

Abstract
Cancer stem-like cells (CSCs) have been implicated in recurrence and treatment resistance in many human cancers. Thus, a CSC-targeted drug delivery strategy to eliminate CSCs is a desirable approach for developing a more effective anticancer therapy. We have developed a tumor-targeting nanodelivery platform (scL) for systemic administration of molecular medicines. Following treatment with the scL nanocomplex carrying various payloads, we have observed exquisite tumor-targeting specificity and significant antitumor response with long-term survival benefit in numerous animal models. We hypothesized that this observed efficacy might be attributed, at least in part, to elimination of CSCs. Here, we demonstrate the ability of scL to target both CSCs and differentiated nonstem cancer cells (non-CSCs) in various mouse models including subcutaneous and intracranial xenografts, syngeneic, and chemically induced tumors. We also show that systemic administration of scL carrying the wtp53 gene was able to induce tumor growth inhibition and the death of both CSCs and non-CSCs in subcutaneous colorectal cancer xenografts suggesting that this could be an effective method to reduce cancer recurrence and treatment resistance. This scL nanocomplex is being evaluated in a number of clinical trials where it has been shown to be well tolerated with indications of anticancer activity.
Introduction
Although current anticancer therapies are effective during the initial phase of treatment, frequently there are recurrences. Such recurrences can often be metastatic and resistant to conventional therapies. Within a tumor, a small population of cells called cancer stem-like cells (CSCs) has stem cell-like properties allowing them to initiate and fuel tumor growth.1 CSCs are thought to be responsible for cancer recurrence after conventional treatments, as well as for tumor initiation and metastasis.1,2 CSCs have been identified in many types of cancer including leukemia,3 breast,4 brain,5 colon,6 lung,7 and prostate.8 However, the majority of conventional cancer therapies—including hormonal, radiation, and chemotherapy—may not efficiently eliminate CSCs. While the details of CSC biology need to be better understood, a great deal of effort is currently focused on the therapeutic targeting of CSCs as a new strategy for drug design for cancer treatment and the prevention of recurrence. However, the most formidable challenge in CSC-specific therapies involves the development of effective means for specifically delivering the therapeutics to the CSCs.
A prerequisite for targeting CSCs is the development of a means to target primary and metastatic tumor cells specifically. Tumor-specific targeting can be accomplished by incorporating affinity ligands, such as RGD,9 iRGD,10 NGR peptide,11 folate,12 transferrin,13,14,15 or antibodies against human epidermal growth factor receptor 2,16 transferrin receptor (TfR),17,18 or aptamers that recognize the prostate-specific membrane antigen19 into delivery vehicles. In each instance, the ligand or antibody is intended to target tumors by interaction with its cognate receptor or antigen.
We have developed a nanotechnology platform for systemic, tumor-targeting delivery of anticancer therapeutics.20,21,22,23,24,25 This scL nanoparticle is a liposomal complex employing an anti-TfR single-chain variable fragment (TfRscFv) as a targeting ligand, taking advantage of the elevated levels of TfR found on most tumor cells26 and the rapid recycling of TfR that serves to accelerate the nanoparticle trafficking through tumor cells. Our scL nanocomplex is designed to bind to TfR on the target cell facilitating transcellular delivery of the payload by receptor-mediated endocytosis.17,18,26 We have shown that the scL nanocomplex can efficiently and specifically deliver various payloads into both primary and metastatic tumors in vivo with a great selectivity over normal cells such as liver hepatocytes.18 When systemically administered, the self-assembled, biodegradable, scL nanocomplex has been shown to have deep tumor penetration in both primary and metastatic disease, resulting in long-term tumor elimination (often without recurrences) and life span prolongation in numerous animal models of human cancer.22,23,24,25 Moreover, in a completed phase I clinical trial, our tumor-targeting nanocomplex (scL) delivering the wild-type p53 (wtp53) gene has shown very low toxicity with indications of anticancer effect in some patients.27
We have hypothesized that the significant efficacies observed with the scL nanocomplex, including complete tumor elimination and lack of tumor recurrence over the life span in some of treated animals, could be attributed to its capability to target CSCs. Here, we report evidences that scL can efficiently target and deliver payloads to both CSCs and differentiated non-CSCs in vitro and in various mouse models of cancer including human brain and colorectal cancer xenografts, syngeneic mouse breast tumor and melanoma, and chemically induced mouse lung and liver cancers. We also report that systemically delivered scL-p53 showed a significant anticancer effect by inhibiting tumor growth and inducing apoptosis in both CSCs and non-CSCs in a mouse model of human colorectal cancer xenografts.
Results
scL-mediated targeting of CSCs in vitro
Since the scL nanocomplex targets TfR overexpressed on the surface of cancer cells, we hypothesized that scL might target CSCs if CSCs also overexpress TfR. To address this, we determined the TfR expression level in CSCs of human colorectal cancer (HT-29, HCT-116) using CSC markers (CD133, CD166, CD44, EpCAM, Msi1, and Nanog). While noncancerous human lung fibroblasts, IMR-90, had low TfR expression, all of the human colorectal cancer cell lines were found to express elevated levels of TfR, which were similar in CSCs (CD133+CD166+CD44+, EpCAMhighCD166+CD44+, Msi+Nanog+, or CD133+) and unselected populations (Figure 1a,b). Western blot analysis of immunomagnetically sorted CD133+ and CD133− populations of HT-29 cells confirmed the increased expression of TfR in both populations compared to that seen in noncancerous IMR-90 cells (Figure 1c). Expression of TfR in CSCs of human brain cancer cells (U251, U87, LN-18, and T98G) was also tested using multiple CSC markers (CD133, Nestin, SSEA-1, Nanog, and Msi1). As was the case with the colorectal cancer cells, all brain cancer cell lines exhibited a similar shift indicating overexpression of TfR that was observed in both CSCs (CD133+Nestin+, CD133+Msi1+, or SSEA-1+Nanog+) and unselected populations (Figure 1d). The observed cosegration of e.g., CD133/CD166/CD44 or SSEA-1/Nanog, indicates the simultaneous expression of different CSC markers in these cells and helps support that the populations isolated are indeed CSCs. These results indicate the potential for targeting both of CSCs and non-CSCs in a solid tumor via binding of the scL nanocomplex to the TfR.
