

Abstract
In the past 10 to 15 years, a considerable progress has been made in the treatment of gastrointestinal (GI) related malignancies, as a number of agents expanded from only one in 1995 to seven in 2006. Current review describes the recent role of targeted therapies, specifically EGFR inhibitors in the treatment of GI cancers. Importance of dietary agents in the treatment and prevention of GI cancers is also reviewed.
Keywords: EGFR inhibitors, colon cancer, apoptosis
INTRODUCTION
In the last decade great strides have been made in improving the survival outcome of patients with gastrointestinal (GI) cancers. Among the GI cancers, colorectal cancer is the most common malignancy worldwide [1]. Even with a significant improvement in conventional chemotherapy, mortality from different GI cancers, including colorectal cancer remains unacceptably high. Our understanding of why individuals respond differently to therapeutics and why some patients relapse whereas, others do not, is still at the rudimentary level. Thus, a better understanding of the reasons for treatment failure and developing an ability to predict those who would benefit the most (and least) remain important aims in the management of GI cancers.
One of the most promising new targets in the treatment of GI cancers is the epithelial growth factor receptor (EGFR). Indeed, considerable progress has been made in the past 10 to 15 years, as number of agents for the treatment of GI related malignancies expanded from only one in 1995 to seven in 2006; the most recent additions being the EGFR targeting agents [2]. Agents that inhibit EGFR (such as monoclonal anti-bodies and small molecule tyrosine kinase inhibitors) have demonstrated clinical efficacy as single agents and in combination with chemotherapy. Although initially, these newer drugs appeared to be very promising, time has begun to show us that these advances are more modest than we had expected. The expectation that the median progression-free survival duration would reach 1 year or more with first-line combination treatments has not been realized. Recent findings that expression of EGFR did not correlate with clinical benefit indicate that we had virtually no understanding of individual responses to therapy. The recent discovery of the K-Ras mutation as a marker of probable failure of EGFR-targeted therapy is the first step in tailoring of treatment of individuals [3, 4]. Furthermore, the issue of de-novo resistance in a large number of patients and the development of acquired resistance among the responders remains a subject of extensive investigation [5]. Therefore, a greater understanding of the molecular underpinnings of a patient’s tumor and his/her genetic makeup may lead to a more careful and scientifically elegant selection of therapies. Thus, new paradigms and the identification of new vulnerable molecular targets are needed. In this review we summarize the current state of clinical effectiveness of EGFR-targeted therapies for GI cancers with a focus on colorectal cancer along with future strategies for the management of GI cancers.
BACKGROUND OF GI CANCERS
The fight against cancer today in general and gastrointestinal (GI) cancer in particular, stands at a turning point in its history. The explosion of information and progress in the understanding of the cellular and molecular biology of cancer in recent years presents tremendous opportunities for the development of new therapeutic strategies for different malignancies, including GI cancers. Over the last three decades, numerous studies have been performed regarding the genetics, diagnosis, staging and therapeutic modalities of GI cancers. Even though surgery remains the cornerstone of treatment of GI cancers, new guidelines have been established for a multimodality treatment resulting in improved survival rate and quality of life. The key challenge, however, remains the translation of the basic knowledge generated in the laboratories into more efficacious, preventative, diagnostic and therapeutic products.
Epidemiology
Despite the tremendous advances in medicine, cancer still poses a huge human and economic burden across the world. According to WHO statistics, 7.4 million people worldwide (13% of all deaths) died from cancer in 2004 [6]. According to WHO projections, cancer will result in 12 million of all deaths across the globe. Different forms of cancer incidences as well as mortality vary among different regions of the world, 9.4% for North America to 49.9 % for Asia [6]. According to the data compiled by International Agency for Research on Cancer for the year 2002, the most common forms of cancer worldwide are lung (12.4%), breast (10.6%) and colorectal (9.2%), while the top three causes of death from cancer are lung (17.6%), gastric (10.4%) and liver (8.9%) [7, 8], Gastric Cancer, the second most frequent cause of cancer deaths shows a high geographical variation [9–11]. The incidence of gastric cancer may range from 4–10 cases per 100,000 people (in North America, Africa and Oceania) to 69 cases per 100,000 people (in North East Asia) [9]. The global incidence of gastric cancer has declined over the past few decades [8]. Until 1980s gastric cancer was the leading cause of cancer related deaths when it was taken over by lung cancer [8, 12]. Few risk factors for development of gastric neoplasia are traditional salt-preserved foods, low consumption of fresh fruits and vegetables, H pylori infections and smoking [13–16].
Likewise, colorectal cancer which is third most common cancer worldwide show significant variations in the distribution globally [17, 18]. Incidences of CRC may vary markedly worldwide, with 4.1 cases per 100,000 males in India to 59.1 cases in Czech Republic. While among females, it ranges from 3.6 in India to 39.5 in New Zealand [17]. Some of the risk factors for colorectal cancers include obesity, a diet low in fruits and vegetables, physical inactivity and smoking [19]. There has been a decrease in the CRC mortality worldwide whereas the incidences have been going up [17]. The decline in CRC deaths is attributed to an advanced diagnostic and prognostic technology, while, the “Westernized” life style in developing countries as well as improved longevity in developed countries, contributes to a greater incidence of CRC [17].
Dynamics of the GI Tract
Gastrointestinal cell proliferation plays an important role in the maintenance of the integrity of the gastrointestinal system. The study of gastrointestinal proliferation kinetics allows a better understanding of the complexity of the system, and also has important implications for the study of gastrointestinal carcinogenesis. Cells of the GI mucosa are subject to a constant process of renewal, which in healthy being reflects a balance between proliferation of precursor cells and exfoliation of surface cells [20, 21]. The epithelium of the GI tract proliferates, matures, and recycles constantly throughout the life of an individual and actually has one of the most rapid cell turnover rates of any tissues in the body. The continuous cell renewal is maintained by the sustained proliferative activity of a small number of mucosal stem cells. The specialized epithelial cells of the gut arise from populations of pluripotential stem cells residing in specific locations along the GI tract i.e., at the origin of the cell flux or the base of the intestinal crypt. In the stomach, the stem cells are located in the isthmus/neck region of the gastric glands. In the small intestine and colon, the stem cells are found in the base region of the crypts. Pluripotential stem cells supply progeny to the proliferative compartment of dividing transit cells which divide asymmetrically, and the daughter cells migrate up the crypt, undergoing a series of divisions before leaving the cell cycle to begin differentiation towards the top of the crypt. The migration kinetics in the gastric glands is assumed to be similar, except that this occurs in two directions, upwards towards the foveola and downwards to the base of the gland [22].
The proliferative zone is comprised of actively dividing cells that are distinct from stem cells, while in a larger nonproliferative zone, cells are undergoing progressive differentiation before being extruded into the lumen [21, 23]. Four primary differentiated cell types found in small intestine include absorptive enterocytes, which make up the predominant population of differentiated cells; goblet cells, paneth cells which are located at the base of the crypts; and enteroendocrine cells [24]. Most noted cell types present in colonic epithelium are columnar absorptive cells and goblet cells. The entire process of proliferation, differentiation and shedding occurs over a period of 3- to 6- day in the small intestine, depending on the species; in the colon it takes 3 to 8 days [25]. Cell production not only compensates for cellular shedding from the surface epithelium but also assures the renewal of certain specialized cells in glandular tubes. Any dysfunction in this process may accelerate or diminish the growth rate resulting in either hyperplasia/neoplasia or atrophy of the organ, respectively.
Biology of GI Cancers
Life may be considered as an internal homeostatic milieu wherein the visceral components of an organism have the functional capacity to adjust or compensate within clearly defined limitations, for any metabolic or physiologic imbalance to which the organism is subjected [26]. Carcinogenesis results from interplay between environmental factors and susceptibility of genes that sets off a complex series of neoplastic events. Diverse molecular events are integrated in the development and progression of GI cancers which are leading causes of cancer related death worldwide [27]. A complex combination of genetic, epigenetic, and postgenetic (e.g., posttranslational) alterations are involved in the multistage development of GI cancers (involving the activation of oncogenes, inactivation of tumor suppressor genes) and paralleling these genetic events, cancer cells also induce profound changes in the normal neighboring tissue [28–30]. This altered tissue which is referred to as tumor stroma, provides an environment for tumor growth, invasion and metastatic spreading. Derangements of key molecular processes, including disruption of signal transduction, cell division, apoptosis, angiogenesis, and compartmental boundaries (e.g., tissue invasion and metastasis), contribute to neoplastic progression [31].
Most GI cancers develop via characteristic series of pathological steps preceded by benign dysplastic intermediates (Fig. (1)). In the colon, epithelial cells acquire abnormal growth and morphological characteristics and form an adenoma (adenomatous polyps), a tumor mass often protruding into the lumen of the GI tract. The earliest distinct lesions in this sequence are aberrant crypt foci (ACF). ACF are microscopic lesions (stained deep blue with methylene-blue) in which crypts appear larger and thicker, with an increased luminal diameter having serrated (slitlike) opening [32]. In time, these lesions enlarge and a subset of cells can acquire additional abnormal growth behaviors, which allow them to invade into the GI wall and metastasize. At this point, the tumors are classified as adenocarcinomas and can be lethal. In the course of this typical multistep process, somatic mutations develop in key genes such as the adenomatous polyposis coli (APC), p53 tumor suppressor genes; K-Ras oncogene; and various genes that mediate DNA mismatch repair [33]. Similar multistep progression from normal tissue through dysplastic intermediates to cancer has been well described for human esophageal, gastric, and pancreatic cancers [34–36]. During these step wise process of neoplastic transformation, DNA mutations and clonal selection of the cells that express certain cardinal features (such as higher proliferative signals, insensitive to antiproliferative signals, evasive to normal apoptotic signals, limitless capacity for replication, tissue invasion and metastasis) are necessary for the development and progression of carcinogenesis [37]. The multistep nature of carcinogenesis provides opportunities for the intervention with agents targeted at specific mechanisms involved in the initiation, promotion, and progression stages of carcinogenesis.
Fig. 1.
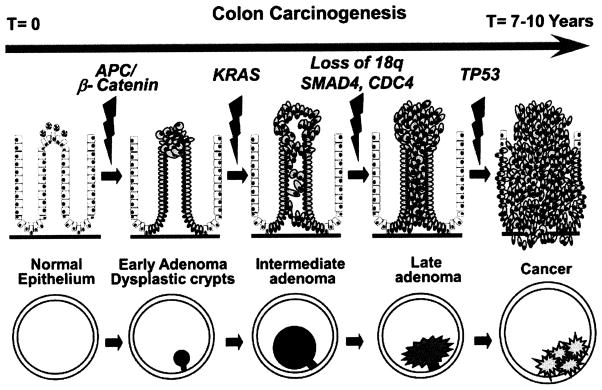
Schematic representation of adenoma-to-carcinoma sequence.
CELLULAR HOMEOSTASIS
In multicellular organisms total number of cells is the balance between cells undergoing mitosis and death. The process by which a cell undergoes natural death or induces to commit suicide is a well regulated process called programmed cell death or apoptosis. Kerr, Wyllie and Currie were the first to coin the term apoptosis and characterized this form of cell death [38]. The first morphological description of apoptosis was provided by Walther Flemming [39], whose elegant drawings show cells shrinkage, nuclear fragmentation and apoptotic body formation, all of which are now accepted hallmarks of apoptosis. Apoptotic bodies are then engulfed by neighboring phagocytotic cells. Internuc-leosomal DNA cleavage yields a ladder pattern in electrophoretic gel, which is a biochemical hallmark of apoptosis. Another hallmark of apoptosis is the movement of the membrane lipid phosphatidylserine from the inner to the outer side of the plasma membrane, where it then functions as a recognition signal for phagocytic cells to engulf apoptotic cells [40, 41], Apoptosis is an evolutionary conserved process and is essential for organ development, tissue remodelling, immune response, and tumor suppression. It is known that the adult human body generates approximately 60 billion cells per day and as a consequence an equal number of cells must die by apoptosis to maintain homeostasis. Therefore it is not surprising that deregulation of apoptosis can lead to an accumulation of live cells and contribute to tumor development. Thus, apoptosis is an important process required for homeostasis [42].
