

Abstract
Two-dimensional 15N chemical shift/1H chemical shift and three-dimensional 1H-15N dipolar coupling/15N chemical shift/1H chemical shift MAS solid-state NMR correlation spectra of the filamentous bacteriophage Pf1 major coat protein show single-site resolution in noncrystalline, intact-phage preparations. The high sensitivity and resolution result from 1H detection at 600 MHz under 50 kHz magic angle spinning using ~ 0.5 mg of perdeuterated and uniformly 15N-labeled protein in which the exchangeable amide sites are partially or completely back-exchanged (reprotonated). Notably, the heteronuclear 1H-15N dipolar coupling frequency dimension is shown to select among 15N resonances, which will be useful in structural studies of larger proteins where the resonances exhibit a high degree of overlap in multidimensional chemical shift correlation spectra.
Keywords: perdeuteration, proton detection, variable contact time cross-polarization, Pf1 bacteriophage, separated local field spectroscopy, two-dimensional NMR, three-dimensional NMR, fast MAS
1. Introduction
Solid-state NMR spectroscopy can be used to characterize the structure and dynamics of proteins and other biopolymers that are inaccessible to x-ray crystallography and solution-state NMR spectroscopy, including membrane proteins and supramolecular assemblies, such as non-crystalline virus particles [1, 2]. The dominant anisotropic nuclear spin interactions, such as heteronuclear 1H-15N and 1H-13C dipolar couplings, provide valuable information about molecular structure as well as backbone and side-chain dynamics. Multiple NMR techniques have been developed that measure heteronuclear dipolar couplings; these include oriented sample (OS) solid-state NMR of aligned, stationary samples [3, 4] and magic angle spinning (MAS) solid-state NMR experiments of unoriented ‘powder’ samples [5]. Applications of MAS solid-state NMR experiments have expanded significantly in scope and utility with the development of methods for reintroducing and measuring specific dipolar coupling frequencies, although most currently available recoupling techniques are designed for moderate spinning rates (< 20 kHz) [5].
Proton-detection in fast (> 50 kHz) MAS solid-state NMR experiments is emerging as an advantageous approach to studying the structure and dynamics of biomolecules. Dramatic improvements in resolution and sensitivity have been obtained, especially in combination with 1H spin dilution via perdeuteration, where all carbon, nitrogen, and oxygen sites in the protein are bonded to 2H instead of 1H [6–8]. Few or many 1H nuclei can be reintroduced into protein sites through back-exchange with H2O or during biosynthesis. Nonetheless, measurements of heteronuclear dipolar couplings from single sites in biological solids remains challenging, since not only are all homonuclear but also all heteronuclear dipolar interactions are averaged out by the fast magic angle spinning [9]. R-symmetry based approaches have been shown to recouple heteronuclear dipolar couplings under MAS frequencies of 40 kHz [10] and 65 kHz [11]. Recently, a family of simple two-dimensional pulse sequences, based on cross-polarization (CP) with variable contact times and direct 13C or 15N detection have been applied successfully to the measurement of 1H-13C and 1H-15N dipolar couplings on single amino acids and tripeptides under 60 kHz MAS [12].
There is a long history of variable contact time cross-polarization experiments applied to both stationary and spinning samples. Here, we build upon both the older and more recent background to demonstrate two- and three- dimensional proton-detected experiments that provide high spectral resolution and accurate measurements of 1H-15N heteronuclear dipolar coupling frequencies under fast MAS on a noncrystalline sample of the coat protein in intact Pf1 bacteriophage particles.
2. Experimental
2.1. Sample preparation
Pf1 bacteriophage is a filament with its DNA enclosed in a sheath of several thousand copies of coat protein monomers. Uniformly 15N-labeled and 13C/15N-doubly labeled Pf1 bacteriophage were prepared and purified as described previously [13]. Perdeuterated and uniformly 15N-labeled Pf1 phage samples were obtained by infecting the host cell Pseudomonas aeruginosa in Bioexpress® cell growth media (U-2H, 98%; U-15N, 98%) and deuterium oxide (2H, 99.9%) (both from Cambridge Isotope Laboratories, Inc.(www.isotope.com)). Remarkably, the protein yield was not affected by perdeuteration under these growth conditions. The extent of perdeuteration was verified by comparing the 1H solution NMR spectra of the detergent solubilized sample of the perdeuterated Pf1 coat protein to that of a regular, fully protonated sample (Figure S1). As indicated by the lack of signals in the aliphatic region of the spectrum, the deuteration level of the protons bonded to carbons appears to be >90%. Two samples are considered below. The first, referred to as the partially protonated sample, maintained significant levels of deuteration at the slowly exchanging amide protons (NH) in the coat proteins even after purification in protonated aqueous solution. The second, completely protonated sample, was generated by placing the partially-protonated bacteriophage particles in 1H2O in a 60°C water bath for 30 min at pH 8, and then slowly cooling the sample to room temperature [14].
