

Abstract
Allopatric speciation is a mechanism to evolve reproductive isolation; it is caused by the accumulation of genetic differences between populations while they are geographically isolated. Here, we studied a simple stochastic model for the time until speciation caused by geographical isolation in fragmented populations that experience recurrent but infrequent migration between subpopulations. We assumed that mating incompatibility is controlled by a number of loci that behave as neutral characters in the accumulation of novel mutations within each population. Genetic distance between populations was defined as the number of incompatibility-controlling loci that differ between them. Genetic distance increases through the separate accumulation of mutations in different populations, but decreases after a successful migration event followed by genetic mixing between migrants and residents. We calculated the time to allopatric speciation, which occurs when the genetic distance exceeds a specified threshold. If the number of invasive individuals relative to the resident population is not very large, diffusion approximation provides an accurate prediction. There is an intermediate optimal rate of migration that maximizes the rate of species creation by recurrent invasion and diversification. We also examined cases that involved more than two populations.
Keywords: allopatric speciation, waiting time, genetic drift, neutral loci, rare migration
1. Introduction
Species diversity is determined by the balance between speciation and extinction. The mechanism of speciation has been a central topic of evolutionary sciences. However, questions that are rarely asked include how the speed at which novel species are created depends on the life history of the organism, geographical structure, habitat stability, mating system, species interactions, etc.
One of the simplest mechanisms that can lead to the creation of novel species is allopatric speciation [1,2]. If two or more populations of the same species are isolated geographically, they will accumulate different mutations and become quite different from each other after many generations. When individuals from these populations encounter each other, they may no longer be able to perform proper sexual reproduction if they have become too different. Then, the two populations can be considered different species. Allopatric speciation is regarded as a major process for species creation [3,4].
However, very few theoretical studies have examined the process of allopatric speciation because it tends to be regarded as ‘theoretically trivial’ [5,6]. As a consequence, almost all of the theoretical studies of speciation conducted within the last few decades have focused on sympatric speciation in which species diverge spontaneously as a result of adaptation to different niches, habitats or mate choice [7–15].
Nei et al. [16] studied a simple population genetics model for allopatric speciation. They assumed that the possibility of sexual reproduction for a pair of individuals was controlled by one or two loci. For example, suppose that there are three alleles (labelled 1, 2 and 3) in a single compatibility-controlling locus. All homozygotes (A1A1, A2A2 and A3A3) are viable and have the same fitness. Heterozygotes A1A2 and A2A3 are also viable. But heterozygotes A1A3 are not viable (they have zero fitness). Initially, the two populations are both composed of A2A2 only. One population might experience mutation to A1, and ultimately be replaced by A1A1, which takes place through neutral evolution because their heterozygotes A1A2 have the same fitness as the homozygotes. Similarly, the other population might be replaced by A3A3 by neutral evolution. After these changes, the two populations are no longer compatible with each other because any breeding between individuals from different populations ends in reproductive failure. Nei et al. [16] also studied a two-locus version of this model (cf. [17]). A simple two-locus model for incompatibility was proposed earlier by Dobzhansky [18] and Muller [19], where the original genotype is aabb and is replaced by AAbb in one population, which is possible because Aabb is viable. In the second population, the original population is dominated by aabb and is replaced by aaBB, where the evolution is also neutral as aaBb is viable. However, the two populations do not cross each other because offspring between AAbb and aaBB are AaBb, which is non-viable. Many theoretical works on allopatric speciation have studied various extensions of these simple models (cf. [20–23]).
Orr & Orr [24] studied the effect of population size on the waiting time until two populations would be considered different species, assuming the Dobzhansky model. They concluded that the waiting time to speciation caused by genetic drift was independent of the sizes of the populations. Gavrilets & Hastings [25] concluded that the founder effect promotes speciation, based on the Dobzhansky model. They measured the degree of reproductive isolation in terms of the proportion of non-viable hybrids when randomly chosen pairs from the two populations are crossed.
In these studies, it was assumed that no migration occurred between the populations when calculating the time to speciation. Gavrilets [26] extended the model to incorporate recurrent migration between two populations. He examined several models, including selection to the local environment for each population. If we focus on the case without selection, Gavrilets [26] assumed that the incompatibility was controlled by multiple loci. As a population is monomorphic with respect to these loci, the state of a population is characterized by the ‘distance’ i between two populations, which indicates the number of incompatibility-controlling loci that differ between them. When K+1 loci are different (K is a ‘threshold’ specifying the genetic architecture of reproductive isolation), the two populations become different species. Gavrilets calculated the waiting time assuming a birth-and-death process in which the distance i could increase by increments of one at a rate that was equal to the mutation rate. He also modelled the effect of migration between two populations as a decrease in distance i at a rate that was proportional to the product of the current distance i and the migration rate.
The time to speciation should increase as the migration between populations becomes larger. In the complete absence of migration, two populations will become different species in the shortest period of time. However, no additional species will be created by this mechanism. If instead there is infrequent but recurrent migration between islands, the populations on the islands will become different species during the intervals between migration events, and the next migration (colonization) by one of the species will cause both islands to contain the same species in addition to another species. This may be followed by genetic differentiation. Repeated events of migration and genetic differentiation that occur on different islands will increase the number of novel species recurrently. Endler [27] reviewed empirical studies and concluded that the most general mode of speciation is parapatric speciation, in which different populations are isolated but not perfectly. Avise [28] also concluded that the geographical structure of most species is consistent with a meta-population, being composed of many local populations that are connected by infrequent migration (see also [29] for mathematical modelling of speciation and diversification in a meta-population).
In the work reported in this paper, we examined a simple stochastic model that determined the time until speciation as a result of geographical isolation in fragmented populations that experience recurrent but infrequent migration among them. We assumed that mating incompatibility was controlled by a number of loci that behave as neutral characters in the accumulation of novel mutations within each population [30]. Genetic distance between populations was defined as the number of incompatibility-controlling loci that differ between two populations. Genetic distance increases through the accumulation of mutations, which occurs separately in different populations, but is reduced after a successful migration event followed by genetic mixing of migrants and residents (cf. [31]). We calculated the time to allopatric speciation, which occurred when the genetic distance exceeded a threshold [32]. If the number of immigrating individuals relative to the resident population is not very large, diffusion approximation provides an accurate prediction. There is an intermediate optimal rate of migration that maximizes the rate of species creation by recurrent invasion and diversification. We also examined cases that included more than two populations.
2. Dynamics of genetic distance between populations
Consider two populations of a sexual haploid species with non-overlapping generations. The populations live on two islands or island-like habitats. For simplicity, we assume that the two populations are of the same size, N. Successful migration of individuals between populations occurs very infrequently. Most migration attempts result in failure. However, when an attempt is successful, a certain number of individuals (instead of a single individual) from one population arrive in the other population.
