

Abstract
One of the powerful tools of adaptive dynamics is its so-called canonical equation (CE), a differential equation describing how the prevailing trait vector changes over evolutionary time. The derivation of the CE is based on two simplifying assumptions, separation of population dynamical and mutational time scales and small mutational steps. (It may appear that these two conditions rarely go together. However, for small step sizes the time-scale separation need not be very strict.) The CE was derived in 1996, with mathematical rigour being added in 2003. Both papers consider only well-mixed clonal populations with the simplest possible life histories. In 2008, the CE's reach was heuristically extended to locally well-mixed populations with general life histories. We, again heuristically, extend it further to Mendelian diploids and haplo-diploids. Away from strict time-scale separation the CE does an even better approximation job in the Mendelian than in the clonal case owing to gene substitutions occurring effectively in parallel, which obviates slowing down by clonal interference.
Keywords: meso-evolution, adaptive dynamics, canonical equation, haplo-diploids, invasion probability, effective reproductive variance
1. Introduction
For context, it is useful to distinguish between micro-, meso- and macro-evolution. The term micro-evolution customarily refers to changes in gene frequencies on a population dynamical time-scale. We will refer to the evolution of quantitative traits through the repeated substitution of novel mutants, including the splitting of the evolutionary path into separate evolutionary lines, as meso-evolution. The term macro-evolution then becomes restricted to larger scale changes such as anatomical innovations, where one cannot even speak in terms of a fixed set of traits.
Adaptive dynamics (AD) was devised as a mathematical framework for dealing with meso-evolution in ‘realistic’ ecological settings. It differs from more classical approaches to modelling evolutionary change, which generally assume constant fitnesses, by its focus on the population dynamical basis for those fitnesses, and hence on their inevitable change over evolutionary time. Of course, the greater realism at the ecological end is brought about by making different simplifying assumptions, this time genetically unrealistic ones. The main simplification is (i) separation of the population dynamical and mutational time scales. In order to concentrate on ecological aspects, unencumbered by the complexities of the genetic architecture and genotype to phenotype map, most AD research moreover assumes (ii) clonal inheritance.
One of the powerful tools of AD is its so-called canonical equation (CE), a differential equation describing how the prevailing trait vector changes over evolutionary time. The derivation of the CE is based on a subsequent further simplifying assumption: (iii) small mutational steps. As the speed at which a mutant substitutes is proportional to its effect, conditions (i) and (iii) will only rarely be met together. In §5, we give arguments why for small step sizes the time-scale separation need not be very strict.
The CE was first derived in [1] for well-mixed clonal populations with the simplest possible life histories. This was underpinned by a level of rigour sufficient to satisfy hard probabilists in [2] and [3]. The initial results were extended in [4] to, possibly spatially distributed, locally well-mixed populations with general life histories, with [5] giving a probabilistically rigorous underpinning for the case of simple age dependence. In this paper, we describe the extension to Mendelian diploids and haplo-diploids, once again on a physicist level of rigour. As it turns out, the CE for the simplest and most complex life histories differs only in a scalar factor summarizing how the intricacies of the life history feed through to the invasion probabilities of advantageous mutants. Mendelian diploidy brings in an additional factor 2, owing to the doubling of the number of mutant alleles per individual over a substitution.
Collet et al. [6] also derives a CE for Mendelian diploids, with full rigour. However, this in essence is a CE for a single locus trait, selected by a two-tiered ecology, within and between diploid bodies, the latter with the simplest possible life history. ([6] also lists the early applications of AD to Mendelian models.) We consider general life histories and phenotypic traits that are underlain by many loci.
In §2, we give a general heuristic derivation of the CE for clonal and for haploid and diploid Mendelian populations. In §3, we work out the details, and in §4 we consider haplo-diploids. The technicalities can be found in a suite of appendices. In the final §5, we discuss the strengths and weaknesses of the CE as a tool for evolutionary understanding.
2. Deriving the canonical equation for the textbook genetic scenarios
Mathematically, the CE is derived by taking two subsequent limits: (i) letting the system size K (and hence the average population size 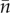 ) go to infinity (to make the community dynamics deterministic) and the mutation probability per birth event ɛ go to zero in such a manner that (a) ɛK ln(K) → 0 (to make the time for a substitution shorter than the time between mutations) and (b) ɛK exp(αK) → ∞ for sufficiently small positive α (to keep the population from going extinct on the time scale of the trait changes), followed by (ii) letting the mutational step sizes go to zero, all the while keeping the trait changes in view by rescaling time. Biologically, the CE is best seen as an approximation. From that perspective, it is expedient to express the result in the original time scale so that the basic biological parameters are kept in view.
) go to infinity (to make the community dynamics deterministic) and the mutation probability per birth event ɛ go to zero in such a manner that (a) ɛK ln(K) → 0 (to make the time for a substitution shorter than the time between mutations) and (b) ɛK exp(αK) → ∞ for sufficiently small positive α (to keep the population from going extinct on the time scale of the trait changes), followed by (ii) letting the mutational step sizes go to zero, all the while keeping the trait changes in view by rescaling time. Biologically, the CE is best seen as an approximation. From that perspective, it is expedient to express the result in the original time scale so that the basic biological parameters are kept in view.
Under the assumptions that K is large, ɛK ln(K) is small and ɛK exp(αK) is large for a sufficiently small positive α, that mutations are unbiased and that the environment as perceived by the individuals does not fluctuate (note that this implies a non-fluctuating resident population), the rate of change of a trait vector X can in the clonal case be expressed approximately as
 |
2.1 |
with s(Y|X) the invasion fitness of Y mutants in the environment generated by X residents (see [7,8]), ∂1s the derivative of s for its first argument, a row vector, and 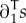 the corresponding column vector, Ts the mean survival time of the residents, Tr their average age at reproduction,
the corresponding column vector, Ts the mean survival time of the residents, Tr their average age at reproduction, 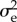 a measure for the variability of their lifetime offspring production (detailed in §3) and C the covariance matrix of the mutational steps. (In the AD literature, the quantity
a measure for the variability of their lifetime offspring production (detailed in §3) and C the covariance matrix of the mutational steps. (In the AD literature, the quantity 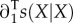 is known as the selection gradient.)
is known as the selection gradient.)
The first term in square brackets after the second equals sign in (2.1) comes from multiplying the probability of a mutation per birth by the population birth rate, 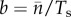 , a formula derived from the consistency relation
, a formula derived from the consistency relation 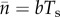 (the average number of particles in a ‘reservoir’ equals the average entrance rate multiplied by the average residence time).
(the average number of particles in a ‘reservoir’ equals the average entrance rate multiplied by the average residence time).