For the NMR experiments, intact isotopically labeled Pf1 bacteriophage particles were concentrated to 150 mg/ml – 200 mg/ml in 5 mM borate solution at pH 8 by ultracentrifugation at 645,000 × g for 20 hr at 15°C. Approximately 2 μl of the concentrated solution of Pf1 bacteriophage particles was transferred into a 1.3 mm outer diameter (OD) rotor for subsequent placement in the stator assembly.
2.2. NMR spectroscopy
Solid-state NMR experiments were performed at 14.1 T (600.01 MHz 1H, 60.8 MHz 15N) on a Bruker AV600 spectrometer equipped with a triple resonance 1.3 mm MAS probe. The sample spinning rate was controlled to 50 kHz (± 2 Hz). The probe temperature was lowered to 14°C using dry-air cooling gas at −36°C and a flow rate of 800 l/h; the actual effective sample temperature based on calibration with KBr [15] was estimated to be 29°C due to frictional heating.
Two-dimensional proton-detected 15N chemical shift/1H chemical shift correlation spectra and three-dimensional proton-detected 1H-15N heteronuclear dipolar coupling/15N chemical shift/1H chemical shift correlation spectra were acquired using the pulse sequence diagrammed in Figure 1, which was adapted from Marchetti et al [16] to include variable contact time (VCT) cross-polarization (CP) in the manner of Paluch et al [12]. In these sequences, hard π/2 pulses were used with nutation frequencies of 83 kHz and 50 kHz for 1H and 15N, respectively. CP was achieved using constant amplitude RF spin-lock pulses with nutation frequencies of 125 kHz for 1H and 75 kHz for 15N (+1 match condition) [17]. The contact time was 2 ms for constant-time CP transfers, and varied between 60 μs and 3840 μs during VCT experiments. XiX 1H decoupling [18], with a nutation frequency of 125 kHz and decoupling pulse width of 57 μs (2.85 τ), was applied during evolution on 15N. MISSISSIPPI water suppression [19] (without homospoil pulses) was implemented during τws on the proton channel using four 75 ms, 9.6 kHz RF saturation pulses. 15N GARP decoupling [20] with irradiation of 22.6 kHz was applied during 1H acquisition.
Figure 1.
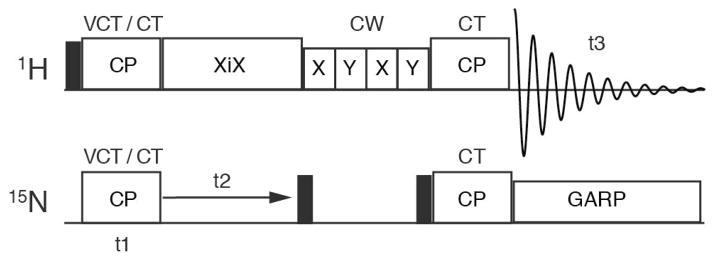
Diagram of the pulse sequence used in the correlation experiments. The two-dimensional experiment utilized constant time (CT) cross polarization (CP) for both magnetization transfer steps. The three-dimensional experiment utilized a variable contact time (VCT) during the first CP. Other aspects are the same for both experiments. Narrow filled rectangles indicated 90° pulses, and blank rectangles are noted with text above or inside.
Correlation spectra were acquired using 64 complex-valued time-domain points with a dwell of 250 μs (spectral width 4 kHz, total data acquisition time 16 ms) in the indirect nitrogen shift dimension, and 256 complex time-domain points with a dwell of 40 μs (spectral width 25 kHz, total data acquisition time 10.2 ms) in the directly detected proton shift dimension. For three-dimensional variable-contact-time experiments, 64 real-valued time-domain points were acquired with an increment of 60 μs. 128 scans per t1 point were averaged for the two-dimensional 15N chemical shift-1H chemical shift correlation experiments; 4 scans per transient were co-added for the three-dimensional correlation experiments. The relaxation delay for all experiments was 2.5 s. The data were zero filled to yield a 1024 × 1024 data matrix for two-dimensional and a 1024 × 128 × 128 data matrix for three-dimensional experiments. Polynomial correction and solvent filter [21] window functions were used in the proton chemical shift and dipolar dimensions, respectively, before the application of a 30° phase shifted sine bell function and an exponential filter (30 Hz – 60 Hz). The NMR data were processed using the program NMRPipe/NMRDraw [22].