The high turnover of epithelial cells in the GI tract necessitates a method to eliminate cells so that the delicate balance of proliferation can be maintained. Alterations in proliferation or apoptosis resulting in an imbalance of the effector protein(s) can lead to carcinogenesis in intestine [43] (Fig. (2)). Also, wealth of information available suggests apoptosis to be a normal mechanism of controling proliferation of epithelial cells in the GI mucosa [44] and therefore proves to be the dominant end effect in any anticancer strategy. The idea that the role of apoptosis can be utilized for cancer cure goes back to the report by Kerr, Wyllie and Currie’s original 1972 paper [38] where they suggested that the regression of tumor growth could be due to increased tumor cell apoptosis. Since then selectively inducing apoptosis in cancer cells has been increasingly recognized as a promising therapeutic approach for many cancers.
Fig. 2.

The induction of carcinogenesis through imbalance in apoptosis and cell cycle regulation. (A) Under disease free condition tissue homeostasis is achieved through a balance between cell proliferation and apoptosis. (B) Dysregulation in cell cycle process leads to increased activation or decreased inhibition resulting in uncontrolled proliferation. (C) The loss of proapoptotic and gain of anti-apoptotic regulators result in increased cell survival and cell number. These events are implicated during the complete imbalance in tissue homeostasis leading to the formation of carcinoma.
There are various stimuli which can trigger apoptosis in mammalian cells viz., growth factor withdrawal, UV light, irradiation or chemicals and the latter two have been significantly utilized in cancer therapy [45]. Apoptosis occurs through two major signaling pathways: the intrinsic pathway (mitochondrial pathway) and extrinsic pathway (death receptor pathway) [42](Fig. (3)). The intrinsic pathway is initiated by any stimuli that can cause oxidative stress, mitochondrial and DNA damage, such as chemotherapy or radiation. In the intrinsic or mitochondrial pathway caspases are key players. This involves the permeabilization of the outer mitochondrial membrane which facilitates Cytochrome c release into the cytoplasm. The released Cytochrome c then binds the caspase adaptor Apaf-1 (apoptotic protease-activating factor-1), thereby triggering the apoptotic cascade by activating procaspase 9 and forming a complex termed the “apoptosome”. This complex in turn activates several downstream effector caspases that include caspases 3, 6 and 7, leading to DNA fragmentation and cell death [46, 47]. Other group of key players involved in mitochondrial pathway of apoptosis is the Bcl-2 (B-cell leukemia/lymphoma 2) family, which consists of more than 20 members of pro-apoptotic proteins (including Bax, Bak, Bok, Bad, Bid, Bik, Bim, Bcl-Xs, Krk, Mtd, Nip3, Nix, Noxa, and Bcl-B), and anti-apoptotic proteins (including Bcl-2, Bcl-XL, Mcl-1, Bfl-1/A1, Bcl-W, and Bcl-G). Pro-apoptotic members induce the release of cytochrome c from mitochondria, whereas anti-apoptotic members can bind and inactivate Apaf-1. In addition, pro-apoptotic members can dissociate the Bcl-XL-Apaf-1 complex, allowing Apaf-1 to activate caspases 9 which leads to subsequent apoptotic process [48]. The cellular homeostasis as governed by the intricate balance between the cellular levels of pro-apoptotic and anti-apoptotic proteins is exploited by drugs for targeting cancer cells.
Fig. 3.

Schematic representation of apoptosis signaling depicting intrinsic and extrinsic pathways.
In contrast, the extrinsic death pathway is initiated through death receptor mediated signals on the cell surface. The most important ligand-death receptor system include tumor necrosis factor (TNF)-tumor necrosis factor receptor 1 (TNFR1), Fas ligand-Fas (CD95 or Apol), TRAIL-TRAIL receptors (including TRAIL-R1, also termed DR4, and TRAIL-R2, also termed DR5). Binding of receptors by their respective ligands leads to receptor oligomerization and recruitment of death signaling adapter proteins. For example, binding of Fas ligand (Fas-L) to Fas, or TRAIL to TRAIL-R1 [49] leads to recruitment of FADD (Fas-associated death domain), and binding of TNF to TNFR1 leads to recruitment of TRADD (TNFR-associated death domain) [46]. The oligomerized receptors and recruited FADD or TRADD form a complex termed DISC (death-inducing signaling complex), which bind to initiator caspases (caspases 8 and caspase 10), thereby triggering the caspases cascade such as activation of caspases 3, 7, and 9, and leading to apoptotic events.
EGFR INHIBITION AND ACTIVATION OF APOPTOSIS
Since apoptosis has an essential role in the maintenance of dynamic balance of cell survival and cell death signaling, its deregulation may lead to the development of cancer. Thus, induction of apoptosis in tumor cell forms the basis of many cancer therapies. As the concept of apoptosis in the management of cancer was getting evolved, so was the identification of various cytotoxic agents to induce cell death in cancer tissues. In the past, various cytotoxic agents were employed in the treatment of GI cancers. Most of these agents were nonselective that killed cancer cells through DNA damage, interfering with DNA repair mechanisms and/or disturbance of metabolic pathways [50]. Though many of these agents caused tumor regression but cellular toxicity caused by them posed a serious problems. With increased understanding of the mechanisms of apoptosis in cancer and signaling pathways, focus was directed towards the development of targeted therapies. In GI cancer therapy apoptosis can be selectively induced by: 1) Inhibiting the cell survival signaling via EGFR, MAPK and PI3K (intrinsic pathway of apoptosis); 2) activating the cell surface death receptors Fas, TRAIL and TNF receptors (extrinsic pathway of apoptosis); 3) altering the balance between pro-apoptotic and anti-apoptotic members of the Bcl-2 family; and/or down regulating the antiapoptotic proteins such as Bcl-XL, XIAP, survivin and c-IAP2. In the last decade a large body of preclinical and clinical findings has highlighted the role of epidermal growth factor receptor (EGFR) family in tumor formation and maintenance. Thus, targeting the EGFR could represent a significant contribution to cancer therapy.
Epidermal Growth Factor Receptor Biology and Signaling
The epidermal growth factor (EGF) is a growth factor that plays an important role in the regulation of cell growth, proliferation, and differentiation by binding to its EGF receptor (EGFR). The EGF family of growth factors consist of at least ten different members that bind and activate four receptors, namely ErbB1 (EGF receptor/HER1), ErbB2 (Her2/Neu), ErbB3 and ErbB4. EGFR(s) are transmembrane glycoproteins that possess intrinsic tyrosine kinase activity [51]. The EGFR family ligands, including EGF, TGF-α, betacelulin, amphiregulin, neuregulins, and heparin binding EGF, represent a major growth factor family that has a profound impact on intestinal epithelial cell restitution and proliferation [52]. The well established textbook dogma for the mechanism by which EGFR(s) ligands is believed to promote carcinogenesis could be summarized as follows (Fig. (4)). EGFR monomers on the cell surface bind a single molecule of EGF. EGFR molecules then dimerize and undergo autophosphorylation of multiple tyrosine residues in their cytoplasmic tails. These residues become docking sites for cytosolic and membrane-associated effectors containing phosphotyrosine-binding SH2 or PTB domains, such as phosphoinositide 3-kinase (PI3K), phospholipase C (PLC) and the adapter molecule Grb2. The ensuing assembly of specific target proteins on the inner leaflet of the plasma membrane culminates in the formation of second messengers such as polyphosphoinositides and inositol polyphosphates, and the activation of key mitogenic pathways, such as the Ras/MAPK cascade. The downstream targets of these pathways include transcription factors, which translocate from the cytosol to the nucleus when stimulated by their respective upstream kinases or phosphatases [53]. Other molecules are enzymes that are activated on EGFR-TK-dependent phosphorylation, including son of sevenless (SOS), PI3K and Grb 2-associated binder-1 (Gab-1). Multiple major signal transduction pathways are initiated by EGFR autophosphorylation, including the Ras-MAPK signaling cascade, Src, and the signal transducers and activators of transcription (STAT) pathways, which are widely used by growth signals to induce gene transcription and promote diverse cell responses. The particular dimer combinations that form at the cell surface after ligand binding determines which signaling molecules will be recruited to the surface [54]. The activation of these pathways downstream of the EGFR leads to cell proliferation, differentiation, and migration or motility and adhesion, protection from apoptosis, enhanced survival, and gene transcription [55].
Fig. 4.
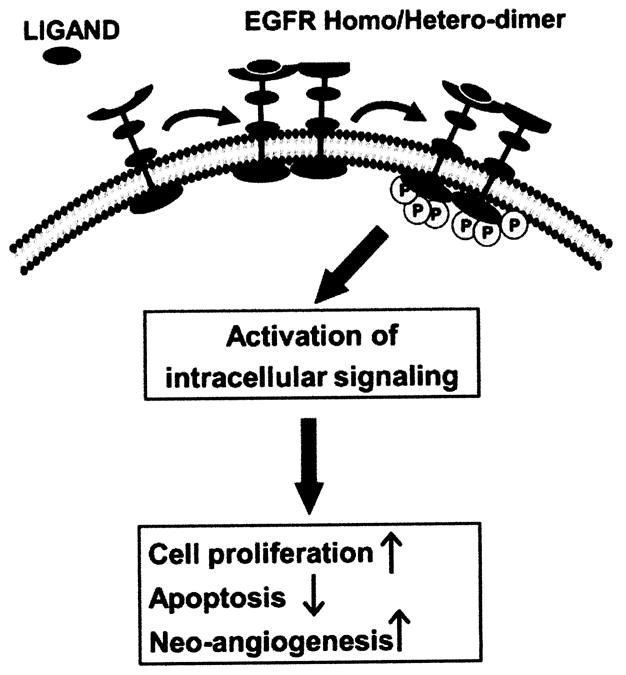
Schematic representation of ligand induced activation of EGFR(s) signalling pathway. Ligand binding to the receptor results in dimerization and auto-/trans-phosphorylation that initiate the signaling cascade.
Although all four EGFR(s) have the same essential domains, the functional activity of each domain varies. EGFR (ErbB-1) and ErbB-4 have active tyrosine kinase domains and known ligands, ErbB-3 can bind to several ligands but lack intrinsic tyrosine kinase activity as it is unable to bind ATP. By contrast, ErbB-2 possesses an active tyrosine kinase domain but no direct ligand has been identified [55–56]. It exists in the active confirmation state, rendering ErbB-2 constitutively available for dimerization [57]. ErbB-3 cannot form homodimers, and only forms heterodimers. The dimers of EGFR family have different signaling potencies; homodimers having weaker signals compared to heterodimers. EGFR, ErbB2- and ErbB-3 are all implicated in the development and progression of cancer. Over-expression of the EGFR tyrosine kinase has been documented across all stages of disease, including precancerous lesions, early cancers, and advanced cancers. Enhanced activity of EGFR has been found to be associated with tumor progression and poor survival in various malignancies, including GI, head and neck, lung, breast and bladder cancers [58–62]. While the role of ErbB-4 in carcinogenesis remains less well defined, the ErbB2-ErbB-3 heterodimer is considered as the most potent pair with respect to strength of interaction, ligand-induced tyrosine phosphorylation and down-stream signaling and functions as an oncogenic unit [63]. ErbB-2 overexpression in cancer cells facilitates the formation of ErbB-2 heterodimers and the spontaneous formation of ErbB-2 homodimers. Consequently, this leads to an excessive ErbB2-mediated signaling, which drives oncogenic cell survival and proliferation [64]. Recent reports suggest that in gastric cancers ErbB-2 overexpression is associated with a poor prognosis [65,66].
Though the impact of EGFR(s) expression on survival remains controversial, the same is linked to an advanced stage colon cancer and can be developed as potential metastatic marker [67]. Aberrant activation of EGFR(s) signaling may be a consequence of mutations, constitutive activation, over-expression of receptors and/or its ligand(s) or cross-talk with other amplified receptors and signaling pathways [63]. The resulting enhanced receptor activity plays a pivotal role in malignant transformation and in maintaining the growth of cancers in model systems [5]. Therefore strategies targeting EGFR(s) expression and/or inhibiting downstream signaling cascade(s) hold great promise in the management of solid cancers, including GI cancers pathways [68]. In addition to suppression of cell proliferation, inhibition of EGFR signaling provides extra benefit of inducing apoptosis in cancer cells [69]. The down-regulation of EGFR specific ligands heparin-binding EGF-like growth factor and amphiregulin whose expression were elevated in gastric and colon cancers cell has been reported to induce significant apoptosis in these cell [70].
Strategy for Targeting the EGFR Network: EGFR Inhibitors
A better understanding of structure and function of EGFR and its family members has led to development of several strategies targeting intracellular or extracellular domain of the receptor to inhibit the signal transduction. From GI cancer therapy point of view, currently, two different EGFR targeted approaches are farthest in development. These utilize monoclonal antibodies (targeting ligand-binding domain) and small molecule (targeting tyrosine kinase enzymatic activity) of EGFR.