We assume that the possibility of successful mating between immigrants and residents is controlled by differences in a set of l loci that control incompatibility. To be specific, the possibility of interbreeding between two individuals is determined by the number of loci that differ between them, denoted by x. Two individuals cannot reproduce sexually if x exceeds a threshold value xc. By contrast, they can mate and produce offspring if x is less than xc.
We assume that the size of each population N is clearly smaller than the inverse of the mutation rate u for these incompatibility-controlling loci (weak mutation limit). Under this assumption, each population is monomorphic most of the time with respect to these loci, except for relatively short periods of replacement following a successful migration event (e.g. [33–42]; see [43] for review). Then, we can define the genetic distance between the two populations in terms of the number of incompatibility-controlling loci that differ between them. If a population is polymorphic with respect to these loci, then we can use the average genetic distance between the two populations [44].
Between successive migration events, the genetic differences between the two populations increase owing to the accumulation of different mutations in different populations. Hence, their genetic distance, or the degree of genetic differentiation, increases with time. With population size N, the mutation rate per loci u and the neutrality of the mutations in the process of their accumulation, the rate of accumulation of novel mutations is k = ul. The time required for fixation is of the order of N generations [45], which is much shorter than the time scale on which the speciation process is occurring. As each population would accumulate novel mutations independently, the rate of divergence is 2k.
Note that the average genetic distance increases only if the replacement of an allele occurs at a locus for which the two populations have the same allele. If replacement occurs in one population at a locus for which the two populations have different fixed alleles, the change does not increase their genetic distance. Hence, the rate of increase in genetic distance owing to replacement by novel mutations should slow down as the distance between two populations increases. Considering this effect, the mean rate of divergence per generation is given by the following equation:
| 2.1 |
between successive migration events.
The timing of successful migrations from one population to the other follows a Poisson point process at a rate m per generation. Here m is very small because the interval between successful invasions is very long.
From the assumption of incompatibility, if immigrants and residents have different alleles at x loci, and if x is greater than the threshold xc, then the immigrants and residents do not mix sexually and they should be treated as different species.
By contrast, if x is smaller than the threshold xc, then individuals from the two populations can freely exchange their genomes through sexual reproduction. According to the stochastic theory of population genetics for neutral loci, a population eventually becomes monomorphic where one allele dominates and the other goes extinct. The probability of fixation for two alleles is proportional to their initial frequencies [46,47]. If the population size of the immigrants is N’ and the population size of the residents is N, then immediately after the invasion, there are x loci that are polymorphic, where immigrants and residents have different alleles. After some number of generations, which is of the order of N [45], the population again becomes monomorphic, and the expectation of loci that show the immigrant allele is equal to the fraction of immigrants 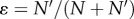 . We here assume that the loci are unlinked for simplicity. If the loci behave independently, the number of loci that still differ after the fixation process follows a binomial distribution
. We here assume that the loci are unlinked for simplicity. If the loci behave independently, the number of loci that still differ after the fixation process follows a binomial distribution  This quantity is equal to the decrease in the distance: xbefore − xafter. Considering the situation in which the number of loci controlling the incompatibility is very large, the distance after mixing is well approximated by the mean value. Hence, we have the genetic distance after one successful migration event as
This quantity is equal to the decrease in the distance: xbefore − xafter. Considering the situation in which the number of loci controlling the incompatibility is very large, the distance after mixing is well approximated by the mean value. Hence, we have the genetic distance after one successful migration event as
| 2.2 |
Normally, 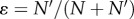 is much smaller than 1.
is much smaller than 1.
The model that we explored in this paper was the stochastic dynamics of genetic distance x, which was given by equations (2.1) and (2.2). We focus on the situation in which the number of loci controlling the incompatibility is large (l and xc are large). In addition, concerning the magnitude of rate parameters, we assume: 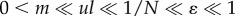 . Successful migrations are less frequent than mutation accumulation. We here focus on the stochasticity caused by the random timing of successful migration events, while we treat other processes as deterministic: namely, the increase of the genetic distance between populations, equation (2.1), and the reduction of the distance after successful migration events, equation (2.2). We name this the ‘population-based model’. In the following, we study several different approximations of the population-based model. We also ran direct computer simulations of an individual-based model of population genetic dynamics, which is described in appendix A. As will be explained later, most of the conclusions of the simplified stochastic dynamics of genetic distance model and the individual-based model were very similar, although the latter model required considerably more computational time to run.
. Successful migrations are less frequent than mutation accumulation. We here focus on the stochasticity caused by the random timing of successful migration events, while we treat other processes as deterministic: namely, the increase of the genetic distance between populations, equation (2.1), and the reduction of the distance after successful migration events, equation (2.2). We name this the ‘population-based model’. In the following, we study several different approximations of the population-based model. We also ran direct computer simulations of an individual-based model of population genetic dynamics, which is described in appendix A. As will be explained later, most of the conclusions of the simplified stochastic dynamics of genetic distance model and the individual-based model were very similar, although the latter model required considerably more computational time to run.
3. Evolutionary trajectories
Figure 1 provides an example of changes in the genetic distance between two populations with time. Among several curves in figure 1, the black curve, labelled population-based model, was generated by direct computer simulations of the stochastic model. Grey curves and the broken line are for the individual-based model and the deterministic model, which is explained later. The black curve indicates that genetic distance increases with time and the rate of increase slows down and eventually converges to a final level (cf. [48–51]) around which it continues to fluctuate. This fluctuation is caused by the stochasticity of the timing of successful migration events.
Figure 1.

Dynamics in the genetic distance (average genetic distance) between two populations in each model. The horizontal axis is time expressed in terms of the number of generations. The dashed line corresponds to the deterministic model. Regarding the two lines with oscillations along the dashed line (upper part of the figure), the black jagged line corresponds to the population-based simulation and the grey jagged line represents the individual-based simulation. The other two lines near the horizontal axis (black and grey) correspond to genetic distances within a subpopulation in the individual-based simulation. Two different threshold levels are indicated by the horizontal lines. Open circles indicate the time to speciation. Parameters are N = 100, k = 0.01, μ = 0.0001, l = 100, m = 0.01 and ɛ = 0.01.
Two different threshold levels are indicated by horizontal lines. Open circles show the time at which genetic distance first reaches the threshold level. The two populations are no longer compatible and when migrants from one population arrive in the other population they cannot mate successfully. Hence, we regard the two populations as two separate species. We can consider that speciation occurs at τ when the genetic distance x reaches the threshold xc, which is indicated by open circles in figure 1. Here, we focus on τ, the time of this first passage to speciation.
After genetic distance exceeds the threshold, migrants from one population can immigrate to the other population and they may quickly evolve mating isolation mechanisms to avoid reproductive interference [52–54].