3. Results and discussion
We previously determined the three-dimensional structure of Pf1 coat protein subunits in their structural form in intact virus particles and in their membrane-bound form in phospholipid bilayers by Oriented Sample (OS) solid-state NMR spectroscopy [13, 23]. The samples used in the experiments described here are highly concentrated aqueous solutions containing noncrystalline Pf1 bacteriophage particles in which the proteins are immobilized on the NMR timescale by their interactions both within and between the large virus particles; this is reflected by the breadth of the powder patterns (~104 Hz) observed under these sample conditions. With only 46-residues, the coat protein provides an excellent model system for the development of new experimental methods that eventually can be applied to larger, more complex proteins and in different supramolecular assemblies, such as membranes.
Figure 2 compares the glycine resonance regions of the proton-detected two-dimensional 15N chemical shift/1H chemical shift correlation spectra of three different samples of Pf1 bacteriophage. The highest sensitivity and proton chemical shift resolution were obtained by applying 50 kHz MAS on a perdeuterated protein in which the exchangeable amide NH sites are completely back-exchanged (Figure 2A). Individual signals from all six glycines are resolved and assigned (Table S1). 33 out of 45 amide signals were resolved in the two-dimensional spectrum of the completely reprotonated sample (Figure S2A), the majority of which have been assigned in (unpublished) MAS experiments at 11 kHz, independently, and through the 15N resonance assignments of McDermott and colleagues [24]. In principle, complete assignment of the spectrum could be accomplished using proton-detected three-dimensional triple-resonance experiments [16]. 13 resonances are missing in the spectrum of the partially reprotonated sample compared to that of the completely reprotonated sample (Figure S2B). Most of the missing resonances correspond to the residues between L26 and Y43 except for glycines and A36 in the membrane-bound form of the coat protein of Pf1 phage in detergents, suggesting the hydrogen bonding in the transmembrane helix is highly resistant to reprotonation (Figure S1).
Figure 2.

Expanded spectral region of two-dimensional proton-detected HN correlation solid-state NMR spectra of the major coat protein of Pf1 bacteriophage at an MAS rate of 50 kHz. (a) 2H, 15N-labeled sample with complete reprotonation of exchangeable NH sites. (b) 2H, 15N-labeled sample with partial reprotonation of exchangeable NH sites. (c) Regular, protonated, uniformly 15N-labeled sample. The assignments of the Gly residues are marked and the linewidths from the slices through the resonance for residue Gly24 are shown.
The proton resonance linewidths of the six glycines of the perdeuterated sample (113 ± 15 Hz) (Figure 2A) are reduced by a factor of approximately 3 compared to those of the fully protonated sample (302 ± 48 Hz) (Figure 2C) under 50 kHz MAS, but only small isotope effects on the chemical shifts were observed. No significant changes in proton linewidiths were observed between the completely reprotonated sample (Figure 2A) and the partially reprotonated sample (Figure 2B) under our experimental conditions, since the deuterated amides in the partially reprotonated sample which are arrayed in the helix rather than randomly dispersed do not affect the network of the dipolar coupled protons. The chemical shifts, 1H linewidths, and 1H-15N dipolar couplings of glycine residues in three different sample conditions are compared in Table S1. Narrower linewidths are potentially obtainable with faster MAS (> 60 kHz) and higher magnetic fields (> 900 MHz) [16], and by further optimization of labeling and of the sample conditions [6].
In general, perdeuteration of proteins in heterologous expression system requires adaptation protocols in deuterated minimal media that often affects protein yield. However, we note that Pseudomonas aeruginosa is very tolerant to deuterium oxide in the growth media and grows well in the presence of 75% deuterium oxide without adaptation, thereby minimal adaptation was required for high levels of perdeuteration of Pf1 bacteriophage. In Figure 2B, the spectrum was obtained on a sample purified following isolation from the growth media that contained only the most rapidly exchanging amide protons. The missing resonances are associated with sites whose amide protons exchange very slowly due to involvement in hydrogen bonding and/or shielding from the solvent in the interior of the virus particles [13, 14, 23].