Figure 1.

Overexpression of transferrin receptor (TfR) in solid tumor cancer stem-like cells (CSCs) and their in vitro targeting by the scL nanocomplex. (a) Expression of TfR was assessed in CSCs of human colorectal cancer cell line HT-29 using multiple stem cell markers (CD133+CD166+CD44+, EpCAMhighCD166+CD44+, or Msi+Nanog+). Black histograms represent isotype control. Red histograms represent marker-positive CSCs. Blue histograms represent unselected cancer cells. (b) TfR expression in CD133+ and CD133− populations in another human colorectal cancer cell line (HCT-116) was compared with TfR expression in a noncancerous human lung fibroblast cell line (IMR-90). Closed gray histograms represent isotype control. (c) Expression of TfR in CD133+ and CD133− populations of HT-29 and IMR-90 was assessed by western blot. (d) Expression of TfR was tested in CSCs of human brain cancer cell lines (U251, U87, LN-18, and T98G) using multiple stem cell markers (CD133+CD166+CD44+, EpCAMhighCD166+CD44+, or Msi+Nanog+). Black histograms represent isotype control. Red histograms represent marker positive CSCs. Blue histograms represent unselected cancer cells. (e) Uptake of fluorescently labeled ODN in CD133+ and CD133− populations 24 hours after transfection of HT-29, HCT-116, and U251 cells with uncomplexed free (black histogram) and scL-complexed (red histogram) 6FAM-ODN. Gray histograms represent untreated control. (f) Time lapse images of subcellular localization of 6FAM-ODN in HT-29 cells after scL-mediated transfection using total internal reflection fluorescence (TIRF) microscopy. HT-29 cells were prelabeled with anti-CD133 antibody (red fluorescence) in culture and transfected with scL-6FAM-ODN (green fluorescence) at 0 hour. Images were obtained over a 17-hour period. Scale bars indicate 10 μm. (g) Fluorescence intensity in the CD133+ HT-29 cells were measured in cell 1 and cell 2 along the scanning line denoted by the arrow in f. Change in fluorescence intensity over time is shown at 7, 8, 9, 10, 13, and 16 hours after transfection. The dotted boxes indicate each cell.
Next, we tested whether scL targets and transfects both populations of CSCs and non-CSCs in vitro using fluorescently labeled oligonucleotide (ODN) as a model payload. We transfected HT-29, HCT-116, and U251 cells with scL-delivered 6FAM-labeled ODN (6FAM-ODN) in vitro. Treatment with free 6FAM-ODN (without scL delivery) resulted in no significant cellular uptake (Figure 1e). In contrast, use of scL-6FAM-ODN resulted in similar high level of transfection in both CD133+ and CD133− populations of HT-29, HCT-116, and U251 cells confirming the ability of scL to target both populations.
We also examined the intracellular uptake and subcellular localization of the payload in CSCs with time lapse imaging of in vitro transfection using total internal reflection fluorescence microscopy (Figure 1f,g). CSCs from HT-29 were prelabeled with anti-CD133 antibody (red fluorescence) and transfected with scL-6FAM-ODN. Significant accumulation of 6FAM-ODN (green fluorescence) in the cytoplasm was observed as early as 3 hours after transfection, and the signal intensified over time. Translocation of 6FAM-ODN into the nucleus, where ODN are expected to act in modulating gene expression, was also observed 9 hours after transfection (Figure 1f,g). Taken together, these results substantiate the ability of the scL nanocomplex to target CSCs in vitro.
scL-mediated transgene expression in CSCs in vitro
To quantify transfection efficiency and evaluate functionality of the delivered payload in CSCs, we transfected HT-29, HCT-116, U251, and U87 cells with scL carrying green fluorescent protein (GFP) plasmid DNA (scL-GFP). Forty-eight hours after transfection, we used Texas Red-labeled anti-CD133 antibody to detect CSCs. Localization of GFP in the cytoplasm of Texas Red-labeled cells was observed in HT-29, U251, and U87 cells (Figure 2a). Using flow cytometry, expression of GFP was observed in more than 70% of both HCT-116 CD133+ and CD133− cells (Figure 2b). In contrast, transfection with GFP DNA alone resulted in no GFP expression in any of the cells. These results demonstrate high transfection efficiency with scL and confirm that scL can deliver payloads to both CSCs and non-CSCs in vitro with similar efficiency.
Figure 2.

scL-mediated exogenous gene expression in cancer stem-like cells (CSCs) in vitro. (a) Fluorescent and differential interference contrast (DIC) images of HT-29, U251, and U87 cells in monolayer culture transfected with scL-GFP in vitro for 48 hours. GFP-expressing cells were stained with an anti-CD133 antibody (red fluorescence). Scale bars indicate 10 μm. (b) Flow cytometry analysis of HCT-116 cells 48 hours after transfection with free GFP plasmid DNA (black open histograms) or scL-GFP (red open histograms). The cells were stained with CD133-PE antibody. GFP positivity in CD133+- and CD133−-gated cells are shown. Gray histograms represent untreated control. (c) Western blot analysis of HT-29 cells 24 hours after transfection with scL-GFP-p53. Expression of GFP and p53 fusion proteins in unselected, FACS-sorted CSCs (CD133+CD166+CD44+) and non-CSCs was assessed on two individual membranes. (−), untransfected cells; (+), scL-GFP-p53 transfected cells. FACS, flow-activated cell sorting.