The second term comes from combining three approximation formulae to calculate the probability p that a mutant invades into the resident population, all coming from a branching process approximation for the invasion phase of the mutant dynamics, followed by averaging the product of the resulting expression and the mutational effect 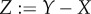 over the distribution g of Z.
over the distribution g of Z.
| 2.2 |
with 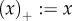 if x ≥ 0 and
if x ≥ 0 and 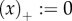 if x ≤ 0, and R0 the average lifetime offspring number of the mutant. (Note that the general R0 concept allows for multiple birth states as, for example, in spatially distributed populations [9].) Equation (2.2) is derived through a perturbation expansion from an equation for the invasion probabilities of a branching process ([10–18], appendix B).
if x ≤ 0, and R0 the average lifetime offspring number of the mutant. (Note that the general R0 concept allows for multiple birth states as, for example, in spatially distributed populations [9].) Equation (2.2) is derived through a perturbation expansion from an equation for the invasion probabilities of a branching process ([10–18], appendix B).
| 2.3 |
Equation (2.3) follows from R0(X|X) = 1.
| 2.4 |
Equation (2.4) is derived through a perturbation expansion from the characteristic equation for the Malthusian parameter of a branching process ([4,19,20], appendix A). (Note that dim(R0) = 1 and dim(s) = 1/time.) Together (i)–(iii) result in
| 2.5 |
When multiplying (2.5) by the mutational effect, it pays first to take transposes to make use of
| 2.6 |
Equation (2.6) follows from the facts that for unbiased mutational effects ∫ZZTg(Z)dZ = C and that the ( )+ means that we effectively integrate over only the half space where ∂1s(X|X)Z > 0.
In the derivation of (2.6), we have used the very strong interpretation of ‘unbiased’ that Z not only has mean zero but also is distributed symmetrically around that mean. Relaxing these assumptions gives an expression with considerably less appeal [1–3]. As relaxing them would make the following arguments less easy to follow while their essence stays the same, we have chosen to stick to the time-honoured simplification.
Moreover, in writing down (2.1), we have tacitly assumed that an invading mutant that makes it through the stochastic boundary layer, where its population dynamics can be approximated by a branching process, also makes it to a full substitution. This ‘invasion implies substitution’ rule presumably holds good for small mutational steps, away from population dynamical bifurcation points and evolutionarily singular strategies (characterized by ∂1s(X|X) = 0). A hard proof is available for the case where the community dynamics allows a finite dimensional representation [21–23]. However, it looks as if with the right mathematical expertise the rule should be extendable to the required generality by combining the approaches in [24] and [25,26].
The two terms in square brackets in (2.1) connect to the simplifying assumptions in the following manner. The first term is contingent on the time-scale separation assumption. Only when substitutions do not interfere does the speed of trait movement become proportional to the number of invasion attempts per time unit. The derivation of the second term is contingent on the smallness of the mutational steps and also on the time-scale separation to produce the constant environment underlying the branching process calculations (which assume fixed probability distributions for the lifetime offspring numbers). More in particular, time-scale separation means that in between the negligibly short substitution events the environment is stationary and resident populations are genetically homogeneous. (More about the latter, seemingly unrealistic, consequence in §5.)
We now consider the Mendelian case. For chromosomal sex determination, we focus on the autosomes, deferring the allosomal contributions to §4.
Thanks to the genetic homogeneity of the residents, the argument in (2.1) extends seamlessly to haploids. The only difference from the clonal case is that thinking genetics points one to the concept of genotype to phenotype map and mutations that occur on multiple loci. However, when substitutions occur singly the latter multiplicity becomes phenotypically inconsequential.
Moving on to diploids, we first take a closer look at genotype to phenotype maps. The prevalent view in Evo-Devo nowadays is that the trait changes that AD attempts to model are mostly caused not by changes in the coding regions of genes but at their regulatory regions (e.g. [27,28]). The latter determine the activity of the genes in different parts of the body, at different times during development and under different micro-environmental conditions. This scenario allows us to look at the genotype as a sequence of vectors 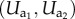 of expression levels, with a (ai) a placeholder for the name of (an allele on) a generic locus. The genotype to phenotype map Φ transforms this sequence into phenotypic traits. It is from this perspective that one should judge the assumption of smallness of mutational steps: the influence of any specific regulatory site among its many colleagues tends to be relatively minor. And it is this perspective that allows us to assume that (iv) genotype to phenotype maps are smooth.
of expression levels, with a (ai) a placeholder for the name of (an allele on) a generic locus. The genotype to phenotype map Φ transforms this sequence into phenotypic traits. It is from this perspective that one should judge the assumption of smallness of mutational steps: the influence of any specific regulatory site among its many colleagues tends to be relatively minor. And it is this perspective that allows us to assume that (iv) genotype to phenotype maps are smooth.
Lemma —
([29], A. Pugliese 1996, personal communication). When there are no parental effects, smooth genotype to phenotype maps are locally additive, i.e. if at a the expression vector Ua mutates to UA, then
2.7
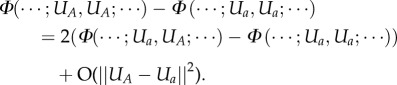
Proof. —
Without parental effects
2.8
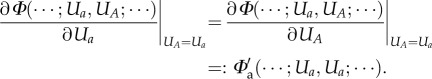
Hence,
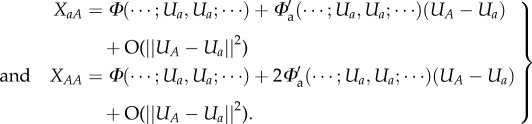 |
2.9 |
▪
In diploids, an invading mutant allele A practically always shares a body with a resident allele a and this aA reproduces through backcrossing with a resident aa. Hence, the allele population initially grows as clonally reproducing aAs (producing aas on the side), and its invasion fitness corresponds to the asymptotic average per capita growth of that clonal population in an environmental background provided exclusively by, also seemingly clonally reproducing (for homogeneous), residents. The invasion implies that substitution theorem also applies unchanged (thanks to the local additivity). However, after substitution the population consists of mutant homozygotes, making the resulting step in phenotype space twice as large as in the clonal and haploid cases. We thus conclude that the CE for Mendelian diploids reads
| 2.10 |
On the right-hand side, we have put first the ecologically determined number of resident haplotypes (sets of chromosomes as present in gametes), followed by the life-history statistics controlling the initial demographic stochasticity of allelic substitutions, followed by an expression quantifying the per birth mutational variability generated by the genetic architecture and genotype to phenotype map, to conclude with the selection gradient, summarizing the ecology's current tendency for filtering novel genetic variation. In Metz & Jansen [30], it is argued that  times the second group of quantities precisely equals the effective population size from population genetics.
times the second group of quantities precisely equals the effective population size from population genetics.
3. Filling in the details
It may seem that with (2.10) we are done. However, the devil is in the detail, to wit the calculation of s, Ts, Tr,  and C. For background material on the ecological models covered, see [20,31] for the resident and [32,33] for the invader dynamics.
and C. For background material on the ecological models covered, see [20,31] for the resident and [32,33] for the invader dynamics.
In principle  also presents a problem, although only when the resident population fluctuates. This is even so in the clonal case as
also presents a problem, although only when the resident population fluctuates. This is even so in the clonal case as 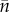 is not just a time average but a peculiarly weighted one (e.g. [34,35]). To keep things simple, we have confined the argument to non-fluctuating residents.
is not just a time average but a peculiarly weighted one (e.g. [34,35]). To keep things simple, we have confined the argument to non-fluctuating residents.
We first consider R0, although this quantity does not appear in our (2.1)-derived list. The reason that it does not do so is that we wanted to write the CE in the form customary in the literature. However, for most structured populations R0 is far easier to calculate than s. Therefore we might just as well in (2.1) and (2.10) drop Tr and replace s with R0.
R0 equals the dominant eigenvalue of the next-generation matrix L(Y|X) (or operator in the case of infinitely many birth states; e.g. [9]). For the clonal case, L is constructed by calculating from a model for the behaviour of individuals how many offspring in different birth states they produce on average, dependent on their own birth state.