The experimental three-dimensional spectrum presented as a cube in Figure 3A was obtained by applying the pulse sequence (Fig. 1) derived from the two-dimensional correlation experiment by systematically varying the length of the first CP contact time. Under the fast (50 kHz) MAS conditions used here, the individual 15N-1H spin pairs along the protein backbone can be considered as isolated two-spin systems [12] and the dipolar transfer of magnetization from 1H to 15N during the spin lock follows the analytical form derived by Vogt et al [25]
Figure 3.

(a) Cube representation of three-dimensional proton-detected solid-state MAS spectrum of 2H, 15N-labeled major coat protein of Pf1 bacteriophage at an MAS rate of 50 kHz obtained by using the pulse sequence in Figure 1. (b) Representative two-dimensional 1H-15N dipolar coupling - 1H chemical shift planes taken from the three-dimensional spectrum in panel (a) at 15N chemical shifts of 128.8 ppm, 114.5 ppm, and 99.7 ppm. The values for 1H-15N dipolar couplings of A46, S41, and G37 are 7.25 kHz, 7.34 kHz, and 7.36 kHz, respectively.
where the dipolar coupling, D, is given in Hz and Jk are Bessel functions of the first kind. The experimental signal is typically shifted so that it decays to zero before Fourier transformation. This can be accomplished by subtracting a constant from the experimental data or, as we do here, using the solvent suppression method of Marion et al [21], that removes additional low-frequency components (that can arise from T1ρ decay during the CP period, for example) along with the zero-frequency term. The Fourier transform of the resulting signal
can be performed analytically [26] to give the frequency-domain signal
Figure 4 shows the theoretical frequency-domain signals for a single NH spin-pair and for two NH spin-pairs with different dipolar couplings. Although the frequency-domain signals show broad powder-pattern responses, multiple spin pairs can clearly be distinguished by their dipolar couplings. In both cases, the dipolar coupling constant can be extracted as √2 times the frequency difference between the discontinuities [12].
Figure 4.
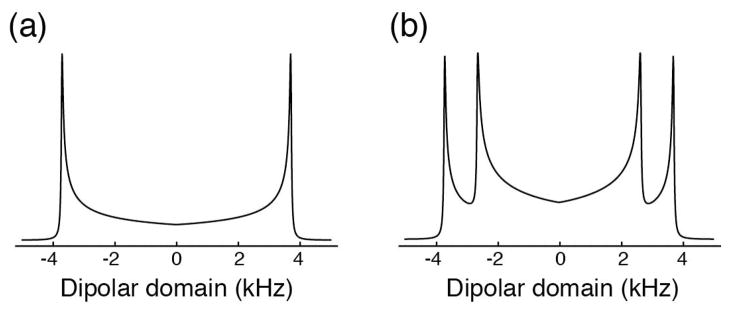
Theoretical lineshapes for the dipolar domain in variable contact time CP experiments. (a) A single spin pair with a dipolar coupling of 10.5 kHz and a line width of 50 Hz. (b) Two spin pairs with dipolar couplings of 10.5 kHz and 7.5 kHz, respectively, and inherent line widths of 50 Hz.
The sample of uniformly 15N and perdeuterated Pf1 bacteriophage yielded the two-dimensional 1H-15N dipolar coupling/1H chemical shift planes taken from the three-dimensional spectrum at 15N chemical shift frequencies of 128.8 ppm, 114.5 ppm, and 99.7 ppm; these spectra show single-site resolution of 1H-15N dipolar couplings for A46, S41, and G37, respectively (Figure 3B). The average 1H-15N dipolar coupling for six glycine residues is 10.49 ± 0.05 kHz, which is close to the rigid lattice value of 10.5 kHz, corresponding to an NH bond length of 1.05 Å, indicating the backbone nitrogen is rigid on time scales of 106 Hz under our sample conditions. The sample has such a high virus particle concentration that rotational motion is suppressed, and the dipolar couplings are effectively those for a rigid lattice. More dilute samples undergo fast diffusion about the major axis of the virus particle, yielding an array of different values for the projected dipolar couplings, which depend on the tertiary structure of the coat protein, and provide input for structure calculations. The range of dipolar couplings provides improved resolution in three-dimensional experiments on samples where the chemical shift frequencies are overlapped but the NH bond vector orientations relative to the rotational axis are different.
The two-dimensional spectra in Figure 2A and B show that high-resolution spectra can be obtained with proton-detection at 50 kHz MAS. They also show that it is possible to count signals and account for all 6 Gly sites in sample that had been ‘back exchanged’ with H2O; it is also possible to differentiate between the very slowly exchanging sites involved in hydrogen bonding and/or shielding from the solvent by the protein coat. This could be verified by comparison with the previously determined structure and assembly of the virus particles. The comparison of the spectra in Figure 2A and C demonstrate the crucial role of perdeuteration in obtaining high-resolution spectra at 50 kHz MAS, which is achievable with a commercial 1.3 mm OD rotor.