From a therapeutic standpoint, the ability to deliver and express wtp53 in CSCs and non-CSCs is the key issue. To demonstrate the ability of the scL nanocomplex to deliver p53 to both CSCs and non-CSCs, we transfected HT-29 cells with the scL nanocomplex carrying a GFP-tagged wtp53 expression plasmid encoding GFP-p53 fusion protein as the payload.28 Twenty-four hours after transfection, the cells were harvested and FACS-sorted using antibodies against CSC markers (CD133, CD166, and CD44). Untransfected cells were also sorted for use as a control. Western blot analysis of both sorted CSCs (CD133+CD166+CD44+) and non-CSCs (flow through cells) showed the expression of the GFP-p53 fusion protein in CSCs as well as in non-CSCs (Figure 2c). A population of unselected cells was also included on the gels. To eliminate cross-reactivity, separate membranes were probed for the expression of GFP or p53 proteins using antibodies against each protein. As expected with a fusion protein, the signal detected with both the anti-GFP and anti-p53 antibodies was evident at the same molecular weight of ˜78 kDa. Although some nonspecific banding was detected in the untreated cells with the anti-p53 antibody (not endogenous p53 which was present at 53 kDa as expected), no expression of GFP was evident in untreated cells in the unselected, CSC or the non-CSC populations. These results indicate that the observed exogeneous p53 and GFP expression is a result of scL delivery to, and expression of, the payload in both CSCs and non-CSCs. Moreover, using fluorescence microscopy, we have also observed a time-dependent increase in the level of green fluorescence in the transfected cells at 16, 24, and 48 hours after transfection (data not shown). Thus, these data demonstrate that the scL-GFP-p53 nanocomplex can target, and deliver exogeneous wtp53 to CSCs (as well as non-CSCs).
Effect of scL-delivered wtp53 on CSC/non-CSC survival in vitro
Next, we sought to evaluate the effectiveness of scL-mediated delivery of therapeutics on CSCs. We examined in vitro the effect of scL-delivered wtp53 DNA on CD133+ CSCs and CD133− non-CSCs from colorectal (HT-29, HCT-116) and brain (U251, U87) cancer cells using the XTT cell survival assay (Figure 3). The graphs represent the mean of triplicate samples. Treatment with liposome only (Lip) and scL carrying empty plasmid vector (scL-vector) served as controls. Transfection with scL-p53 resulted in a statistically significant increase in cell death in the all cell lines tested. More importantly, scL-p53 induced similar responses in both CD133+ CSCs and CD133− non-CSCs (similar IC50 values) (Figure 3e). In contrast, transfection with the controls, at what would be clinically relevant doses, did not result in significant cell death demonstrating that the response to scL-p53 is not a result of nonspecific cytotoxicity.
Figure 3.

Effect of scL-delivered wtp53 on cancer stem-like cell (CSC) and non-CSC survival in vitro. The XTT-based in vitro cell viability assay was performed to compare the anticancer effect of scL-mediated delivery of wtp53 on CD133+ CSC and CD133− non-CSC cell populations from human: (a) HT-29 colorectal cancer; (b) HCT-119 colorectal cancer; (c) U251 brain cancer; (d) U87 brain cancer cells. The XTT assay was performed 48 hours after transfection. (e) Compilation of the IC50 values, the drug concentration resulting in 50% cell kill, for CD133+ and CD133− populations in the tumor cell lines after transfection with scL-p53. Data are represented as mean ± SD of triplicate samples. Statistical significance was determined using analysis of variance (*P < 0.05; **P < 0.001).
CSC targeting in vivo via systemically delivered scL in human brain and colorectal cancer xenografts
While the in vitro studies described above serve as proof-of-principle, it is critical to assess the ability of scL to deliver its payload to CSCs in vivo after systemic administration. We employed three different animal models of human cancer xenografts: colorectal (HT-29) and brain (U251 and U87).
Mice bearing HT-29 subcutaneous xenograft tumors were injected intravenously (i.v.) three times over 24 hours with either scL-6FAM-ODN, the complex minus the targeting moiety (Lip-6FAM-ODN), or free 6FAM-ODN (all at 100 μg of 6FAM-ODN/mouse/injection, 4–5 mice/group). Twenty-four hours after the last injection, the level of fluorescence in the tumor and normal tissues (liver, kidney, lung, heart, and spleen) was determined. In the tumors from mice injected with scL-6FAM-ODN, strong fluorescence was observed throughout the tumor (on the surface and also in the center), except in necrotic regions. However, only weak fluorescence was detected in tumors from mice treated with Lip-6FAM-ODN or free 6FAM-ODN. The fluorescence intensity was quantified and plotted (Figure 4a). While the majority of fluorescence observed after injection of either free 6FAM-ODN or Lip-6FAM-ODN was present in the liver, with only low levels in the tumor, the opposite was true after injection with scL-6FAM-ODN. In this case, the majority of the signal was in the tumor. Tumor fluorescence with scL-6FAM-ODN was 4.5 and 6.8 times higher than after treatment with Lip-6FAM-ODN or with free 6FAM-ODN, respectively. Tumor specificity of scL-6FAM-ODN was also supported by the differences in fluorescence intensity in normal tissues between groups. Fluorescence in liver, kidney, and spleen from the free 6FAM-ODN treated animals were 2.5, 3.8, and 9.2 times higher, respectively, than the fluorescence seen in those treated with scL-6FAM-ODN (Figure 4a). Similar tumor specificity was observed in the subcutaneous U251 brain tumor xenograft model after a single i.v. injection of the three reagents described above (4–5 mice/group) (Figure 4c).
Figure 4.