If the threshold to speciation xc is below the equilibrium, the first passage time to speciation is rather short. If the threshold xc is above the equilibrium level, stochastic fluctuations above equilibrium in the genetic difference allow the threshold to be reached in finite time.
In the following section, we analyse the stochastic model of genetic distance by introducing approximations and compare the results with direct computer simulations and an individual-based model of the corresponding population genetic process.
3.1. Deterministic approximation
For the two islands population-based model, under approximations that the migration rate m is large and the impact of migration ɛ is small, we can approximate the decrease in genetic distance as a deterministic process. Then, the ordinary differential equation for genetic distance is given by
| 3.1 |
where t is time measured in terms of generations. Here, we neglect terms of the order of m2 because m is assumed to be small.
In figure 1, the dashed curve indicates the change in genetic distance predicted by the deterministic model. It increases smoothly with time and converges to a final level. Given the initial condition x(0) = 0, the solution of equation (3.1) is
| 3.2 |
At equilibrium, the mean genetic distance 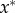 is
is
| 3.3 |
which increases with the rate of mutation accumulation k but decreases with the migration rate m and the impact of migration events ε. The time until speciation is
| 3.4a |
and
| 3.4b |
Equation (3.4b) implies that speciation never occurs if the threshold genetic distance is greater than the saturation level in the deterministic model.
3.2. Diffusion approximation
Note that the stochastic model for genetic distance is a Markov process, which implies that once the value of x at time t is known, the behaviour of the model after t becomes independent of any additional information from the past trajectory. If, in addition, the trajectory is continuous with a probability of one, the model is equivalent to a diffusion process [55,56]. In the present model, there is a discontinuous decline in x after every successful migration event, as is shown by equation (2.2). Hence, the diffusion approximation is accurate if the magnitude of each drop is small—that is the impact of invasion ɛ is not large.
The change that occurs in a short time interval of length Δt is given as follows:
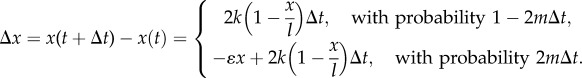 |
3.5 |
Then, the mean rate of increase in x, 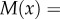
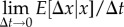 , and the rate of variance generation,
, and the rate of variance generation, 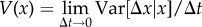 , are given by
, are given by
| 3.6a |
and
| 3.6b |
The probability density function of genetic distance is defined by
| 3.7a |
which satisfies the following forward equation (or Fokker–Planck equation):
| 3.7b |
The stationary distribution 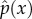 at equilibrium can be derived from
at equilibrium can be derived from 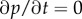 , which is
, which is
| 3.8 |
where C is a normalization constant that makes 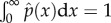 hold. Here a is a constant given by
hold. Here a is a constant given by 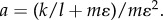
Figure 2 shows the distribution predicted by equation (3.8) and the histogram of genetic distance from the direct computer simulation using a population-based model. The two distributions have the same mean genetic distance but differ in variance. If the impact of migration event ɛ is not very large (say less than 0.1), the diffusion approximation is quite accurate. However, when ɛ is larger, the distribution from the diffusion approximation becomes deformed and has a fat tail to the right.
Figure 2.
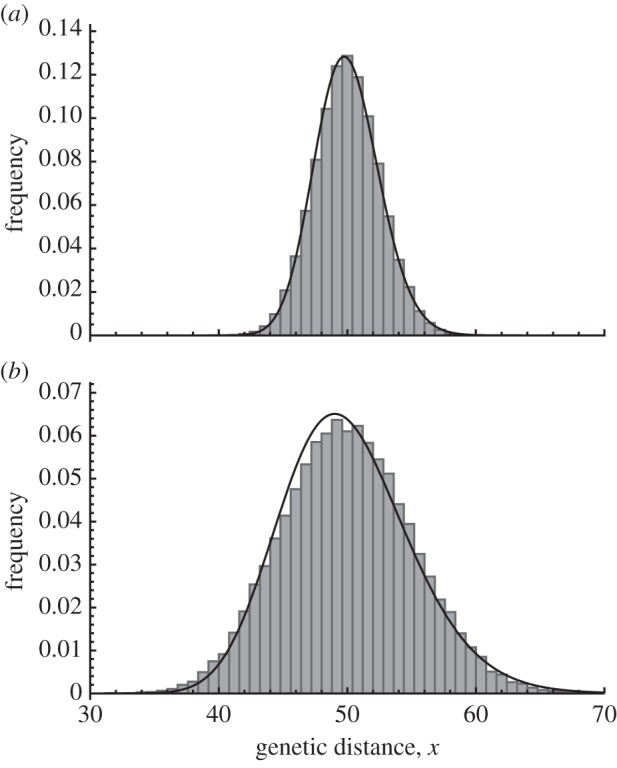
Stationary distribution of genetic distance with two parameter sets. The horizontal axes are genetic distance and the vertical axes are its frequency at equilibrium. The histograms show the results of simulations with the population-based model, with 50 000 samples of genetic distance per simulation. The solid black lines show solutions from the diffusion approximation to the model. Parameters are (a) k = 0.01, l = 100, m = 0.01 and ɛ = 0.01 and (b) k = 0.01, l = 100, m = 0.0025 and ɛ = 0.04.
4. Average time to speciation
Speciation between two subpopulations occurs when the genetic distance between them reaches the threshold xc. If the genetic distance x is smaller than the threshold, immigrants can mate with residents without any disadvantage. On the other hand, if genetic distance x exceeds the threshold, individuals from different subpopulations cannot mate. The speciation threshold xc may differ between species; moreover, here we assumed that all of the neutral loci contributed equally to reproductive isolation. The initial value of genetic distance is x0 and the probability that the genetic distance from x0 reaches the threshold xc within time t is given by 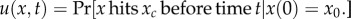 . The initial and boundary conditions are
. The initial and boundary conditions are 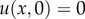
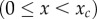 and
and 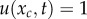 (t > 0), respectively. These conditions satisfy the backward equation
(t > 0), respectively. These conditions satisfy the backward equation
| 4.1 |
Solving the above equation (see appendix B for the derivation), we finally obtain the average time to speciation T, as follows:
| 4.2a |
where
| 4.2b |
and 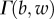 is an upper incomplete gamma function
is an upper incomplete gamma function 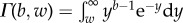 .
.
The deterministic approximation predicts that speciation will never occur if the equilibrium value x* given by equation (3.3) is smaller than the threshold xc. In reality, owing to stochastic fluctuations, the genetic distance becomes higher than x* by chance and might reach xc within a finite period of time. The diffusion approximation can take this into consideration. It predicts that the mean time to speciation is finite even when xc > x*.