Given the next-generation matrix, we can introduce two further quantities for later use: the stationary birth state distribution, i.e. normalized positive right eigenvector of L(X|X) going with R0 = 1, 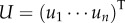 , 1TU = 1 with
, 1TU = 1 with 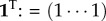 , and the corresponding reproductive values, i.e. co-normalized left eigenvector,
, and the corresponding reproductive values, i.e. co-normalized left eigenvector, 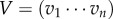 , VU = 1. vi equals the expected contribution of a newborn of type i to future birth rates.
, VU = 1. vi equals the expected contribution of a newborn of type i to future birth rates.
How can we define R0 for evolving sexual diploids? The first answer is that in hermaphrodites one can just add the numbers of offspring that individuals father and mother over their life (i.e. produce through the micro- and macro-gametic routes) and divide by 2. The factor 1/2 comes from the fact that in the Mendelian process each allele is only transferred with that probability. When hermaphrodites are born stochastically equal (i.e. their birth states have the same probability distribution),
| 3.1 |
with m and f the average number of offspring fathered or mothered by a randomly chosen individual. For later use, we moreover note that for the resident
| 3.2 |
as resident densities are supposedly constant and every individual has one father and one mother.
Equations (3.1) and (3.2) also hold good for dioecious organisms, but with a twist. For later use, we note that we then can rewrite (3.1) and (3.2) by letting pf and pm denote the fractions of newly produced females and males and f+ and m+ the average lifetime numbers of offspring begotten by a female or male. Then f = pff+ and m = pmm+, so that
| 3.3 |
and for the residents
| 3.4 |
The above results are not completely trivial, as, in contrast to hermaphrodites, individuals of dioecious species are born in different flavours. To account for this, L(Y|X) should be properly extended. If no other birth state distinctions are needed
| 3.5 |
with ℓff, ℓfm the average lifetime numbers of daughters of a female, male, and ℓmf, ℓmm the corresponding average lifetime numbers of sons, all for mutant heterozygotes in the environmental and genetic background provided by the resident. The simplest case is when there is no connection between the traits and sex determination so that ℓff = pff+, ℓmf = pmf+, ℓfm = pfm+ and ℓmm = pmm+. Then L has rank one and R0 = 1/2(pff+ + pmm+). Another way of getting the latter result is by observing that in this case all offspring are born stochastically equal, with each having the same probability of being born female. We can then proceed as if sex is determined after birth and calculate R0 by averaging over the possible courses of a life. When there is a connection, we end up with the same formula by defining pm and pf as the asymptotic probabilities of being born male or female, i.e. by choosing for pm and pf the components of the right eigenvector U of L, and defining m+ and f+ again as the average numbers of offspring fathered or mothered over a lifetime given the parental sex, f+ = ℓff + ℓmf, m+ = ℓfm + ℓmm. By using R0 = 1TLU, we again get R0 = 1/2(pff+ + pmm+). However, only the similarity of the expression is pleasing as this time it is not explicit, as to calculate pm and pf one first needs to calculate R0.
Appendix C indicates how the preceding considerations extend in the presence of additional birth state distinctions.
Next we consider Ts, the quantity that had to be combined with 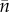 to get the population birth rate.
to get the population birth rate.
| 3.6 |
with 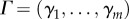 , γi being the probability density of time until death of a resident born in state i. In the dioecious case, with all offspring born stochastically equal
, γi being the probability density of time until death of a resident born in state i. In the dioecious case, with all offspring born stochastically equal
| 3.7 |
The mean age at reproduction Tr needs more thought as the offspring may also differ in their birth states. The perturbation expansion for s in appendix A tells us that we should weight those offspring with their reproductive values.
| 3.8 |
with Λ(a) composed of 1/2 times the average per capita parenting rates of age a residents split according to the birth states of offspring and parent. This formula generalizes the usual definition of the age at reproduction for all offspring born equal. (Note that 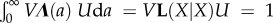 so that VΛ(a)U is a probability density.) When all offspring are born stochastically equal
so that VΛ(a)U is a probability density.) When all offspring are born stochastically equal 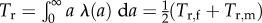 with λ = λf + λm, λf and λm being half the average per capita mothering and fathering rates of the residents. For dioecious organisms, λf = pfλf+, λm = pmλm+ with λf+, λm+ being half the average female, male parenting rates. Yet, Tr,f+ = Tr,f, Tr,m+ = Tr,m, thanks to the normalization of λf+ and λm+ before calculating the mean parenting ages.
with λ = λf + λm, λf and λm being half the average per capita mothering and fathering rates of the residents. For dioecious organisms, λf = pfλf+, λm = pmλm+ with λf+, λm+ being half the average female, male parenting rates. Yet, Tr,f+ = Tr,f, Tr,m+ = Tr,m, thanks to the normalization of λf+ and λm+ before calculating the mean parenting ages.
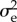 , appearing as a coefficient in the approximation formula for the invasion probability p, is the most complicated beast. We first give its formula for the clonal case
, appearing as a coefficient in the approximation formula for the invasion probability p, is the most complicated beast. We first give its formula for the clonal case
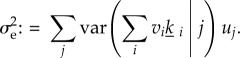 |
3.9 |
Here, the ki are the lifetime numbers of i-offspring of residents and var(h|j) means that the variance of the random quantity h for an individual born in state j. Equation (3.9) emerges from the perturbation expansion (appendix B).
To calculate 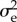 for the Mendelian diploid case, we apply (3.9) to the alleles. We only consider the case with all offspring born stochastically equal.
for the Mendelian diploid case, we apply (3.9) to the alleles. We only consider the case with all offspring born stochastically equal.
| 3.10 |
with 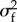 and
and 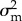 the variances of the lifetime offspring numbers kf and km produced, respectively, through the macro- and micro-gametic routes, and c their covariance (appendix D). The 1 inside the inner brackets comes from the Mendelian sampling of alleles.
the variances of the lifetime offspring numbers kf and km produced, respectively, through the macro- and micro-gametic routes, and c their covariance (appendix D). The 1 inside the inner brackets comes from the Mendelian sampling of alleles.
For dioecious organisms, c = −1 (as Ekfkm = 0 and Ekf = Ekm = 1), and 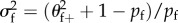 ,
, 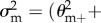
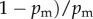 , θf+ = σf+/f+ and θm+ = σm+/m+. The latter result is obtained from the following lemma together with the observation that E + kf = f+ and E +
km = m+, with E + the expectation conditional on the parental sex.
, θf+ = σf+/f+ and θm+ = σm+/m+. The latter result is obtained from the following lemma together with the observation that E + kf = f+ and E +
km = m+, with E + the expectation conditional on the parental sex.
Lemma. —
Let the discrete non-negative random variable k be constructed from a random variable k+ by setting k = k+ with probability p and k = 0 with probability 1 − p, then Ek = pEk+ and var(k) = Ek2 − (Ek)2 = p[var(k+) + (Ek+)2] − (pEk+)2. When moreover Ek = 1 so that Ek+ = p−1,
3.11
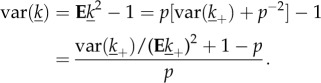
The mutational covariance matrix C is primarily a phenomenological quantity, although in principle it can be expressed in terms of per locus statistics and the genotype to phenotype map,
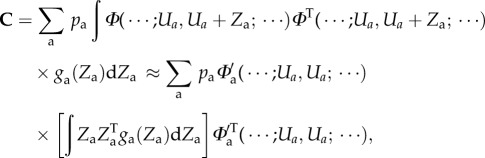 |
3.12 |
with pa the relative frequency with which a mutation occurs at locus a and ga the probability density of mutational steps in the allelic trait vector of that locus.