Tumor specificity and cancer stem-like cell (CSC)–targeting ability of the scL nanocomplex in vivo after systemic administration. (a) Quantitative analysis of the intensity of green fluorescence signal, representing uptake of 6FAM-ODN, in HT-29 subcutaneous tumors and normal tissues using the Maestro in vivo fluorescence imaging system. Signal intensity is expressed as photons/cm2/second. (b) Flow-activated cell sorting (FACS) analysis of 6FAM-ODN uptake in CD133+ CSCs and CD133− non-CSCs isolated from the HT-29 tumors after imaging. (c) Ex vivo fluorescence imaging of U251 subcutaneous tumors after a single i.v. injection with 100 µg of 6FAM-ODN/mouse. (d) FACS analysis of 6FAM-ODN uptake in CD133+ CSCs and CD133− non-CSCs isolated from U251 tumors. Analysis was performed 24 hours after injection. In b and d, the values listed above the bar in each histogram indicate the percentage of the cells positive for 6FAM-ODN. Gray histograms represent untreated control. (e) FACS analysis of 6FAM-ODN uptake in stem cell markers (CD133, SSEA-1, Nestin, Nanog, and Msi1)-positive populations isolated from U87 intracranial tumor xenografts 24 hours after a single i.v. injection with 100 µg of 6FAM-ODN/mouse administered as scL-6FAM-ODN. Black histograms represent untreated control. The values listed above the bar in each histogram indicate the percentage of the cells positive for 6FAM-ODN. Statistical significance was determined using analysis of variance (*P < 0.05).
We sought to determine if the tumor-targeting described above included targeting to CSCs in these in vivo models. After imaging the tumors, CSCs were isolated and uptake of 6FAM-ODN was analyzed by flow cytometry. As in the in vitro studies, 6FAM-ODN uptake was evident in both CD133− non-CSCs and CD133+ CSCs isolated from tumors of mice that received scL-6FAM-ODN (Figure 4b). Furthermore, this analysis demonstrated that a high level of in vivo uptake efficiency was obtained after systemic administration of the scL nanodelivery system. After three i.v. injections, the scL nanocomplex was taken up by ˜90% of CD133+ CSCs and 83% of CD133− non-CSCs in the HT-29 subcutaneous xenografts confirming its ability to efficiently target and deliver the payloads to both populations in vivo. This level of tumor targeting was three to six times greater than that observed with the controls. Free 6FAM-ODN was present in only ˜15% of the tumor cells, while the 6FAM-ODN was present in ˜30% of tumor cells after injection of untargeted Lip-6FAM-ODN. The uptake observed with Lip-6FAM-ODN may be due to nonspecific (nonreceptor mediated) uptake of the liposome particle which may be attributed to what has been termed the enhanced permeability and retention effect.29
The CSC-targeting capability of the scL nanodelivery system in vivo was also confirmed in a subcutaneous tumor model of U251 human brain cancer. Consistent with the results obtained with HT-29, a systemic single injection of scL-6FAM-ODN resulted in an uptake efficiency of ˜65% in both CD133+ CSCs and CD133− non-CSCs. This was 7–10 times greater than the efficiency observed with the controls. Free 6FAM-ODN was present in only ˜6% of the tumor cells, while ˜10% showed the presence of the 6FAM-ODN after injection of untargeted Lip-6FAM-ODN (Figure 4d).
The CSC-targeting capability of the scL nanocomplex in vivo was further confirmed in an intracranial tumor model of U87 human brain cancer using multiple CSC markers (Figure 4e). Systemic injection of scL-6FAM-ODN resulted in an uptake efficiency of 37–60% of CSC marker-positive cells (CD133+, SSEA-1+, Nestin+, Nanog+, and Msi1+) after a single injection.
Taken together, these results confirm the tumor specificity and CSC-targeting in vivo of the scL nanocomplex in a wide range of tumor types after systemic administration in mice bearing these human tumor xenografts.
scL-mediated transgene expression in CSCs in xenografts of human colorectal cancer
The ability of systemically administered scL to deliver its payload to CSCs was further evaluated in vivo using GFP plasmid DNA as a payload. Mice bearing HT-29 subcutaneous tumors were injected i.v. once with either scL-GFP, the complex minus the targeting moiety (Lip-GFP), or free GFP (all at 100 μg of GFP plasmid DNA/mouse, 3 mice/group). Sixty-four hours after the injection, the level of fluorescence in the tumor was determined (Figure 5a). Strong green fluorescence was observed in the tumors from mice injected with scL-GFP while only weak fluorescence was detected in tumors from mice that received Lip-GFP or free GFP. The fluorescence intensity was quantified and plotted (Figure 5b). Using fluorescence microscopy, localization of GFP and CSC marker CD133 in the same cells was observed in the tumor sections from scL-GFP treated mice (Figure 5c). These results demonstrate the scL can also deliver this plasmid payload to CSCs with a high efficiency in vivo and more significantly, that the delivered payload functions normally within the target cells.
Figure 5.

scL-mediated exogenous gene expression in cancer stem-like cells (CSCs) in vivo. (a) Ex vivo fluorescence imaging of GFP in subcutaneous HT-29 tumor xenografts 64 hours after i.v. injection with 100 µg of GFP plasmid DNA/mouse as in Figure 4. (b) Quantitative analysis of the intensity of GFP signal in the tumors expressed as photons/cm2/second. (c) Fluorescent images of HT-29 tumor treated with scL-GFP. GFP-expressing tumor slice was stained with an anti-CD133 antibody (red fluorescence) and analyzed using confocal microscope. Scale bar indicates 25 μm. Statistical significance was determined using analysis of variance (*P < 0.05).
Systemically delivered scL-mediated targeting of CSCs in syngeneic mouse tumors
In vivo tumor-targeting and CSC-targeting capability of scL was further studied in immune competent syngeneic mouse models of lung metastases of B16F10 melanoma (3 mice/group) and subcutaneous breast tumor (EO771, 3 mice/group). Systemic administration of scL-6FAM-ODN resulted in tumor-specific delivery of 6FAM-ODN to the lung metastases. Compared to uncomplexed (free) or untargeted complex (Lip) controls, scL-mediated delivery of 6FAM-ODN showed significantly stronger fluorescence signal in the lung tumors (Figure 6a), with great targeting efficiency 24 hours after i.v. injection (Figure 6b). Flow-activated cell sorting (FACS) analysis of cancer cells isolated from the tumor nodules demonstrated a high degree of targeting (˜51%) in vivo in CD44+CD24+CD133+ mouse melanoma CSC populations after a single injection (Figure 6c). In contrast, animals treated with free or Lip controls resulted in very low uptake of 6FAM-ODN in CSCs.