Figure 3 shows the average time to speciation T from the initial genetic distance under four different thresholds. The four groups of curves represent the different threshold values xc. Within each group, curves show the predictions from the diffusion approximation and from the deterministic approximation. Solid symbols indicate the results from direct computer simulations using a population-based model, where different symbols indicate the results for different threshold levels. When the speciation threshold was below the equilibrium level (xc < x*), all of the values were similar, which suggests that the deterministic model was simple but provided relatively accurate predictions. However, when the speciation threshold was above or equal to the equilibrium level (xc ≥ x*), the deterministic model predicted that speciation was impossible, as is shown by equation (3.4a,b), but the direct computer simulation (filled symbols) provided a threshold at a finite time, which was not very different from the prediction using the diffusion approximation (solid curve).
Figure 3.
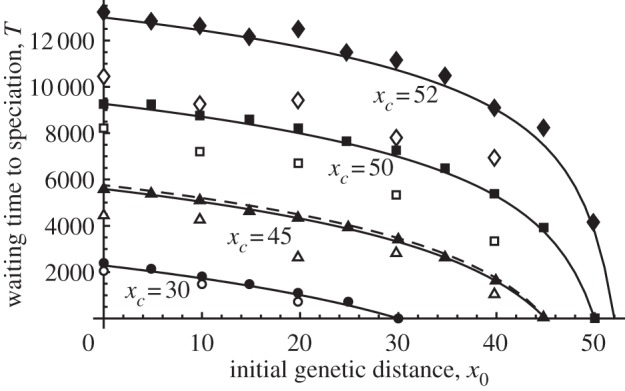
Average waiting time to speciation, expressed in terms of the number of generations, from an initial genetic distance x0 to four speciation thresholds, xc = 30, 45, 50 and 52. The horizontal axis is the initial genetic distance between the two populations. A filled symbol shows the average waiting time to speciation calculated from 200 population-based simulations and an open symbol shows the average time calculated from 50 individual-based simulations. Different symbols distinguish results for different threshold levels: diamonds for xc = 52, squares for xc = 50, triangles for xc = 45 and circles for xc = 30. Solid lines indicate solutions from the diffusion approximation and the dashed line shows the time calculated from the deterministic model. At xc = 30, the solid line and the dashed line overlap completely. Parameters are N = 100, k = 0.01, l = 100, m = 0.01 and ɛ = 0.01. This parameter set is the same as in figure 1 and we can easily suppose the threshold that is shown there.
In contrast to these results, the individual-based model produced a shorter estimated time to speciation in all cases, as is indicated by the open symbols that are located below the others. Intuitively, the individual-based model shows greater stochastic fluctuations in genetic distance, which reduces the time to reach the threshold relative to the population-based model that showed weaker stochasticity.
Figure 4 shows the average time to speciation, where the horizontal axis is the threshold level for speciation xc. The average waiting time increased with the threshold level, but the results of the computer simulation of the population-based model were close to the predictions of the diffusion approximation. The deterministic model was able to predict divergence time at equilibrium.
Figure 4.
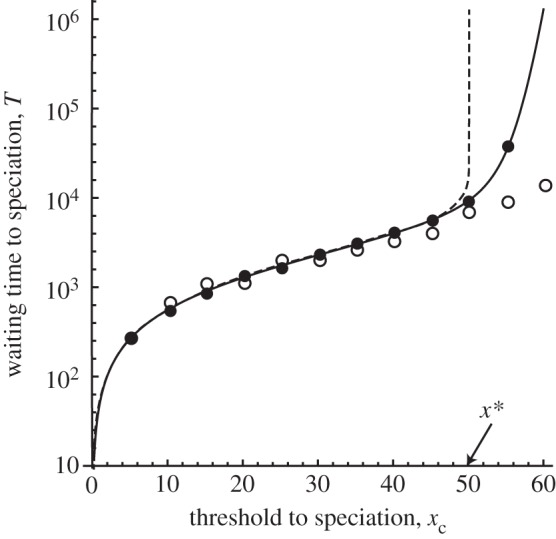
Average waiting time to speciation from an initial genetic distance of 0. The horizontal axis is the threshold to speciation xc. The dashed line corresponds to the deterministic model and the solid line corresponds to the diffusion approximation. A filled circle shows the average waiting time from 200 simulations with the population-based model and an open circle shows the average time from 50 individual-based simulations. Parameters are N = 100, k = 0.01, l = 100, m = 0.01 and ɛ = 0.01.
5. Migration between two populations affects their genetic distances with other populations
Migration between two populations can affect the genetic distance between one of them and another population. This effect is quite important when considering the effect of migration when there are more than two populations.
Suppose that there are three islands (A, B and C), all of which are connected with each other equally. There exist six directions of migration and three genetic distances xij provided that 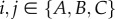 ,
, 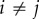 and xij = xji. For this model, the reduction in genetic distance because of migration from island A to B is described by the two following formulae:
and xij = xji. For this model, the reduction in genetic distance because of migration from island A to B is described by the two following formulae:
| 5.1a |
and
| 5.1b |
Equation (5.1a) is the same as equation (2.2), which means that there is a direct effect of migration and it invariably reduces genetic distance. However, equation (5.1b) is peculiar to situations when there are three or more islands. This equation represents indirect effects on genetic distance that are connected with the migrating population; the effect is caused by changes in the genetic composition of the migrating population. In this case, after migration, the population on the island B now shares a fraction (ɛ) of alleles with the residents of A on average. Hence, the genetic distance caused by this event is calculated by comparing the relationship between B and C. This effect sometimes causes a reduction in genetic distance and at other times it causes an increase. This rule can be applied to situations with different numbers of islands and different structures among them because we can decompose all of the island structures into closed relationships among two or three (a triangle) islands.
5.1. Reduction in variance in genetic distance and increased waiting time
Here, we consider a situation in which there are more than two populations that are connected to each other by migration events. It is clear that the average waiting time is strongly dependent on the value of genetic distance and the threshold for speciation. Therefore, using a population-based model we simulated dynamics in genetic distance for different numbers and structures of subpopulations. We considered Wright's island model [57] in which there are n populations, each of which is connected by migration with all of the other populations. The graph of connections among the populations forms a ‘complete graph’ with equal migration rates and migration impacts. Figure 5a shows the distribution of genetic distance at equilibrium with the number of islands by sampling a random pair of populations and then calculating their genetic distances. As the number of islands increases, the average genetic distance between islands remains unchanged, but the variance between pairs of islands decreases. This result might be caused by cancellation between direct and indirect effects on genetic distance, which intensifies when the number of islands increases.
Figure 5.