Anti-EGFR Monoclonal Antibodies (Extracellular Blockade)
This strategy exploits antibodies to block the extracellular ligand-binding region of the receptor. The monoclonal antibodies bind to the extracellular binding domain of the EGFR and competes with the EGFR ligands (e.g., EGF and TGF-α) to inhibit both phosphorylation and activation of EGFR-associated kinases, causing internalization of the receptor, inhibition of cellular growth, induction (i.e. restoration) of apoptosis, and decreased production of growth factors (e.g., proinflammatory cytokines, vascular endothelial growth factor [VEGF]) [71]. The immunogenicity of a monoclonal antibody is determined by the species of the antibody source, and currently there are four types of monoclonal antibodies based on its source (murine, chimeric, humanized, and fully human) [72]. The earliest type of monoclonal antibodies developed were murine monoclonal antibodies which were derived entirely from mice (i.e. they contain 100% mouse protein). These were associated with a high incidence of human anti-mouse antibody (HAMA) reactions. Failed efforts with murine monoclonal antibodies led to the development of monoclonal antibodies with human components. Chimeric monoclonal antibodies (having human constant region and a murine variable region) were next generation of monoclonal antibodies which showed much lower incidence of HAMA than those experienced with murine monoclonal antibodies. It resulted in wide range of antigen specificities and enhanced efficacy. Continued efforts to improve monoclonal antibodies led to the development of humanized antibodies, which were 90% human with 10% mouse protein. They were found to be even less immunogenic than chimeric monoclonal antibodies. Further progress led to the development of fully human monoclonal antibodies, which were produced in a mouse whose murine genes were inactivated and replaced by human sequences. These monoclonal antibodies showed less plasma clearance and demonstrated beneficial pharmacokinetic and pharmacodynamics than chimeric and humanized monoclonal antibodies due to lack of mouse components [73].
Most of the 20th century elapsed before a monoclonal anti-body with therapeutic applications in the treatment of cancer became available in the United States. The first fully human anti-EGFR monoclonal anti-body for the treatment of cancer was approved by the Food and Drug Administration (FDA) in 2006, and numbers of other fully human monoclonal antibodies for treatment of cancer are under investigation in clinical trials. Cytotoxic monoclonal antibodies in use for the treatment of cancer can be classified into three categories:
Unconjugated monoclonal antibodies
Conjugated monoclonal antibodies
Radioimmunoconjugates (RICs)
Of these only unconjugated monoclonal antibodies are in use for the treatment of GI cancers, whereas, conjugated monoclonal antibodies or immunotoxins, which are monoclonal antibodies conjugated with a potent toxin, as well as radioimmunoconjugates (RICs), which are monoclonal antibodies conjugated with radioisotopes, have not yet been approved for GI cancer therapy [73]. In GI cancer therapy, EGFR is one the important target antigen for monoclonal antibodies (other important target antigen being VEGF). Currently there are three monoclonal antibody products directed at these targets (two as anti-EGFR and one as anti-VEGF) approved by FDA while many others are in development. The two anti-EGFR monoclonal antibodies for the GI cancer therapy are Cetuximab and Panitumumab.
Cetuximab
Cetuximab is the first human-mouse chimeric immunoglobulin (Ig) G1 monoclonal antibody targeting EGFR with 5–10 fold greater affinity than natural ligands [74]. It has been approved by FDA for the treatment of EGFR-expressing metastatic colorectal carcinoma in patients who are refractory to or intolerant of irinotecan-based chemotherapy [75].
Preclinical studies show that cetuximab inhibits the proliferation of cell lines expressing EGFR, and increases the cytotoxic activity of chemotherapy and radiation [76]. Cetuximab treatment leads to cell-cycle arrest and accumulation of cells in the G1 phase by increasing the expression of p27Kip1 and p15INK4B, which are cyclin-dependent kinase (CDK) inhibitors. This prevents the cell entry into the S phase by downregulating the activities of CDK2, CDK4 and CDK6. Cetuximab causes more accumulation of pro-apoptotic protein Bax thereby inducing cancer cell apoptosis [77]. Cetuximab has demonstrated activity, both as a single agent and in combination with irinotecan, in patients with metastatic colorectal cancer whose disease has progressed after receiving irinotecan-, oxaliplatin-, and/or fluorouracil-based chemotherapy [78–80].
Cetuximab monotherapy has been found to improve the overall survival time, response rate, time to disease progression and progression-free survival time compared with single agent irinotecan treatment in patients with metastatic colorectal cancer who have failed standard therapies. Cetuximab augments the susceptibility of cancer cells to the chemotherapy by blocking EGFR nuclear import and the associated activation of DNA protein kinase enzymes necessary for repairing chemotherapy-induced DNA damage [77]. Results of phase III trial of both Bowel Oncology and Cetuximab Antibody (BOND)-1 study and Erbitux Plus Irinotecan for Metastatic Colorectal Cancer (EPIC) study, which compared the cetuximab monotherapy with cetuximab plus irinotecan-based therapies, revealed a significantly improved overall response rate, progression-free survival time and preserved quality of life with the combination therapy [81, 82]. Cetuximab has also produced promising results in phase II trials evaluating its combination with standard therapies such as irinotecan plus infusional fluorouracil-leucovorin (FOLFIRI) and capecitabine plus oxaliplatin (CAPOX), in previously treated patients with metastatic colorectal cancer [83].
The evaluation of cetuximab as the first-line therapy for the treatment of metastatic colorectal cancer has undergone intense investigation. The ACROBAT phase II trial conducted in Europe evaluated the efficacy of cetuximab plus FOLFOX-4 in the first line setting [84]. Cetuximab in combination showed promising clinical activity which was well tolerated and did not exacerbate the toxicities typically associated with FOLFOX-4 chemotherapy. A randomized phase III trial (CRYSTAL) conducted by the van Custem et al. [85] evaluated the benefits of cetuximab plus FOLFIRI in comparison to FOLFIRI alone in the first-line therapy for metastatic colorectal cancer overexpressing EGFR. They found that the cetuximab-FOLFIRI combination produced significantly longer progression-free survival and increased response rates than that noted with FOLFIRI alone. However, the treatment related toxicities such as neutropenia, diarrhea, and skin reactions were more frequent with the combination [71]. CRYSTAL study showed that cetuximab could be safely and effectively combined with FOLFIRI but combining cetuximab with irinotecan-based chemotherapy did not alter its pharmacokinetic profile [86, 87]. Thus, for GI cancers, cetuximab could be considered as an appropriate first-line and neoadjuvant therapy.
Panitumumab
Panitumumab, also known as ABX-EGF, is a fully human IgG2 monoclonal antibody targeting EGFR approved by FDA for the treatment of EGFR-expressing metastatic colorectal carcinoma with disease progression or following fluoropyrimidine-, oxaliplatin-, and irinotecan-containing chemotherapy regimens [88]. Although their target is same, the pharmacokinetics and pharmacodynamics of panitumumab differ from cetuximab as these bind to different epitopes on EGFR [84]. Moreover, being IgG2 isotype, panitumumab does not exert its cytotoxic effects through immunologic-mediated mechanisms. Like cetuximab, panitumumab induces cell-cycle arrest and panitumumab causes arrest in the G0–G1 interphase, whereas cetuximab causes arrest in the G1 phase [89]. Panitumumab has a longer half-life (7.5 days) than cetuximab (4.7 days) which has been attributed to differences in murine protein content of the antibody [75]. Panitumumab shows lesser immunogenicity and causes fewer infusion reactions than cetuximab. It has also higher binding affinity for EGFR and is more potent as an inhibitor of signaling through EGFR than cetuximab [90].
Panitumumab monotherapy treatment results of phase II trial in which patients with refractory metastatic colorectal cancer received 2.5 mg/kg intravenously dose of Panitumumab for 8 weeks showed a response rate similar to that noted with cetuximab monotherapy. Based on the encouraging clinical outcomes, a randomized, controlled phase III study compared panitumumab 6 mg/kg intravenously every 2 weeks plus best supportive care with best supportive care alone [85]. Patients enrolled had metastatic colorectal cancer and documented progressive disease during the treatment period or within 6 months of completing the treatment with a fluoro-pyrimidine, irinotecan, and oxaliplatin. The overall survival time was not significantly different between the two arms, but the similarity was likely confounded by the high rate of crossover in the best supportive care arm and the comparable efficacy of panitumumab in the crossover population [91]. Overall, this treatment was well-tolerated but showed skin toxicities, diarrhea, and hypomagnesaemia which were the most common toxicities. In contrast to cetuximab, panitumumab had a low frequency of infusion-related reactions and no antibody formation. Based on these results, panitumumab monotherapy was approved by the FDA in September 2006 for the treatment of metastatic colorectal cancer with disease progression while receiving or after receiving fluoropyrimidine, oxaliplatin, and irinotecan chemotherapy regimens.
The use of panitumumab as first-line therapy was initially investigated in combination with irinotecan-based chemotherapy in a 2-part phase II trial [84]. In the first part of the trial panitumumab at a dose of 2.5 mg/kg weekly was combined with the bolus weekly schedule of fluorouracil-leucovorin (IFL) whereas in the 2nd part panitumumab at the same dose and schedule was combined with FOLFIRI. Due to unacceptable toxicity in part 1 (with 58% incidence of diarrhea vs. 25% in part2) the study was modified to evaluate panitumumab in combination with FOLFIRI (part2). Although the panitumumab/IFL combination was not well tolerated, the panitumumab/FOLFIRI regimen showed promising clinical efficacy with a manageable safety profile. An international, randomized phase III trial was carried out for safety analysis of the panitumumab in combination with chemotherapy for metastatic colorectal cancer and compared it with panitumumab plus FOLFOX4 chemotherapy versus FOLFOX4 alone in treatment-naïve patients [71]. The toxicities associated with this adjuvant therapy mainly comprised skin reaction and increased incidence of grade 3 diarrhea and dehydration. However, in a pre-planned interim efficacy analysis of Panitumumab Advanced Colorectal Cancer evaluation (PACCE) trial demonstrated a significant difference in both progression free as well as overall survival in the favor of the control arm, which led to the discontinuation of panitumumab treatment in this trial [92]. Though both cetuximab and panitumumab are limited by their effectiveness with regards to K-ras status, clinical impact of the relative utility of these drugs remains to be demonstrated because no head-to-head comparisons have been performed so far. One such randomized phase 3 study called ASPECCT is scheduled to begin in 2010 with an estimated completion in 2013. This study is designed to compare the efficacy and safety of panitumumab to cetuximab in subjects with K-ras wild-type metastatic colorectal cancer [6]. It is highly premature to speculate a better clinical outcome for one of the therapy, nonetheless, the revelations of this study is expected to facilitate the development of individually tailored treatments of colorectal cancers.
Small Molecule EGFR Tyrosine Kinase Inhibitors (Intracellular Blockade)
This anti-EGFR approach is based on the observation of mutations in the ATP-kinase of the receptor leading to its dysregulation. Abnormal activation of tyrosine kinase produces a gain of function that results in proliferation and survival and in some circumstances resistance to anticancer chemotherapies [93] indicating that the receptor’s tyrosine kinase is critical for EGFR-mediated tumor progression. Small molecule tyrosine kinase inhibitors (TKIs) were designed to bind to the ATP binding cleft in the kinase domain producing their effect in a competitive manner with ATP, thereby, preventing phosphorylation and subsequent downstream signaling. Many of these small molecules are being investigated in clinical trials. Tyrosine kinase inhibitors have the theoretical advantage of also blocking activating cytoplasmic signals when compared to agents that block activation at an extracellular level (i.e., receptor MAbs). The potentially favorable feature is that these agents are orally active which makes them suitable for long-term therapy. The two EGFR targeting compounds extensively investigated for GI cancers in preclinical and clinical settings are gefitinib and erlotinib.
Gefitinib (ZD1839, Iressa) is an oral EGFR-specific anilinoquinazoline which reversibly inhibits autophosphorylation, resulting in reduced c-FOS mRNA—a transcription factor forming part of the AP1 complex—and a shift of cells from S phase into G0/G1 [94]. Gefitinib is currently approved for the treatment of locally advanced or metastatic non-small cell lung cancer (NSCLC) in patients who have previously received chemotherapy. While gefitinib has yet to be proven to be effective in other cancers, there is potential for its use in the treatment of other cancers including GI cancers where EGFR over expression is involved. Erlotinib hydrochloride, [6,7-Bis(2-methoxy-ethoxy)-quinazolin-4-yl]-(3-ethynyl-phenyl) amine hydrochloride, also known as CP-358,774, OSI-774 and Tarceva, is an orally active potent selective inhibitor of the EGFR tyrosine kinase (TK). It competes for the ATP-binding site in the intracellular TK domain of EGFR with an IC50 of 2 nM against the kinase. Erlotinib induces apoptosis in selective, in vitro cancer cell lines and has antiproliferative activity against numerous human cancer xenografts in vivo [95].