Figure 6.

scL-mediated in vivo targeting of cancer stem-like cells (CSCs) in syngeneic tumors. (a) Ex vivo fluorescence imaging of syngeneic B16F10 melanoma (lung metastatic tumors) 24 hours after i.v. injection with 100 µg of 6FAM-ODN. (b) Green fluorescence representing uptake of 6FAM-ODN in lung metastatic tumors was quantitatively determined as in Figure 4. (c) Flow-activated cell sorting (FACS) analysis of 6FAM-ODN uptake in mouse melanoma CSCs (CD44+CD24+CD133+) isolated from B16F10 lung metastatic tumors. (d) FACS analysis of 6FAM-ODN uptake in unselected tumor cell population (top line) and mouse breast tumor CSCs (CD44+CD24−/low or CD133+) isolated from subcutaneous syngeneic EO771 breast tumors 24 hours after i.v. injection with 100 µg of 6FAM-ODN/mouse. Statistical significance was determined using analysis of variance (*P < 0.05).
In the EO771 syngeneic breast tumor model, systemic administration of scL-6FAM-ODN also resulted in significant percentage of the tumor cells positive for the presence of the payload 24 hours after i.v. injection (Figure 6d). In these tumors, the percentage of cells exhibiting 6FAM-ODN uptake was 13.9 or 8.5-fold higher with scL-mediated delivery than with free or Lip controls, respectively. Further analysis using CSC markers demonstrated ˜40 % of both CD44+CD24− or CD133+ breast CSC populations had taken up the payload after a single scL-6FAM-ODN injection (Figure 6d). In contrast, CSCs in tumors from animals treated with free or Lip controls showed only ˜4 to 8.5% of CSCs contained 6FAM-ODN. These results confirm the tumor specificity and CSC-targeting of the scL nanodelivery system after systemic administration in mouse syngeneic models of primary and metastatic tumors.
CSC-targeting via systemically delivered scL in chemically induced mouse tumors
Tumor-targeting and more specifically CSC-targeting by scL was further tested in vivo in a chemically induced mouse tumor model (3–4 mice/group). This model more closely resembles the human clinical situation. Systemic administration of scL-6FAM-ODN into A/J mice bearing chemically induced lung tumors resulted in tumor-specific delivery of 6FAM-ODN (Figure 7a), achieving a high level of targeting efficiency in vivo (˜40% of lung CSCs) after a single injection (Figure 7b,c). In contrast, treatment with uncomplexed (free 6FAM-ODN) or untargeted control complexes (Lip-6FAM-ODN) did not result in significant levels of uptake of 6FAM-ODN in CSCs in any of the animals.
Figure 7.
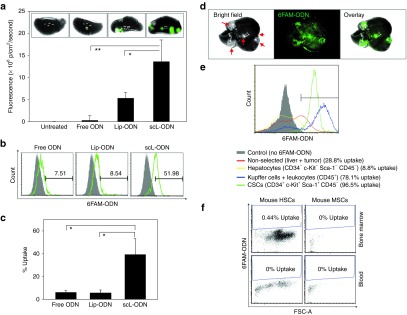
scL-mediated in vivo targeting of cancer stem-like cells (CSCs) in chemically induced mouse tumors. (a) Ex vivo fluorescence imaging of chemically induced lung tumors in A/J mice 24 hours after i.v. injection with 100 µg of 6FAM-ODN. Green fluorescence representing uptake of 6FAM-ODN in lung metastatic tumors was quantitatively determined as in Figure 4. (b) 6FAM-ODN uptake in lung CSCs (CD34+c-Kit+Sca-1+CD45−) was determined by flow-activated cell sorting (FACS) analysis and compared in graph c. (d) Ex vivo fluorescence imaging and (e) FACS analysis of liver tumors in A/J mice i.v. treated with scL-6FAM-ODN for 24 hours. Arrows indicate chemically induced liver tumors. (f) 6FAM-ODN uptake in mouse hematopoietic stem cells (mHSCs, CD34−c-Kit+Sca-1+CD45+Lin−) and mouse mesenchymal stem cells (mMSCs, CD34−c-Kit−Sca-1+CD45−Lin−) were monitored in the bone marrow and blood by FACS. Statistical significance was determined using analysis of variance (*P < 0.05; **P < 0.001).
Systemically administered scL-6FAM-ODN also resulted in tumor-specific delivery to chemically induced tumors found in the liver with a high efficiency of targeting. Over 90% of CSCs in liver showed 6FAM-ODN uptake (Figure 7d,e). Of note, no significant 6FAM-ODN uptake was observed in the nonhematopoietic subpopulation (CD45−) in the liver including hepatocytes which are negative for CSC surface markers (CD34−c-Kit−Sca-1−CD45−). In the CD45+ hematopoietic fraction of liver, including Kupffer cells and circulating leukocytes, uptake of 6FAM-ODN was observed in about 78% of the cells. Importantly, very low levels of 6FAM-ODN uptake was observed in mouse hematopoietic stem cells (mHSCs, CD34−c-Kit+Sca-1+CD45+Lin−) and mouse mesenchymal stem cells (mMSCs, CD34−c-Kit−Sca-1+CD45−Lin−) from bone marrow and peripheral blood of scL-6FAM-ODN treated mice (Figure 7f). Taken together, these results confirm the in vivo tumor specificity and CSC-targeting ability of the systemically administered scL nanodelivery system in this chemically induced syngeneic mouse tumor model.
