

Abstract
Purpose.
To compare in vivo retinal nerve fiber layer thickness (RNFLT) and axonal transport at 1 and 2 weeks after an 8-hour acute IOP elevation in rats.
Methods.
Forty-seven adult male Brown Norway rats were used. Procedures were performed under anesthesia. The IOP was manometrically elevated to 50 mm Hg or held at 15 mm Hg (sham) for 8 hours unilaterally. The RNFLT was measured by spectral-domain optical coherence tomography. Anterograde and retrograde axonal transport was assessed from confocal scanning laser ophthalmoscopy imaging 24 hours after bilateral injections of 2 μL 1% cholera toxin B-subunit conjugated to AlexaFluor 488 into the vitreous or superior colliculi, respectively. Retinal ganglion cell (RGC) and microglial densities were determined using antibodies against Brn3a and Iba-1.
Results.
The RNFLT in experimental eyes increased from baseline by 11% at 1 day (P < 0.001), peaked at 19% at 1 week (P < 0.0001), remained 11% thicker at 2 weeks (P < 0.001), recovered at 3 weeks (P > 0.05), and showed no sign of thinning at 6 weeks (P > 0.05). There was no disruption of anterograde transport at 1 week (superior colliculi fluorescence intensity, 75.3 ± 7.9 arbitrary units [AU] for the experimental eyes and 77.1 ± 6.7 AU for the control eyes) (P = 0.438) or 2 weeks (P = 0.188). There was no obstruction of retrograde transport at 1 week (RCG density, 1651 ± 153 per mm2 for the experimental eyes and 1615 ± 135 per mm2 for the control eyes) (P = 0.63) or 2 weeks (P = 0.25). There was no loss of Brn3a-positive RGC density at 6 weeks (P = 0.74) and no increase in microglial density (P = 0.92).
Conclusions.
Acute IOP elevation to 50 mm Hg for 8 hours does not cause a persisting axonal transport deficit at 1 or 2 weeks or a detectable RNFLT or RGC loss by 6 weeks but does lead to transient RNFL thickening that resolves by 3 weeks.
Keywords: glaucoma, retinal ganglion cell, retinal nerve fiber layer, axonal transport, optical coherence tomography, confocal scanning laser ophthalmoscope
Acute IOP elevation to 50 mm Hg for 8 hours in young rats does not cause a persisting axonal transport deficit at 1 or 2 weeks or a detectable RNFLT or RGC loss by 6 weeks but does lead to a substantial, transient RNFL thickening that peaks at 1 week (15%–19%) and resolves by 3 weeks.
Introduction
Glaucoma is a progressive, chronic optic neuropathy that results in the degeneration of retinal ganglion cell (RGC) axons, apoptosis of RGC somas, and corresponding loss of visual function.1,2 Glaucoma is also specifically characterized by conformational changes within the optic nerve head (ONH) that distinguish it from other optic neuropathies.1,3 Although the precise sequence of pathophysiological events leading to vision loss is only partially understood, it is thought to begin with injury to axons within the ONH, perhaps as a consequence of its characteristic ONH deformations.1,4–6 In a collective effort to understand the details of this sequence and injury process, numerous experimental models of glaucoma have been developed over the last several decades.7–13 Because IOP is the most well-known risk factor and currently the only target for therapeutic intervention, almost all of these experimental models are based on elevated IOP. Many aspects of the structural and functional changes observed clinically in human eyes with chronic glaucoma have been recapitulated in various species of experimental animal models based on chronic (long-term) IOP elevation such as progressive cupping of the optic disc (which includes axon loss within the neuroretinal rim and deformation and remodeling of the ONH and peripapillary scleral connective tissues),14–21 selective loss of RGCs and their axons from the retina with associated loss of function,14,17,22–26 alterations in ONH blood flow and its autoregulation,27–30 and impaired axonal transport.31–36
Notably, similar alterations have been observed after brief periods (usually <4 hours) of acute IOP elevation to subischemic levels (≤50 mm Hg), including the following: deformation of the ONH and peripapillary tissues in monkey37–41 and rat42; reduced ocular blood flow with autoregulation dysfunction (if systemic blood pressure is also low) in human43 and monkey44,45; reduced ocular blood flow (primarily from decreased flow velocity rather than reduced vessel diameter)42,46 without hypoxia47 in rat; RGC-specific functional abnormalities in monkey,48 cat,49 and rodent50–54; and impaired axonal transport in monkey,55–59 rat,60–62 and pig.63,64 However, these alterations are completely reversible within a short duration of IOP normalization, with no permanent changes in structure or function (e.g., ONH and peripapillary deformation,37–39,42 RGC dysfunction,48,49,51,52,54 reduced ocular blood flow,43,44,46 and axonal transport disruption56,58,61). This raises an important question about how these short-term, reversible abnormalities lead to the permanent changes observed in the chronic conditions (glaucoma and its experimental models). One possibility is that repeated insults have compounding effects that activate mechanisms, which then lead to chronic changes, including permanent ONH deformations, lethal axonal injury, and subsequent RGC death.65–68 Another possibility is that longer-duration acute episodes exceed some tolerance threshold (IOP × time integral69) and thus trigger the mechanisms that lead to these chronic changes and permanent loss.
In this study, we examined the IOP tolerance threshold in rat eyes by extending the period of subischemic (50 mm Hg) acute IOP elevation to 8 hours. We investigated whether a longer-duration (8 hour) single episode of IOP elevation to 50 mm Hg would result in axonal transport abnormalities that persist at 1 or 2 weeks after IOP normalization. Second, we assessed whether this IOP elevation caused permanent injury to RGCs by measuring both in vivo retinal nerve fiber layer thickness (RNFLT) and postmortem RGC densities using the immunohistochemical marker Brn3a for up to 6 weeks of follow-up.
Methods
Subjects
The subjects of this study were 47 adult male Brown Norway rats (Rattus norvegicus; Charles River Laboratories, Inc., Wilmington, MA) that were aged 8 to 12 weeks and weighed 168 to 240 g at baseline. Rats were maintained under a 12-hour light and 12-hour dark cycle with normal rat chow and water available ad libitum. All experimental methods and animal care procedures adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved and monitored by the Institutional Animal Care and Use Committee at Legacy Health.
Anesthesia
During the episode of acute IOP elevation, as well as for all imaging and intravitreal injection procedures, animals were anesthetized with an intramuscular injection of a rodent cocktail containing ketamine (55 mg/kg, Ketaset; Fort Dodge Animal Health, Fort Dodge, IA), xylazine (5 mg/kg, AnaSed; Lloyd, Inc., Shenandoah, IA), and acepromazine maleate (1 mg/kg; Vedco, Inc., St. Joseph, MO). This dosage provided general anesthesia for 45 to 90 minutes. Half of the original dose was used for repeated injections as necessary for longer durations. For stereotactic injection procedures, animals were anesthetized with 2% isofluorane gas at 2 mL/min in a 2:1 ratio of nitrous oxide to oxygen. For all procedures under anesthesia, body temperature was maintained with a heat mat. For procedures lasting longer than 90 minutes, lactated Ringer's solution (Baxter Healthcare Corporation, Toronto, ON, Canada) was injected subcutaneously at regular intervals to maintain hydration.
Eight-Hour Acute IOP Elevation Protocol
The anterior chamber of the right eye of each rat was cannulated through the peripheral nasal cornea with a 30G needle. The needle was connected by polyethylene tubing to a reservoir filled with sterile balanced salt solution (Alcon Laboratories, Inc., Fort Worth, TX) so that IOP could be manometrically controlled by positioning the height of the reservoir. The cannula remained inserted in the anterior chamber for 8 hours in all animals. The cannulation site was visually examined for leakage regularly during the period of IOP elevation, and the cornea was tested gently for firmness with the tip of a WECK-CEL eye spear (Medtronic, Minneapolis, MN). As a secondary confirmation, IOP was monitored indirectly during cannulation with a rodent tonometer (Tonolab; Icare Finland Oy, Helsinki, Finland). Topical anesthetic (0.5% proparacaine hydrochloride; Alcon Laboratories, Inc.) was instilled before cannulation. Ocular lubricants were applied at regular intervals to both eyes (Celluvisc; Allergan, Irvine, CA). After cannula removal, antibiotic ointment was applied (neomycin, polymyxin B sulfates, and dexamethasone; Falcon Pharmaceuticals Ltd., Fort Worth, TX).
In Vivo Spectral-Domain Optical Coherence Tomography Measurements
Peripapillary RNFLT and total retinal thickness (RT) were measured longitudinally in vivo using spectral-domain optical coherence tomography (SD-OCT) (Spectralis; Heidelberg Engineering GmbH) as previously described.42 Topical anesthesia and mydriasis (tropicamide 0.5% [Alcon Laboratories, Inc.] and phenylephrine 2.5% [Bausch & Lomb, Incorporated, Tampa, FL]) were instilled, custom rigid gas permeable contact lenses were inserted, and rats were placed on a custom-built imaging stage. Average and sectorial (superior, inferior, nasal, and temporal) peripapillary RNFLT and RT were determined from a circular B-scan (12° diameter) centered on the optic disc. The digital axial resolution was 3.9 μm. The B-scan was composed of 1536 A-scans and consisted of an average of 100 individual sweeps using automatic real-time eye tracking software to reduce speckle noise. Follow-up scans were collected using the eye tracking software so that they were in identical positions relative to baseline. The SD-OCT data were exported for performing retinal layer segmentations and deriving thickness values using custom software.42 Retinal thickness was defined as the thickness from the inner limiting membrane to Bruch's membrane–RPE complex, as is consistent with previous studies.42,70–72
Anterograde Axonal Transport Assay
Anterograde axonal transport within RGCs was assessed with the tracer cholera toxin B-subunit conjugated to AlexaFluor 488 (CTB) dissolved in sterile PBS as previously described.73 In brief, at 24 hours after bilateral intravitreal injection of 2 μL 1% CTB, the ocular fundi were imaged in vivo by confocal scanning laser ophthalmoscopy in fluorescence mode (CSLO-FL) (Spectralis HRA; Heidelberg Engineering GmbH) to confirm successful injections. Rats were then overdosed with an intraperitoneal injection of pentobarbital sodium and phenytoin sodium (0.7–1.4 mL/kg, Euthasol Solution; Virbac Animal Health, Inc., Fort Worth, TX). After enucleating the eyes, animals were transcardially perfused with 0.1 mL heparin sodium (10,000 United States Pharmacopeia U/mL; APP Pharmaceuticals, LLC, Schaumburg, IL), followed by 125 mL cold 4% paraformaldehyde in 0.5 M phosphate buffer (PB [pH 7.35]). The retinas were dissected from the globes, and the brain was dissected from the skull; they were then immersion fixed in 4% paraformaldehyde in 0.5 M PB for 30 minutes in preparation for immunohistochemistry and postmortem CSLO, respectively.