A phase I/II study of the evaluation of gefitinib in combination with 5-fluorouracil (5-FU)/folinic acid and irinotecan (FOLFIRIAIO) in patients with 5-FU/oxaliplatin-refractory colorectal cancer had to be stopped early because of gastrointestinal toxicity and fatigue. This treatment regimen did not seem advantageous with respect to efficacy in comparison with FOLFIRIAIO alone. Recently, a phase II randomized multicenter trial of gefitinib plus FOLFIRI and FOLFIRI alone in patients with metastatic colorectal cancer [96] concluded that adding gefitinib to FOLFIRI did not improve the efficacy of FOLFIRI regimen. These disappointing results could be related to the high toxicity and led to significant dose reductions and delays. In clinical trials, erlotinib has shown anticancer activity in several malignancies, including lung, pancreas, ovarian, head and neck, endometrial and biliary tract cancers [97–101]. It has been found to be safe and well tolerated with the most common side effects being diarrhea, rash, nausea, headache, emesis and fatigue. A phase II study of erlotinib in patients with metastatic colorectal cancer was conducted by Townsley et al. [102] to evaluate its efficacy as a single agent in patients with recurrent or metastatic colorectal cancer. Results showed that only a portion of the population responded to the targeted therapy. Meyerhardt et al. [103] recently conducted a Phase II study evaluating the efficacy of erlotinib in combination with FOLFOX and bevacizumab as first-line therapy for patients with metastatic colorectal cancer. The combination of FOLFOX, bevacizumab and erlotinib led to higher numbers of early withdrawal than expected due to toxicity, limiting conclusions regarding the efficacy. These findings raise concern regarding the tolerability of adding more agents to already complex combination regimens for metastatic colorectal cancer.
Interestingly, limited preclinical data suggest that erlotinib, but not gefitinib, can inhibit EGFR [104, 105]. However, these preliminary data require confirmation as the observed differences in the efficacies of the two agents could be due to differences in structure or potency or cell type–dependent factors. Extensive preclinical studies with erlotinib and gefitinib showed that both agents effectively inhibit tumor growth when used alone or in combination with various chemotherapeutic agents, and both regimens were well tolerated [2, 67]. However, both these EGFR TKIs have so far demonstrated disappointing response rate in metastatic colorectal cancer and, when combined with the conventional metastatic colorectal cancer chemotherapy [106]. These new small molecules have had success in a number of solid tumors including GIST, non small cell lung cancer, renal cancer, hepatocellular carcinoma, pancreatic cancer and breast cancer and in some of these cases the success has been paradigm changing (GIST, renal cell cancer and hepatocellular carcinoma). But so far, no TKI have been approved for the treatment of colorectal cancer, despite the fact that monoclonal antibodies and TKI have been made available for clinical testing at approximately the same time. This may be due to the reasons that both erlotinib and gefitinib (unlike cetuximab and panitumumab) failed to produce substantial efficacy as monotherapy [106] and these agents have failed to provide a strong additional anticancer activity when combined with chemotherapy without adding to increase in toxicity [107]. Along the line of toxicities, the small molecules present an additional peculiarity: although they generally do not cause the classical severe toxicities of chemotherapy, other toxicities that are low on the objective scales, but hard on the patients, are quite common (asthenia, myalgia, arthralgia, edema, altered taste etc.). The jury is still out about the role of small molecules in the management of colorectal cancer. The growing number of these compounds and the challenge that drug companies are taking by launching risky, but justified randomized phase III trials on several of these drugs allow a cautious optimism to further enrich our therapeutic armamentarium against this disease.
TARGETED THERAPIES: WHERE ARE WE NOW?
Targeting multiple pathways involved in cancer development and progression is an attractive area of research. For patients with colorectal cancer, bevacizumab has potentiated the efficacy of standard cytotoxic chemotherapies, though the exact mechanism is unknown [108, 109]. EGFR appears to play an important role in the progression of colorectal cancer, and inhibition of receptor activity has proven beneficial in some patients [110, 111]. Combining these multiple strategies (traditional cytotoxic chemotherapies and EGFR inhibitors) is a logical step in clinical investigations.
To date, in the clinical setting, only the monoclonal antibodies have really shown activity, and not the small molecules. One issue requiring particular attention involves the predictive response markers that could help select those patients most likely to respond to EGFR-targeting agents. Some investigators have suggested that cutaneous side-effects might be predictors of efficacy. Extensive skin rashes or acne, for instance, seem to be associated with improved survival following anti-EGFR therapy [112, 113]. Although EGFR is not considered to be a prognostic factor in colorectal cancer patients, it plays a major role in tumor cell proliferation. Convincing preclinical and clinical studies have already demonstrated the efficacy of EGFR inhibitors in advanced colorectal carcinomas and their potential synergistic combinations with chemo- and radiation therapy. It is becoming increasingly clear that toxicity is the main drawback of these combination strategies. Additional efforts and trials are needed to evaluate the role of EGFR inhibitors in the adjuvant setting so as to establish better predictors of the efficacy of such agents. Since EGFR expression in cancerous tissues does not seem to be a valuable predictor of response to anti-EGR therapy, other biomarkers need to be evaluated besides EGFR mutated forms, p-Akt expression or EGFR amplification. The gain in the knowledge gathered from clinical studies has led to identification of new key areas regarding the role of EGFR family in cancer formation, progression and maintenance.
Gene Mutations and Resistance to EGFR Inhibitors
Although anti-EGFR therapies, as discussed above, may lead to partial response or disease stabilization in some patients, many patients do not benefit from anti-EGFR therapy, and those who do eventually develop resistance to that therapy. There could be several reasons for cancer cell resistance to EGFR antagonists. One of the possible mechanism by which tumor cells become resistant to EGFR antagonists in GI cancer therapy is EGFR gene mutations and the loss of the target gene. Among the first genetic alterations of the EGFR studied is the EGFR variant (EGFR-vIII), which is the result of an inframe deletion from exons 2 through 7 in the extracellular domain that inhibits EGFR-vIII from binding to its ligands, leading to a constitutive signaling and to resistance to EGFR-targeted therapy [5]. This ligand-independent mutant EGFR-vIII is constitutively activated in many human cancers. Cell lines expressing the mutant variant EGFR-vIII appear to be relatively resistant to gefitinib since higher doses and longer exposure to gefitinib are necessary to significantly decrease EGFR-vIII phosphorylation. Reports suggest that this may be due, in part, to the phosphorylation of Akt, which is inhibited in EGFR expressing cells after treatment with gefitinib, but is unaffected in cells expressing EGFR-vIII [114].
Additionally, reports have shown the occurrence of specific mutations in the kinase domain of ErbB-2 in many human cancers such as lung, gastric, breast, and colorectal cancers, and suggested that alterations of ErbB-2-mediated signaling pathway by ErbB-2 mutations alone or together with K-ras mutations may contribute to the development of resistance to anti-EGFR(s) therapies [115]. Other studies have revealed the occurrence of oncogenic mutations of K-Ras in approximately 35–40% of colorectal cancer cases, arising early during colorectal carcinogenesis [116]. K-Ras protein is a downstream component to the EGFR that links growth promoting signals from the cell surface to the nucleus. K-ras gene mutations are single nucleotide point mutations which occur mostly in codons 12 and 13 of exon 2, with mutations in codon 12 accounting for about 80% of the total K-ras gene mutations observed and have been found to be responsible for EGFR-independent activation of K-Ras signaling in tumor cells [5, 117]. For this reason, EGFR-antagonists may have little effect on the activation of K-ras dependent downstream signaling in cancer cells carrying the mutated form of proto-oncogene. Substantial evidences have been generated from phase I to HI studies suggesting the correlation of activating K-ras gene mutations and lack of efficacy of anti-EGFR drugs. The strongest clinical evidence in this direction was derived from the results of CRYSTAL study (a phase III trial which evaluated the combination of cetuximab with FOLFIRI chemotherapy in previously untreated metastatic colorectal cancer) in which K-ras gene was observed to be mutated in 36% cases [85]. Cetuximab addition to FOLFIRI had no effect in patients with a mutated K-ras tumor when compared to patients whose tumors were carrying a wild-type K-ras gene. On the basis of these findings, in July 2008 the European Medicines Agency (EMEA) has extended approval for cetuximab to any line of treatment and any fluoropyrimidine-based chemotherapy combination in metastatic colorectal cancer, but only in patients with a wild-type KRAS tumor [118]. Further, in January 2009, the American Society of Clinical Oncology (ASCO) in its Provisional Clinical Opinion strongly recommended the use of anti-EGFR drugs in metastatic colorectal cancer only in patients with a wild-type KRAS tumor [119]. Recently Di Nicolantonio and colleagues [120] reported that wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. BRAF gene encodes for a protein that acts downstream of KRAS in the EGFR-activated intracellular signaling machinery and could be responsible for the lack of efficacy of EGFR inhibitors in wild-type KRAS metastatic colorectal cancer. Mutations in the two genes are mutually exclusive and BRAF mutations may account for another 12% of resistant cases [120]. Inspite of the predictive value of both K-Ras and BRAF mutations, these mutations do not account for the entire spectrum of resistance to EGFR targeted therapies.
More recently, it has been reported that existence of certain EGFR mutations may render tumors susceptible to tyrosine kinase inhibitors of this receptor [113, 121]. These EGFR kinase mutations alter the three dimensional confirmation and activity of receptor such that the tumor is rendered susceptible/sensitive to tyrosine kinase inhibitors like gefitinib and erlotinib. Among these mutations, EGFR exon 19 deletions or L858R mutations are the best in providing therapeutic advantage to patients with advanced lung cancer. Phase II studies have shown that these two mutations are major markers of response to treatment in advance lung cancer with >80% response with increased progression-free survival [122–126]. Though the efficacy of anti-EGFR antibodies in lung cancer cell lines seems to be independent of the presence of EGFR mutations [127], predicting the efficacy of other EGFR targeted therapies is a challenge for the next years. Achieving this goal relies on a better characterization and understanding of secondary events, which may involve re-biopsy of tumor tissue on acquisition of resistance. However, the prevalence of EGFR mutations is very low or even absent in other cancers. With particular reference to colorectal cancers, an in vitro study testing 11 different colorectal cell lines reported complete absence of such mutations [128]. A study examining 239 colorectal specimens reported the presence of only one mutation which was found to be activating mutation as described in non small cell lung cancer (NSCLC) [129]. Similarly, in several clinical trials, no EGFR mutations were found in evaluable responder as well as non-responder patients [110, 130, 131]. Only 1 in 31 patients in two phase I/II trials of FOLFIRI chemotherapy in combination with gefitinib was found to have an EGFR mutation, and this patient was a non-responder [107]. Taken together, it appears that presence of EGFR mutations is not a significant determinant of clinical benefit with EGFR targeted therapies in colorectal cancers.
In addition to the EGFR mutations, EGFR activation can be affected by the formation of receptor homo- or heterodimers within each of the other the three members of the EGFR family (such as ErbB-2 and ErbB-3) and increased expression of different ligands. Increased co-expression of both ErbB-2/ErbB-3 dimer complex and EGFR has been indicated to be responsible for the development of enhanced drug resistance in many cancers [132]. Other mechanisms of resistance to EGFR inhibitors includes: increased levels of soluble proteins acting as a ligands such as insulin-like growth factor-binding proteins (IGFBPs) and amplification of Met gene both of which have been demonstrated to be correlated to the resistant phenotype [133] and has been found to be associated with the acquired resistance to anti-EGFR monoclonal antibodies as well as EGFR-TKIs [134]. Activation of other alternative pathways such as IGF-IR, counteracts the ability of EGFR kinase inhibitors to block EGFR activity and therefore, confers the resistance to anti-EGFR therapy. Constitutive activation of EGFR-independent downstream pathways involving PI3k/Akt and MAPK pathways, has been reported to be insensitive to the EGFR inhibition, and therefore represents an important cause of resistance to EGFR-targeted therapy in solid tumors including GI cancers [5].