Efficient tumor growth inhibition and induction of apoptosis in CSCs/non-CSCs in colorectal xenografts
To directly evaluate the therapeutic potential of the tumor-targeted scL nanodelivery system, tumor and CSC responses to scL-delivered wtp53 was tested in subcutaneous xenograft tumors established by injecting CD133+ HT-29 cells (10–11 mice/group). Tumor-bearing mice were i.v. treated with scL-p53 nanocomplex (30 µg DNA/mouse/injection) twice per week for 6 weeks. Untreated mice were used as controls. Systemic administration of scL-p53 resulted in significant tumor growth inhibition (Figure 8a). Three weeks after the first treatment, tumor size was only 30.3% of untreated control mice. Completing the 6-week treatments, 63.2% of tumor growth inhibition was observed in scL-p53 treated tumors compared to untreated control. The antitumor effect persisted even after treatment had ended. At 7 weeks, tumor size in the scL-p53 treatment group was 18.3% of untreated control, representing an 81.7% inhibition. Not unexpectedly with scL-p53 as a single-agent, tumor regression was not observed. However, scL-p53 was very effective in controlling the tumor growth as the mean tumor growth time (time required to reach a fivefold increase of tumor volume from the size at the start of treatment) was 41.3 days for scL-p53 treated animals while only 12.4 days for untreated control mice. This represents a 28.9-day growth delay (Figure 8b). Antitumor efficacy was further determined using cell cycle analysis of cancer cells isolated from the tumor (Figure 8c). Compared to untreated tumors, FACS analysis showed a significantly increased sub-G1 population (approximately threefold) after scL-p53 treatment, indicating a strong inhibition of cell cycle progression and induction of apoptosis. A change in number of CSCs in the tumors during scL-p53 treatment was monitored over time (Figure 8d). The relative number of CSCs was calculated from the tumor size shown in Figure 8a and the percentage of CD133+ CSCs analyzed by FACS (data not shown). Compared to the untreated tumors, the relative number of CD133+ CSCs in the tumors from the scL-p53 treated mice was significantly lower with a 65.7% reduction 18 days after the first treatment. At the end of 6 weeks treatment, the relative number of CD133+ CSCs in the scL-p53 treatment group was only 28.8% of those from untreated control mice showing significant increase of apoptosis (cell kill) in these CSCs.
Figure 8.
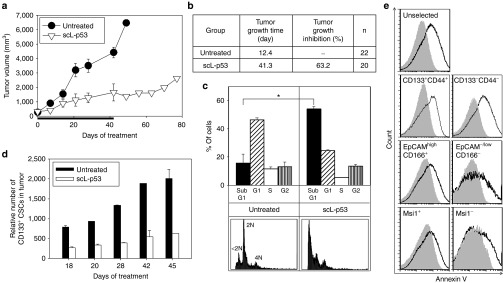
Efficient tumor growth inhibition and induction of apoptosis in cancer stem-like cells (CSCs)/non-CSCs in colorectal xenografts. (a) Changes in tumor sizes with treatments. Mice were treated i.v. with scL-p53 (30 µg DNA/mouse/injection) twice per week for 6 weeks and tumor sizes were monitored weekly. Tumor sizes were plotted versus the number of days after initiation of the treatment. Black bar indicates the duration of treatment. (b) Summary of tumor growth. Tumor growth time: time in days required to reach a fivefold increase in tumor volume from the size at the start of treatment. Tumor growth inhibition: calculated as the ratio of tumor volume between scL-p53 treated tumors and untreated control tumors after completing treatment (6 weeks). (c) Cell cycle analysis of tumor cells after treatment. Sub-G1 peaks (<2N) represent apoptotic cells. Apoptotic response was determined by quantifying the sub-G1 population. Representative histograms are shown. (d) Changes of the relative numbers of CD133+ CSCs in tumors. The relative number of CSCs was calculated from tumor size in a and percentage of CD133+ CSCs by flow-activated cell sorting (FACS) analysis. (e) Apoptosis in CSCs after treatment in a second in vivo experiment. The ratio of Annexin V positive cells in tumors after treatment was evaluated by FACS analysis. Tumor cells were colabeled with CSC markers (CD133+CD44+, EpCAMhighCD166+, or Msi1+) for gating. Representative histograms are shown. Gray solid histogram: untreated cells. Black histogram: scL-p53 treated cells. Statistical significance was determined using analysis of variance (*P < 0.05).
To confirm this apoptotic response in CSCs, and examine the effect of scL-p53 in non-CSCs, induction of apoptosis was determined using antibodies against Annexin V and surface antigens of CSCs (CD133+CD44+, EpCAMhighCD166+, or Msi1+) in a second in vivo experiment. With the CSC markers tested, treatment with scL-p53 increased positive staining for Annexin V in the both CSC marker-positive and -negative cells suggesting that scL-p53 was able to target and induce apoptosis not only in the CSCs but also in the non-CSCs (Figure 8e). In addition, the observed cosegration of e.g., CD133/CD166/CD44 or SSEA-1/Nanog, indicates the simultaneous expression of different CSC markers in these cells further and again indicates that this response was observed in CSCs. Moreover, the level of increase in Annexin V positive staining was similar between the CSC marker-positive and -negative populations.
Discussion
Recent identification of putative CSC surface markers has led to a quest for methods to deliver therapeutic agents to CSCs. However, while there is no current consensus on the optimal markers for CSCs, numerous studies employ surface antigens (i.e., CD133, CD44, CD24, etc) as markers for CSCs.30,31 While the relevance of putative CSC surface markers remains controversial,32 a few studies have reported targeting of CSCs based on these distinct stem cell markers.33 Since it is also plausible that the depletion of the CSCs may not be sufficient for complete tumor regression if the remaining differentiated tumor cells are still capable of sustaining growth of a tumor mass,34 a more practical approach would be to target both CSC and non-CSC populations using a common target, such as the TfR.