Axonal transport was assessed from postmortem CSLO-FL (Spectralis HRA; Heidelberg Engineering GmbH) of the optic nerves and superior colliculi. The CSLO-FL images were obtained with an additional +25-diopter lens mounted to the camera objective. The BluePeak (Heidelberg Engineering GmbH) blue laser (488 nm) autofluorescence imaging mode was used with the standard contrast setting (i.e., without contrast normalization), and 100 frames were averaged to produce the final recorded image. Analysis of the relative fluorescence intensity ratio of the experimental to control superior colliculus was performed in ImageJ software (National Institutes of Health, Bethesda, MD) using a colliculus-shaped polygon to define the region of interest. Note that approximately 95% of axons cross at the chiasm in pigmented rats,74,75 so the experimental superior colliculus is of opposite laterality to the experimental prechiasmal optic nerve and eye.
Retrograde Axonal Transport Assay
The retrograde transport assay involved bilateral injections of 2 μL 1% CTB into the superior colliculi with the rat held in a stereotactic device (Kopf Instruments, Tujunga, CA) as previously described.73 The stereotactic coordinates used were as follows: −5.5 mm anterior-posterior and ±1.25 mm medial-lateral (both relative to the bregma skull landmark) and −4.5, −4.25, −4.0, and −3.75 mm dorsal-ventral (from the skull surface). At each dorsoventral location, 0.5 μL CTB was injected to optimize diffusion of CTB throughout the superior colliculi. Intramuscular buprenorphine (0.3 mg/mL) analgesic was administered after surgery. Retrograde transport was assessed from in vivo CSLO-FL images of the retinas at 24 hours after injection. Relative RGC density between experimental and control eyes was determined for an area of 4.57 mm2 centered on the optic disc using ImageJ software (National Institutes of Health). Rats were overdosed, and the retinas and brain were dissected and immersion fixed as described for the anterograde assay. Whole-mounted retinas were imaged with fluorescence microscopy (×5 air objective, DMRXE; Leica, Wetzlar, Germany) using filter set 513808 (FITC, excitation of 450–490 nm, 515-nm long-pass emission; Chroma GmbH & Co., Münster, Germany). Relative RGC density between experimental and control eyes for an area of 5.79 mm2 was determined using ImageJ software (National Institutes of Health) for comparison with the in vivo CSLO results. Postmortem CSLO-FL of the superior colliculi was performed to confirm successful CTB injections.
Follow-up IOP Measurements
Although IOP was controlled manometrically during the 8-hour episode of acute elevation, additional IOP measurements were made using a rodent tonometer immediately before and after anterior chamber cannulation for up to 30 minutes to determine whether a decrease in IOP occurred due to persistent leakage of aqueous humor after cannula removal. The IOP was also measured by tonometry at follow-up imaging sessions. All IOP recordings were measured under general and topical anesthesia as the average of five consecutive measurements.
Experimental Design
Rats were assigned to one of four groups. In group 1 (n = 34), rats had the IOP of the right eye acutely elevated to 50 mm Hg for 8 hours, with the left eye as an untouched control, and were observed for 1 or 2 weeks. Four of these rats were excluded from all analyses because of unreliable IOP elevation due to either leakage at the cannulation site or raised IOP after cannulation removal from a presumed angle-closure event. The remaining 30 rats had the anterograde or retrograde transport assay performed at 1 or 2 weeks after IOP elevation, although 10 were excluded because of failed injections. Failed intravitreal injections were due to retinal detachment and/or vitreous hemorrhage and resulted in uneven uptake of CTB by RGCs across the central retina. Failed stereotactic injections were classified as having less than 50% of the central superior colliculus surface filled by CTB on postmortem CSLO-FL. Hence, transport data were analyzed for 20 rats, with five in each subgroup (anterograde at 1 week, retrograde at 1 week, anterograde at 2 weeks, and retrograde at 2 weeks). For SD-OCT analysis, an additional two rats were excluded from the 30 rats with successful IOP elevation due to poor imaging because of corneal opacities following cannulation. Thus, SD-OCT was evaluated at baseline and at the 1-week (n = 13) or 1-week and 2-week (n = 15) follow-up in a total of 28 animals with unilateral IOP elevation to 50 mm Hg for 8 hours. The 1-week and 2-week time points for simultaneous assessment of RNFLT and transport were chosen for the possibility that there may be axon transport deficits without accompanying structural loss of RGC axons.
In group 2 (n = 4), rats were assigned to longer-term follow-up duration, in which they were observed for 6 weeks after the IOP of the right eye was acutely elevated to 50 mm Hg for 8 hours, with the left eye as an untouched control. Group 3 rats (n = 4) were assigned to a sham control group, in which the right eye was cannulated and held at an IOP of 15 mm Hg for 8 hours, while the left eye served as an untouched control, and were observed for 6 weeks. The 6-week follow-up period was chosen because it can take 1 to 2 months for RGCs to degenerate and die after injury.76 All eight rats in groups 2 and 3 had successful IOP elevation without subsequent complications and were included in the analyses. The SD-OCT was performed at baseline; at follow-up time points of 1, 3, and 7 days; and thereafter weekly to 6 weeks. There was no axonal transport assay conducted in group 2 or group 3. Group 4 rats (n = 5) were healthy, naive animals sacrificed specifically as a control group for retinal immunohistochemistry and did not undergo anterior chamber cannulation.
Our laboratory has previously reported a mean ± SD femoral artery blood pressure of 97.6 ± 10.7 mm Hg (range, 80–120 mm Hg) in a group of naive normal Brown Norway rats under ketamine anesthesia.50 Therefore, the perfusion pressure is expected to be at least 30 mm Hg in the group 1 and group 2 experimental eyes elevated to IOPs of 50 mm Hg, which should be well above the threshold for ischemia.47,54,69 We have previously monitored systemic blood pressure for as long as 5 hours and found (unpublished data) that it decreases by approximately 5 to 15 mm Hg for 15 to 20 minutes immediately after each anesthetic dose is administered; it then stabilizes at a level (approximately 100 mm Hg) near what is observed in awake animals (by noninvasive measurements) until the subsequent dose is administered.
Retinas from follow-up periods (1, 2, and 6 weeks) were processed with immunohistochemical markers against Brn3a and Iba-1 to assess RGC density77 and microglial activation in the retinal nerve fiber layer (RNFL),78 respectively. Measurement of RGC density allows assessment of whether the acute IOP elevation resulted in permanent RGC loss. Determining microglial activation alongside RNFLT and axonal transport may give insight into the sequence of the retinal response to IOP elevation because there is evidence that microglial activation has a role in disease progression and that microglia phagocytose dying or degenerating RGCs.79–85
Immunohistochemistry
Table 1 gives a summary of the antibodies used in this study, Brn3a for labeling RGC somas and Iba-1 for labeling microglia. Brn3a is a 47-kilodalton (kDa) class IV POU domain–containing protein (transcription factor) specific for RGCs in vertebrate retinas,86 including rats.77,87,88 Iba-1 is a 17-kDa EF hand protein specifically expressed in microglia and upregulates during microglia activation.78,89
Table 1.
Primary Antibodies
|
Antibody |
Immunogen |
Supplier and Catalog No. |
Species and Type |
Dilution |
| Brn3a | Amino acids 186–224 of Brn3a fused to t7 gene protein 10 | Millipore, Billerica, MA, MAB1585 (provided by Catherine Morgans, Oregon Health Sciences University, Portland, OR) | Mouse, monoclonal | 1:250 |
| Brn3a (C-20) | N-terminus synthetic peptide of human Brn3a | Santa Cruz Biotechnology, Inc., Santa Cruz CA, sc-31984 | Goat, polyclonal | 1:50 |
| Iba-1 | C-terminus synthetic peptide of human, rat, and mouse Iba-1 | Wako Pure Chemical Industries Ltd., Richmond, VA, 019-19741 | Rabbit, polyclonal | 1:500 |
Retinas were rinsed in PBS (pH 7.4), immersed in 3% Triton X-100 overnight, and rinsed again in PBS before preincubation for 2 days at 4°C in either 5% goat or donkey blocking serum (Life Technologies, Carlsbad, CA) as appropriate. Primary antibodies were diluted in either 5% goat or donkey blocking serum, and retinas were incubated in the primary antibody solution for 3 days at 4°C. Retinas from group 1 animals sacrificed at 1 or 2 weeks contained CTB, so they were single-labeled with mouse anti-Brn3a (n = 12 rats), goat anti-Brn3a (n = 8), or rabbit anti–Iba-1 (n = 8). Retinas from group 2 and group 3 animals sacrificed at 6 weeks (n = 8 rats) and from group 4 naive rats (n = 5) did not have CTB, so they were doubled-labeled with mouse anti-Brn3a and rabbit anti–Iba-1. Retinas were rinsed in PBS before incubation in secondary antibody solution for 24 hours. Secondary antibodies were goat anti-rabbit IgG coupled to AlexaFluor 594 diluted 1:400 in 5% goat blocking serum, donkey anti-goat IgG coupled to AlexaFluor 568 diluted 1:500 in 5% donkey blocking serum, goat anti-mouse IgG coupled to AlexaFluor 594 diluted 1:400 in 5% goat blocking serum, and goat anti-rabbit IgG coupled to AlexaFluor 488 diluted 1:400 in 5% goat blocking serum. Preparations were coverslipped in Vectorshield with DAPI (Vector Laboratories, Burlingame, CA).
RGC and Microglia Density Analysis
In experimental and fellow control retinas from all four groups of rats, the densities of RGCs and microglia in the RNFL were determined by counting somas labeled with antibodies to Brn3a and Iba-1, respectively, and dividing by the area of the region counted. Micrographs were acquired with the following: (1) a digital camera (QICAM 12-bit; QImaging, Surrey, BC, Canada) attached to a DMIRB inverted microscope (Leica) with ×20 air objective, (2) filter sets 513828 (FITC, excitation of 450–490 nm, 515-nm long-pass emission) and 513832 (CY3, excitation of 512–560 nm, 590-nm long-pass emission) (Chroma GmbH & Co.), (3) imaging software (Qcapture; QImaging), and (4) a computer-controlled (X-Y-Z) stage (Applied Scientific Instrumentation, Inc., Eugene OR). Micrographs were obtained in a tiled array centered on the optic disc covering most of the central retina (total areas analyzed were 8.1 ± 1.0 mm2 for RGCs and 7.6 ± 1.8 mm2 for microglia). Stacks of 15 to 45 images with 1-μm axial spacing through the RGC layer (for Brn3a) or the RNFL (for Iba-1) were taken at each tile location within the array, with a 10% overlap between each tile. Each tile was composed of 1392 × 1040 pixels, covering an area of 0.17 mm2. Custom software was used to compress the image stack at each tile location using only the pixels in focus from the z stack. Images were montaged with custom software, and somas were counted with a semiautomated method using ImageJ software (National Institutes of Health).