The horizontal axes are the number of islands. The following data come from population-based simulations with parameters k = 0.01, l = 100, m = 0.01 and ɛ = 0.01. The bold horizontal line in each box is the median and the top and bottom of the box are one quartile above and below the median, respectively. The whiskers extend to the most extreme data within the simulation range. (a) Box-plots showing genetic distance between two populations at equilibrium (100 000 samples in each). (b) Waiting time until the first genetic distance reaches the threshold xc = 55 from an initial genetic distance of x0 = 0. This corresponds to the situation in which (a) has a threshold of xc = 55. Each distribution was created from 200 samples. Average waiting times to first speciation, indicated by filled circles, from 100 samples when the threshold was (c) xc = 49 and (d) xc = 51. Box-plots show distributions of waiting time, as in (b).
The variance in a stationary distribution of genetic distance is important when considering the average waiting time to speciation, when the threshold is higher than the equilibrium value. In such situations, we can intuitively expect a large variance to lead to a high potential for speciation. This pattern was shown in the distribution of average waiting times to first speciation with an increasing number of islands under the assumption that all of the islands had the same genetic distance at equilibrium (figure 5). Later we will discuss the relationship between waiting time to speciation and the number of islands in more detail. A stationary distribution of genetic distance that has low variance requires a longer period of time to undergo speciation when the threshold is larger than the equilibrium value. Note that the distribution of waiting times has a heavy tail towards longer times because some populations seem simply unable to reach the threshold through chance migration events. This pattern is not observed when the threshold is lower than the equilibrium value.
6. Discussion
In this paper, we present a simple model of allopatric speciation in which the possibility of sexual reproduction is controlled by the number of loci. Suppose that two individuals have different alleles at x loci that control incompatibility. They cannot engage in successful sexual reproduction if x exceeds a threshold value xc, but they mate and breed without problems if x is smaller than xc. Assuming that incompatibility-controlling loci are monomorphic within populations, we can define the genetic distance between populations. Genetic distance increases with time through the accumulation of mutations, but decreases when migration from one population to the other occurs. When successful migration events occur very infrequently but many individuals immigrate during each successful migration event, the stochasticity of the migration events causes genetic distance to fluctuate and the time until speciation can be obtained from stochastic process calculations.
6.1. Dynamics of genetic distance
The distance between two populations tends to increase rapidly when the distance is short, but the rate of increase tends to slow and eventually saturate. There are two reasons for this saturation. First, there is a maximum number of loci l and the speed of the increase in genetic distance between two populations slows down as the number of loci with alleles that are common to both populations becomes smaller. Second, successful migrations should cause a drop in genetic distance with a magnitude that is proportional to the genetic distance immediately before the migration event. The process of saturation can even be represented by the simplest deterministic model, equation (3.2). The equilibrium level x* is given by equation (3.3).
If the threshold for speciation is below the equilibrium, speciation occurs rather quickly and the waiting time can be accurately predicted by the deterministic model, equation (3.4) (figure 3). By contrast, if the threshold for speciation is above the equilibrium level x*, then the time to speciation is critically dependent on the magnitude of the fluctuations. In fact, in the absence of fluctuations, speciation will not occur. By contrast, in the stochastic model, which we termed a ‘population-based model’, owing to stochasticity in the timing of migration, genetic distance fluctuates after it reaches the equilibrium and it can cross the speciation threshold after a finite period of time. The mean time to speciation in this stochastic model can be accurately calculated using a diffusion approximation, as shown in figures 3 and 4.
In the model that considered population processes, which we termed an ‘individual-based model’, in figures 1, 3 and 4, genetic distance fluctuated with a greater variance than in the corresponding population-based model. This was caused by stochasticity in the mutation accumulation process and stochasticity in the fixation fraction after migration events, which were neglected in the simple population-based model.
As figures 3 and 4 show, the individual-based model resulted in shorter times to speciation than the population-based model with corresponding parameters.
We believe that the population-based model is worthy of study by itself. First, even if the individual-based model is a true one, the corresponding population-based model can be a reasonably accurate approximation when the number of loci controlling incompatibility is sufficiently large owing to the law of large numbers. Traditionally most theoretical models of speciation, such as Dobzhansky's and Nei et al.'s models, assumed that the incompatibility is controlled by a small number of loci (such as one or two); in this paper, we study the opposite case with a large number of loci controlling incompatibility. Second, a useful approach of analysing a given model's property is to first study a simplified version, and then to compare the original complex model with the simplified model. In a similar manner, we do believe that it is worth studying the population-based model and its property carefully, and then study the individual-based model which includes additional stochasticity by comparing their difference and similarity. Third, the population-based model represents an idea that different populations while separated accumulate the difference with time at a constant rate. Accumulation of neutral mutations is one of the candidate processes causing this increase in the distance, but it is also possible that different populations may change their genetic structure by experiencing different environment, which is not covered by the individual-based model.
6.2. Three or more populations
We also studied a case in which more than two populations were connected by migration. Figure 5a shows the stationary distribution of genetic distance x. We can see that the mean distance remains unchanged as the number of populations increases. However, the variance of genetic distance becomes smaller. If we think of a pair of populations, each population is connected with many others through migration, and migration events that occur through these additional bonds would have impacts that could increase or decrease the genetic distance between the focal pair of populations. Figure 5b shows the earliest time for one of the bonds to reach the threshold xc. As there are multiple bonds, the graph actually includes n(n − 1)/2 bonds in total and when one of these many bonds reaches the threshold xc, it is counted as the event of interest. If the genetic distances for the different bonds are independent of each other and they have the same probability distribution, then the average time to the first event in any one bond that exceeds the threshold should become shorter as the number of populations increases. In fact, the average time increases over time. If we focus on any one of the bonds and estimate the time until it reaches the threshold, the estimate should be much longer as the number of populations increases. This is owing to the smaller variance in genetic distance, as indicated in figure 5a.
Figure 5c,d shows the waiting time until n islands are split into two groups between which no bond has a genetic distance that is shorter than the threshold. The waiting time clearly increases with the number of populations.
From these simulations, increases in the number of populations would make it more difficult to complete speciation. This suggests a relationship between biodiversity and the spatial structure of the habitats, which may give testable hypotheses.
6.3. Optimal migration rate for species creation
Migration events reduced the genetic distance between populations, and hence the time to speciation increased with the migration rate m. By contrast, after genetic distance reaches the threshold, we must wait for the next successful colonization event to observe the creation of a novel species. The waiting time to the next colonization event is longer when the migration rate is smaller. We may ask, ‘What is the possible rate of species creation in multiple populations between which there are recurrent migration events and where genetic divergence between two populations occurs through the accumulation of mutations in incompatibility loci?’ We suspect that there is an intermediate optimal migration rate between two islands that achieves the maximum rate of species creation.