As a final point, we need to say something about the traits and the concept of phenotype. In general, the phenotypes of AD should be seen as reaction norms, i.e. maps from micro-environmental conditions to characteristics of individuals (another term is conditional strategies). Only in the simplest cases a reaction norm is degenerate, taking only a single value. The dioecious case is similar in that the development of the sexes need not be, and in fact rarely is, equal. Hence, trait vectors will generally consist of two components, traits of the male, Xm, and of the female, Xf. In general, the traits of the two sexes evolve dependently as they are coupled by their mutational covariances. In the extreme case that the mutational covariances between male and female traits are all zero the female and male coevolve as if they are separate species. At the opposite end of the spectrum Xm = Gm(Xf), or Xf = Gf(Xm), and the mutational variation in the male and female traits is fully correlated. The upshot is that, except in the fully correlated case, we cannot work with a monolithic trait vector influencing both macro- and micro-gametic reproduction, as in hermaphrodites. Instead, we should take into account the fact that when the sexes are separate they can evolve in their own ways.
4. Haplo-diploids: a not uncommon, but often-neglected, reproductive mode
In addition to the haploid and diploid ones, there exist all sorts of other lifestyles. One common type is where haploid and diploid phases alternate (as in, for example, ferns, mosses and a great variety of algae). It is then necessary, as in dioecious diploids, to introduce trait vectors for each separate phase. We can then consider a diploid plus its haploid offspring as a single generalized individual ([36] gives a relaxed introduction to this useful concept) and apply the theory of the previous two sections, where for the diploid phase traits we again have to put in a factor 2.
Still another lifestyle is the so-called haplo-diploid one, where one sex is diploid and the other haploid (the supplementary material to [37] lists the many known haplo-diploid taxa). Although this case can also be treated through the mental construction of appropriate generalized individuals, we follow the strategy of §3, and treat the two sexes as different birth states, as this is simpler and allows us to illustrate further tricks of the trade.
Although the opposite also occurs, we shall for definiteness take the hymenopteran situation as reference and assume that females are diploid. In that case only the female reproductive output needs to be discounted by 1/2. We allow any physiological structure and only assume that within each sex all offspring are born equal. The next-generation matrix then becomes
| 4.1 |
with ff and fm the average lifetime numbers of female and male offspring of a female, and m the average lifetime number of (female) offspring of a male. ff and fm depend only on the female traits, m only on the male traits.
For the resident ff = 1 (as the density of females is constant) and hence fm = r, the sex ratio at birth (the number of males born relative to females), and m = r−1 (since all females have a father and one female is born for r males). Hence, the resident's next-generation matrix is
| 4.2 |
with co-normalized eigenvectors
| 4.3 |
As is commonly the case in models with more than one birth state, the resulting expression for R0 does not excel in transparency. However, we do not need R0 but its derivative, for which there exists a simple formula given in the following lemma, derived by differentiating through the characteristic equation and solving for ∂R0/∂Y (see [38,39] and in particular [40]).
Lemma. —
Let
and
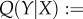
, then
4.4
So far, we have listed the additional ideas. The rest is work, following the route outlined in §§2 and 3.
We begin with the selection gradient
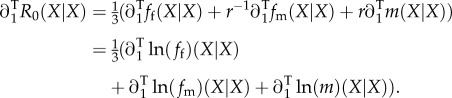 |
4.5 |
As we start from R0 instead of s, there is no need for us to calculate Tr, and as Ts has nothing to do with the reproductive system, (3.7) holds good as for any other sex-differentiated population. There remains
| 4.6 |
where the |f, |m in the indices indicates that the quantity is calculated conditional on the parent being female or male. There are nicer expressions than (4.6). If you wish, you can check it by working through appendix E. The term 1 + r inside the inner brackets derives from the Mendelian sampling in the females. Furthermore, for the male traits the factor 2 in front of 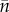 in (2.10) should be removed. Lastly, a factor accounting for the reduced number of haplotypes per individual has to be inserted—for sex-independent per haplotype mutation frequencies (1 + (1/2)r)/(1 + r).
in (2.10) should be removed. Lastly, a factor accounting for the reduced number of haplotypes per individual has to be inserted—for sex-independent per haplotype mutation frequencies (1 + (1/2)r)/(1 + r).
Our arguments for diploids only considered autosomes. To obtain the full CE in the case of chromosomal sex determination, it suffices to add the allosomal contributions to the autosomal one. The contribution of the chromosome occurring only in the heterogametic sex follows the rules for clonal reproduction, that of the chromosome of the homogametic sex follows those for haplo-diploids.
5. Discussion
Away from evolutionarily singular strategies (where ∂1s(X|X) = 0), the time-scale separation assumption combined with the assumption of small mutational steps guarantees practically permanent genetic homogeneity of the resident population. More in particular, a population with sufficiently restricted variability will become homogeneous on the time scale of the substitutions, thanks to the close to additive genetics, and will effectively stay so until the next substitution. The exceptions to this homogeneity occur when an evolutionary trajectory comes close to an evolutionarily singular strategy. The reason is that, where everywhere else we have an approximately linear selection regime, near the singular strategies the quadratic terms in the local expansion of the fitness landscape start to dominate, creating epistasis (non-additivity) at the level of the fitnesses.
Genetic homogeneity of the resident population lies at the base of the approximations made to reach the CE and of its easy extension to a Mendelian world. At first sight this seems a strong argument against the CE's applicability, as genetic variability appears to be rampant. There are two arguments for yet thinking that the CE may often be a fair description of reality. The first one is that we need mutation limitation only for genes affecting the trait. The thrust of our theoretical deductions is not affected by selectively protected variability that is developmentally and selectively unconnected to the focal traits or variability on neutral loci subjected to mutation, drift and draft (hitchhiking with loci under selection). The only effect that selectively kept variability at unconnected loci may have is that it makes the lifetime offspring numbers of the substituting alleles more variable, enlarging 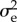 . However, as we treated the components of
. However, as we treated the components of 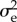 as empirical quantities, any genetic causes of this variability are automatically included.
as empirical quantities, any genetic causes of this variability are automatically included.
The second argument has a bearing on variability owing to a lack of strict time-scale separation. As long as that variability stays sufficiently limited, it should in Mendelian populations have little effect on the quality of the CE approximation. The reason is that the effect of such variability on the invasion fitness is of higher order in the mutational step size than the terms retained in the derivation of the CE. (The argument for the latter statement may be found in [41]. This paper admittedly only considers unstructured populations. However, the nature of the argument suggests that with the right mathematical expertise it should be extendable to structured ones.) In clonal populations, a lack of mutation limitation yet presents an obstacle to the quantitative applicability of the CE owing to clonal interference: a substituting clone may be ousted en route by a better mutant so that only a fraction of the mutants that invade contribute to longer run evolutionary change. We may thus expect ‘reality’ to be slower than the CE. Mendelian populations do not suffer this slowing down as mutants on different loci effectively substitute in parallel without interfering thanks to recombination (and the approximate additivity coming from the smallness of mutational effects).