TfR was shown in the studies described above to be overexpressed on CSCs as well as non-CSCs. More importantly, targeting studies in vitro and in vivo demonstrated that our TfR-targeting nanocomplex (scL) can efficiently target and deliver payloads into both populations in brain and colorectal cancers. Transfection of brain and colorectal cancer cells with a scL-6FAM-ODN nanocomplex in vitro resulted in a high level of transfection efficiency in both CSCs and non-CSCs with similar delivery efficiencies in the two populations. The ability to target CSCs using the scL nanocomplex was further confirmed in vivo using chemically induced, syngeneic, and xenograft mouse tumor models. In all of these different mouse models of various types of cancer, high levels of CSC-targeting were observed. It is significant that targeting was not limited to the subcutaneous and intracranial xenograft tumors, or one tumor type, but was also observed in the syngeneic and, most importantly, the chemically induced tumors. Although xenograft tumors are the most frequently used models to assess tumor response, they might not be the most representative of human cancer since they are induced in animals that have impaired immune systems. While syngeneic tumors are more representative of the natural situation as they are produced in fully immune competent animals, they are still somewhat artificial as the tumor location often does not closely mirror the microenvironment of the organ from which the original tumor was derived. However, the tumors that arose naturally and spontaneously after treatment with chemical carcinogens, rather than introduction of exogenous tumor cells, are likely the most representative of the actual clinical situation. The demonstration of CSC targeting in these tumors is an indication that there is potential to reproduce this finding in human patients.
Furthermore, the data obtained also demonstrated that the targeting observed was truly specific for CSCs and not a generalized binding to “stem cells.” Uptake of the scL-delivered 6FAM-ODN payload was not significantly detected in either normal mouse hematopoietic stem cells or mesenchymal stem cells obtained from bone marrow and blood. This was shown to be the case with normal stem cells isolated not only from mice bearing chemically induced tumors, but more importantly, from nontumor bearing mice (data now shown). Normal stem cells possess very low levels of the TfR. Thus, due to the TfR-targeting moiety on the nanocomplex, the specificity of this systemically administered scL nanocomplex should also prevent deleterious and potentially dangerous side effects resulting from nonspecific toxicity in normal stem cells. This CSC specificity is another significant advantage of this nanodelivery system with respect to potential clinical applications.
Previously published studies have demonstrated not only tumor growth inhibition, but also complete tumor regression, in animal models of cancer after treatment with the combination of molecular medicines systemically delivered via scL, and conventional chemo/radio therapies.14,17,22,25 Such complete tumor responses likely reflect the elimination of CSCs in addition to the elimination of differentiated cancer cells. Thus having demonstrated efficient targeting and delivery of the payload to CSCs by the scL nanocomplex, we hypothesize that therapeutic targeting of CSCs using a scL nanoparticle will be able to exercise a significant antitumor efficacy and CSC death. The exquisite specificity of scL to target tumors and CSCs resulted in a significant antitumor effect demonstrated in in vitro and in vivo efficacy studies presented above. Considering that mutation in the tumor-suppressor gene p53 is the most common genetic alterations in advanced colorectal cancer, a therapeutic approach restoring p53 function could greatly improve the treatment outcomes. Both mutant (HT-29 and U251) and wild-type p53 (HCT-116 and U87) cell lines were assessed in vitro for the effect of scL-delivered wtp53 on the survival of CSC and non-CSC. As shown in Figure 3, a p53 specific increase in cell death was evident in CSCs and non-CSCs of all four of the cell lines irrespective of their p53 status indicating the broad applicability of this anticancer approach. It should be noted that wtp53 status does not necessarily mean that the p53 signaling pathway is functional.35 It has been suggested that the overexpression of wtp53 could overcome downstream defects through an as yet unknown mechanism.36 In addition, in vitro transfection experiment with scL nanocomplex carrying GFP-tagged p53 plasmid (scL-GFP-p53) resulted in the expression of exogeneous wtp53 and GFP fusion protein in CSCs (CD133+CD166+CD44+) and non-CSCs of HT-29 human colrectal cancer cells. Moreover, scL-GFP-p53 treatment induced the expression of p21 (CIP1/WAF1), a downstream effector of p53, and cleaved Caspase-3 and cleaved PARP, indicators of apoptosis demonstrating the CSC-killing efficacy of scL-delivered wtp53 (data not shown). In in vivo efficacy studies using a colorectal cancer xenograft model, the systemic administration of scL-encapsulated wtp53 plasmid DNA restored a functional apoptotic pathway in p53 mutated HT-29 cells, resulting in an 81.7% inhibition of tumor growth in the scL-p53 treated mice compared to untreated controls. Although we have not assessed if there was a functional reduction in CSC frequency using serial transplants following treatment with scL-p53, the minimal increase in the relative number of CSCs in the tumors over time shown in Figure 8d suggests that scL-p53 treatment eliminated a significant percentage of CSCs. The increase in the sub-G1 population indicates the induction of apoptosis plays a role. scL-p53 was also able to target and induce similar responses in both CD133+ CSCs and CD133− non-CSCs. Taken together, these results confirm the tumor-targeting and CSC-targeting ability as well as antitumor efficacy of the scL nanodelivery system.
Single-agent scL-p53 treatment was shown to be very effective in controlling the tumor size and eliminating CSCs (about 81.7% inhibition of tumor growth). However, since in this particular experiment we initiated treatment when the tumors were a relatively large size (˜300 mm3), we did not observe tumor regression. We envision use of scL-p53 in the clinic as one component of a combination therapy resulting in sensitization to standard radiation/chemotherapy leading to improved tumor response, including regression.