In figure 6, the dashed line shows the mean time interval for migration events between two islands and the dashed-and-dotted line indicates the waiting time to first speciation starting from two populations that have a genetic distance of zero. The horizontal axis is the migration rate m. The bars indicate the number of species that are created between two islands after 10 000 generations have passed (see appendix C for details of the simulation). The initial condition for the two islands is that there is only one species that is common to both immediately after geographical isolation. We can clearly see that the rate of creation of novel species by this mechanism occurs most frequently around an intermediate rate of migration m. If the migration rate is too low, the waiting time until the next migration event that results in the two islands having populations of the same species is longer. By contrast, if the migration rate is too fast, genetic divergence slows down and it takes longer for the genetic distance to reach the speciation threshold. The optimal rate of migration that achieves the maximum chance of species creation was close to the rate that was predicted by the shortest sum of waiting time to migration and speciation.
Figure 6.
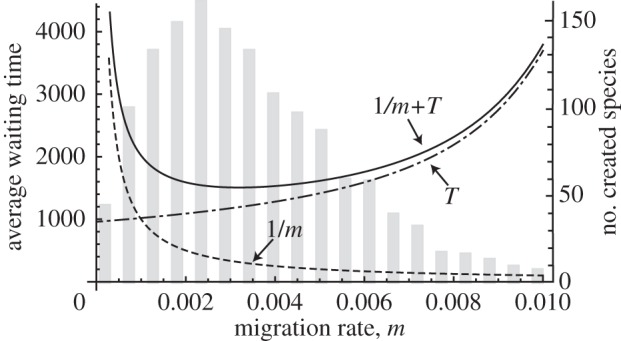
Optimal rate of migration that maximizes the rate of species creation. The horizontal axis is the migration rate. The dashed line shows the mean time interval between migration events and the dashed-and-dotted line shows the average waiting time until speciation. The solid line represents their sum. The bar chart shows the average number of species created between two populations after 10 000 generations passed during five simulations. One species existed at time 0. Parameters are k = 0.01, l = 1000, ɛ = 0.06 and xc = 19.
Incompatibility may be achieved by postzygotic mechanisms (e.g. lower viability of hybrids), by prezygotic mechanisms (e.g. mate choice) or both [58]. Most models of allopatric speciation that have calculated waiting time to speciation can be interpreted in any of these situations, although they explain the results in terms of the lower viability of hybrids [16]. Our discussion of the rate of species creation (e.g. figure 6) is valid for prezygotic mechanisms. However, if a prezygotic mechanism is not effective and a postzygotic mechanism is important, then a newly arrived population would suffer severe reproductive interference by the resident species, which could result in the extinction of the invader. Yamaguchi & Iwasa [54] analysed quick evolution in prezygotic isolation (the evolution of female mate choice to avoid mating with males from different species) and discussed the possibility of extinction caused by reproductive interference. This idea is supported by the observation that the degree of prezygotic isolation is stronger between sympatric species than between allopatric species [59,60].
In this paper, we considered a case in which incompatibility was controlled by multiple loci and could be treated as a quantitative trait. By contrast, most theoretical studies of allopatric speciation have assumed a small number of incompatibility loci [16,18,24,25], except for Gavrilets [26]. Coyne & Orr [61] reviewed studies of the number of genes that controls incompatibility (or isolation mechanism) and concluded that the number of incompatibility-controlling loci ranges from few to many.
Although most theoretical studies of allopatric speciation have assumed that no migration occurs between populations, Gavrilets [26] studied a case that involved some migration as parapatric speciation without local adaptation. He assumed that migration caused a stochastic decrease in the genetic distance between populations. As he adopted a birth-and-death process, genetic distance could decrease in only single steps. However, migrants from one population and residents in another should differ in a number of loci, and it is possible for multiple loci to fix in a population, which would result in a population with substitutions at multiple loci.
6.4. Future work
As shown in figure 1, the population-based model had a smaller variance than the individual-based model. There are two reasons for this difference. First, the mutation accumulation process was treated as a deterministic increase in genetic distance. However, the time points for replacements should occur as a Poisson point process; therefore there is variance in terms of the number of replacements that occurred within a fixed length of time. Second, after a successful migration event that is followed by genetic mixing between migrants and residents, there is stochasticity concerning which of the two alleles will become fixed. If different segregating loci evolve completely independently, the number of different loci after migration xafter is a stochastic variable that follows a binomial distribution with mean (1 − ɛ)xbefore and variance (1 − ɛ)ɛxbefore. The maximum recombination rate per generation is one-half, and there might be some correlation between different loci concerning which one will eventually be fixed. This could further increase the variance after migration events. This represents an interesting future theoretical problem.
Funding statement
This work was performed under the support of the Environment Research and Technology Development Fund (S9) of the Ministry of the Environment, Japan; and also supported by a Grant-in-Aid for Scientific Research to Y.I.
Appendix A
Consider two populations, each of which is composed of N individuals of a species that is sexual and haploid. Each individual has l loci that are not linked (i.e. located on different chromosomes). During sexual reproduction, two individuals are paired and each offspring has a genome that includes one copy of an allele from each of the two parents at the corresponding locus. During sexual reproduction, there is also a chance of mutation. The mutation rate per locus per generation is u. When a mutation occurs in a locus, it is a novel mutant that is different from any other alleles that have been created before.
In the initial population, all of the individuals in the two subpopulations have the same allele for all loci. The total mutation rate per individual per generation is k = lu, and the total number of mutants in a population per generation is Nlu. As mutants are neutral within a population, the fixation probability is 1/N.
We define xαβ, the genetic distance between individuals α and β, as the number of loci that have different alleles between these two haploid individuals. It should satisfy 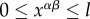 .
.
If each population is close to monomorphic, we can define the genetic distance between these populations in terms of the average value of xαβ, where α and β are randomly chosen from each population. The average value of xαβ when α and β are chosen from the same population is the within-population distance, which indicates the degree of polymorphism.
During a migration event, N’ individuals from population i successfully arrive in population j. These immigrants are randomly selected from population i and participate in mating in population j. The selected immigrants are removed from population i. We adopted a Wright–Fisher model [57,62] for random sampling in each generation. The number of offspring was kept at a constant N during every generation.