The picture of multiple substituting mutants with small additive effect may seem close to the one considered by Lande [42,43]. However, there is a difference as in our picture only a small and variable number of loci are simultaneously substituting, with new mutants continually coming and old ones going. By contrast, the quantitative genetics picture underlying Lande's work has its basis in Fisher's picture of evolutionary change coming from changes in the allele frequencies on a large number of loci with all alleles present from the start [44]. (An argument against that picture is that the genetic differences between, say, a choanoflagellate and a human are so many that the attendant polymorphism would contain more genotypes than the number of atoms in the Earth, invalidating the classical population genetic calculations.) Of course, Lande did not subscribe to this simplified picture (e.g. [45,46]), and neither did Fisher, at least not wholeheartedly [47, chapter IV]. However, the mathematical details of the connection between the approximations introduced in Lande's various papers remains to be worked out.
One of the consequences of having only a few loci substituting at any one time is that the within-population variance of the trait fluctuates stochastically and appears to be highly variable (J. Ripa 2005, personal communication). This notwithstanding a negative feedback: when the standing variability is high, mutant alleles will find it more difficult to pass through the stochastic boundary layer because of the larger fluctuations in their offspring numbers. Unfortunately, this picture does not easily lead to explicit expressions, as the fluctuations in variability occur on a time scale similar to that of the substitutions so that we cannot use our earlier branching process calculation. However, we surmise that in reality the allelic reproductive variability coming from co-substituting alleles can usually be neglected relative to the variability in offspring numbers caused by alleles not affecting the traits and by micro-environmental variability such as some individuals running into a predator and others not.
Although the previous paragraphs may give the impression that we are quite optimistic about the applicability of the CE, there is one problem that in applications is usually ignored: there is no need for C to stay constant. It may perhaps seem, when the mutations with small and hence additive effect all occur on different loci, that also their cumulative effect will be additive. However, this is not the case. This can be seen by looking at the developmental process as a sequence of maps, each of which transforms the genetic information plus environmental inputs further towards the final phenotype. Even if linearity were to prevail early on, the accumulated change will be appreciable, and when the output from those early stages is fed into a nonlinear map to get to the next stage, approximate linearity is lost and the derivatives of Φ that appear in (3.12) change over evolutionary time.
In principle, it is possible to write down a CE for the extended trait vector (X, C) to obtain the change in C as correlated response to selection on X. However, going through the calculations in the manner of (3.12) shows that the expression for the covariances between the mutational steps in X and C generically involves higher moments. We should thus see the CE as the first step in a truncated moment expansion.
All this does not mean that using the CE with fixed C is never more than a heuristic tool, with no strong connection to reality. There are scenarios where one may with impunity assume C to be constant, in particular when investigating the behaviour of evolutionary trajectories close to evolutionarily singular strategies. In a linearized stability analysis as in [48] or in the analysis of scenarios for initial divergence close to an adaptive branching point (as defined in [49–51]) as in [4,52–54], only those situations are considered where the change in X, and hence the associated change in C, is small. (Such arguments involve two trait scales, a small one for linearizing the CE and a smaller one for the mutational steps.)
Acknowledgements
We thank Jacques van Alphen for inviting the first author to a workshop on insect parasitoids, thus sparking the research reported in §4.
Appendix A. Approximating a mutant's invasion fitness
For the sake of the exposition, we first consider the case of a single birth state. Then s satisfies the characteristic equation (Lotka's equation)
| A1 |
depending on whether we are dealing with continuous or discrete time community dynamics. Here, λ(a) is the average birth rate or ratio at age a, where the average is taken over whatever stochastic trajectories individuals may follow during their life.
From here on, we concentrate on the continuous time case. Rewriting (A 1) by introducing the probability density ℓ of the age at reproduction a, and its cumulant generating function ϕ,
| A2 |
gives
| A3 |
Expanding ϕ as
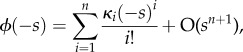 |
A4 |
with κi the ith cumulant, 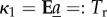 ,
, 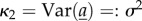 , and solving subsequently for the first- and second-order terms (on the assumption that |ln(R0)| is small) gives
, and solving subsequently for the first- and second-order terms (on the assumption that |ln(R0)| is small) gives
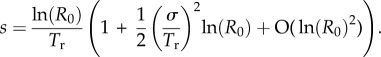 |
A5 |
Equation (A 5) often performs far better than the estimate of the error term suggests. The reason is that a similar result appears for birth kernels with potentially large R0 but narrow spread. When the ith central moment μi of a scales like σn, (A 1) can be written as
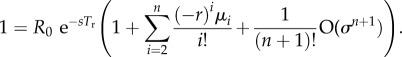 |
A6 |
Taking logarithms and solving subsequently for first- and second-order terms gives
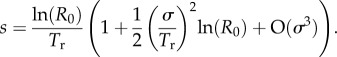 |
A7 |
The two approximations agree up to their second-order terms (but not in higher order ones).
For more than one birth state, we introduce Λ(a) = λij(a), with λij(a) the average rate at which an individual born in state j gives birth to offspring in state i. Then s can be calculated from the characteristic equation
| A8 |
with
| A9 |
or, equivalently, as the rightmost solution of
| A10 |
Let λd stand for ‘dominant eigenvalue of’ and 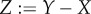 be the mutational step. Then (A 8) can be rewritten as
be the mutational step. Then (A 8) can be rewritten as
| A11 |
Expanding ψ as a function of its first argument gives
| A12 |
On the assumption that s = O(||Z||) for small ||Z|| we can rewrite (A 12) as
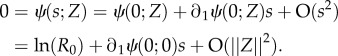 |
A13 |
Hence,
| A14 |
It remains to calculate
| A15 |
The denominator in the first factor equals R0(X|X) = 1. The second factor equals (e.g. [55])
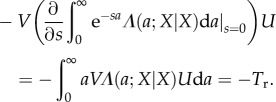 |
A16 |
Hence,
| A17 |
In Appendix B of [4] it is moreover proved that when ln(R0) = O(||Z||2), as is the case around evolutionarily singular strategies, it is still possible to use (A 17) with O(||Z||2) replaced by O(||Z||3).
Appendix B. Approximating a mutant's invasion probability
Equation (3.9) has a long history, starting with [10,11] for single birth states, with refinements in [12–14]. The work of Eshel [15] starts the treatment for multiple birth states, with refinements in [16–18]. All later papers are rather complicated as they aim at the strongest possible results. However, when the offspring numbers have third moments a standard perturbation expansion suffices.