FUTURE DIRECTIONS
Targeting Cancer Stem Cells
Recently, cancer stem cells (CSCs), which represent a subpopulation of cells within the tumor possessing the ability to self-renew and give rise to all the cells within a tumor, have been implicated in resistance to conventional chemotherapy and subsequent relapse of many malignancies, including GI cancers [135, 136]. CSCs are the earliest undifferentiated progenitors with an unlimited capacity to propagate. CSCs have three unique properties that make them particularly important for tumor initiation, growth, and metastasis: self renewal; multipotency, and a high proliferative capacity upon differentiation [137]. Characterization of cancer stem cells has led to the identification of key cellular activities that may make cancer stem cells vulnerable to therapeutic strategies that target drug-effluxing capabilities, stem cell pathways, anti-apoptotic mechanisms, and induction of differentiation. If for a given tumor, a CSC is responsible for disease manifestation, future therapies that are not effective against CSCs may fall short of providing long term disease management. Recent results from our laboratory and others support a role of CSCs in many cancers [138, 139]. CSCs have been mainly characterized by a distinctive profile of surface markers that can be differentially and reproducibly isolated from the heterogeneous tumor cell population by flow cytometry. For GI cancers, they include CD44, CD133, CD 166, EpCAM,. Their function in promoting CSC maintenance and activity is largely unknown [140]. To date, CSCs have been studied in a relatively small number of patient samples and cancer types. It is not fully understood whether the CSC model is universal to all human cancers [141, 142]. Many research groups including us have reported the presence of CSCs in GI cancer [138, 139, 143–147].
Of note, reports from many research groups indicate a close association between increased expression of CSC markers and multi drug resistance (MDR) in various cancers [148–150]. To examine a potential association between the CSC and resistance to chemotherapeutic drugs, we evaluated the presence of CSC population in FOLFOX resistant human colon cancer cells. The CSC population was found to be increased manifold in the chemoresistant cells along with the marked increase in the expression of CSC specific markers such as CD133, CD166 and CD44, accompanied by increased EGFR expression [138, 151]. Recently, we demonstrated that CSC population in the colonic mucosa increases with aging which is further increased in response to the colonic carcinogen dimethylhydrazine [151]. In addition, we have demonstrated that in humans, the age-related rise in colonic polyps was accompanied by a concomitant rise in CSCs in macroscopically normal colonic mucosa [139]. Moreover, the age-related increase in CSC was found to be associated with a parallel rise in EGFR expression, indicating a relationship between the two events. The latter raises the possibility that EGFR and its family members may potentially be important for regulating the CSC mediated drug-resistance in GI cancers. Future studies will certainly address the role of EGFR(s) and the resistance mechanisms in CSCs as important steps in the development of targeted therapeutics. However, the biologic relevance of CSCs in human cancer will be established by concentrating on the following research endeavors: improving the assay and purification of CSC and non-CSC subsets, carrying out detailed genomic or proteomic analysis on these subsets to identify CSC-specific signatures, and obtaining such signatures from a large number and wide range of tumors. Of course, the most direct proof for the relevance of CSCs to cancer would be to use the CSC-specific signatures to elucidate the key molecular pathways involved, develop the means to target them effectively, and establish in clinical trials whether targeting them enhances patients’ disease free survival.
Chemoprevention by Natural Agents
The most practical approach to reduce the morbidity and mortality of GI cancer, is to delay the process of carcinogenesis through the use of chemopreventive natural agents. The use of nontoxic natural agents & phytochemicals in the dietary form for preventing cancer dates back to antiquity. Positive associations between dietary habits and increased risk of GI cancer have been alleged for many years. Various foods and dietary agents have been suggested to have potent anticancer activities. This necessitates that safer compounds, especially those derived from natural sources must be critically examined for chemoprevention. Indeed, there are number of clinical trials that have evaluated natural agents for their efficacy in treatment of GI cancers (Table 1).
Table 1.
Evaluation of Phytochemicals Targeting GI Cancers in Clinical Trials
| Phytochemical | GI Cancer Target | Phase | Result | * Reference |
|---|---|---|---|---|
| Carotenoids | Colon | - | In progress | NCT00475722 |
| Epigallocatechin gallate (EGCG) or green tea extracts | Esophageal | I | In progress | NCT00233935 |
| Colon | II | In progress | NCT00718094 | |
| Resveratrol | Colon | I | In progress | NCT00433576 |
| Colon | I | In progress | NCT00578396 | |
| Colon | I and II | In progress | NCT00256334 | |
| Curcumin | Colon | I | In progress | NCT00973869 |
| Colon | I | Completed | NCT00027495 | |
| Colon | II | In progress | NCT00118989 | |
| Colon | - | In progress | NCT00927485 | |
| Colon | - | Completed | NCT00176618 | |
| Colon | II | In progress | NCT00745134 | |
| Colon | III | In progress | NCT00706121 | |
| Colon | II | In progress | NCT00365209 | |
| Pancreatic | II | Completed | NCT00094445 |
Indicates reference found at www.clinicaltrials.gov with corresponding identifier code (NCT).
Among the most extensively studied natural compounds include plant polyphenols and phytochemical curcumin (the active compound in the spice turmeric). Polyphenol compounds are active agents in green tea and include EGCG and epigallocatechin (EGC), the most biologically active catechins. The ability of these compounds to inhibit tumor formation both in vitro and in vivo is well documented [152–154]. EGCG alters the expression of cell cycle regulatory proteins that are critical for cell survival and was effective in increasing apoptosis, mitochondrial membrane depolarization, and cytosolic cytochrome C expression. In animal studies EGCG has been reported to be effective in decreasing the growth of intestinal tumors and inhibits angiogenesis in mice inoculated with human colon cancer [155].
Curcumin affects multiple cell signaling pathways. It has been to inhibit cell cycle progression, cell proliferation via blocking the EGFR, MAPK and PI3K/AKT pathways, suppress angiogenesis by blocking VEGF, antagonizing inflammation via inhibiting NF-κB, TNFα and COX-2 and induce apoptosis by activating caspases [42, 152, 156]. Curcumin has also been shown to improve the histology of intestinal metaplasia of the stomach in 17% of patients [157]. Curcumin is considered non-toxic since a dose of 8 g daily for three months and Phase 1 clinical trials showed no apparent signs toxicity; therefore, curcumin can be considered a promising chemopreventive agent for GI cancers, specifically colon cancer [158–161]. Recent studies have further demonstrated that the combinatorial treatment of curcumin and resveratrol causes a greater inhibition of colon tumors in a SCID mice xenograft model, which could be attributed to decreased inhibition of activation of EGFR and its family members as well as IGF-1R [152]. In addition, curcumin has been shown to synergize with FOLFOX, the mainstay of colon cancer chemotherapy, to inhibit the growth of colon cancer cells in vitro (Patel et al. 2009 – Anticancer Research-In Press). Furthermore, curcumin has also been shown synergize with dasatanib, a specific inhibitor of c-Src tyrosine kinases, to inhibit the growth of colon cancer cells as well as intestinal adenomas in APCmin−/+ mice [162]. Curcumin alone or in combination with FOLFOX has been found to be effective in eliminating the colon cancer-stem like cells [138].
Of particular importance are the recent findings showing that natural agents (like curcumin and EGCG) interact with ABC transport proteins and reverse multidrug resistance (MDR) phenotype in vitro as well as in vivo [155, 163–166]. Various independent studies have shown that combination treatment of curcumin or EGCG with a variety of chemotherapy drugs (i.e., cisplatin, danorubicin, doxorubicin, and vinscristine) enhances the cellular accumulation of these drugs thereby increasing the cells sensitivity to the chemotherapeutics [167, 168]. These findings strongly indicate that natural products hold a great promise as anti-MDR agents and given a strong link between the CSCs and MDR this could be utilized to target CSCs [140]. Given the clinical implications and the widespread use, phytochemicals represents a promising addition to standard therapy in the treatment of GI cancers.
Combinatorial Therapies
It is now well established that EGFR signaling is important to both normal development and neoplastic transformation and that EGFR inhibition represents a valid anticancer approach. However, constitutive and acquired resistance to EGFR inhibitors represents a major and unsolved clinical problem in treating GI cancer patients who show high EGFR expression in tumors. The elucidation of the molecular mechanisms of resistance to anti-EGFR therapies has stimulated novel combined molecularly targeted therapeutic approaches aimed at blocking the EGFR(s) and their downstream events. Angiogenesis is an important prerequisite for cancer development and progression and strategies aiming at inhibiting angiogenesis are of potential value in cancer therapy. Therefore, strong rationale exists for using EGFR(s) targeted therapies in combination with anti-angiogenic treatment modalities or with other drugs that induces apoptosis. Hence, there are many novel targets that are currently being evaluated in clinical trials [169]. As discussed above the evaluation of anti-EGFR monoclonal antibodies and anti VEGF inhibitor (bevacizumab) in combination with each other and with chemotherapeutics other than irinotecan is being evaluated to assess the best regimens in colorectal cancer patients [170]. Regimens combining nontoxic natural chemopreventive agents like curcumin and EGCG with the chemotherapy or biologic agents are also being evaluated in the preclinical and clinical settings. However, important clinical and pharmacological work need to be conducted in the areas of patient selection, selection of appropriate dose and schedules with new agents, and implementation of strategies to study the appropriate combinations. Future clinical trials with strong correlative markers will allow us to delineate some of these issues.
Acknowledgments
A part of the work presented in this communication has been supported by grants to Dr. Majumdar by NIH/NIA (AG014343) and the Department of Veterans Affairs (VA Merit Review).
References
- 1.Gravalos C, Cassinello J, Fernandez-Ranada I, Holgado E. Role of tyrosine kinase inhibitors in the treatment of advanced colorectal cancer. Clin Colorectal Cancer. 2007;6:691–9. doi: 10.3816/CCC.2007.n.038. [DOI] [PubMed] [Google Scholar]
- 2.Saltz L. Colorectal Cancer Treatment: What’s Next? (or: Is There Life After EGFR and VEGF?) Gastrointest Cancer Res. 2008;2:S20–2. [PMC free article] [PubMed] [Google Scholar]
- 3.Amado RG, Wolf M, Peeters M, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 4.Lievre A, Bachet JB, Boige V, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008;26:374–9. doi: 10.1200/JCO.2007.12.5906. [DOI] [PubMed] [Google Scholar]
- 5.Morgillo F, Cantile F, Fasano M, Troiani T, Martinelli E, Ciardiello F. Resistance mechanisms of tumour cells to EGFR inhibitors. Clin Transl Oncol. 2009;11:270–5. doi: 10.1007/s12094-009-0354-6. [DOI] [PubMed] [Google Scholar]
- 6.World health organization. [accessed December 14, 2009];Media Center- Fact sheet N° 297, February 2009- Cancer. Available at: http://www.who.int/mediacentre/factsheets/fs297/en/index.html.