Statistical Analysis
Two-way repeated-measures ANOVAs with Bonferroni-corrected post hoc tests were performed on the SD-OCT measurements of RNFLT and RT, Brn3a cell densities, and Iba-1 cell densities. Wilcoxon nonparametric paired t-tests were applied to compare relative fluorescence intensity (experimental versus control) across hemispheres of the superior colliculus for the anterograde axonal transport assay, as well as to compare the relative RGC density between experimental versus control retinas for the retrograde axonal transport assay. A two-way ANOVA with Bonferroni-corrected post hoc tests was used to analyze IOP data. All statistical analyses were performed using commercial software (Prism, version 5; GraphPad Software, Inc., San Diego, CA).
Results
RNFL Thickness
Figure 1 shows a representative example of longitudinal RNFLT measurements derived from peripapillary, circular SD-OCT B-scans at baseline and 1 and 2 weeks after an 8-hour episode of acute IOP elevation. The RNFLT results for the entire group 1 cohort are plotted in Figure 2A (for the peripapillary average) and Figure 2B (for individual quadrants). In group 1, there was no difference in RNFLT at baseline between experimental eyes (45.9 ± 2.5 μm) and fellow control eyes (46.3 ± 2.8 μm) (P > 0.05). One week after the 8-hour episode of IOP elevation to 50 mm Hg, RNFLT increased in the experimental eyes to 54.4 ± 5.7 μm (18.7 ± 11.1% increase relative to baseline, P < 0.001) and then partially receded by the 2-week follow-up time point to 50.4 ± 4.4 μm (10.5% ± 7.6% increase relative to baseline, P < 0.001). RNFLT in the fellow control eyes remained equivalent to baseline values at follow-up, averaging 45.9 ± 2.9 μm (−0.6% ± 4.7% decrease relative to baseline, P > 0.05) at week 1 and 46.3 ± 2.2 μm (0.5 ± 4.3% increase relative to baseline, P > 0.05) at week 2. There were no differences in the magnitude of RNFLT changes from baseline across superior, inferior, nasal, or temporal quadrants at either the 1-week (P = 0.11) or 2-week (P = 0.62) follow-up time points, although there was a trend in experimental eyes for the nasal quadrant to show less thickening than other quadrants (Fig. 2B).
Figure 1.
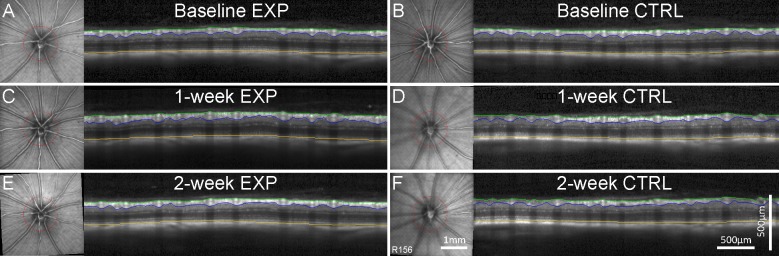
Longitudinal measurements of RNFLT and RT obtained by SD-OCT are shown for a single representative rat at baseline (A, B) and at 1 week (C, D) and 2 weeks (E, F) after acute, unilateral IOP elevation to 50 mm Hg for 8 hours. Data from the experimental eye (Exp) appear in the left column and from the fellow control eye (Ctrl) in the right column. In each panel, the SD-OCT B-scan is shown alongside the infrared ocular fundus CSLO image. The B-scan position is indicated by the red circle in the CSLO image and is maintained at follow-up with eye tracking software. B-scan images show the segmentations of the inner limiting membrane (green), outer border of the nerve fiber layer (blue), and Bruch's membrane–RPE complex (yellow), used to calculate peripapillary RNFLT (green to blue) and RT (green to yellow). The RNFLT increased at the 1-week and 2-week follow-up (C, E) compared with baseline (A) for the experimental eye, while the fellow control eye remains consistent (B, D, F). Scale bars apply to (A) through (F).
Figure 2.

Percentage change in RNFLT at follow-up time points relative to baseline (BL) for each group of experimental (Exp) and fellow control (Ctrl) eyes. (A) Group 1 (50 mm Hg) rats show an increase in RNFLT at both the 1-week (n = 28) and 2-week (n = 15) follow-up in the experimental eyes but not in their fellow control eyes. (B) The change in peripapillary RNFLT for group 1 eyes from baseline was similar across superior, temporal, inferior, and nasal quadrants at both the 1-week (P = 0.11) and 2-week (P = 0.62) follow-up. (C) Data from group 2 with a 6-week follow-up (50 mm Hg [n = 4]) show that the increase in RNFLT is transient, recovering to baseline by the 3-week follow-up, with a peak observed at 3 days and no loss evident at 6 weeks. (D) Data from group 3 with a 6-week follow-up (sham, 15 mm Hg [n = 4]) show a smaller, transient increase in RNFLT in the experimental eyes at the 1-day follow-up only. Error bars denote ±SEM. *P < 0.05 (Bonferroni post hoc test).
The increase in RNFLT was examined over a more detailed time course out to 6 weeks in group 2 (50–mm Hg IOP elevation) and group 3 (sham, 15–mm Hg IOP) as shown in Figure 2C and Figure 2D, respectively. For group 2 experimental eyes, RNFLT increased from a baseline of 50.1 ± 2.2 μm to 55.6 ± 2.1 μm (11.1 ± 1.9%) at 1 day after the IOP elevation episode (baseline P < 0.001) and continued to increase, reaching a peak of 57.5 ± 4.7 μm (14.6 ± 5.6% above baseline) at the 3-day follow-up (P < 0.001). These eyes recovered back to the mean (SD) baseline and control levels (P > 0.05) by 3 weeks (52.5 ± 3.9 μm), with no loss evident at 6 weeks (50.8 ± 1.5 μm). As observed in group 1, the quadrant analysis of group 2 also showed no differences in the magnitude of RNFLT changes from baseline across superior, temporal, inferior, or nasal quadrants at any time point (P = 0.12). For group 3 sham experimental eyes, RNFLT increased from a baseline of 47.2 ± 2.7 μm to 50.1 ± 3.9 μm (6.2 ± 2.6%) at 1 day (P < 0.05) but had returned to baseline and fellow control values (48.3 ± 2.6 μm) by 3 days (P > 0.05). This result in the sham group indicates that some of the initial (1 day) increase of RNFLT in group 1 and group 2 is likely due to the cannulation procedure (insertion or removal) itself. Most important, there was no indication of RNFL thinning at any follow-up time point for either group 1 or group 2 after the 8-hour episode of IOP elevation to 50 mm Hg even for time points out to 6 weeks (Figs. 2A, 2C).
Retinal Thickness
Figure 1 shows a representative example of longitudinal RT segmentations in peripapillary SD-OCT B-scans at baseline and the 1-week and 2-week follow-up after an 8-hour episode of acute IOP elevation. For group 1, the RT at baseline were 224.9 ± 7.7 μm in the experimental (right) eye and 225.6 ± 7.7 μm in the fellow control (left) eye (P > 0.05). At the 1-week follow-up, the mean (SD) RT had increased by 7.3 ± 7.9 μm (3.3 ± 3.5%) compared with baseline (P < 0.001) in the experimental eye and at 2 weeks remained thicker by 5.2 ± 8.4 μm (2.3 ± 3.6%) compared with baseline (P < 0.01). The retinal layers minus the RNFL (i.e., RGC layer to Bruch's membrane–RPE complex) showed negligible changes in the experimental eyes compared with baseline: the mean ± SD values were −1.2 ± 5.2 μm (− 0.7 ± 2.9%, P > 0.05) at 1 week and 2.5 ± 3.9 μm (0.2 ± 2.9%, P > 0.05) at 2 weeks. These findings indicate that only the RNFL changed in thickness, while the other retinal layers did not, and show that the increase in total RT is attributable to the increase in RNFLT.
Anterograde Axonal Transport Assay
Figure 3 shows a representative example of anterograde axonal transport assay results. Fluorescence of the RNFL with in vivo CSLO imaging of retinas 24 hours after bilateral intravitreal CTB injections (1 week after the acute IOP elevation event) demonstrates that injections were successful (Figs. 3A, 3B). Postmortem CSLO imaging of the optic nerves (ventral midbrain [Fig. 3C]) and superior colliculi (dorsal midbrain [Figs. 3D, 3E]) shows equivalent CTB fluorescence between experimental and fellow control pathways. This indicates that anterograde transport of CTB is intact and symmetric all the way along both optic nerves and optic tracts to the superior colliculi at 1 week after the unilateral acute IOP elevation event. Quantitative results of the anterograde axonal transport assay data set at both 1 and 2 weeks after the acute IOP elevation episode are listed in Table 2. There was no difference in the intensity of fluorescence between experimental and fellow control optic nerves or superior colliculi. Hence, there was no evidence of anterograde transport disruption persisting at 1 or 2 weeks after acute IOP elevation to 50 mm Hg for 8 hours with this assay. Axonal transport assays were not performed on animals observed for 6 weeks (group 2 and group 3) because there was no persisting transport obstruction at either the 1-week or 2-week follow-up and no evidence of decreased RNFLT at the 6-week follow-up.
Figure 3.
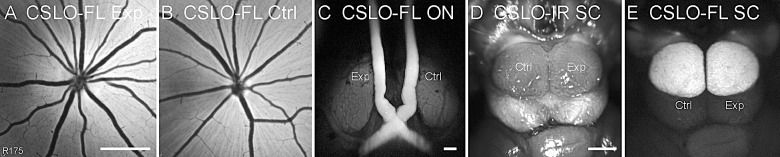
Results of the anterograde axonal transport assay 1 week after acute IOP elevation to 50 mm Hg for 8 hours in the experimental eye in one representative animal. (A, B) The CSLO-FL images of the ocular fundi obtained in vivo from the experimental (Exp [A]) and control (Ctrl [B]) eyes 24 hours after bilateral intravitreal injections of CTB demonstrate successful CTB uptake and transport by RGCs along their axons to the optic disc. (C) Postmortem CSLO-FL of the optic nerves and chiasm shows equivalent fluorescence along both nerves, indicating intact anterograde axonal transport along experimental and control pathways of the optic nerve. (D) Postmortem CSLO infrared (IR) image of the dorsal midbrain shows the position of the superior colliculi for orientation. (E) Postmortem CSLO-FL image of the superior colliculi shows equivalent intensity and coverage of CTB fluorescence between colliculi, indicating that anterograde axonal transport to the colliculi is intact along both experimental and control pathways. Quantitative results are summarized in Table 2. Scale bars: 1 mm (A applies to A and B; D applies to D and E).
Table 2.