Appendix B
We define the average waiting time to speciation:
| B1 |
Then, from equation (4.1) with the initial condition u(x, 0) = 0 (0 ≤ x < xc) and boundary condition u(xc, t) = 1 (t > 0), we can derive the following ordinary differential equation:
| B2 |
Here, we use the replacement 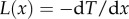 for equation (B 2) and the equation becomes
for equation (B 2) and the equation becomes
| B3 |
In addition, if we write 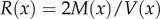 and multiply both sides by
and multiply both sides by 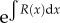 , we obtain
, we obtain
| B4 |
The solution with 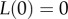 , which comes from the assumption that the average waiting time to speciation T does not change with slight increases in initial genetic distance from 0, is
, which comes from the assumption that the average waiting time to speciation T does not change with slight increases in initial genetic distance from 0, is
| B5 |
Next, we substitute 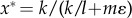 and
and 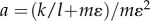 into equation (B 5). In addition, let us replace the lower limit of the outer integration with 0 = δ to allow us to obtain
into equation (B 5). In addition, let us replace the lower limit of the outer integration with 0 = δ to allow us to obtain
| B6 |
Using the variable transformation 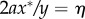 , the domain of integration is changed. Hence, equation (B 6) becomes
, the domain of integration is changed. Hence, equation (B 6) becomes
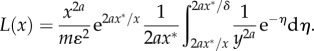 |
B7 |
Taking the limit when δ → 0, equation (B 7) becomes equation (5.1) in the text.
Appendix C
Consider two populations of a haploid and sexual species with non-overlapping generations. These populations live on two islands or island-like habitats. Initially, both islands include the same species and the genetic distance between them is x0 = 0. The genetic distance increases under the rules of equations (2.1) and (2.2). After some number of generations passes, the genetic distance exceeds the threshold (x > xc). Then, we treat the two populations as different species. Subsequently, migrants from a population on one island successfully arrive on the other island. As the latter island does not contain the same species as the migrant, the migrants do not mix with the residents. The migrants become established as a new population. Then, we redefine the genetic distance between the two islands as the difference between the new population on the second island and the original population on the first island and the same process proceeds as before. We assume that there is no extinction because after the genetic distance exceeds the threshold, immigrants that move from one island to the other quickly evolve mating isolation mechanisms to avoid reproductive interference [54]. Hence, we can count the number of species that are created by this mechanism whenever the distance between two populations reaches the threshold.
References
- 1.Mayr E. 1942. Systematics and the origin of species. New York, NY: Columbia University Press. [Google Scholar]
- 2.Mayr E. 1963. Animal species and evolution. Cambridge, MA: Harvard University Press. [Google Scholar]
- 3.Brooks DR, McLennan DA. 1991. Phylogeny, ecology, and behavior: a research program in comparative biology. Chicago, IL: University of Chicago Press. [Google Scholar]
- 4.Rosenzweig ML. 1995. Species diversity in space and time. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 5.Kondrashov AS. 2001. Speciation: Darwin revisited. Trends Ecol. Evol. 16, 412 ( 10.1016/S0169-5347(01)02216-9)11403875 [DOI] [Google Scholar]
- 6.Thomas NS, Davis MW. 2009. Big questions in ecology and evolution. Oxford, UK: Oxford University Press. [Google Scholar]
- 7.Kondrashov AS. 1986. Multilocus model of sympatric speciation III. Computer simulations. Theor. Popul. Biol. 29, 1–15. ( 10.1016/0040-5809(86)90002-X) [DOI] [PubMed] [Google Scholar]
- 8.Wagner A, Wagner GP, Similion P. 1994. Epistasis can facilitate the evolution of reproductive isolation by peak shifts: a two-locus two-allele model. Genetics 138, 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turner GF, Burrows MT. 1995. A model of sympatric speciation by sexual selection. Proc. R. Soc. Lond. B 260, 287–292. ( 10.1098/rspb.1995.0093) [DOI] [Google Scholar]
- 10.Johnson PA, Hoppenstaedt FC, Smith JJ, Bush GL. 1996. Conditions for sympatric speciation: a diploid model incorporating habitat fidelity and non-habit assortative mating. Evol. Ecol. 10, 187–205. ( 10.1007/BF01241784) [DOI] [Google Scholar]
- 11.Kondrashov AS, Kondrashov FA. 1999. Interactions among quantitative traits in the course of sympatric speciation. Nature 400, 351–354. ( 10.1038/22514) [DOI] [PubMed] [Google Scholar]
- 12.Dieckmann U, Doebeli M. 1999. On the origin of species by sympatric speciation. Nature 400, 354–357. ( 10.1038/22521) [DOI] [PubMed] [Google Scholar]
- 13.Doebeli M, Dieckmann U. 2000. Evolutionary branching and sympatric speciation caused by different types of ecological interactions. Am. Nat. 156, S77–S101. ( 10.1086/303417) [DOI] [PubMed] [Google Scholar]
- 14.Doebeli M, Dieckmann U. 2003. Speciation along environmental gradients. Nature 421, 259–264. ( 10.1038/nature01247) [DOI] [PubMed] [Google Scholar]
- 15.van Doorn GS, Dieckmann U, Weissing FJ. 2004. Sympatric speciation by sexual selection: a critical reevaluation. Am. Nat. 163, 709–725. ( 10.1086/383619) [DOI] [PubMed] [Google Scholar]
- 16.Nei M, Maruyama T, Wu CI. 1983. Models of evolution of reproductive isolation. Genetics 103, 557–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu CI. 1985. A stochastic simulation study on speciation by sexual selection. Evolution 39, 66–82. ( 10.2307/7408517) [DOI] [PubMed] [Google Scholar]
- 18.Dobzhansky TG, Dobzhansky T. 1937. Genetics and the origin of species. New York, NY: Columbia University Press. [Google Scholar]
- 19.Muller HJ. 1942. Isolating mechanisms, evolution and temperature. Biol. Symp. 811, 71–125. [Google Scholar]
- 20.Higgs PG, Derrida B. 1992. Genetic distance and species formation in evolving populations. J. Mol. Evol. 35, 454–465. ( 10.1007/BF00171824) [DOI] [PubMed] [Google Scholar]
- 21.Manzo F, Peliti L. 1994. Geographic speciation in the Derrida–Higgs model of species formation. J. Phys. A 27, 7079–7086. ( 10.1088/0305-4470/27/21/022) [DOI] [Google Scholar]
- 22.Gavrilets S, Gravner J. 1997. Percolation on the fitness hypercube and the evolution of reproductive isolation. J. Theor. Biol. 184, 51–64. ( 10.1006/jtbi.1996.0242) [DOI] [PubMed] [Google Scholar]