- 7.Parkin DM, Bray F, Ferlay J, Pisani P. Global Cancer Statistics 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 8. [[accessed December 14, 2009].];International agency for reearch on cancer (World health organization)- camcer mondial. Available at: http://www-dep.iarc.fr/
- 9.Yamaoka Y, Kato M, Asaka M. Geographic differences in gastric cancer incidence can be explained by differences between Helicobacter pylori strains. Intern Med. 2008;47:1077–83. doi: 10.2169/internalmedicine.47.0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamangar F, Dores GM, Anderson WF. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. J Clin Oncol. 2006;24:2137–50. doi: 10.1200/JCO.2005.05.2308. [DOI] [PubMed] [Google Scholar]
- 11.Kurie JM, Shin HJ, Lee JS, et al. Increased epidermal growth factor receptor expression in metaplastic bronchial epithelium. Clin Cancer Res. 1996;2:1787–93. [PubMed] [Google Scholar]
- 12.Bosetti C, Malvezzi M, Chatenoud L, Negri E, Levi F, La Vecchia C. Trends in cancer mortality in the Americas, 1970–2000. Ann Oncol. 2005;16:489–511. doi: 10.1093/humrep/mdi086. [DOI] [PubMed] [Google Scholar]
- 13.Bae JM, Lee EJ, Guyatt G. Citrus fruit intake and stomach cancer risk: a quantitative systematic review. Gastric Cancer. 2008;11:23–32. doi: 10.1007/s10120-007-0447-2. [DOI] [PubMed] [Google Scholar]
- 14.Ladeiras-Lopes R, Pereira AK, Nogueira A, et al. Smoking and gastric cancer: systematic review and meta-analysis of cohort studies. Cancer Causes Control. 2008;19:689–701. doi: 10.1007/s10552-008-9132-y. [DOI] [PubMed] [Google Scholar]
- 15.Tsugane S, Sasazuki S. Diet and the risk of gastric cancer: review of epidemiological evidence. Gastric Cancer. 2007;10:75–83. doi: 10.1007/s10120-007-0420-0. [DOI] [PubMed] [Google Scholar]
- 16.Group HaCC. Gastric cancer and Helicobacter pylori: a combined analysis of 12 case control studies nested within prospective cohorts. Gut. 2001;49:347–53. doi: 10.1136/gut.49.3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Center MM, Jemal A, Ward E. International trends in colorectal cancer incidence rates. Cancer Epidemiol Biomarkers Prev. 2009;18:1688–94. doi: 10.1158/1055-9965.EPI-09-0090. [DOI] [PubMed] [Google Scholar]
- 18.Parkin DM. International variation. Oncogene. 2004;23:6329–40. doi: 10.1038/sj.onc.1207726. [DOI] [PubMed] [Google Scholar]
- 19.Botteri E, Iodice S, Bagnardi V, Raimondi S, Lowenfels AB, Maisonneuve P. Smoking and colorectal cancer: a meta-analysis. JAMA. 2008;300:2765–78. doi: 10.1001/jama.2008.839. [DOI] [PubMed] [Google Scholar]
- 20.Leblond CP, Stevens CE. The constant renewal of the intestinal epithelium in the albino rat. Anat Rec. 1948;100:357–77. doi: 10.1002/ar.1091000306. [DOI] [PubMed] [Google Scholar]
- 21.Lipkin M. In: Physiology of the gastrointestinal tract. Johnson LR, editor. New York: Raven Press; 1987. pp. 255–84. [Google Scholar]
- 22.Wong WM, Wright NA. Cell proliferation in gastrointestinal mucosa. J Clin Pathol. 1999;52:321–33. doi: 10.1136/jcp.52.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leblond CP, Messier B. Renewal of chief cells and goblet cells in the small intestine as shown by radioautography after injection of thymidine-H3 into mice. Anat Rec. 1958;132:247–59. doi: 10.1002/ar.1091320303. [DOI] [PubMed] [Google Scholar]
- 24.Radtke F, Clevers H. Self-renewal and cancer of the gut: two sides of a coin. Science. 2005;307:1904–9. doi: 10.1126/science.1104815. [DOI] [PubMed] [Google Scholar]
- 25.Podolsky DK. Regulation of intestinal epithelial proliferation: a few answers, many questions. Am J Physiol. 1993;264:G179–86. doi: 10.1152/ajpgi.1993.264.2.G179. [DOI] [PubMed] [Google Scholar]
- 26.Jaszewski R, Ehrinpreis MN, Majumdar AP. Aging and cancer of the stomach and colon. Front Biosci. 1999;4:D322–8. doi: 10.2741/jaszewsk. [DOI] [PubMed] [Google Scholar]
- 27.Jemal A, Thomas A, Murray T, Thun M. Cancer statistics, 2002. CA Cancer J Clin. 2002;52:23–47. doi: 10.3322/canjclin.52.1.23. [DOI] [PubMed] [Google Scholar]
- 28.Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer. 2009;9:489–99. doi: 10.1038/nrc2645. [DOI] [PubMed] [Google Scholar]
- 29.Chung DC. The genetic basis of colorectal cancer: insights into critical pathways of tumorigenesis. Gastroenterology. 2000;119:854–65. doi: 10.1053/gast.2000.16507. [DOI] [PubMed] [Google Scholar]
- 30.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 31.Liotta LA, Clair T. Cancer. Checkpoint for invasion. Nature. 2000;405:287–8. doi: 10.1038/35012728. [DOI] [PubMed] [Google Scholar]
- 32.Roncucci L, Pedroni M, Vaccina F, Benatti P, Marzona L, De Pol A. Aberrant crypt foci in colorectal carcinogenesis. Cell Crypt dynamics Cell Prolif. 2000;33:1–18. doi: 10.1046/j.1365-2184.2000.00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–70. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 34.Hruban RH, Takaori K, Klimstra DS, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004;28:977–87. doi: 10.1097/01.pas.0000126675.59108.80. [DOI] [PubMed] [Google Scholar]
- 35.Lin J, Beerm DG. Molecular biology of upper gastrointestinal malignancies. Semin Oncol. 2004;31:476–86. doi: 10.1053/j.seminoncol.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 36.Yuasa Y. Control of gut differentiation and intestinal-type gastric carcinogenesis. Nat Rev Cancer. 2003;3:592–600. doi: 10.1038/nrc1141. [DOI] [PubMed] [Google Scholar]
- 37.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 38.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flemming W. Über die Bildung von Richtungsfiguren in Säugethiereiern beim Untergang Graaf’ scher Follikel. Arch Anat Physiol. 1885:221–44.
- 40.Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–16. [PubMed] [Google Scholar]
- 41.Martin SJ, Reutelingsperger CP, McGahon AJ, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–56. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qiao L, Wong BC. Targeting apoptosis as an approach for gastrointestinal cancer therapy. Drug Resist Updat. 2009;12:55–64. doi: 10.1016/j.drup.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 43.Ramachandran A, Madesh M, Balasubramanian KA. Apoptosis in the intestinal epithelium: Its relevance in normal and pathophysiological conditions. J Gastroenterol Hepatol. 2000;15:109–20. doi: 10.1046/j.1440-1746.2000.02059.x. [DOI] [PubMed] [Google Scholar]
- 44.Majumdar APN, Basson MD. In: Physiology of the gastrointestinal tract. Johnson LR, editor. Vol. 1. New York: Academic Press; 2006. pp. 405–33. [Google Scholar]
- 45.Takeda K, Stagg J, Yagita H, Okumura K, Smyth MJ. Targeting death-inducing receptors in cancer therapy. Oncogene. 2007;26:3745–57. doi: 10.1038/sj.onc.1210374. [DOI] [PubMed] [Google Scholar]
- 46.Iannolo G, Conticello C, Memeo L, De Maria R. Apoptosis in normal and cancer stem cells. Crit Rev Oncol Hematol. 2008;66:42–51. doi: 10.1016/j.critrevonc.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 47.Oliver L, Vallette FM. The role of caspases in cell death and differentiation. Drug Resist Updat. 2005;8:163–70. doi: 10.1016/j.drup.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 48.Reed JC. Mechanisms of apoptosis avoidance in cancer. Curr Opin Oncol. 1999;11:68–75. doi: 10.1097/00001622-199901000-00014. [DOI] [PubMed] [Google Scholar]
- 49.Thorburn A, Behbakht K, Ford H. TRAIL receptor-targeted therapeutics: resistance mechanisms and strategies to avoid them. Drug Resist Updat. 2008;11:17–24. doi: 10.1016/j.drup.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Letai AG. Diagnosing and exploiting cancer’s addiction to blocks in apoptosis. Nat Rev Cancer. 2008;8:121–32. doi: 10.1038/nrc2297. [DOI] [PubMed] [Google Scholar]
- 51.Cohen S, Ushiro H, Stoscheck C, Chinkers M. A native 170,000 epidermal growth factor receptor-kinase complex from shed plasma membrane vesicles. J Biol Chem. 1982;257:1523–31. [PubMed] [Google Scholar]
- 52.Jorissen RN, Walker F, Pouliot N, Garrett TPJ, Ward CW, Burgess AW. Epidermal growth factor receptor: mechanisms of activation and signalling. Exper Cell Res. 2003;284:31–53. doi: 10.1016/s0014-4827(02)00098-8. [DOI] [PubMed] [Google Scholar]
- 53.Nautiyal J, Rishi AK, Majumdar AP. Emerging therapies in gastrointestinal cancers. World J Gastroenterol. 2006;12:7440–50. doi: 10.3748/wjg.v12.i46.7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olayioye MA, Neve RM, Lane HA, Hynes NE. The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 2000;19:3159–67. doi: 10.1093/emboj/19.13.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burgess AW, Cho HS, Eigenbrot C, et al. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol Cell. 2003;12:541–52. doi: 10.1016/s1097-2765(03)00350-2. [DOI] [PubMed] [Google Scholar]
- 56.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–54. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 57.Garrett TP, McKern NM, Lou M, et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol Cell. 2003;11:495–505. doi: 10.1016/s1097-2765(03)00048-0. [DOI] [PubMed] [Google Scholar]
- 58.Yasui W, Sumiyoshi H, Hata J, et al. Expression of epidermal growth factor receptor in human gastric and colonic carcinomas. Cancer Res. 1988;48:137–41. [PubMed] [Google Scholar]
- 59.Kaklamanis L, Gatter KC, Mortensen N, Harris AL. Interleukin-4 receptor and epidermal growth factor receptor expression in colorectal cancer. Br J Cancer. 1992;66:712–6. doi: 10.1038/bjc.1992.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zimmermann M, Zouhair A, Azria D, Ozsahin M. The epidermal growth factor receptor (EGFR) in head and neck cancer: its role and treatment implications. Radiat Oncol. 2006;1:11. doi: 10.1186/1748-717X-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dimri M, Naramura M, Duan L, et al. Modeling breast cancer-associated c-Src and EGFR overexpression in human MECs: c-Src and EGFR cooperatively promote aberrant three-dimensional acinar structure and invasive behavior. Cancer Res. 2007;67:4164–72. doi: 10.1158/0008-5472.CAN-06-2580. [DOI] [PubMed] [Google Scholar]
- 62.Villares GJ, Zigler M, Blehm K, et al. Targeting EGFR in bladder cancer. World J Urol. 2007;25:573–9. doi: 10.1007/s00345-007-0202-7. [DOI] [PubMed] [Google Scholar]
- 63.Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009;9:463–75. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- 64.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–37. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 65.Uchino S, Tsuda H, Maruyama K, et al. Overexpression of c-erbB-2 protein in gastric cancer. Its correlation with long-term survival of patients. Cancer. 1993;72:3179–84. doi: 10.1002/1097-0142(19931201)72:11<3179::aid-cncr2820721108>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 66.Vermeij J, Teugels E, Bourgain C, et al. Genomic activation of the EGFR and HER2-neu genes in a significant proportion of invasive epithelial ovarian cancers. BMC Cancer. 2008;8:3. doi: 10.1186/1471-2407-8-3. Available from: http://www.biomedcentral.com/1471-2407/8/3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giaccone G. HER1/EGFR-targeted agents: predicting the future for patients with unpredictable outcomes to therapy. Ann Oncol. 2005;16:538–48. doi: 10.1093/annonc/mdi129. [DOI] [PubMed] [Google Scholar]
- 68.Tedesco KL, Lockhart AC, Berlin JD. The epidermal growth factor receptor as a target for gastrointestinal cancer therapy. Curr Treat Options Oncol. 2004;5:393–403. doi: 10.1007/s11864-004-0029-z. [DOI] [PubMed] [Google Scholar]