Results of Anterograde Axonal Transport Assay
|
Structure and Time Point |
Experimental Intensity, Mean (SD), AU |
Control Intensity, Mean (SD), AU |
Experimental Minus Control, Divided by Control, Mean (SD), % |
P
Value, Wilcoxon Nonparametric Paired
t-Test |
n |
| Optic nerves, 1 wk | 48.6 (6.9) | 49.7 (4.5) | −1.7 (−15.7) | 0.773 | 5 |
| Optic nerves, 2 wk | 55.7 (8.3) | 57.4 (8.7) | −2.7 (−7.3) | 0.313 | 5 |
| Superior colliculi, 1 wk | 75.3 (7.9) | 77.1 (6.7) | −2.4 (−5.2) | 0.438 | 5 |
| Superior colliculi, 2 wk | 86.6 (19.5) | 95.2 (16.2) | −9.6 (−9.9) | 0.188 | 5 |
Retrograde Axonal Transport Assay
Figure 4 shows a representative example of the retrograde axonal transport assay results. Twenty-four hours after bilateral CTB injections into the superior colliculi, punctate CTB fluorescence of RGC somas is visible at equivalent brightness and density in both experimental and fellow control retinas as measured either in vivo by CSLO (Figs. 4A, 4B) or postmortem by microscopy (Figs. 4C, 4D). Symmetric fluorescence of both hemispheres of the superior colliculus as imaged by postmortem CSLO demonstrates that both CTB injections were successful (Fig. 4E). These results indicate uninterrupted retrograde transport along RGC axons from the superior colliculi to RGC somas. Quantitative results of the full retrograde axonal transport assay data set at the 1-week and 2-week follow-up are listed in Table 3. There was no difference in the density of fluorescent RGC somas between experimental and fellow control eyes. Hence, there was no evidence of retrograde transport disruption persisting at either 1 or 2 weeks after an 8-hour episode of acute IOP elevation to 50 mm Hg with this assay.
Figure 4.
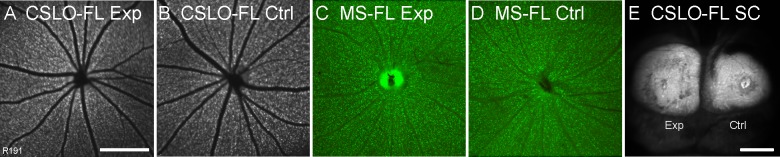
Results of the retrograde axonal transport assay 2 weeks after the 8-hour episode of acute IOP elevation to 50 mm Hg in the experimental eye in one representative animal. (A, B) In vivo CSLO-FL images of the experimental (A) and fellow control (B) ocular fundi showing equivalent fluorescence (intensity and density) of RGC somas 24 hours after bilateral injections of CTB into the superior colliculi. (C, D) Postmortem fluorescence microscopy (MS-FL) of experimental (C) and fellow control (D) retinal whole mounts also shows equivalent fluorescence between eyes. The fluorescence of the RGCs is similar between the CSLO and microscopy images, although the optic disc is usually brighter in the microscopy image. Note that the disc in (D) has been partially removed during the retina dissection, so it is not as bright as the disc in C. These results indicate uninterrupted retrograde axonal transport of CTB along experimental and control pathways. Quantitative results are summarized in Table 3. (E) Postmortem CSLO-FL image of the superior colliculi shows that CTB injections were successful. Scale bars: 1 mm (A applies to A–D).
Table 3.
Results of Retrograde Axonal Transport Assay
|
Method and Time Point |
Experimental Density, Mean (SD), cells/mm2 |
Control Density, Mean (SD), cells/mm2 |
Experimental Minus Control, Divided by Control, Mean (SD), % |
P
Value, Wilcoxon Nonparametric Paired
t-Test |
n |
| In vivo CSLO, 1 wk | 1651 (153) | 1615 (135) | 2.4 (8.2) | 0.625 | 5 |
| In vivo CSLO, 2 wk | 1628 (143) | 1571 (185) | 3.9 (4.4) | 0.250 | 4 |
| Microscope, 1 wk | 2277 (183) | 2219 (178) | 2.7 (5.3) | 0.438 | 5 |
| Microscope, 2 wk | 2260 (294) | 2194 (340) | 3.7 (10.6) | 0.313 | 5 |
Follow-up IOP
To determine if the RNFL thickening might have been caused by a transient IOP decrease due to leakage of aqueous humor after cannula removal, IOP was measured by tonometry in a subset of animals immediately before and after cannulation and at follow-up time points. In group 1 experimental eyes (n = 15), IOP decreased from a baseline of 11.3 ± 1.5 mm Hg to 7.2 ± 2.6 mm Hg (P < 0.001) at approximately 15 minutes after cannula removal and had recovered to 11.1 ± 3.0 mm Hg by the 1-day follow-up (Fig. 5). Fellow control eyes remained similar to baseline after cannulation. The IOP was measured under general anesthesia and was therefore lower than the IOP in awake rats due to the known effect of anesthesia on IOP.90 Group 2 and group 3 experimental eyes also showed a similar pattern of IOP decrease immediately after cannula removal that recovered by the 1-day follow-up (Fig. 5). The transient decrease in IOP in all experimental eyes after cannula removal correlates with the transient 1-day increase in RNFLT in experimental eyes of sham animals (group 3). Hence, the cannulation procedure may be the source of the 1-day RNFL thickening in sham animals and may contribute to the initial RNFL thickening in group 2 and group 3 animals at the 1-day follow-up only.
Figure 5.

The IOP of experimental (Exp) and fellow control (Ctrl) eyes under general anesthesia at baseline immediately before cannulation (BL), at 0 to 5 minutes and 6 to 20 minutes after cannula removal, and at the 1-day, 2-day, 3-day, 7-day, and 14-day follow-up. Data are shown for group 1 (n = 15) and group 2 (n = 4), which both had IOP elevation to 50 mm Hg for 8 hours, and for group 3 (n = 4, sham), which had IOP held at 15 mm Hg for 8 hours. The IOP in the experimental eyes decreased immediately after cannula removal but recovered relative to baseline and fellow control by the 1-day follow-up. Error bars = SEM. *P < 0.05 in experimental eyes compared with fellow control at that time point. §P < 0.05 in experimental eyes compared with baseline (Bonferroni post hoc test).
RGC Density
To assess whether there was any change in RGC density after IOP elevation, retinas from all four groups groups were stained with an antibody against Brn3a. Antibodies against Brn3a label most (92%–95%) RGCs, and it is known that Brn3a is a reliable marker to identify and quantify RGC density in both normal retinas and retinas with RGC loss.77,86,87 The Brn3a-positive RGC density (Fig. 6) was equivalent in experimental and control retinas (P = 0.74) and across all groups (P = 0.34). This indicates that there was no loss of RGCs at even 6 weeks (2998 ± 347 cells/mm2 in the experimental eyes and 2844 ± 282 cells/mm2 in the control eyes of group 2) after the 8-hour IOP elevation event relative to normal naive rats (2772 ± 250 cells/mm2 in the right eyes and 2823 ± 139 cells/mm2 in the left eyes of group 4) and sham rats (2574 ± 300 cells/mm2 in the experimental eyes and 2538 ± 391 cells/mm2 in the control eyes of group 3). The RGC densities determined by Brn3a labeling over a large region of central retina in this study are slightly higher than those reported for pigmented rats in a previous study (1331 ± 129 cells/mm2),87 which were also determined by Brn3a immunohistochemistry. This is likely because the previous study determined the average density for the entire retina, whereas estimates in the present study were derived without including the far periphery, where RGC density is lower. However, RGC densities derived from retrograde Fluorogold labeling for a similar area of retina as the present study are comparable in another study91 (2113 ± 144 cells/mm2) to those found in this study using Brn3a. Although the densities derived in the present study by Brn3a analysis are comparable to those derived by retrograde transport of CTB for a similar retinal sample area and imaged by microscopy, the densities derived in vivo by CSLO imaging are slightly lower (Table 3), despite being based on a more central portion of the retina, probably because of a combination of lower resolution and signal-to-noise ratio compared with postmortem, microscopy-derived estimates.
Figure 6.
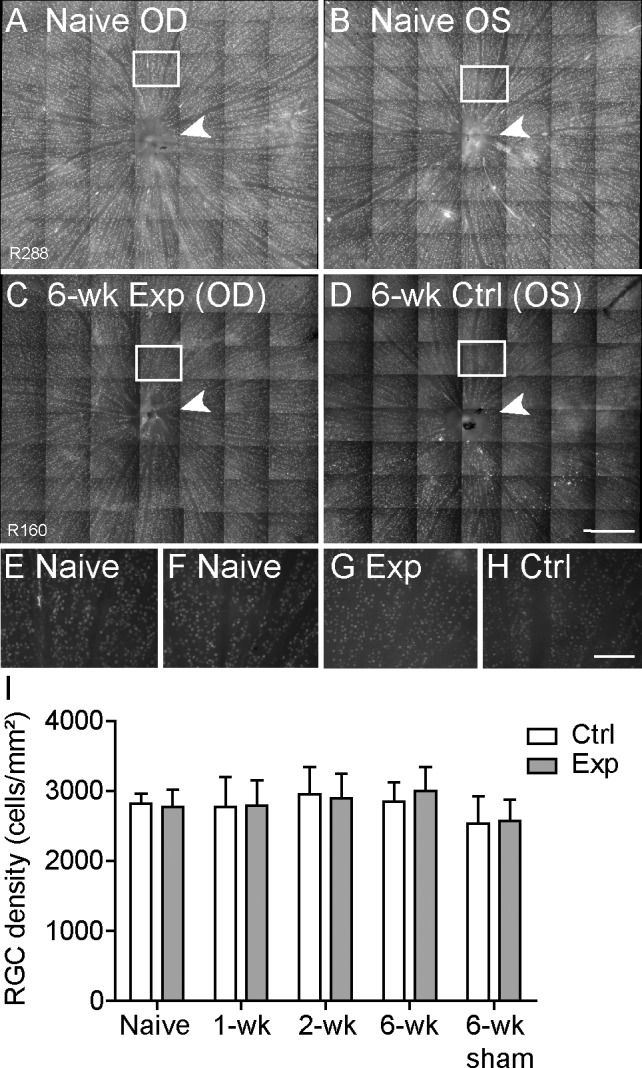
Density of Brn3a-labeled RGCs across 8.1 ± 1.0 mm2 of central retina. (A–D) Representative example of RGC somas seen in a montage of micrographs (×20) from right (A) and left (B) naive normal retinas and in experimental (C) and fellow control (D) retinas from a group 2 rat sacrificed 6 weeks after the IOP elevation episode. Arrowheads indicate the optic disc. (E–H) Higher-resolution micrographs from boxed regions in (A–D) are shown in (E–H), respectively. (I) Quantification of RGC density for experimental (Exp) and fellow control (Ctrl) eyes from rats sacrificed 1 and 2 weeks (group 1 [n = 9 and n = 11, respectively]) and 6 weeks (group 2 [n = 4]) after IOP elevation to 50 mm Hg for 8 hours, along with rats sacrificed 6 weeks (group 3 [n = 4]) after sham cannulation to 15 mm Hg for 8 hours and naive normal rats (group 4 [n = 5]). There was no loss of RGCs in experimental eyes relative to fellow control eyes (P = 0.74) or between groups (P = 0.34). Error bars = SEM. Scale bars: 0.5 mm (A–D); 100 μm (E–H).