To begin, we consider the case of a single birth state. The following calculation can be found in any textbook devoting space to branching processes. Denote the lifetime number of offspring of a representative individual as k where the underlining signifies that k is a random variable, and its so-called generating function as
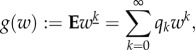 |
B1 |
with qk the probability that an individual begets k offspring. By differentiating one finds for n = 0,1,…
| B2 |
with 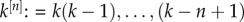 and
and 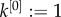 . Ek[n] is known as the nth factorial moment. Let p denote the probability of invasion. When an individual is known to beget k offspring, the chance that its line goes extinct is (1 − p)k. Hence,
. Ek[n] is known as the nth factorial moment. Let p denote the probability of invasion. When an individual is known to beget k offspring, the chance that its line goes extinct is (1 − p)k. Hence,
| B3 |
We know that p = 0 for R0 ≤ 1. For small R0 − 1, and hence small p
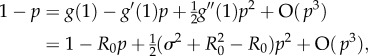 |
B4 |
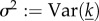 . Substituting the ansatz p = c(R0 − 1) + O((R0 − 1)2) and solving for c gives
. Substituting the ansatz p = c(R0 − 1) + O((R0 − 1)2) and solving for c gives
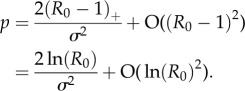 |
B5 |
The calculation for more than one birth state starts from the vector of generating functions 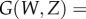
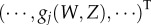 with
with
| B6 |
j the birth state of the parent. By a similar argument as before (e.g. [33])
| B7 |
with 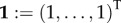 and a prime denoting the derivative for Z (
and a prime denoting the derivative for Z (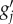 is thus a row vector with components ∂g/∂zi and G’ a matrix), and
is thus a row vector with components ∂g/∂zi and G’ a matrix), and
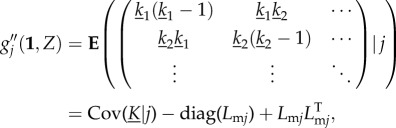 |
B8 |
with
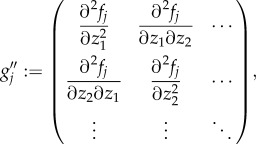 |
and Lm j the jth column of L m.
Let P denote the vector of invasion probabilities depending on the birth state of the newly arrived mutant. By a similar argument as before, P can be shown to satisfy
| B9 |
[33]. Expanding and making the ansatz that P = CZ + O(||Z||2) gives
| B10 |
with 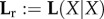 ,
, 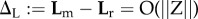 and
and 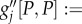
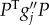 . Collecting the first-order terms gives
. Collecting the first-order terms gives
| B11 |
Hence,
| B12 |
with κ = O(||Z||). Substituting this in the equation that results from collecting the second-order terms gives
| B13 |
which, on dividing by κ and right multiplying with U to get the projection on the dominant left eigenspace, gives
| B14 |
Finally use that VΔLU = ln(R0) + O(||Z||2) to arrive at
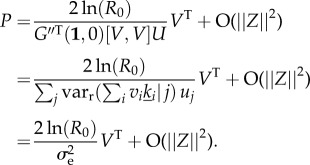 |
B15 |
(The step from the factorial moments in G’’T(1, 0)[V, V]U to variances is made by observing that 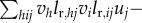
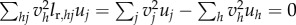 .)
.)
Now suppose that there is no relation between birth state and mutation propensity. Then the birth state distribution of a newly appeared mutant is U, and the probability that a random mutant invades can be approximated as
| B16 |
As in the invasion fitness case, (B 16), with O(||Z||2) replaced by O(||Z||3), also applies near evolutionarily singular strategies where ln(R0) = O(||Z||2).
Appendix C. Calculating R0 for a population in two patches with separate sexes
The proper next-generation matrix for this situation is
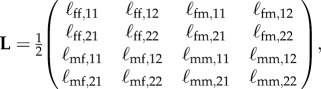 |
C1 |
with ℓfm,12 the average lifetime number of female offspring produced in patch 1 by a mutant male born in patch 2 (through fertilizing resident type females), etc.
Now assume that an individual's sex is at most dependent on its birth patch, as is, for example, the case when sex determination is fully environmental or when fathers have no influence on the sex of their offspring, mothers let the sex of their offspring depend at most on where those offspring are born, and the traits are not connected with sex allocation. Then L can be written as
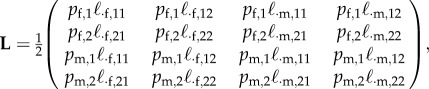 |
C2 |
with 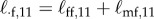 , etc. and
, etc. and 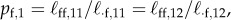 , etc., and thus has rank 2. After one generation, the births can be written as
, etc., and thus has rank 2. After one generation, the births can be written as 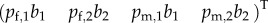 . From then on multiplication with L gives
. From then on multiplication with L gives
| C3 |
Hence, R0 can be calculated as the dominant eigenvalue of
| C4 |
When in the general case we write the dominant eigenvector of L as 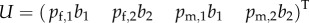 , with
, with 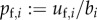 ,
, 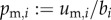 ,
, 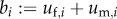 , multiplying U with L also gives (C 3). Hence, in this case R0 also corresponds to the dominant eigenvalue of a matrix constructed by adding the average numbers of offspring that individuals father or mother over their life and dividing by 2. However, in general these quantities are no more than population averages that can be determined only after establishing the stable birth state distribution of the full next-generation operator.
, multiplying U with L also gives (C 3). Hence, in this case R0 also corresponds to the dominant eigenvalue of a matrix constructed by adding the average numbers of offspring that individuals father or mother over their life and dividing by 2. However, in general these quantities are no more than population averages that can be determined only after establishing the stable birth state distribution of the full next-generation operator.
Appendix D. The effective offspring variance for sexual diploid residents
We only consider the case where except for genetic and possibly sex differences all individuals are born equal. In sexually reproducing individuals, an allele is born in either of two states, with potentially different futures, to wit in a macro-gamete or micro-gamete. In the simplest case, these states are randomly allotted, independent of which route put the parental allele in the parental body. This happens, for example, in hermaphrodites or when sex is determined environmentally or by alleles on a different chromosome. We concentrate on that simplest case. Then 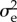 is the variance of the number of allelic copies reaching the next generation of zygotes from a copy that has just ended up in a zygote. Let km be the number of alleles that does so micro-gametically, and kf the number that does so macro-gametically. Generally, km and kf are dependent. For example, for a seed, and hence for the two alleles in it, the size of the plant it engenders is a random variable, with larger plants usually producing more ovules as well as pollen so that km and kf are positively correlated. As the extreme opposite, in dioecious organisms a new zygote will go on to produce either micro- or macro-gametes, i.e. kmkf = 0. Hence, the starting point of the calculation is the generating function of the pair (kf, km)
is the variance of the number of allelic copies reaching the next generation of zygotes from a copy that has just ended up in a zygote. Let km be the number of alleles that does so micro-gametically, and kf the number that does so macro-gametically. Generally, km and kf are dependent. For example, for a seed, and hence for the two alleles in it, the size of the plant it engenders is a random variable, with larger plants usually producing more ovules as well as pollen so that km and kf are positively correlated. As the extreme opposite, in dioecious organisms a new zygote will go on to produce either micro- or macro-gametes, i.e. kmkf = 0. Hence, the starting point of the calculation is the generating function of the pair (kf, km)
| D1 |
The generating function of the number k of aA offspring of an aA individual reproducing in an aa population background is then
| D2 |
with ½ + ½w the generating function of the number of A alleles, 0 or 1, in a gamete. For the calculation of 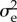 the allele A is supposed to have no phenotypic effect. Hence, at population dynamical equilibrium both ∂1g(1, 1) = ∂2g(1, 1) = 1 because the number of individuals stays constant over the generations and each offspring has one father and one mother. Therefore,
the allele A is supposed to have no phenotypic effect. Hence, at population dynamical equilibrium both ∂1g(1, 1) = ∂2g(1, 1) = 1 because the number of individuals stays constant over the generations and each offspring has one father and one mother. Therefore,
| D3 |
and
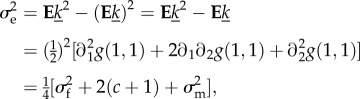 |
D4 |
with
| D5 |
the covariance between macro- and micro-gametically produced offspring.
