integration of microRNAs in the regulatory programs. However, our results suggest that microRNAs do not explain additional expression variation at the module level compared with cancer driver genes based on our observation that they are not selected in the regulatory programs. This is most likely to be caused by the observation that many microRNAs are located in introns of so-called host genes, confounding gene expression. Accordingly, microRNAs do not seem to explain more expression variation compared with candidate cancer drivers. We also investigated DNA mutation data from sequencing technology, but two issues emerged. First, a mutation does not necessarily have to affect gene expression. For example, mutations that constitutively activate protein function may not result in increased expression, but in increased function. Second, mutation data are notoriously sparse, thereby limiting our ability to find statistically significant relationships between DNA mutations and gene expression. Further work is necessary on the most optimal way of integrating microRNAs, mutations and other omics data in AMARETTO.
The main limitation of methods such as AMARETTO is the difficulty in evaluating the resulting network models. The most accepted route is experimental validation, a laborious and time-consuming process. In silico validation by comparison with other modelling strategies or networks is a more efficient validation strategy. However, the lack of standardized databases that store computational network models of cancer further complicate this level of validation. Most models are now hidden in supplementary files in non-standardized formats. A community effort in this area could create a comprehensive resource of computational models that can serve as a resource for experimental biologists.
In summary, we developed AMARETTO as a biocomputational approach for integrating multi-dimensional cancer data in a manner that enables the identification and analysis of genomic and epigenomic features that influence disease-specific gene expression. Using this method, we identified several novel oncogenic drivers in ovarian cancer and developed several new biological and clinical hypotheses. We identified potential drivers of the mesenchymal and proliferative ovarian cancer subtypes. Finally, we identified modules predictive of good versus poor outcome that implicate DNA repair pathways as a marker of ovarian cancer outcome as well as response to platinum therapy.
References
- 1.Sotiriou C. 2009. Molecular biology in oncology and its influence on clinical practice: gene expression profiling. Ann. Oncol. 20, 1019454421 [Google Scholar]
- 2.Pao W, et al. 2009. Integration of molecular profiling into the lung cancer clinic. Clin. Cancer Res. 15, 5317–5322 10.1158/1078-0432.CCR-09-0913 (doi:10.1158/1078-0432.CCR-09-0913) [DOI] [PubMed] [Google Scholar]
- 3.Gevaert O, De Moor B. 2009. Prediction of cancer outcome using DNA microarray technology: past, present and future. Expert Opin. Med. Diagn. 3, 157–165 10.1517/17530050802680172 (doi:10.1517/17530050802680172) [DOI] [PubMed] [Google Scholar]
- 4.Ciriello G, Cerami E, Sander C, Schultz N. 2011. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 22, 398–406 10.1101/gr.125567.111 (doi:10.1101/gr.125567.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vandin F, Upfal E, Raphael BJ. 2011. De novo discovery of mutated driver pathways in cancer. Genome Res. 22, 375–385 10.1101/gr.120477.111 (doi:10.1101/gr.120477.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akavia UD, et al. 2010. An integrated approach to uncover drivers of cancer. Cell 143, 1005–1017 10.1016/j.cell.2010.11.013 (doi:10.1016/j.cell.2010.11.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bell D, et al. 2011. Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615 10.1038/nature10166 (doi:10.1038/nature10166) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gevaert O, Plevritis S. 2013. Identifying master regulators of cancer and their downstream targets by integrating genomic and epigenomic features, pp. 123–134 Big Island, HI: Pacific Symposium on Biocomputing; [PMC free article] [PubMed] [Google Scholar]
- 9.Gevaert O, Rao X, Plevritis S. Submitted MethylMix: identifying DNA methylation-driven genes in cancer.