Given the variances observed in this study, there was 80% power to detect an 11% reduction in the mean RGC (Brn3a+) density for a group of four (e.g., group 2 observed for 6 weeks). This evidence supports the findings that there was no RNFL thinning at 6 weeks and that the initial RNFL thickening seen in this study was a transient event (i.e., the normalization of group 2 RNFLT at the 3-week time point was not masking long-standing RNFL thickening offset by concurrent RGC loss).
Microglial Density
To determine whether RNFL microglial activation and potential increase in cell density could have contributed to the transient increase in RNFLT in rats that had the acute IOP elevation event, retinas from all groups were stained with an antibody against Iba-1. The microglial density was equivalent (P = 0.92) in experimental and control eyes across all groups (Fig. 7). There was no increase in the RNFL microglia at 1 week (616 ± 40 in the experimental eyes and 662 ± 86 in the control eyes) or 2 weeks (622 ± 59 in the experimental eyes and 663 ± 84 in the control eyes) after the IOP elevation event relative to the normal naive group (668 ± 28 in group 4) (P > 0.05 for all). This suggests that the RNFL thickening that occurs at 1 and 2 weeks after IOP elevation is not due to a significant increase in the number of microglia within the RNFL. Furthermore, there was no evidence of morphologic transformation of the microglia in experimental eyes from a ramified to an amoeboid (large, irregular) shape (Fig. 7E–H), as has been previously noted in retinas with activated microglia.92–95
Figure 7.
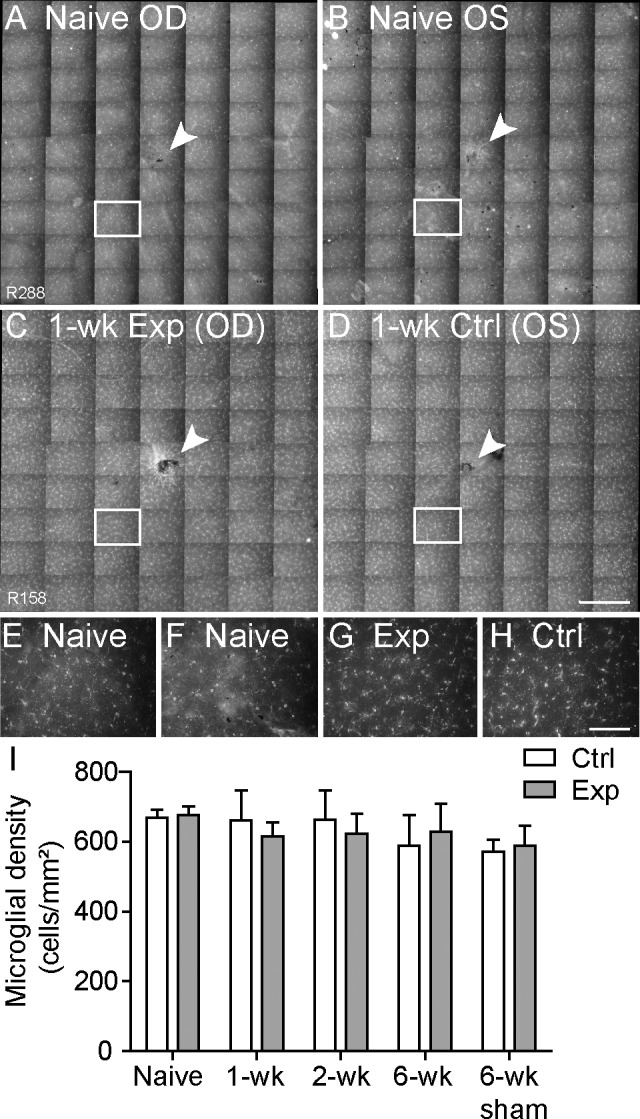
Density of Iba-1–labeled microglia within the RNFL across 7.6 ± 1.8 mm2 of central retina. (A–D) Representative example of microglia seen in a montage of micrographs from right (A) and left (B) naive normal retinas and experimental (Exp [C]) and fellow control (Ctrl [D]) retinas from a rat sacrificed 1 week after IOP elevation, which correlates with the peak RNFL thickening seen in group 1 and group 2 experimental eyes. Arrowheads indicate the optic disc. (E–H) Higher-resolution micrographs from boxed regions in A through D are shown in E through H, respectively. The shape and density of microglia appear similar between retinas, indicating no activation of microglia in experimental eyes. (I) Quantification of RNFL microglia density for experimental and fellow control eyes sacrificed at 1 and 2 weeks (group 1 [n = 4 each]) and 6 weeks (group 2 [n = 4]) after IOP elevation to 50 mm Hg for 8 hours, along with rats sacrificed 6 weeks (group 3 [n = 4]) after sham cannulation to 15 mm Hg for 8 hours and naive normal rats (group 4 [n = 5]). Microglial density was equivalent between experimental relative to fellow control eyes (P = 0.92) and in experimental and sham groups relative to the naive normal group (P > 0.05 for all). This indicates that microglial activation is not responsible for RNFL thickening. Error bars = SEM. Scale bars: 0.5 mm (applies to A–D); 100 μm (applies to E–H).
Discussion
Short-term (≤4 hours) acute elevations of IOP to subischemic levels (≤50 mm Hg) cause deformation of the ONH and peripapillary tissues, dysfunction of retinal neuronal signaling, reduction of ocular blood flow (including within the ONH), and disruption of axonal transport, all of which are reversible within minutes to hours of IOP normalization, without evidence of permanent loss in structure or function. This study used a longer-term acute elevation of IOP (50 mm Hg for 8 hours) and found that it also has no lasting effect on anterograde or retrograde transport of small vesicular cargoes at either 1 or 2 weeks or on RNFLT at 6 weeks. IOP elevation to 50 mm Hg for 6 hours was previously found to reduce retrograde transport by 74%, and IOP elevation to 75 mm Hg for 6 hours reduced transport by 83%.62 Therefore, it is expected that 8 hours of IOP elevation to 50 mm Hg will cause substantial disruption to axonal transport during the period of IOP elevation, although the results herein show no persisting transport deficit by 1 week after the acute IOP event. This is consistent with previous studies60,61 showing recovery of retrograde transport 2 hours after IOP normalization from shorter-term acute IOP elevation events of 50 mm Hg for 2 hours and 180 mm Hg for 10 minutes.
This study used an assay time point of 24 hours after injection, which is approximately 18 hours after CTB is first noticed above background fluorescence at the superior colliculus in the anterograde assay and approximately 21 hours after it is seen in the RGCs in the retrograde assay.73 It is possible that the transport assay used in this study (specifically, the 24-hour postinjection time point) might not be sensitive to subtle delays in transport rate (e.g., due to mildly dysfunctional axons). However, it is sensitive to transport blockade as shown previously.73 Even if only some of the axon bundles in the nerve are persistently affected by transport blockade 1 week after 8 hours of acute IOP elevation, the 24-hour assay will still be able to detect this regional blockade of transport due to retinotopic mapping within the pathway (i.e., patches of missing or reduced fluorescence within the superior colliculus would result in an overall decrease of its total fluorescence). However, if any transport rate reduction persisting to 1 week after acute IOP elevation was subtle enough to go undetected at the 24-hour postinjection time point, it was also evidently not severe enough to fatally injure RGCs (measured by Brn3a+ densities after death) and/or cause RGC axon loss (measured by RNFLT in vivo).
Despite seeing no changes in axonal transport at either 1 or 2 weeks after the acute event or any evidence of RNFL loss at 6 weeks, this study did show substantial RNFL thickening that is seen by 1 day (11% [5 μm]), is greatest at 3 to 7 days (15%–19% [7–9 μm]), and is resolved by 3 weeks. A sham group with IOP of 15 mm Hg for 8 hours also showed some RNFL thickening (6% [3 μm]) at 1 day that had resolved by 3 days. This corresponds to the 1-day period of mild IOP decrease (≤6 mm Hg) seen after cannula removal. Hence, approximately half of the RNFL thickening seen at 1 day in the 50–mm Hg groups is likely due to the transient IOP decrease associated with the cannulation procedure. However, the RNFL thickening in the 50–mm Hg group that persists between 1 day and 3 weeks is due to the 8-hour episode of acute IOP elevation per se because this was not observed in sham eyes. The RT results show that the thickening is only in the RNFL, and quadrant analysis demonstrates no predilection of RNFL thickening for any specific quadrant.
The RNFL thickening is of similar magnitude and sequence to that found in patients observed after treatment for unilateral acute angle-closure glaucoma (IOP >40 mm Hg).96,97 These patients had a thickened RNFL in all quadrants (except temporal) relative to their fellow eyes by 3 days (peak, 17.8% increase) after the treated acute angle-closure episode, which was still partially elevated at 2 weeks (7.1% increase) and had recovered by 1 month. However, eyes that had experienced the angle-closure attack showed progressive RNFL thinning from 1 to 6 months (13.2%–22.7% decrease) suggestive of permanent axon loss, which is inconsistent with the present rat study, which found no RNFL thinning even at the 6-week follow-up. Unfortunately, the level and duration of acute IOP elevation in the human acute angle-closure eyes are not known and cannot be compared with our study. However, it is likely that the magnitude and duration of IOP elevation in the human angle-closure eyes exceeded those of our study and contribute to the difference in reversible (no RNFL thinning or RGC loss in rats after 8 hours of 50 mm Hg at 6 weeks) versus irreversible (progressive RNFL thinning in human eyes treated for acute angle glaucoma >40 mm Hg after 1 month) effects on RGCs. Underlying species differences in susceptibility may also contribute but are beyond the scope of this study.
Aging increases the susceptibility of humans to glaucomatous optic neuropathy, possibly in an exponential relationship.98,99 Rat models have also shown increased RGC susceptibility to permanent injury after IOP elevation and sham cannulation insult in older eyes.100,101 Furthermore, it is known that spontaneous axonal degeneration in the optic nerves of Brown Norway rats increases at an exponential rate with age, showing greater degeneration in 31-month-old rats than in 5-month-old rats.102 This study used young rats (age range, 8–12 weeks); hence, it is possible that repeating this experiment in older animals may show different outcomes for RNFLT and axonal transport and could possibly even produce permanent RGC injury.
A previous study42 in our laboratory assessed the effect of acute IOP elevation to 50 mm Hg for 1 hour on peripapillary RT during and immediately after the event. It found a rapid (within seconds) decrease in peripapillary RT to a maximum of 11% from baseline within the central 10° from the disc. Reversal to baseline RT occurred in approximately 30 minutes, and there was no long-term effect on RT at either 2 or 4 weeks. This indicates that the RNFL thickening seen in the present study likely did not occur until well after cannulation removal and IOP normalization. Furthermore, the longer-term approach of 8 hours of IOP elevation had a greater impact on the RNFL response than the shorter-term 1-hour elevation because RNFL thickening was seen at the 2-week follow-up in the present study but not in the short-term study. A recent study68 of the effect of intermittent IOP elevation in a rat model found permanent RGC injury (22%–25% RNFL thinning, 7%–10% RGC loss, and 6% axon loss) after 6 weeks of daily 1-hour IOP elevation to 35 mm Hg. Given that 8 hours of 50 mm Hg did not cause permanent damage to RGCs at 6 weeks but 1 hour of 35 mm Hg daily for 6 weeks did, a combination of the IOP elevation level and the duration seems to be both important in whether irreversible injury occurs.