Appendix E. The effective offspring variance for haplo-diploid residents
Following the pattern from appendix D, we first express the variances and covariances of the allelic offspring numbers in terms of the variances and covariances of the numbers of offspring of the resident individuals. Let ga denote the generating function of the lifetime offspring numbers of a residing in a female and ending up in female and male children of that female and let g denote the generating function of those numbers of children kf and km irrespective of their genotype, then
| E1 |
Differentiating gives, with i, j = f, m,
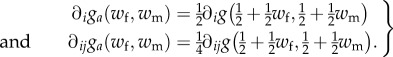 |
E2 |
Hence
| E3 |
and
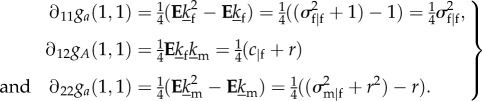 |
E4 |
Therefore,
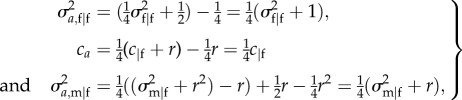 |
E5 |
where the |f in the indices of σ and c indicates that the quantities are calculated conditional on the parent being a female. This gives all the ingredients for calculating 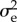 . First, calculate
. First, calculate
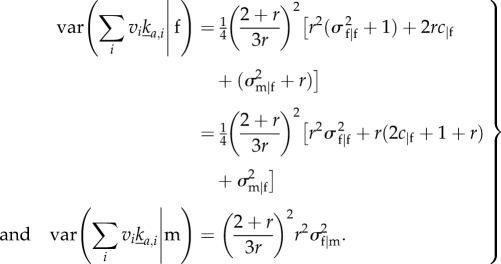 |
E6 |
Substituting (4.3) and (E 6) in (3.9) gives
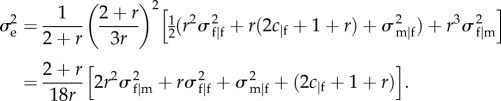 |
E8 |
For good measure we add the formula for the average age at reproduction
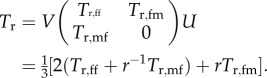 |
E9 |
Funding statement
C.G.F.d.K. was supported by the Netherlands Organization of Scientific Research (NWO), grant 813.04.001. J.A.J.M. benefited from the support of the Chair Modélisation Mathématique et Biodiversité VEOLIA-École Polytechnique-MNHN-F.X.c.
References
- 1.Dieckmann U, Law R. 1996. The dynamical theory of coevolution: a derivation from stochastic ecological processes. J. Math. Biol. 34, 579–612. ( 10.1007/BF02409751) [DOI] [PubMed] [Google Scholar]
- 2.Champagnat N. 2003. Convergence of adaptive dynamics n-morphic jump processes to the canonical equation and degenerate diffusion approximation. Prépublication de l'Université de Nanterre (Paris X) 03/7. [Google Scholar]
- 3.Champagnat N, Méléard S. 2011. Polymorphic evolution sequence and evolutionary branching. Probab. Theory. Relat. Fields 151, 45–94. ( 10.1007/s00440-010-0292-9) [DOI] [Google Scholar]
- 4.Durinx M, Metz JAJ, Meszéna G. 2008. Adaptive dynamics for physiologically structured models. J. Math. Biol. 56, 673–742. ( 10.1007/s00285-007-0134-2) [DOI] [PubMed] [Google Scholar]
- 5.Méléard S, Tran VC. 2009. Trait substitution sequence process and canonical equation for age-structured populations. J. Math. Biol. 58, 881–921. ( 10.1007/s00285-008-0202-2) [DOI] [PubMed] [Google Scholar]
- 6.Collet P, Méléard S, Metz JAJ. 2013. A rigorous model study of the adaptative dynamics of Mendelian diploids. J. Math. Biol. 67, 569–607. ( 10.1007/s00285-012-0562-5) [DOI] [PubMed] [Google Scholar]
- 7.Metz JAJ, Nisbet RM, Geritz SAH. 1992. How should we define ‘fitness’ for general ecological scenarios? TREE 7, 198–202. ( 10.1016/0169-5347(92)90073-K) [DOI] [PubMed] [Google Scholar]
- 8.Metz JAJ. 2008. Fitness. In Encyclopedia of ecology. Vol. 2. Evolutionary ecology (eds Jørgensen SE, Fath BD.), pp. 1599–1612. Oxford, UK: Elsevier. [Google Scholar]
- 9.Diekmann O, Heesterbeek JAP, Metz JAJ. 1990. On the definition and the computation of the basic reproduction ratio R0 in models for infectious diseases in heterogeneous populations. J. Math. Biol. 28, 365–382. ( 10.1007/BF00178324) [DOI] [PubMed] [Google Scholar]
- 10.Haldane JBS. 1927. A mathematical theory of natural and artificial selection. V. Selection and mutation. Proc. Camb. Philos. Soc. 23, 838–844. ( 10.1017/S0305004100015644) [DOI] [Google Scholar]
- 11.Ewens W. 1969. Population genetics. London, UK: Methuen. [Google Scholar]
- 12.Eshel I. 1981. On the survival probability of a slightly advantageous mutant gene with a general distribution of progeny size—a branching process model. J. Math. Biol. 12, 355–362. ( 10.1007/BF00276922) [DOI] [PubMed] [Google Scholar]
- 13.Hoppe F. 1992. Asymptotic rates of growth of the extinction probability of a mutant gene. J. Math. Biol. 30, 547–566. ( 10.1007/BF00948890) [DOI] [PubMed] [Google Scholar]
- 14.Athreya K. 1992. Rates of decay for the survival probability of a mutant gene. J. Math. Biol. 30, 577–581. ( 10.1007/BF00948892) [DOI] [PubMed] [Google Scholar]
- 15.Eshel I. 1984. On the survival probability of a slightly advantageous mutant gene in a multitype population: a multidimensional branching process model. J. Math. Biol. 19, 201–209. ( 10.1007/BF00277746) [DOI] [PubMed] [Google Scholar]
- 16.Hoppe F. 1992. The survival probability of a mutant in a multidimensional population. J. Math. Biol. 30, 567–575. ( 10.1007/BF00948891) [DOI] [PubMed] [Google Scholar]
- 17.Pollak E. 1992. Survival probabilities for some multitype branching processes in genetics. J. Math. Biol. 30, 583–596. ( 10.1007/BF00948893) [DOI] [PubMed] [Google Scholar]
- 18.Athreya K. 1993. Rates of decay for the survival probability of a mutant gene. II. The multitype case. J. Math. Biol. 32, 45–53. ( 10.1007/BF00160373) [DOI] [PubMed] [Google Scholar]
- 19.Lotka AJ. 1939. Théorie analytique des associations biologiques. II. Analyse démographique avec application particulière à l'espèce humaine. Actualités Scientifiques et Industrielles 740. Paris, France: Hermann & Cie. [Google Scholar]
- 20.Metz JAJ, Diekmann O. (eds). 1986. The dynamics of physiologically structured populations. Lecture Notes in Biomathematics, 68 Berlin, Germany: Springer. [Google Scholar]
- 21.Geritz SAH, Gyllenberg M, Jacobs FJA, Parvinen K. 2002. Invasion dynamics and attractor inheritance. J. Math. Biol. 44, 548–560. ( 10.1007/s002850100136) [DOI] [PubMed] [Google Scholar]
- 22.Dercole F, Rinaldi S. 2008. Analysis of evolutionary processes: the adaptive dynamics approach and its applications. Princeton, NJ: Princeton University Press. [Google Scholar]