In summary, high-resolution proton-detected correlation experiments are demonstrated under fast MAS on a protein in noncrystalline filamentous bacteriophage particles. A three-dimensional experiment with cross polarization using a variable contact time enabled accurate measurement of 1H-15N dipolar coupling frequencies. Although chemical shift dispersion is limited in helical proteins, the dipolar couplings can vary widely and provide a mechanism for resolving signals from many sites; in rotationally aligned samples the orientation-dependent heteronuclear dipolar couplings can be measured for structure determination.
Supplementary Material
Highlights.
Heteronuclear dipolar couplings of non-crystalline proteins can be measured with 1H-detected MAS.
Observed resonances from exchangeable sites reflect hydrogen bonding and buried nature of the residues.
A heteronuclear dipolar coupling dimension contributes to resolution.
Acknowledgments
The research was supported by grants RO1GM099986, RO1GM066978, RO1AI065361 and PO1AI074805 (SJO) and R01GM097569 (LJM) from the National Institutes of Health, and utilized the Biomedical Technology Resource for NMR Molecular Imaging of Proteins at the University of California, San Diego, which is supported by grant P41EB002031.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Opella SJ. Structure Determination of Membrane Proteins in Their Native Phospholipid Bilayer Environment by Rotationally Aligned Solid-State NMR Spectroscopy. Accounts of chemical research. 2013 doi: 10.1021/ar400067z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weingarth M, Baldus M. Solid-State NMR-Based Approaches for Supramolecular Structure Elucidation. Accounts of chemical research. 2013 doi: 10.1021/ar300316e. [DOI] [PubMed] [Google Scholar]
- 3.Waugh JS. Uncoupling of local field spectra in nuclear magnetic resonance: determination of atomic positions in solids. Proceedings of the National Academy of Sciences of the United States of America. 1976;73:1394–1397. doi: 10.1073/pnas.73.5.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu CH, Ramamoorthy A, Opella SJ. High-Resolution Heteronuclear Dipolar Solid-State NMR Spectroscopy. Journal of Magnetic Resonance, Series A. 1994;109:270–272. [Google Scholar]
- 5.De Paepe G. Dipolar recoupling in magic angle spinning solid-state nuclear magnetic resonance. Annual review of physical chemistry. 2012;63:661–684. doi: 10.1146/annurev-physchem-032511-143726. [DOI] [PubMed] [Google Scholar]
- 6.Akbey U, Lange S, Trent Franks W, Linser R, Rehbein K, Diehl A, van Rossum BJ, Reif B, Oschkinat H. Optimum levels of exchangeable protons in perdeuterated proteins for proton detection in MAS solid-state NMR spectroscopy. Journal of biomolecular NMR. 2010;46:67–73. doi: 10.1007/s10858-009-9369-0. [DOI] [PubMed] [Google Scholar]
- 7.Reif B. Ultra-high resolution in MAS solid-state NMR of perdeuterated proteins: implications for structure and dynamics. Journal of magnetic resonance. 2012;216:1–12. doi: 10.1016/j.jmr.2011.12.017. [DOI] [PubMed] [Google Scholar]
- 8.Zhou DH, Nieuwkoop AJ, Berthold DA, Comellas G, Sperling LJ, Tang M, Shah GJ, Brea EJ, Lemkau LR, Rienstra CM. Solid-state NMR analysis of membrane proteins and protein aggregates by proton detected spectroscopy. Journal of biomolecular NMR. 2012;54:291–305. doi: 10.1007/s10858-012-9672-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saalwachter K, Graf R, Spiess HW. Recoupled polarization-transfer methods for solid-state (1)H--(13)C heteronuclear correlation in the limit of fast MAS. Journal of magnetic resonance. 2001;148:398–418. doi: 10.1006/jmre.2000.2259. [DOI] [PubMed] [Google Scholar]