The RNFL thickening before thinning was not noted in two chronic IOP elevation studies19,71 that observed RNFLT longitudinally with SD-OCT, although a study103 in monkeys did show a small (2.4%) increase in RNFLT immediately before ONH topography change and subsequent RNFL thinning. The chronic models of IOP elevation generally have a lower IOP (peak and sustained) than the 50–mm Hg elevation used in the present study but have a longer duration of elevation. Hence, it appears again that both the level and duration of IOP elevation in this study have contributed to the 3-week course of RNFL thickening. Because there was no loss of Brn3a-labeled RGCs at the 6-week follow-up, the RNFL thickening effect seems to be transient and is not simply a consequence of factors other than RGC loss masking corresponding axon loss from the RNFL at later time points. However, we cannot confirm that there was no peripheral cell loss because we only measured RGC density in the central 8.1 mm2 of retina.
Studies of the ocular blood flow response to acute IOP elevation have shown a progressive reduction in blood flow at the ONH and in the central retinal artery with increasing IOP. Elevating IOP to 45 mm Hg in human patients showed a reduction of blood flow in the retinal arterioles and the ONH, without corresponding change in the ophthalmic artery, indicating that blood flow in the central retinal artery was highly dependent on IOP.104,105 In nonhuman primates, the blood flow in the ONH reduced within seconds of the IOP being acutely elevated from 10 to 30 mm Hg at low and medium blood pressures but did not change at high blood pressure.44 When the IOP was lowered from 30 mm Hg back to 10 mm Hg, the ONH blood flow returned to baseline within minutes. In response to acute IOP elevation, the ONH blood flow starts recovering as the IOP is still increasing (i.e., the time taken for blood flow to change is shorter than the time taken for IOP to change), indicating that autoregulation responses activate immediately after IOP elevation.45 Furthermore, oxidative stress and mitochondrial dysfunction from reduced ocular blood flow have been associated with RGC death and glaucoma pathogenesis.106–108 Hence, it appears that reduced blood flow is rapidly reversed following IOP normalization after acute IOP elevation to nonischemic levels, occurring much faster than the 3 weeks it took for RNFL thickening to recover. Hypoxia and subsequent permanent RGC damage occur at IOPs above 70 mm Hg.47,54
Electroretinogram studies of the acute IOP model in rat found functional changes in RGC components with IOPs of 30 to 50 mm Hg50,53 and observed for a fixed IOP elevation that functional recovery is linearly related to IOP duration time.51 Extrapolating from these data51 predicts RGC-specific functional recovery by 11.2 hours after an IOP insult of 50 mm Hg for 8 hours. Moreover, permanent loss of RGC function and a decrease in histologic RGC layer density at 4 weeks occurs with an IOP of 60 to 70 mm Hg sustained for at least 105 minutes, while complete long-term functional recovery (and no RGC density loss) occurs at IOPs less than 60 mm Hg.54 Integrating our results with the previous studies shows that recovery of blood flow (within minutes), RGC function (by 1 day), axonal transport (by 7 days), and RNFL thickening (by 3 weeks) occurs readily after IOP normalization from 8 hours at 50 mm Hg, despite their all being highly affected during the IOP elevation episode itself. Given that transport disruption is potentially the causative event,16,109 it is notable that the RNFL thickening response peaks (3–7 days) and remains present (3 weeks) for longer than the presumed axonal transport disruption (<1 week).
Immunohistochemical labeling of microglia with an antibody against Iba-1 showed that there was no increased density of microglia in the RNFL in response to the 8 hours of 50 mm Hg at 1, 2, or 6 weeks compared with naive normal control retinas. This suggests that the RNFL thickening response (maximal at 3–7 days) was not due to microglial activation. This contrasts with a previous acute IOP elevation study110 (50 mm Hg for 30 minutes) that found an increased density of Iba-1–positive hyalocytes and subretinal macrophages at 1 week, although the study also found equivalent changes in fellow sham eyes. The sham eye effect suggests that those results were primarily due to the cannulation procedure itself and agrees with another study111 finding that eye puncture activates retinal microglia. Studies using chronic models of IOP elevation have shown microglia clustering early in the progression of DBA/2J inherited glaucoma (bilateral)84 and microglial activation in both experimental and control eyes of a laser-induced IOP elevation model.85 However, the reason for microglial activation in these studies is also difficult to interpret due to a lack of internal control or a control eye effect. Future studies are required to fully understand the pathophysiological sequence and cause of the transient RNFL thickening.
In summary, this study found that acute IOP elevation to 50 mm Hg for 8 hours in rats has no permanent effect on anterograde or retrograde axonal transport at either 1 or 2 weeks but does result in substantial, transient RNFL thickening (peak, 15%–19%) that lasts for up to 3 weeks. Both axonal transport and RNFL thickening are reversible events that do not result in loss of RGC density at 6 weeks. Future studies will assess axonal transport alongside in vivo longitudinal measures of RNFLT in a rat model of longer-duration IOP elevation in both young and old rats to further elucidate reversible and irreversible aspects of the pathophysiological response to IOP elevation.
Acknowledgments
The authors thank Catherine Morgans for providing the mouse anti-Brn3a antibody and Grant Cull for technical assistance.
Supported by Grant R21-EY021311 from the National Institutes of Health (BF), Legacy Good Samaritan Foundation, and Heidelberg Engineering GmbH (equipment).
Disclosure: C.J. Abbott, None; T.E. Choe, None; T.A. Lusardi, None; C.F. Burgoyne, Heidelberg Engineering GmbH (F, C); L. Wang, None; B. Fortune, Heidelberg Engineering GmbH (F)
References
- 1. Quigley HA. Neuronal death in glaucoma. Prog Retin Eye Res. 1999; 18: 39–57 [DOI] [PubMed] [Google Scholar]
- 2. Weinreb RN, Khaw PT. Primary open-angle glaucoma. Lancet. 2004; 363: 1711–1720 [DOI] [PubMed] [Google Scholar]
- 3. Danesh-Meyer HV, Boland MV, Savino PJ, et al. Optic disc morphology in open-angle glaucoma compared with anterior ischemic optic neuropathies. Invest Ophthalmol Vis Sci. 2010; 51: 2003–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Emery JM, Landis D, Paton D, Boniuk M, Craig JM. The lamina cribrosa in normal and glaucomatous human eyes. Trans Am Acad Ophthalmol Otolaryngol. 1973; 78: OP290–OP297 [PubMed] [Google Scholar]
- 5. Vrabec F. Glaucomatous cupping of the human optic disk. Arch Klin Exp Ophthalmol. 1976; 198: 223–234 [DOI] [PubMed] [Google Scholar]
- 6. Quigley HA, Addicks EM, Green WR, Maumenee AE. Optic nerve damage in human glaucoma, II: the site of injury and susceptibility to damage. Arch Ophthalmol. 1981; 99: 635–649 [DOI] [PubMed] [Google Scholar]
- 7. Gelatt K. Animal models for glaucoma. Invest Ophthalmol Vis Sci. 1977; 16: 592–596 [PubMed] [Google Scholar]
- 8. Goldblum D, Mittag T. Prospects for relevant glaucoma models with retinal ganglion cell damage in the rodent eye. Vision Res. 2002; 42: 471–478 [DOI] [PubMed] [Google Scholar]
- 9. Levkovitch-Verbin H. Animal models of optic nerve diseases. Eye. 2004; 18: 1066–1074 [DOI] [PubMed] [Google Scholar]
- 10. Libby RT, Anderson MG, Pang IH, et al. Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis Neurosci. 2005; 22: 637–648 [DOI] [PubMed] [Google Scholar]
- 11. Weinreb RN, Lindsey JD. The importance of models in glaucoma research. J Glaucoma. 2005; 14: 302–304 [DOI] [PubMed] [Google Scholar]
- 12. Pang IH, Clark AF. Rodent models for glaucoma retinopathy and optic neuropathy. J Glaucoma. 2007; 16: 483–505 [DOI] [PubMed] [Google Scholar]
- 13. Morrison JC, Johnson E, Cepurna WO. Rat models for glaucoma research. Prog Brain Res. 2008; 173: 285–301 [DOI] [PubMed] [Google Scholar]
- 14. Chauhan BC, Pan J, Archibald ML, LeVatte TL, Kelly ME, Tremblay F. Effect of intraocular pressure on optic disc topography, electroretinography, and axonal loss in a chronic pressure-induced rat model of optic nerve damage. Invest Ophthalmol Vis Sci. 2002; 43: 2969–2976 [PubMed] [Google Scholar]
- 15. Burgoyne CF, Downs JC, Bellezza AJ, Suh JK, Hart RT. The optic nerve head as a biomechanical structure: a new paradigm for understanding the role of IOP-related stress and strain in the pathophysiology of glaucomatous optic nerve head damage. Prog Retin Eye Res. 2005; 24: 39–73 [DOI] [PubMed] [Google Scholar]
- 16. Yang H, Downs JC, Bellezza A, Thompson H, Burgoyne CF. 3-D histomorphometry of the normal and early glaucomatous monkey optic nerve head: prelaminar neural tissues and cupping. Invest Ophthalmol Vis Sci. 2007; 48: 5068–5084 [DOI] [PubMed] [Google Scholar]
- 17. Howell GR, Libby RT, Jakobs TC, et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007; 179: 1523–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roberts MD, Grau V, Grimm J, et al. Remodeling of the connective tissue microarchitecture of the lamina cribrosa in early experimental glaucoma. Invest Ophthalmol Vis Sci. 2009; 50: 681–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Strouthidis NG, Fortune B, Yang H, Sigal IA, Burgoyne CF. Longitudinal change detected by spectral domain optical coherence tomography in the optic nerve head and peripapillary retina in experimental glaucoma. Invest Ophthalmol Vis Sci. 2011; 52: 1206–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang H, Williams G, Downs JC, et al. Posterior (outward) migration of the lamina cribrosa and early cupping in monkey experimental glaucoma. Invest Ophthalmol Vis Sci. 2011; 52: 7109–7121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Burgoyne CF. A biomechanical paradigm for axonal insult within the optic nerve head in aging and glaucoma. Exp Eye Res. 2011; 93: 120–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marx MS, Podos SM, Bodis-Wollner I, Lee PY, Wang RF, Severin C. Signs of early damage in glaucomatous monkey eyes: low spatial frequency losses in the pattern ERG and VEP. Exp Eye Res. 1988; 46: 173–184 [DOI] [PubMed] [Google Scholar]