- 23.Geritz SAH. 2005. Resident-invader dynamics and the coexistence of similar strategies. J. Math. Biol. 50, 67–82. ( 10.1007/s00285-004-0280-8) [DOI] [PubMed] [Google Scholar]
- 24.Greiner G, Heesterbeek JAP, Metz JAJ. 1994. A singular perturbation theorem for evolution equations and time-scale arguments for structured population models. Can. Appl. Math. Quart. 2, 435–459. [Google Scholar]
- 25.Diekmann O, Getto P, Gyllenberg M. 2007. Stability and bifurcation analysis of Volterra functional equations in the light of suns and stars. SIAM J. Math. Anal. 39, 1023–1069. ( 10.1137/060659211) [DOI] [Google Scholar]
- 26.Diekmann O, Gyllenberg M. 2012. Equations with infinite delay: blending the abstract and the concrete. J. Diff. Equ. 252, 819–851. ( 10.1016/j.jde.2011.09.038) [DOI] [Google Scholar]
- 27.Carroll SB. 2005. Endless forms most beautiful: the new science of Evo Devo. New York, NY: W. W. Norton & Cy. [Google Scholar]
- 28.Davidson EH. 2006. The regulatory genome: gene regulatory networks in development and evolution. Amsterdam, The Netherlands: Academic Press. [Google Scholar]
- 29.Van Dooren TJM. 2006. Protected polymorphism and evolutionary stability in pleiotropic models with trait-specific dominance. Evolution 60, 1991–2003. ( 10.1554/05-259.1) [DOI] [PubMed] [Google Scholar]
- 30.Metz JAJ, Jansen VAA. In preparation Adaptive dynamics for Mendelian genetics: relating the speed of evolution to the effective population size. [Google Scholar]
- 31.Diekmann O, Gyllenberg M, Metz JAJ. 2003. Steady state analysis of structured population models. Theor. Popul. Biol. 63, 309–338. ( 10.1016/S0040-5809(02)00058-8) [DOI] [PubMed] [Google Scholar]
- 32.Jagers P. 1975. Branching processes with biological applications. London, UK: Wiley. [Google Scholar]
- 33.Haccou P, Jagers P, Vatutin VA. 2005. Branching processes: variation, growth, and extinction of populations. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 34.Ripa J, Dieckmann U. 2013. Mutant invasions and adaptive dynamics in variable environments. Evolution 67, 1279–1290. ( 10.1111/evo.12046) [DOI] [PubMed] [Google Scholar]
- 35.Metz JAJ, Stanková K, Johansson J. In preparation. The adaptive dynamics of life-histories: from fitness-returns to selection gradients and Pontryagin's maximum principle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Metz JAJ. 2013. On the concept of individual in ecology and evolution. J. Math. Biol. 66, 635–647. ( 10.1007/s00285-012-0610-1) [DOI] [PubMed] [Google Scholar]
- 37.Otto SP, Jarne P. 2001. Evolution: haploids hapless or happening? Science 292, 2441–2443. ( 10.1126/science.1062890) [DOI] [PubMed] [Google Scholar]
- 38.Taylor PD, Bulmer MG. 1980. Local mate competition and the sex ratio. J. Theor. Biol. 86, 409–419. ( 10.1016/0022-5193(80)90342-2) [DOI] [PubMed] [Google Scholar]
- 39.Courteau J, Lessard S. 2000. Optimal sex ratios in structured populations. J. Theor. Biol. 207, 159–175. ( 10.1006/jtbi.2000.2160) [DOI] [PubMed] [Google Scholar]
- 40.Metz JAJ, Leimar O. 2011. A simple fitness proxy for ESS calculations in structured populations with continuous traits, with applications to the evolution of haplo-diploids and genetic dimorphisms. J. Biol. Dyn. 5, 163–190. ( 10.1080/17513758.2010.502256) [DOI] [PubMed] [Google Scholar]
- 41.Meszéna G, Gyllenberg M, Jacobs FJ, Metz JAJ. 2005. Link between population dynamics and dynamics of Darwinian evolution. Phys. Rev. Lett. 95, 078105 ( 10.1103/PhysRevLett.95.078105) [DOI] [PubMed] [Google Scholar]
- 42.Lande R. 1979. Quantitative genetic analysis of multivariate evolution, applied to brain:body size allometry. Evolution 33, 402–416. ( 10.2307/2407630) [DOI] [PubMed] [Google Scholar]
- 43.Lande R. 1982. A quantitative genetic theory of life history evolution. Ecology 63, 607–615. ( 10.2307/1936778) [DOI] [Google Scholar]
- 44.Bulmer MG. 1980. The mathematical theory of quantitative genetics. Oxford, UK: Clarendon Press. [Google Scholar]
- 45.Lande R. 1976. Natural selection and random genetic drift in phenotypic evolution. Evolution 30, 314–334. ( 10.2307/2407703) [DOI] [PubMed] [Google Scholar]
- 46.Lande R. 1980. The genetic covariance between characters maintained by pleiotropic mutations. Genetics 94, 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fisher RA. 1958. The genetical theory of natural selection, 2nd edn. New York, NY: Dover. [Google Scholar]
- 48.Leimar O. 2009. Multidimensional convergence stability. Evol. Ecol. Res. 11, 191–208. [Google Scholar]
- 49.Metz JAJ, Geritz SAH, Meszéna G, Jacobs FJA, van Heerwaarden JS. 1996. Adaptive dynamics, a geometrical study of the consequences of nearly faithful reproduction. In Stochastic and spatial structures of dynamical systems (eds van Strien SJ, Verduyn Lunel SM.), pp. 183–231. Amsterdam, The Netherlands: North-Holland. [Google Scholar]
- 50.Geritz SAH, Metz JAJ, Kisdi É, Meszéna G. 1997. Dynamics of adaptation and evolutionary branching. Phys. Rev. Lett. 78, 2024–2027. ( 10.1103/PhysRevLett.78.2024) [DOI] [Google Scholar]
- 51.Geritz SAH, Kisdi É, Meszéna G, Metz JAJ. 1998. Evolutionarily singular strategies and the adaptive growth and branching of the evolutionary tree. Evol. Ecol. 12, 35–57. ( 10.1023/A:1006554906681) [DOI] [Google Scholar]
- 52.Van Dooren TJM, Durinx M, Demon I. 2004. Sexual dimorphism or evolutionary branching? Evol. Ecol. Res. 6, 857–871. [Google Scholar]
- 53.Durinx M, van Dooren TJM. 2009. Assortative mate choice and dominance modification: alternative ways of removing heterozygote disadvantage. Evolution 63, 334–352. ( 10.1111/j.1558-5646.2008.0578.x) [DOI] [PubMed] [Google Scholar]
- 54.Geritz SAH, Metz JAJ. In preparation. Evolutionary branching of multi-dimensional strategies. [Google Scholar]
- 55.Caswell H. 2001. Matrix population models, 2nd edn Sunderland, MA: Sinauer. [Google Scholar]