- 69.Tortora G, Bianco R, Daniele G, et al. Overcoming resistance to molecularly targeted anticancer therapies: Rational drug combinations based on EGFR and MAPK inhibition for solid tumours and haematologic malignancies. Drug Resist Updat. 2007;10:81–100. doi: 10.1016/j.drup.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yotsumoto F, Yagi H, Suzuki SO, et al. Validation of HB-EGF and amphiregulin as targets for human cancer therapy. Biochem Biophys Res Commun. 2008;365:555–61. doi: 10.1016/j.bbrc.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 71.Patel DK. Clinical use of anti-epidermal growth factor receptor monoclonal antibodies in metastatic colorectal cancer. Pharmacotherapy. 2008;28:31S–41S. doi: 10.1592/phco.28.11-supp.31S. [DOI] [PubMed] [Google Scholar]
- 72.Weiner LM. Fully human therapeutic monoclonal antibodies. J Immunother. 2006;29:1–9. doi: 10.1097/01.cji.0000192105.24583.83. [DOI] [PubMed] [Google Scholar]
- 73.Goodin S. Development of monoclonal antibodies for the treatment of colorectal cancer. Am J Health Syst Pharm. 2008;65:S3–7. doi: 10.2146/ajhp080100. [DOI] [PubMed] [Google Scholar]
- 74.Galizia G, Lieto E, De Vita F, et al. Cetuximab, a chimeric human mouse anti-epidermal growth factor receptor monoclonal antibody, in the treatment of human colorectal cancer. Oncogene. 2007;26:3654–60. doi: 10.1038/sj.onc.1210381. [DOI] [PubMed] [Google Scholar]
- 75.Jean GW, Shah SR. Epidermal growth factor receptor monoclonal antibodies for the treatment of metastatic colorectal cancer. Pharmacotherapy. 2008;28:742–54. doi: 10.1592/phco.28.6.742. [DOI] [PubMed] [Google Scholar]
- 76.Needle MN. Safety experience with IMC-C225, an anti-epidermal growth factor receptor antibody. Semin Oncol. 2002;29:55–60. doi: 10.1053/sonc.2002.35648. [DOI] [PubMed] [Google Scholar]
- 77.Mahtani RL, Macdonald JS. Synergy between cetuximab and chemotherapy in tumors of the gastrointestinal tract. Oncologist. 2008;13:39–50. doi: 10.1634/theoncologist.2006-0049. [DOI] [PubMed] [Google Scholar]
- 78.Raoul JL, Van Laethem JL, Peeters M, et al. Cetuximab in combination with irinotecan/5-fluorouracil/folinic acid (FOLFIRI) in the initial treatment of metastatic colorectal cancer: a multicentre two-part phase I/II study. BMC Cancer. 2009;9:112. doi: 10.1186/1471-2407-9-112. Avaialble from: http://www.biomedcentral.com/1471-2407/9/112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saltz LB, Douillard JY, Pirotta N, et al. Irinotecan plus fluorouracil/leucovorin for metastatic colorectal cancer: a new survival standard. Oncologist. 2001;6:81–91. doi: 10.1634/theoncologist.6-1-81. [DOI] [PubMed] [Google Scholar]
- 80.Sobrero A, Andretta V. Small molecule inhibitors of tyrosine kinase: waiting for the good news in colorectal cancer as well. Onkologie. 2008;31:224–5. doi: 10.1159/000122265. [DOI] [PubMed] [Google Scholar]
- 81.Cunningham D, Humblet Y, Siena S, et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med. 2004;351:337–45. doi: 10.1056/NEJMoa033025. [DOI] [PubMed] [Google Scholar]
- 82.Sobrero AF, Maurel J, Fehrenbacher L, et al. EPIC: phase III trial of cetuximab plus irinotecan after fluoropyrimidine and oxaliplatin failure in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:2311–9. doi: 10.1200/JCO.2007.13.1193. [DOI] [PubMed] [Google Scholar]
- 83.Souglakos J, Kalykaki A, Vamvakas L, et al. Phase II trial of capecitabine and oxaliplatin (CAPOX) plus cetuximab in patients with metastatic colorectal cancer who progressed after oxaliplatin-based chemotherapy. Ann Oncol. 2007;18:305–10. doi: 10.1093/annonc/mdl392. [DOI] [PubMed] [Google Scholar]
- 84.Lee JJ, Chu E. An update on treatment advances for the first-line therapy of metastatic colorectal cancer. Cancer J. 2007;13:276–81. doi: 10.1097/PPO.0b013e3181570062. [DOI] [PubMed] [Google Scholar]
- 85.Van Cutsem E, Nowacki M, Lang I, et al. Randomized phase III study of irinotecan and 5-FU/FA with or without cetuximab in the first-line treatment of patients with metastatic colorectal cancer (mCRC): The CRYSTAL trial. J Clin Oncol (Meeting Abstracts) 2007;25:4000. Available from: http://meeting.ascopubs.org/cgi/content/abstract/25/18_suppl/4000. [Google Scholar]
- 86.Delbaldo C, Pierga JY, Dieras V, et al. Pharmacokinetic profile of cetuximab (Erbitux) alone and in combination with irinotecan in patients with advanced EGFR-positive adenocarcinoma. Eur J Cancer. 2005;41:1739–45. doi: 10.1016/j.ejca.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 87.Folprecht G, Lutz MP, Schoffski P, et al. Cetuximab and irinotecan/5-fluorouracil/folinic acid is a safe combination for the first-line treatment of patients with epidermal growth factor receptor expressing metastatic colorectal carcinoma. Ann Oncol. 2006;17:450–6. doi: 10.1093/annonc/mdj084. [DOI] [PubMed] [Google Scholar]
- 88.Saadeh CE, Lee HS. Panitumumab: a fully human monoclonal antibody with activity in metastatic colorectal cancer. Ann Pharmacother. 2007;41:606–13. doi: 10.1345/aph.1H492. [DOI] [PubMed] [Google Scholar]
- 89.Carteni G, Fiorentino R, Vecchione L, Chiurazzi B, Battista C. Panitumumab a novel drug in cancer treatment. Ann Oncol. 2007;18:vi16–21. doi: 10.1093/annonc/mdm218. [DOI] [PubMed] [Google Scholar]
- 90.Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005;23:1147–57. doi: 10.1038/nbt1137. [DOI] [PubMed] [Google Scholar]
- 91.Van Cutsem E, Siena S, Humblet Y, et al. An open-label, single-arm study assessing safety and efficacy of panitumumab in patients with metastatic colorectal cancer refractory to standard chemotherapy. Ann Oncol. 2008;19:92–8. doi: 10.1093/annonc/mdm399. [DOI] [PubMed] [Google Scholar]
- 92.Berlin J, Posey J, Tchekmedyian S, et al. Panitumumab with Irinotecan/Leucovorin/5-Fluorouracil for first-line treatment of metastatic colorectal cancer. Clin Colorect Cancer. 2007;6:427–32. doi: 10.3816/CCC.2007.n.011. [DOI] [PubMed] [Google Scholar]
- 93.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–25. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 94.Pao W, Miller VA, Kris MG. ‘Targeting’ the epidermal growth factor receptor tyrosine kinase with gefitinib (Iressa®) in non-small cell lung cancer (NSCLC) Semin Cancer Biol. 2004;14:33–40. doi: 10.1016/j.semcancer.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 95.Pollack VA, Savage DM, Baker DA, et al. Inhibition of epidermal growth factor receptor-associated tyrosine phosphorylation in human carcinomas with CP-358,774: dynamics of receptor inhibition in situ and antitumor effects in athymic mice. J Pharmacol Exp Ther. 1999;291:739–48. [PubMed] [Google Scholar]
- 96.Santoro A, Comandone A, Rimassa L, et al. A phase II randomized multicenter trial of gefitinib plus FOLFIRI and FOLFIRI alone in patients with metastatic colorectal cancer. Annals of Oncology. 2008;19:1888–93. doi: 10.1093/annonc/mdn401. [DOI] [PubMed] [Google Scholar]
- 97.Jasas KV, Fyles A, Elit L, et al. Phase II study of erlotinib (OSI 774) in women with recurrent or metastatic endometrial cancer: NCIC CTG IND-148. Proc Am Soc Clin Oncol. 2004;22:14S. doi: 10.1200/JCO.2007.15.8808. [DOI] [PubMed] [Google Scholar]
- 98.Perez-Soler R. Phase II clinical trial data with the epidermal growth factor receptor tyrosine kinase inhibitor erlotinib (OSI-774) in non-small-cell lung cancer. Clin Lung Cancer. 2004;6:S20–3. doi: 10.3816/clc.2004.s.010. [DOI] [PubMed] [Google Scholar]
- 99.Philip PA, Mahoney MR, Allmer C, et al. Phase II study of Erlotinib (OSI-774) in patients with advanced hepatocellular cancer. J Clin Oncol. 2005;23:6657–63. doi: 10.1200/JCO.2005.14.696. [DOI] [PubMed] [Google Scholar]
- 100.Soulieres D, Senzer NN, Vokes EE, Hidalgo M, Agarwala SS, Siu LL. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J Clin Oncol. 2004;22:77–85. doi: 10.1200/JCO.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 101.Gordon AN, Finkler N, Edwards RP, et al. Efficacy and safety of erlotinib HCl, an epidermal growth factor receptor (HER1/EGFR) tyrosine kinase inhibitor, in patients with advanced ovarian carcinoma: results from a phase II multicenter study. Int J Gynecol Cancer. 2005;15:785–92. doi: 10.1111/j.1525-1438.2005.00137.x. [DOI] [PubMed] [Google Scholar]
- 102.Townsley CA, Major P, Siu LL, et al. Phase II study of erlotinib (OSI-774) in patients with metastatic colorectal cancer. Br J Cancer. 2006;94:1136–43. doi: 10.1038/sj.bjc.6603055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Meyerhardt JA, Stuart K, Fuchs CS, et al. Phase II study of FOLFOX, bevacizumab and erlotinib as first-line therapy for patients with metastatic colorectal cancer. Ann Oncol. 2007;18:1185–9. doi: 10.1093/annonc/mdm124. [DOI] [PubMed] [Google Scholar]
- 104.Chen J, Smith M, Kolinsky K, et al. Antitumor activity of HER1/EGFR tyrosine kinase inhibitor erlotinib, alone and in combination with CPT-11 (irinotecan) in human colorectal cancer xenograft models. Cancer Chemother Pharmacol. 2007;59:651–9. doi: 10.1007/s00280-006-0320-8. [DOI] [PubMed] [Google Scholar]
- 105.Kelley RK, Ko AH. Erlotinib in the treatment of advanced pancreatic cancer. Biologies. 2008;2:83–95. doi: 10.2147/btt.s1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rothenberg ML, LaFleur B, Levy DE, et al. Randomized phase II trial of the clinical and biological effects of two dose levels of gefitinib in patients with recurrent colorectal adenocarcinoma. J Clin Oncol. 2005;23:9265–74. doi: 10.1200/JCO.2005.03.0536. [DOI] [PubMed] [Google Scholar]
- 107.Ogino S, Meyerhardt JA, Cantor M, et al. Molecular alterations in tumors and response to combination chemotherapy with gefitinib for advanced colorectal cancer. Clin Cancer Res. 2005;11:6650–6. doi: 10.1158/1078-0432.CCR-05-0738. [DOI] [PubMed] [Google Scholar]
- 108.Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 109.Kabbinavar FF, Schulz J, McCleod M, et al. Addition of bevacizumab to bolus fluorouracil and leucovorin in first-line metastatic colorectal cancer: results of a randomized phase II trial. J Clin Oncol. 2005;23:3697–705. doi: 10.1200/JCO.2005.05.112. [DOI] [PubMed] [Google Scholar]
- 110.Saltz LB, Meropol NJ, Loehrer PJ, Sr, Needle MN, Kopit J, Mayer RJ. Phase II trial of cetuximab in patients with refractory colorectal cancer that expresses the epidermal growth factor receptor. J Clin Oncol. 2004;22:1201–8. doi: 10.1200/JCO.2004.10.182. [DOI] [PubMed] [Google Scholar]
- 111.Hecht JR. Improved imaging and the clinician: the role of positron emission tomography in the management of colorectal cancer. Mol Imaging Biol. 2004;6:208–13. doi: 10.1016/j.mibio.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 112.Ellis LM, Hoff PM. Targeting the epidermal growth factor receptor: an important incremental step in the battle against colorectal cancer. J Clin Oncol. 2004;22:1177–9. doi: 10.1200/JCO.2004.01.971. [DOI] [PubMed] [Google Scholar]
- 113.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 114.Learn CA, Hartzell TL, Wikstrand CJ, et al. Resistance to tyrosine kinase inhibition by mutant epidermal growth factor receptor variant III contributes to the neoplastic phenotype of glioblastoma multiforme. Clin Cancer Res. 2004;10:3216–24. doi: 10.1158/1078-0432.ccr-03-0521. [DOI] [PubMed] [Google Scholar]
- 115.Bianco R, Troiani T, Tortora G, Ciardiello F. Intrinsic and acquired resistance to EGFR inhibitors in human cancer therapy. Endocr Relat Cancer. 2005;1(Suppl12):S159–71. doi: 10.1677/erc.1.00999. [DOI] [PubMed] [Google Scholar]
- 116.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–67. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 117.Forbes S, Clements J, Dawson E, et al. Cosmic 2005. Br J Cancer. 2006;94:318–22. doi: 10.1038/sj.bjc.6602928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Siena S, Sartore-Bianchi A, Di Nicolantonio F, Balfour J, Bardelli A. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst. 2009;101:1308–24. doi: 10.1093/jnci/djp280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.American Society of Clinical Oncology (ASCO) [[accessed December 14, 2009].]; Available at: http://www.asco.org/pco/kras.