- 23. Frishman LJ, Shen FF, Du L, et al. The scotopic electroretinogram of macaque after retinal ganglion cell loss from experimental glaucoma. Invest Ophthalmol Vis Sci. 1996; 37: 125–141 [PubMed] [Google Scholar]
- 24. Mittag TW, Danias J, Pohorenec G, et al. Retinal damage after 3 to 4 months of elevated intraocular pressure in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2000; 41: 3451–3459 [PubMed] [Google Scholar]
- 25. Fortune B, Bui BV, Morrison JC, et al. Selective ganglion cell functional loss in rats with experimental glaucoma. Invest Ophthalmol Vis Sci. 2004; 45: 1854–1862 [DOI] [PubMed] [Google Scholar]
- 26. Fortune B, Burgoyne CF, Cull GA, Reynaud J, Structural Wang L. and functional abnormalities of retinal ganglion cells measured in vivo at the onset of optic nerve head surface change in experimental glaucoma. Invest Ophthalmol Vis Sci. 2012; 53: 3939–3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schmidl D, Garhofer G, Schmetterer L. The complex interaction between ocular perfusion pressure and ocular blood flow: relevance for glaucoma. Exp Eye Res. 2011; 93: 141–155 [DOI] [PubMed] [Google Scholar]
- 28. Wang L, Cull GA, Piper C, Burgoyne CF, Fortune B. Anterior and posterior optic nerve head blood flow in nonhuman primate experimental glaucoma model measured by laser speckle imaging technique and microsphere method. Invest Ophthalmol Vis Sci. 2012; 53: 8303–8309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lavery WJ, Muir ER, Kiel JW, Duong TQ. Magnetic resonance imaging indicates decreased choroidal and retinal blood flow in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2012; 53: 560–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cull G, Burgoyne CF, Fortune B, Wang L. Longitudinal hemodynamic changes within the optic nerve head in experimental glaucoma. Invest Ophthalmol Vis Sci. 2013; 54: 4271–4277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quigley HA, Addicks EM. Chronic experimental glaucoma in primates, II: effect of extended intraocular pressure elevation on optic nerve head and axonal transport. Invest Ophthalmol Vis Sci. 1980; 19: 137–152 [PubMed] [Google Scholar]
- 32. Martin KRG, Quigley HA, Valenta D, Kielczewski J, Pease ME. Optic nerve dynein motor protein distribution changes with intraocular pressure elevation in a rat model of glaucoma. Exp Eye Res. 2006; 83: 255–262 [DOI] [PubMed] [Google Scholar]
- 33. Buckingham BP, Inman DM, Lambert W, et al. Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J Neurosci. 2008; 28: 2735–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Salinas-Navarro M, Alarcón-Martínez L, Valiente-Soriano FJ, et al. Ocular hypertension impairs optic nerve axonal transport leading to progressive retinal ganglion cell degeneration. Exp Eye Res. 2010; 90: 168–183 [DOI] [PubMed] [Google Scholar]
- 35. Crish SD, Sappington RM, Inman DM, Horner PJ, Calkins DJ. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci U S A. 2010; 107: 5196–5201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chidlow G, Ebneter A, Wood JP, Casson RJ. The optic nerve head is the site of axonal transport disruption, axonal cytoskeleton damage and putative axonal regeneration failure in a rat model of glaucoma. Acta Neuropathol. 2011; 121: 737–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Coleman A, Quigley H, Vitale S, Dunkelberger G. Displacement of the optic nerve head by acute changes in intraocular pressure in monkey eyes. Ophthalmology. 1991; 98: 35–40 [DOI] [PubMed] [Google Scholar]
- 38. Burgoyne CF, Quigley HA, Thompson HW, Vitale S, Varma R. Measurement of optic disc compliance by digitized image analysis in the normal monkey eye. Ophthalmology. 1995; 102: 1790–1799 [DOI] [PubMed] [Google Scholar]
- 39. Quigley HA, Pease ME. Change in the optic disc and nerve fiber layer estimated with the glaucoma-scope in monkey eyes. J Glaucoma. 1996; 5: 106–116 [PubMed] [Google Scholar]
- 40. Fortune B, Yang H, Strouthidis NG, et al. The effect of acute intraocular pressure elevation on peripapillary retinal thickness, retinal nerve fiber layer thickness, and retardance. Invest Ophthalmol Vis Sci. 2009; 50: 4719–4726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Strouthidis NG, Fortune B, Yang H, Sigal IA, Burgoyne CF. Effect of acute intraocular pressure elevation on the monkey optic nerve head as detected by spectral domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2011; 52: 9431–9437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fortune B, Choe TE, Reynaud J, et al. Deformation of the rodent optic nerve head and peripapillary structures during acute intraocular pressure elevation. Invest Ophthalmol Vis Sci. 2011; 52: 6651–6661 [DOI] [PubMed] [Google Scholar]
- 43. Riva C, Grunwald J, Petrig B. Autoregulation of human retinal blood flow: an investigation with laser Doppler velocimetry. Invest Ophthalmol Vis Sci. 1986; 27: 1706–1712 [PubMed] [Google Scholar]
- 44. Liang Y, Downs JC, Fortune B, Cull G, Cioffi GA, Wang L. Impact of systemic blood pressure on the relationship between intraocular pressure and blood flow in the optic nerve head of nonhuman primates. Invest Ophthalmol Vis Sci. 2009; 50: 2154–2160 [DOI] [PubMed] [Google Scholar]
- 45. Liang Y, Fortune B, Cull G, Cioffi GA, Wang L. Quantification of dynamic blood flow autoregulation in optic nerve head of rhesus monkeys. Exp Eye Res. 2010; 90: 203–209 [DOI] [PubMed] [Google Scholar]
- 46. Zhi Z, Cepurna WO, Johnson EC, Morrison JC, Wang RK. Impact of intraocular pressure on changes of blood flow in the retina, choroid, and optic nerve head in rats investigated by optical microangiography. Biomed Opt Express. 2012; 3: 2220–2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Holcombe DJ, Lengefeld N, Gole GA, Barnett NL. The effects of acute intraocular pressure elevation on rat retinal glutamate transport. Acta Ophthalmol. 2008; 86: 408–414 [DOI] [PubMed] [Google Scholar]
- 48. Gerstle CL, Anderson DR, Hamasaki DI. Pressure effect on ERG and optic nerve conduction of visual impulse: short-term effects in owl monkeys. Arch Ophthalmol. 1973; 90: 121–124 [DOI] [PubMed] [Google Scholar]
- 49. Siliprandi R, Bucci M, Canella R, Carmignoto G. Flash and pattern electroretinograms during and after acute intraocular pressure elevation in cats. Invest Ophthalmol Vis Sci. 1988; 29: 558–565 [PubMed] [Google Scholar]
- 50. Bui BV, Edmunds B, Cioffi GA, Fortune B. The gradient of retinal functional changes during acute intraocular pressure elevation. Invest Ophthalmol Vis Sci. 2005; 46: 202–213 [DOI] [PubMed] [Google Scholar]
- 51. He Z, Bui BV, Vingrys AJ. The rate of functional recovery from acute IOP elevation. Invest Ophthalmol Vis Sci. 2006; 47: 4872–4880 [DOI] [PubMed] [Google Scholar]
- 52. Nagaraju M, Saleh M, Porciatti V. IOP-dependent retinal ganglion cell dysfunction in glaucomatous DBA/2J mice. Invest Ophthalmol Vis Sci. 2007; 48: 4573–4579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chrysostomou V, Crowston JG. The photopic negative response of the mouse electroretinogram: reduction by acute elevation of intraocular pressure. Invest Ophthalmol Vis Sci. 2013; 54: 4691–4697 [DOI] [PubMed] [Google Scholar]
- 54. Bui BV, Batcha AH, Fletcher EL, Wong VH, Fortune B. Relationship between the magnitude of intraocular pressure during an episode of acute elevation and retinal damage four weeks later in rats [serial online]. PLoS One. 2013; 8: e70513 Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3726657/. Accessed December 28, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Anderson DR, Hendrickson A. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Invest Ophthalmol. 1974; 13: 771–783 [PubMed] [Google Scholar]
- 56. Quigley H, Anderson DR. The dynamics and location of axonal transport blockade by acute intraocular pressure elevation in primate optic nerve. Invest Ophthalmol. 1976; 15: 606–616 [PubMed] [Google Scholar]
- 57. Quigley HA, Anderson DR. Distribution of axonal transport blockade by acute intraocular pressure elevation in the primate optic nerve head. Invest Ophthalmol Vis Sci. 1977; 16: 640–644 [PubMed] [Google Scholar]
- 58. Minckler DS, Bunt AH, Johanson GW. Orthograde and retrograde axoplasmic transport during acute ocular hypertension in the monkey. Invest Ophthalmol Vis Sci. 1977; 16: 426–441 [PubMed] [Google Scholar]
- 59. Quigley HA, Flower RW, Addicks EM, McLeod DS. The mechanism of optic nerve damage in experimental acute intraocular pressure elevation. Invest Ophthalmol Vis Sci. 1980; 19: 505–517 [PubMed] [Google Scholar]
- 60. Johansson JO. Retrograde axoplasmic transport in rat optic nerve in vivo: what causes blockage at increased intraocular pressure? Exp Eye Res. 1986; 43: 653–660 [DOI] [PubMed] [Google Scholar]
- 61. Johansson JO. Inhibition and recovery of retrograde axoplasmic transport in rat optic nerve during and after elevated IOP in vivo. Exp Eye Res. 1988; 46: 223–227 [DOI] [PubMed] [Google Scholar]
- 62. Quigley HA, McKinnon SJ, Zack DJ, et al. Retrograde axonal transport of BDNF in retinal ganglion cells is blocked by acute IOP elevation in rats. Invest Ophthalmol Vis Sci. 2000; 41: 3460–3466 [PubMed] [Google Scholar]
- 63. Balaratnasingam C, Morgan WH, Bass L, Matich G, Cringle SJ, Yu DY. Axonal transport and cytoskeletal changes in the laminar regions after elevated intraocular pressure. Invest Ophthalmol Vis Sci. 2007; 48: 3632–3644 [DOI] [PubMed] [Google Scholar]
- 64. Balaratnasingam C, Morgan WH, Bass L, Cringle SJ, Yu DY. Time-dependent effects of elevated intraocular pressure on optic nerve head axonal transport and cytoskeleton proteins. Invest Ophthalmol Vis Sci. 2008; 49: 986–999 [DOI] [PubMed] [Google Scholar]
- 65. Kitazawa Y, Horie T. Diurnal variation of intraocular pressure in primary open-angle glaucoma. Am J Ophthalmol. 1975; 79: 557–566 [DOI] [PubMed] [Google Scholar]