- 10.Nicolau M, Tibshirani R, Borresen-Dale AL, Jeffrey SS. 2007. Disease-specific genomic analysis: identifying the signature of pathologic biology. Bioinformatics 23, 957–965 10.1093/bioinformatics/btm033 (doi:10.1093/bioinformatics/btm033) [DOI] [PubMed] [Google Scholar]
- 11.Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. 2011. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41. 10.1186/gb-2011-12-4-r41 (doi:10.1186/gb-2011-12-4-r41) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor BS, Barretina J, Socci ND, Decarolis P, Ladanyi M, Meyerson M, Singer S, Sander C. 2008. Functional copy-number alterations in cancer. PLoS ONE 3, e3179. 10.1371/journal.pone.0003179 (doi:10.1371/journal.pone.0003179) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee S-I, Dudley AE, Drubin D, Silver P, Krogan N, Pe'er D, Koller D. 2009. Learning a prior on regulatory potential from eQTL data. PLoS Genet. 5, e1000358. 10.1371/journal.pgen.1000358 (doi:10.1371/journal.pgen.1000358) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Troyanskaya O, Cantor M, Sherlock G, Brown P, Hastie T, Tibshirani R, Botstein D, Altman RB. 2001. Missing value estimation methods for DNA microarrays. Bioinformatics 17, 520–525 10.1093/bioinformatics/17.6.520 (doi:10.1093/bioinformatics/17.6.520) [DOI] [PubMed] [Google Scholar]
- 15.Johnson WE, Li C, Rabinovic A. 2007. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 8, 118–127 10.1093/biostatistics/kxj037 (doi:10.1093/biostatistics/kxj037) [DOI] [PubMed] [Google Scholar]
- 16.Tone AA, Begley H, Sharma M, Murphy J, Rosen B, Brown TJ, Shaw PA. 2008. Gene expression profiles of luteal phase fallopian tube epithelium from BRCA mutation carriers resemble high-grade serous carcinoma. Clin. Cancer Res. 14, 4067–4078 10.1158/1078-0432.CCR-07-4959 (doi:10.1158/1078-0432.CCR-07-4959) [DOI] [PubMed] [Google Scholar]
- 17.Kim J, Coffey DM, Creighton CJ, Yu Z, Hawkins SM, Matzuk MM. 2012. High-grade serous ovarian cancer arises from fallopian tube in a mouse model. Proc. Natl Acad. Sci. USA 109, 3921–3926 10.1073/pnas.1117135109 (doi:10.1073/pnas.1117135109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subramanian A, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15 545–15 550 10.1073/pnas.0506580102 (doi:10.1073/pnas.0506580102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Culhane AC, et al. 2010. GeneSigDB—a curated database of gene expression signatures. Nucleic Acids Res. 38, D716–D725 10.1093/nar/gkp1015 (doi:10.1093/nar/gkp1015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma'ayan A. 2010. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26, 2438–2444 10.1093/bioinformatics/btq466 (doi:10.1093/bioinformatics/btq466) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B 57, 289–300 [Google Scholar]
- 22.Tibshirani R, Hastie T, Friedman J. 2010. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33, 1–22 [PMC free article] [PubMed] [Google Scholar]
- 23.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. 2004. A census of human cancer genes. Nat. Rev. Cancer 4, 177–183 10.1038/nrc1299 (doi:10.1038/nrc1299) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Velasco-Velazquez M, Jiao X, De La Fuente M, Pestell TG, Ertel A, Lisanti MP, Pestell RG. 2012. CCR5 antagonist blocks metastasis of basal breast cancer cells. Cancer Res. 72, 3839–3850 10.1158/0008-5472.CAN-11-3917 (doi:10.1158/0008-5472.CAN-11-3917) [DOI] [PubMed] [Google Scholar]
- 25.Bignotti E, et al. 2007. Gene expression profile of ovarian serous papillary carcinomas: identification of metastasis-associated genes. Am. J. Obstet. Gynecol. 196, 245.e1–245.e11 10.1016/j.ajog.2006.10.874 (doi:10.1016/j.ajog.2006.10.874) [DOI] [PubMed] [Google Scholar]
- 26.McAuliffe SM, et al. 2012. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc. Natl Acad. Sci. USA 109, E2939–E2948 10.1073/pnas.1206400109 (doi:10.1073/pnas.1206400109) [DOI] [PMC free article] [PubMed] [Google Scholar]