- 10.Hou G, Byeon IJ, Ahn J, Gronenborn AM, Polenova T. 1H-13C/1H-15N heteronuclear dipolar recoupling by R-symmetry sequences under fast magic angle spinning for dynamics analysis of biological and organic solids. Journal of the American Chemical Society. 2011;133:18646–18655. doi: 10.1021/ja203771a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Q, Lu X, Lafon O, Trebosc J, Deng F, Hu B, Chen Q, Amoureux JP. Measurement of 13C-1H dipolar couplings in solids by using ultra-fast magic-angle spinning NMR spectroscopy with symmetry-based sequences. Physical chemistry chemical physics: PCCP. 2011;13:5967–5973. doi: 10.1039/c0cp01907k. [DOI] [PubMed] [Google Scholar]
- 12.Paluch P, Pawlak T, Amoureux JP, Potrzebowski MJ. Simple and accurate determination of X-H distances under ultra-fast MAS NMR. Journal of magnetic resonance. 2013;233:56–63. doi: 10.1016/j.jmr.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 13.Thiriot DS, Nevzorov AA, Zagyanskiy L, Wu CH, Opella SJ. Structure of the coat protein in Pf1 bacteriophage determined by solid-state NMR spectroscopy. Journal of molecular biology. 2004;341:869–879. doi: 10.1016/j.jmb.2004.06.038. [DOI] [PubMed] [Google Scholar]
- 14.Schiksnis RA, Bogusky MJ, Tsang P, Opella SJ. Structure and dynamics of the Pf1 filamentous bacteriophage coat protein in micelles. Biochemistry. 1987;26:1373–1381. doi: 10.1021/bi00379a025. [DOI] [PubMed] [Google Scholar]
- 15.Thurber KR, Tycko R. Measurement of sample temperatures under magic-angle spinning from the chemical shift and spin-lattice relaxation rate of 79Br in KBr powder. Journal of magnetic resonance. 2009;196:84–87. doi: 10.1016/j.jmr.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marchetti A, Jehle S, Felletti M, Knight MJ, Wang Y, Xu ZQ, Park AY, Otting G, Lesage A, Emsley L, Dixon NE, Pintacuda G. Backbone assignment of fully protonated solid proteins by 1H detection and ultrafast magic-angle-spinning NMR spectroscopy. Angewandte Chemie. 2012;51:10756–10759. doi: 10.1002/anie.201203124. [DOI] [PubMed] [Google Scholar]
- 17.Stejskal EO, Schaefer J, Waugh JS. Magic-angle spinning and polarization transfer in proton-enhanced NMR. Journal of Magnetic Resonance (1969) 1977;28:105–112. [Google Scholar]
- 18.Detken A, Hardy EH, Ernst M, Meier BH. Simple and efficient decoupling in magic-angle spinning solid-state NMR: the XiX scheme. Chemical Physics Letters. 2002;356:298–304. [Google Scholar]
- 19.Zhou DH, Shah G, Cormos M, Mullen C, Sandoz D, Rienstra CM. Proton-detected solid-state NMR spectroscopy of fully protonated proteins at 40 kHz magic-angle spinning. Journal of the American Chemical Society. 2007;129:11791–11801. doi: 10.1021/ja073462m. [DOI] [PubMed] [Google Scholar]
- 20.Shaka AJ, Barker PB, Freeman R. Computer-optimized decoupling scheme for wideband applications and low-level operation. Journal of Magnetic Resonance (1969) 1985;64:547–552. [Google Scholar]
- 21.Marion D, Ikura M, Bax A. Improved solvent suppression in one- and two-dimensional NMR spectra by convolution of time-domain data. Journal of Magnetic Resonance (1969) 1989;84:425–430. [Google Scholar]
- 22.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. Journal of biomolecular NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 23.Park SH, Marassi FM, Black D, Opella SJ. Structure and dynamics of the membrane-bound form of Pf1 coat protein: implications of structural rearrangement for virus assembly. Biophysical journal. 2010;99:1465–1474. doi: 10.1016/j.bpj.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldbourt A, Gross BJ, Day LA, McDermott AE. Filamentous phage studied by magic-angle spinning NMR: resonance assignment and secondary structure of the coat protein in Pf1. Journal of the American Chemical Society. 2007;129:2338–2344. doi: 10.1021/ja066928u. [DOI] [PubMed] [Google Scholar]
- 25.Vogt FG, Aurentz DJ, Mueller KT. Determination of internuclear distances from solid-state nuclear magnetic resonance: Dipolar transforms and regularization methods. Molecular Physics. 1998;95:907–919. [Google Scholar]
- 26.van Rossum BJ, de Groot CP, Ladizhansky V, Vega S, de Groot HJM. A Method for Measuring Heteronuclear (1H–13C) Distances in High Speed MAS NMR. Journal of the American Chemical Society. 2000;122:3465–3472. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.