- 66. Brubaker RF. Targeting outflow facility in glaucoma management. Surv Ophthalmol. 2003; 48: S17–S20 [DOI] [PubMed] [Google Scholar]
- 67. Sung VC, Barton K. Management of inflammatory glaucomas. Curr Opin Ophthalmol. 2004; 15: 136–140 [DOI] [PubMed] [Google Scholar]
- 68. Joos KM, Li C, Sappington RM. Morphometric changes in the rat optic nerve following short-term intermittent elevations in intraocular pressure. Invest Ophthalmol Vis Sci. 2010; 51: 6431–6440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bui BV, He Z, Vingrys AJ, Nguyen CT, Wong VH, Fortune B. Using the electroretinogram to understand how intraocular pressure elevation affects the rat retina [serial online]. J Ophthalmol. 2013; 2013: 262467 Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3570935/. Accessed December 28, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nagata A, Higashide T, Ohkubo S, Takeda H, Sugiyama K. In vivo quantitative evaluation of the rat retinal nerve fiber layer with optical coherence tomography. Invest Ophthalmol Vis Sci. 2009; 50: 2809–2815 [DOI] [PubMed] [Google Scholar]
- 71. Guo L, Normando EM, Nizari S, Lara D, Cordeiro MF. Tracking longitudinal retinal changes in experimental ocular hypertension using the cSLO and spectral domain–OCT. Invest Ophthalmol Vis Sci. 2010; 51: 6504–6513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lozano DC, Twa MD. Quantitative evaluation of factors influencing the repeatability of SD-OCT thickness measurements in the rat. Invest Ophthalmol Vis Sci. 2012; 53: 8378–8385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Abbott CJ, Choe TE, Lusardi TA, Burgoyne CF, Wang L, Fortune B. Imaging axonal transport in the rat visual pathway. Biomed Opt Express. 2013; 4: 364–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Moore RY, Lenn NJ. A retinohypothalamic projection in the rat. J Comp Neurol. 1972; 146: 1–14 [DOI] [PubMed] [Google Scholar]
- 75. Land PW, Lund RD. Development of the rat's uncrossed retinotectal pathway and its relation to plasticity studies. Science. 1979; 205: 698–700 [DOI] [PubMed] [Google Scholar]
- 76. Villegas-Pérez MP, Vidal-Sanz M, Rasminsky M, Bray GM, Aguayo AJ. Rapid and protracted phases of retinal ganglion cell loss follow axotomy in the optic nerve of adult rats. J Neurobiol. 1993; 24: 23–36 [DOI] [PubMed] [Google Scholar]
- 77. Nadal-Nicolás FM, Jiménez-López M, Sobrado-Calvo P, et al. Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naive and optic nerve–injured retinas. Invest Ophthalmol Vis Sci. 2009; 50: 3860–3868 [DOI] [PubMed] [Google Scholar]
- 78. Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun. 1996; 224: 855–862 [DOI] [PubMed] [Google Scholar]
- 79. Neufeld AH, Sawada A, Becker B. Inhibition of nitric-oxide synthase 2 by aminoguanidine provides neuroprotection of retinal ganglion cells in a rat model of chronic glaucoma. Proc Natl Acad Sci U S A. 1999; 96: 9944–9948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yuan L, Neufeld AH. Activated microglia in the human glaucomatous optic nerve head. J Neurosci Res. 2001; 64: 523–532 [DOI] [PubMed] [Google Scholar]
- 81. Naskar R, Wissing M, Thanos S. Detection of early neuron degeneration and accompanying microglial responses in the retina of a rat model of glaucoma. Invest Ophthalmol Vis Sci. 2002; 43: 2962–2968 [PubMed] [Google Scholar]
- 82. Bosco A, Inman DM, Steele MR, et al. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Invest Ophthalmol Vis Sci. 2008; 49: 1437–1446 [DOI] [PubMed] [Google Scholar]
- 83. Ebneter A, Casson RJ, Wood JP, Chidlow G. Microglial activation in the visual pathway in experimental glaucoma: spatiotemporal characterization and correlation with axonal injury. Invest Ophthalmol Vis Sci. 2010; 51: 6448–6460 [DOI] [PubMed] [Google Scholar]
- 84. Bosco A, Steele MR, Vetter ML. Early microglia activation in a mouse model of chronic glaucoma. J Comp Neurol. 2011; 519: 599–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gallego BI, Salazar JJ, de Hoz R, et al. IOP induces upregulation of GFAP and MHC-II and microglia reactivity in mice retina contralateral to experimental glaucoma. J Neuroinflammation. 2012; 9: 92–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Xiang M, Zhou L, Macke J, et al. The Brn-3 family of POU-domain factors: primary structure, binding specificity, and expression in subsets of retinal ganglion cells and somatosensory neurons. J Neurosci. 1995; 15: 4762–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Nadal-Nicolás FM, Jiménez-López M, Salinas-Navarro M, et al. Whole number, distribution and co-expression of Brn3 transcription factors in retinal ganglion cells of adult albino and pigmented rats [serial online]. PLoS One. 2012; 7: e49830 Available at: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3500320/. Accessed December 28, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Santos-Carvalho A, Aveleira CA, Elvas F, Ambrósio AF, Cavadas C. Neuropeptide Y receptors Y1 and Y2 are present in neurons and glial cells in rat retinal cells in culture. Invest Ophthalmol Vis Sci. 2013; 54: 429–443 [DOI] [PubMed] [Google Scholar]
- 89. Ito D, Tanaka K, Suzuki S, Dembo T, Fukuuchi Y. Enhanced expression of Iba1, ionized calcium-binding adapter molecule 1, after transient focal cerebral ischemia in rat brain. Stroke. 2001; 32: 1208–1215 [DOI] [PubMed] [Google Scholar]
- 90. Jia L, Cepurna WO, Johnson EC, Morrison JC. Effect of general anesthetics on IOP in rats with experimental aqueous outflow obstruction. Invest Ophthalmol Vis Sci. 2000; 41: 3415–3419 [PubMed] [Google Scholar]
- 91. Parrilla-Reverter G, Agudo M, Sobrado-Calvo P, Salinas-Navarro M, Villegas-Pérez MP, Vidal-Sanz M. Effects of different neurotrophic factors on the survival of retinal ganglion cells after a complete intraorbital nerve crush injury: a quantitative in vivo study. Exp Eye Res. 2009; 89: 32–41 [DOI] [PubMed] [Google Scholar]
- 92. Langmann T. Microglia activation in retinal degeneration. J Leukoc Biol. 2007; 81: 1345–1351 [DOI] [PubMed] [Google Scholar]
- 93. Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009; 27: 119–145 [DOI] [PubMed] [Google Scholar]
- 94. Liang KJ, Lee JE, Wang YD, et al. Regulation of dynamic behavior of retinal microglia by CX3CR1 signaling. Invest Ophthalmol Vis Sci. 2009; 50: 4444–4451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Vessey KA, Greferath U, Jobling AI, et al. Ccl2/Cx3cr1 knockout mice have inner retinal dysfunction but are not an accelerated model of AMD. Invest Ophthalmol Vis Sci. 2012; 53: 7833–7846 [DOI] [PubMed] [Google Scholar]
- 96. Liu X, Li M, Zhong YM, Xiao H, Huang JJ, Kong XY. Damage patterns of retinal nerve fiber layer in acute and chronic intraocular pressure elevation in primary angle closure glaucoma. Int J Ophthalmol. 2010; 3: 152–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Liu X, Li M, Zhong Y, Xiao H, Huang J, Mao Z. The damage patterns of retinal nerve fiber layer in acute and chronic intraocular pressure elevation in primary angle closure glaucoma. Eye Sci. 2011; 26: 154–160 [DOI] [PubMed] [Google Scholar]
- 98. Dielemans I, Vingerling JR, Wolfs RC, Hofman A, Grobbee DE, de Jong P. The prevalence of primary open-angle glaucoma in a population-based study in The Netherlands: the Rotterdam Study. Ophthalmology. 1994; 101: 1851–1855 [DOI] [PubMed] [Google Scholar]
- 99. Quigley HA. Number of people with glaucoma worldwide. Br J Ophthalmol. 1996; 80: 389–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kong YX, van Bergen N, Bui BV, et al. Impact of aging and diet restriction on retinal function during and after acute intraocular pressure injury [serial online]. Neurobiol Aging. 2012; 33: 1126.e15–1126.e25 Available at: http://www.neurobiologyofaging.org/article/S0197-4580(11)00510-0/abstract. Accessed December 28, 2013 [DOI] [PubMed] [Google Scholar]
- 101. Charng J, Nguyen CT, Vingrys AJ, Jobling AI, Bui BV. Increased susceptibility to injury in older eyes. Optom Vis Sci. 2013; 90: 275–281 [DOI] [PubMed] [Google Scholar]
- 102. Cepurna WO, Kayton RJ, Johnson EC, Morrison JC. Age related optic nerve axonal loss in adult brown Norway rats. Exp Eye Res. 2005; 80: 877–884 [DOI] [PubMed] [Google Scholar]
- 103. Fortune B, Burgoyne CF, Cull G, Reynaud J, Onset Wang L. and progression of peripapillary retinal nerve fiber layer (RNFL) retardance changes occur earlier than RNFL thickness changes in experimental glaucoma. Invest Ophthalmol Vis Sci. 2013; 54: 5653–5661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Harris A, Joos K, Kay M, et al. Acute IOP elevation with scleral suction: effects on retrobulbar haemodynamics. Br J Ophthalmol. 1996; 80: 1055–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Michelson G, Groh M, Langhans M. Perfusion of the juxtapapillary retina and optic nerve head in acute ocular hypertension. Ger J Ophthalmol. 1996; 5: 315–321 [PubMed] [Google Scholar]
- 106. Kong GY, Van Bergen NJ, Trounce IA, Crowston JG. Mitochondrial dysfunction and glaucoma. J Glaucoma. 2009; 18: 93–100 [DOI] [PubMed] [Google Scholar]
- 107. Osborne NN. Mitochondria: their role in ganglion cell death and survival in primary open angle glaucoma. Exp Eye Res. 2010; 90: 750–757 [DOI] [PubMed] [Google Scholar]
- 108. Tezel G. Oxidative stress in glaucomatous neurodegeneration: mechanisms and consequences. Prog Retin Eye Res. 2006; 25: 490–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Nickells RW. The cell and molecular biology of glaucoma: mechanisms of retinal ganglion cell death. Invest Ophthalmol Vis Sci. 2012; 53: 2476–2481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Kezic JM, Chrysostomou V, Trounce IA, McMenamin PG, Crowston JG. Effect of anterior chamber cannulation and acute IOP elevation on retinal macrophages in the adult mouse. Invest Ophthalmol Vis Sci. 2013; 54: 3028–3036 [DOI] [PubMed] [Google Scholar]
- 111. Sobrado-Calvo P, Vidal-Sanz M, Villegas-Pérez MP. Rat retinal microglial cells under normal conditions, after optic nerve section, and after optic nerve section and intravitreal injection of trophic factors or macrophage inhibitory factor. J Comp Neurol. 2007; 501: 866–878 [DOI] [PubMed] [Google Scholar]