- 23.Gavrilets S, Hai L, Vose MD. 2000. Patterns of parapatric speciation. Evolution 54, 1126–1134. ( 10.1111/j.0014-3820.2000.tb00548.x) [DOI] [PubMed] [Google Scholar]
- 24.Orr HA, Orr LH. 1996. Waiting for speciation: the effect of population subdivision on the waiting time to speciation. Evolution 50, 1742–1749. ( 10.2307/2410732) [DOI] [PubMed] [Google Scholar]
- 25.Gavrilets S, Hastings A. 1996. Founder effect speciation: a theoretical reassessment. Am. Nat. 147, 466–491. ( 10.1086/285861) [DOI] [Google Scholar]
- 26.Gavrilets S. 2000. Waiting time to parapatric speciation. Proc. R. Soc. Lond. B 267, 2483–2492. ( 10.1098/rspb.2000.1309) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Endler JA. 1977. Geographic variation, speciation and clines. Princeton, NJ: Princeton University Press. [PubMed] [Google Scholar]
- 28.Avise JC. 2000. Phylogeography. Cambridge, MA: Harvard University Press. [Google Scholar]
- 29.Gavrilets S, Acton R, Gravner J. 2000. Dynamics of speciation and diversification in a metapopulation. Evolution 54, 1493–1501. ( 10.1111/j.0014-3820.2000.tb00695.x) [DOI] [PubMed] [Google Scholar]
- 30.Orr HA. 1995. The population genetics of speciation: the evolution of hybrid incompatibilities. Genetics 139, 1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slatkin M. 1987. Gene flow and the geographic structure of natural populations. Science 236, 787–792. ( 10.1126/science.3576198) [DOI] [PubMed] [Google Scholar]
- 32.Turelli M, Orr HA. 1995. The dominance theory of Haldane's rule. Genetics 140, 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maruyama T. 1970. On the fixation probability of mutant genes in a subdivided population. Genet. Res. 15, 221–225. ( 10.1017/S0016672300001543) [DOI] [PubMed] [Google Scholar]
- 34.Maruyama T. 1983. Stochastic theory of population genetics. Bull. Math. Biol. 45, 521–554. ( 10.1007/BF02459586) [DOI] [Google Scholar]
- 35.Slatkin M. 1976. The rate of spread of an advantageous allele in a subdivided population. In Population genetics and ecology (eds S Karlin, E Nevo), pp. 767–780. New York, NY: Academic Press. [Google Scholar]
- 36.Slatkin M. 1981. Fixation probabilities and fixation times in a subdivided population. Evolution 35, 477–488. ( 10.2307/2408196) [DOI] [PubMed] [Google Scholar]
- 37.Lande R. 1979. Effective deme sizes during long-term evolution estimated from rates of chromosomal rearrangement. Evolution 33, 234–251. ( 10.2307/2407380) [DOI] [PubMed] [Google Scholar]
- 38.Lande R. 1985. The fixation of chromosomal rearrangements in a subdivided population with local extinction and colonization. Heredity 54, 323–332. ( 10.1038/hdy.1985.43) [DOI] [PubMed] [Google Scholar]
- 39.Tachida H, Iizuka M. 1991. Fixation probability in spatially changing environments. Genet. Res. 58, 243–251. ( 10.1017/S0016672300029992) [DOI] [PubMed] [Google Scholar]
- 40.Barton NH. 1993. The probability of fixation of a favoured allele in a subdivided population. Genet. Res. 62, 149–157. ( 10.1017/S0016672300031748) [DOI] [Google Scholar]
- 41.Rieseberg LH. 2001. Chromosomal rearrangements and speciation. Trends Ecol. Evol. 16, 351–358. ( 10.1016/S0169-5347(01)02187-5) [DOI] [PubMed] [Google Scholar]
- 42.Whitlock MC. 2003. Fixation probability and time in subdivided populations. Genetics 164, 767–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patwa Z, Wahl LM. 2008. The fixation probability of beneficial mutations. J. R. Soc. Interface 5, 1279–1289. ( 10.1098/rsif.2008.0248) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gavrilets S, Hai L, Vose MD. 1998. Rapid parapatric speciation on holey adaptive landscapes. Proc. R. Soc. Lond. B 265, 1483–1489. ( 10.1098/rspb.1998.0461) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kimura M, Ohta T. 1969. The average number of generations until fixation of a mutant gene in a finite population. Genetics 61, 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kimura M. 1968. Evolutionary rate at the molecular level. Nature 217, 624–626. ( 10.1038/217624a0) [DOI] [PubMed] [Google Scholar]
- 47.Kimura M, Ohta T. 1969. The average number of generations until extinction of an individual mutant gene in a finite population. Genetics 63, 701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nei M, Feldman MW. 1972. Identity of genes by descent within and between populations under mutation and migration pressures. Theor. Popul. Biol. 3, 460–465. ( 10.1016/0040-5809(72)90017-2) [DOI] [PubMed] [Google Scholar]
- 49.Chakraborty R, Nei M. 1974. Dynamics of gene differentiation between incompletely isolated populations of unequal sizes. Theor. Popul. Biol. 5, 460–469. ( 10.1016/0040-5809(74)90064-1) [DOI] [PubMed] [Google Scholar]
- 50.Slatkin M, Maruyama T. 1975. The influence of gene flow on genetic distance. Am. Nat. 109, 597–601. ( 10.1086/283031) [DOI] [Google Scholar]
- 51.Li WH. 1976. Effect of migration on genetic distance. Am. Nat. 110, 841–847. ( 10.1086/283106) [DOI] [Google Scholar]
- 52.Konuma J, Chiba S. 2007. Ecological character displacement caused by reproductive interference. J. Theor. Biol. 247, 354–364. ( 10.1016/j.jtbi.2007.03.013) [DOI] [PubMed] [Google Scholar]
- 53.Thibert-Plante X, Hendry AP. 2009. Five questions on ecological speciation addressed with individual-based simulations. J. Evol. Biol. 22, 109–123. ( 10.1111/j.1420-9101.2008.01627.x) [DOI] [PubMed] [Google Scholar]
- 54.Yamaguchi R, Iwasa Y. 2013. Reproductive character displacement by the evolution of female mate choice. Evol. Ecol. Res. 15, 25–41. [Google Scholar]
- 55.Feller W. 1971. An introduction to probability theory and its applications, vol. 2, p. 669 New York, NY: Wiley. [Google Scholar]
- 56.Karlin S, Taylor HM. 1981. A second course in stochastic processes, p. 542 New York, NY: Academic Press. [Google Scholar]
- 57.Wright S. 1931. Evolution in Mendelian populations. Genetics 16, 97–159. ( 10.1007/BF02459575) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coyne JA, Orr HA. 2004. Speciation. Sunderland, MA: Sinauer Associates. [Google Scholar]
- 59.Coyne JA, Orr HA. 1989. Patterns of speciation in Drosophila. Evolution 43, 362–381. ( 10.2307/2409213) [DOI] [PubMed] [Google Scholar]
- 60.Coyne JA, Orr HA. 1997. Patterns of speciation in Drosophila revisited. Evolution 51, 295–303. ( 10.2307/2410984) [DOI] [PubMed] [Google Scholar]
- 61.Coyne JA, Orr HA. 1998. The evolutionary genetics of speciation. Phil. Trans. R. Soc. Lond. B 353, 287–305. ( 10.1098/rstb.1998.0210) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fisher RA. 1930. The genetical theory of natural selection. Oxford, UK: Clarendon Press. [Google Scholar]