- 120.Di Nicolantonio F, Martini M, Molinari F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26:5705–12. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 121.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 122.Sequist LV, Bell DW, Lynch TJ, Haber DA. Molecular predictors of response to epidermal growth factor receptor antagonists in non-small-cell lung cancer. J Clin Oncol. 2007;25:587–95. doi: 10.1200/JCO.2006.07.3585. [DOI] [PubMed] [Google Scholar]
- 123.Inoue A, Suzuki T, Fukuhara T, et al. Prospective phase II study of gefitinib for chemotherapy-naive patients with advanced non-small-cell lung cancer with epidermal growth factor receptor gene mutations. J Clin Oncol. 2006;24:3340–6. doi: 10.1200/JCO.2005.05.4692. [DOI] [PubMed] [Google Scholar]
- 124.Mitsudomi T, Kosaka T, Endoh H, et al. Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non-small-cell lung cancer with postoperative recurrence. J Clin Oncol. 2005;23:2513–20. doi: 10.1200/JCO.2005.00.992. [DOI] [PubMed] [Google Scholar]
- 125.van Zandwijk N, Mathy A, Boerrigter L, et al. EGFR and KRAS mutations as criteria for treatment with tyrosine kinase inhibitors: retro- and prospective observations in non-small-cell lung cancer. Ann Oncol. 2007;18:99–103. doi: 10.1093/annonc/mdl323. [DOI] [PubMed] [Google Scholar]
- 126.Han SW, Kim TY, Hwang PG, et al. Predictive and prognostic impact of epidermal growth factor receptor mutation in non-small-cell lung cancer patients treated with gefitinib. J Clin Oncol. 2005;23:2493–501. doi: 10.1200/JCO.2005.01.388. [DOI] [PubMed] [Google Scholar]
- 127.Doody JF, Wang Y, Patel SN, et al. Inhibitory activity of cetuximab on epidermal growth factor receptor mutations in non small cell lung cancers. Mol Cancer Ther. 2007;6:2642–51. doi: 10.1158/1535-7163.MCT-06-0506. [DOI] [PubMed] [Google Scholar]
- 128.Nagahara H, Mimori K, Ohta M, et al. Somatic mutations of epidermal growth factor receptor in colorectal carcinoma. Clin Cancer Res. 2005;11:1368–71. doi: 10.1158/1078-0432.CCR-04-1894. [DOI] [PubMed] [Google Scholar]
- 129.Barber TD, Vogelstein B, Kinzler KW, Velculescu VE. Somatic mutations of EGFR in colorectal cancers and glioblastomas. N Engl J Med. 2004;351:2883. doi: 10.1056/NEJM200412303512724. [DOI] [PubMed] [Google Scholar]
- 130.Lenz HJ, Van Cutsem E, Khambata-Ford S, et al. Multicenter phase II and translational study of cetuximab in metastatic colorectal carcinoma refractory to irinotecan, oxaliplatin, and fluoropyrimidines. J Clin Oncol. 2006;24:4914–21. doi: 10.1200/JCO.2006.06.7595. [DOI] [PubMed] [Google Scholar]
- 131.Tsuchihashi Z, Khambata-Ford S, Hanna N, Janne PA. Responsiveness to cetuximab without mutations in EGFR. N Engl J Med. 2005;353:208–9. doi: 10.1056/NEJM200507143530218. [DOI] [PubMed] [Google Scholar]
- 132.Chen X, Yeung TK, Wang Z. Enhanced Drug Resistance in Cells Coexpressing ErbB2 with EGF Receptor or ErbB3. Biochem Biophys Res Commun. 2000;277:757–63. doi: 10.1006/bbrc.2000.3731. [DOI] [PubMed] [Google Scholar]
- 133.Guix M, Faber AC, Wang SE, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest. 2008;118:2609–19. doi: 10.1172/JCI34588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bean J, Riely GJ, Balak M, et al. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res. 2008;14:7519–25. doi: 10.1158/1078-0432.CCR-08-0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ishii H, Haraguchi N, Ieta K, Mimori K, Mori M. In: Stem cells and cancer. Teicher Beverly A, Bagley Rebecca G., editors. Part 4. New York: Springer; 2009. pp. 155–63. [Google Scholar]
- 136.Huang EH, Wicha MS. Colon cancer stem cells: implications for prevention and therapy. Trends Mol Med. 2008;14:503–9. doi: 10.1016/j.molmed.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 138.Yu Y, Kanwar SS, Patel BB, Nautiyal J, Sarkar FH, Majumdar APN. Elimination of Colon Cancer Stem–Like Cells by the Combination of Curcumin and FOLFOX. Transl Oncol. 2009;2:321–8. doi: 10.1593/tlo.09193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Patel BB, Yu Y, Du J, Levi E, Phillip PA, Majumdar AP. Age-related increase in colorectal cancer stem cells in macroscopically normal mucosa of patients with adenomas: a risk factor for colon cancer. Biochem Biophys Res Commun. 2009;378:344–7. doi: 10.1016/j.bbrc.2008.10.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kawasaki BT, Hurt EM, Mistree T, Farrar WL. Targeting cancer stem cells with phytochemicals. Mol Interv. 2008;8:174–84. doi: 10.1124/mi.8.4.9. [DOI] [PubMed] [Google Scholar]
- 141.Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15:494–501. doi: 10.1016/j.tcb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 142.Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–84. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 143.Carpentino JE, Hynes MJ, Appelman HD, et al. Aldehyde dehydrogenase-expressing colon stem cells contribute to tumorigenesis in the transition from colitis to cancer. Cancer Res. 2009;69:8208–15. doi: 10.1158/0008-5472.CAN-09-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ricci-Vitiani L, Fabrizi E, Palio E, De Maria R. Colon cancer stem cells. J Mol Med. 2009;87:1097–104. doi: 10.1007/s00109-009-0518-4. [DOI] [PubMed] [Google Scholar]
- 145.Todaro M, D’Asaro M, Caccamo N, et al. Efficient killing of human colon cancer stem cells by gammadelta T lymphocytes. J Immunol. 2009;182:7287–96. doi: 10.4049/jimmunol.0804288. [DOI] [PubMed] [Google Scholar]
- 146.Chu P, Clanton DJ, Snipas TS, et al. Characterization of a subpopulation of colon cancer cells with stem cell-like properties. Int J Cancer. 2009;124:1312–21. doi: 10.1002/ijc.24061. [DOI] [PubMed] [Google Scholar]
- 147.Boman BM, Huang E. Human colon cancer stem cells: a new paradigm in gastrointestinal oncology. J Clin Oncol. 2008;26:2828–38. doi: 10.1200/JCO.2008.17.6941. [DOI] [PubMed] [Google Scholar]
- 148.Shervington A, Lu C. Expression of multidrug resistance genes in normal and cancer stem cells. Cancer Invest. 2008;26:535–42. doi: 10.1080/07357900801904140. [DOI] [PubMed] [Google Scholar]
- 149.Keshet GI, Goldstein I, Itzhaki O, et al. MDR1 expression identifies human melanoma stem cells. Biochem Biophys Res Commun. 2008;368:930–6. doi: 10.1016/j.bbrc.2008.02.022. [DOI] [PubMed] [Google Scholar]
- 150.Zhou J, Zhang H, Gu P, Bai J, Margolick JB, Zhang Y. NF-kappaB pathway inhibitors preferentially inhibit breast cancer stem-like cells. Breast Cancer Res Treat. 2008;111:419–27. doi: 10.1007/s10549-007-9798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Levi E, Misra S, Du J, Patel BB, Majumdar AP. Combination of aging and dimethylhydrazine treatment causes an increase in cancer-stem cell population of rat colonic crypts. Biochem Biophys Res Commun. 2009;385:430–3. doi: 10.1016/j.bbrc.2009.05.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Majumdar APN, Banerjee S, Nautiyal J, et al. Curcumin Synergizes With Resveratrol to Inhibit Colon Cancer. Nut Cancer. 2009;61:544–53. doi: 10.1080/01635580902752262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chen HF. Computational study of the binding mode of epidermal growth factor receptor kinase inhibitors. Chem Biol Drug Des. 2008;71:434–46. doi: 10.1111/j.1747-0285.2008.00656.x. [DOI] [PubMed] [Google Scholar]
- 154.Ichikawa H, Nakamura Y, Kashiwada Y, Aggarwal BB. Anticancer drugs designed by mother nature: ancient drugs but modern targets. Curr Pharm Des. 2007;13:3400–16. [PubMed] [Google Scholar]
- 155.Chan MM, Fong D. In: Drug Resistance in Cancer Cells. Siddik Zahid H, Mehta Kapil., editors. New York: Springer; 2009. pp. 315–42. [Google Scholar]
- 156.Anand P, Sundaram C, Jhurani S, Kunnumakkara AB, Aggarwal BB. Curcumin and cancer: An “old-age” disease with an “age-old” solution. Cancer Lett. 2008;267:133–64. doi: 10.1016/j.canlet.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 157.Cheng AL, Hsu CH, Lin JK, et al. Phase I clinical trial of curcumin, a chemopreventive agent, in patients with high-risk or pre-malignant lesions. Anticancer Res. 2001;21:2895–900. [PubMed] [Google Scholar]
- 158.Lynch PM. Chemoprevention with special reference to inherited colorectal cancer. Fam Cancer. 2008;7:59–64. doi: 10.1007/s10689-007-9158-4. [DOI] [PubMed] [Google Scholar]
- 159.Hatcher H, Planalp R, Cho J, Torti FM, Torti SV. Curcumin: from ancient medicine to current clinical trials. Cell Mol Life Sci. 2008;65:1631–52. doi: 10.1007/s00018-008-7452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Goel A, Kunnumakkara AB, Aggarwal BB. Curcumin as “Curecumin”: from kitchen to clinic. Biochem Pharmacol. 2008;75:787–809. doi: 10.1016/j.bcp.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 161.Moragoda L, Jaszewski R, Majumdar AP. Curcumin induced modulation of cell cycle and apoptosis in gastric and colon cancer cells. Anticancer Res. 2001;21:873–8. [PubMed] [Google Scholar]
- 162.Patel BB, Sengupta R, Qazi S, et al. Curcumin enhances the effects of 5-fluorouracil and oxaliplatin in mediating growth inhibition of colon cancer cells by modulating EGFR and IGF-1R. Int J Cancer. 2008;122:267–73. doi: 10.1002/ijc.23097. [DOI] [PubMed] [Google Scholar]
- 163.Wu DL, Xu Y, Yin LX, Lu HZ. Reversal of multidrug resistance in vincristine-resistant human gastric cancer cell line SGC7901/VCR by LY980503. World J Gastroenterol. 2007;13:2234–7. doi: 10.3748/wjg.v13.i15.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shukla S, Wu CP, Ambudkar SV. Development of inhibitors of ATP-binding cassette drug transporters: present status and challenges. Expert Opin Drug Metab Toxicol. 2008;4:205–23. doi: 10.1517/17425255.4.2.205. [DOI] [PubMed] [Google Scholar]
- 165.Zhang Q, Wei D, Liu J. In vivo reversal of doxorubicin resistance by (−)-epigallocatechin gallate in a solid human carcinoma xenograft. Cancer Lett. 2004;208:179–86. doi: 10.1016/j.canlet.2004.01.033. [DOI] [PubMed] [Google Scholar]
- 166.Limtrakul P, Khantamat O, Pintha K. Inhibition of P-glycoprotein activity and reversal of cancer multidrug resistance by Momordica charantia extract. Cancer Chemother Pharmacol. 2004;54:525–30. doi: 10.1007/s00280-004-0848-4. [DOI] [PubMed] [Google Scholar]
- 167.Harbottle A, Daly AK, Atherton K, Campbell FC. Role of glutathione S-transferase P1, P-glycoprotein and multidrug resistance-associated protein 1 in acquired doxorubicin resistance. Int J Cancer. 2001;92:777–83. doi: 10.1002/ijc.1283. [DOI] [PubMed] [Google Scholar]
- 168.Choi BH, Kim CG, Lim Y, Shin SY, Lee YH. Curcumin down-regulates the multidrug-resistance mdr1b gene by inhibiting the PI3K/Akt/NF kappa B pathway. Cancer Lett. 2008;259:111–8. doi: 10.1016/j.canlet.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 169.Iqbal S, Lenz H-J. Integration of novel agents in the treatment of colorectal cancer. Cancer Chemother Pharmacol. 2004;54:S32–S9. doi: 10.1007/s00280-004-0884-0. [DOI] [PubMed] [Google Scholar]
- 170.Yau T, Chan P, Ching Chan Y, Wong BC, Liang R, Epstein RJ. Review article: current management of metastatic colorectal cancer - the evolving impact of targeted drug therapies. Aliment Pharmacol Ther. 2008;27:997–1005. doi: 10.1111/j.1365-2036.2008.03684.x. [DOI] [PubMed] [Google Scholar]