In the studies herein presented, different payloads (reporter plasmid DNA and ODN) were incorporated into the scL nanodelivery system to demonstrate targeting. These results indicate the versatility of this delivery system as a modular platform technology adaptable to various payloads. This nanotechnology has been shown to be capable of specifically and efficiently delivering to tumors a wide variety of payloads including nucleic acid-based therapeutics (plasmid DNA,14,17,18,23,25,37 si/miRNA,21,22 antisense oligonucleotides),24 diagnostic imaging probes,38,39,40 small molecules20 and chemotherapeutic agents. These payloads can be incorporated into the nanoparticle individually or in combination for delivery to both CSCs and non-CSCs of tumors. Thus, this CSC-targeting approach has the potential to be applied to multiple CSC-directed treatment strategies. For example, the molecular targeting and ablation of developmental pathways that are preferentially induced or operative in CSCs (e.g., hedgehog, Wnt/β-catenin, and Notch) could be developed.41,42 Targeting self-renewal pathways of CSCs may also be possible as treatment options to reduce the CSC population in the tumor.43,44 Our CSC-targeting method could also be applied to overcome drug resistance and disrupt CSC-niche cell interaction.45,46 Delivery of an appropriate payload could also potentially induce CSC differentiation by upregulating BMP-4 expression,47 or downregulate cytokine (IL-4) production which is an important intermediate in the antiapoptotic mechanism in CSCs of colorectal cancer.48
The prototype of this platform technology (gene therapy with p53) is now in clinical trials where it has been shown to be well tolerated.27 In this phase I trial, there were indications of anticancer activity as a single agent in some patients, even at the lowest dose administered. More than 70% of the patients had stable disease at the end of the treatment cycle.
In summary, our data suggest that systemic delivery of nanomedicines via the anti-TfRscFv–directed cationic liposome nanocomplex (scL) is a promising therapeutic strategy with selectivity for in vivo targeting of, and enhanced delivery to, both differentiated cancer cells and CSCs. In addition, this study suggests that CSC-targeted nanodelivery of the tumor-suppressor gene wtp53 could be an effective curative approach by eliminating CSCs in colorectal cancer. We believe that the tumor-targeting and CSC-targeting capability, in combination with conventional chemo- or radio-therapy, could lead to significant reduction and possibly even elimination of cancer recurrence/resistance. With this great versatility and flexibility of selecting payloads, proven safety, and targeting advantage, our CSC-targeted nanodelivery system has the potential for clinical translation to become a drug delivery platform for diagnosis, prevention, and treatment of cancers. This scL-p53 nanocomplex is currently being evaluated in a number of clinical trials in patients with advanced solid tumors where it has been shown to be well tolerated with indications of anticancer activity.
Materials and Methods
Complex preparations. Cationic liposome 1,2-dioleoyl-3-trimethylammonium propane (DOTAP)/dioleolylphosphatidyl ethanolamine (DOPE) (Avanti Polar Lipids, Alabaster, AL), referred to as Lip in this study, was prepared as described previously.17 scL-DNA and scL-ODN complexes were prepared as previously described.37
Cell lines. Human colorectal cancer cell lines HT-29 and HCT-116 were obtained from the Tissue Culture Shared Resource of the Lombardi Comprehensive Cancer Center at Georgetown University. Human brain cancer cell lines U87, LN-18, and T98G, human melanoma cell line B16F10, and normal human lung fibroblast cell line IMR-90 were obtained from American Type Culture Collection (ATCC; Manassas, VA). Human brain cancer cell line U251 was obtained from the Division of Cancer Treatment and Diagnosis Tumor Repository, National Cancer Institute (NCI)-Frederick (Frederick, MD). Murine breast cancer cell line EO771 was kindly provided by Dr. J. Yan (James Graham Brown Cancer Center, Louisville, KY). Cells were maintained at 37 °C in a 5% CO2 atmosphere in McCoy's 5A medium (Mediatech, Manassas, VA; HT-29 and HCT-116), modified IMEM (Gibco, Grand Island, NY; U87), RPMI 1640 medium (Gibco; U251 and EO771), MEM (Mediatech; T98G), or DMEM (Gibco; LN-18 and B16F10) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Omega Scientific, Tarzana, CA), 2 mmol/l L-glutamine (Mediatech), and 50 μg/ml each of penicillin, streptomycin, and neomycin (PSN). IMR-90 cells were cultured in Eagle's MEM (Mediatech) supplemented with 10% heat-inactivated FBS, 2 mmol/l L-glutamine, 0.1 mmol/l nonessential amino acids (Gibco), 1 mmol/l sodium pyruvate (Gibco), and 50 μg/ml PSN. The p53 status of the cell lines is as follows: HT-29, U251, LN-18, and T98G all carry mtp53; U87, HCT-116, B16F10 and IMR-90 all have wtp53.
Flow cytometry. To determine TfR expression levels and to identify the CSC population, cancer cells were stained with antibodies against TfR and CSC markers and analyzed by flow cytometry. Antibodies used in this study are monoclonal antibodies for human CD71-FITC (anti-TfR; BioLegend, San Diego, CA), human CD133-PE (Miltenyi Biotec, Auburn, CA), human CD166 (Stemgent, San Diego, CA), human CD44-APC (BioLegend), human EpCAM-PerCP/Cy5.5 (BioLegend), human Msi1 (BD Pharmingen, San Diego, CA), human Nanog-Alexa Fluor 647 (Cell Signaling Technology, Danvers, MA), human SSEA-1 (Stemgent), human Nestin (Abcam, Cambridge, MA), goat anti-mouse IgG-PE (Jackson ImmunoResearch Laboratories, West Grove, PA), goat anti-mouse IgG-APC (Jackson ImmunoResearch Laboratories), and goat anti-rabbit IgG-APC (Jackson ImmunoResearch Laboratories). Cells were labeled with a series of primary and secondary antibodies for 30 minutes at 4 °C in the dark. After washing, the labeled cells were analyzed by BD FACS Aria flow cytometer (BD Biosciences, San Jose, CA).
Immunomagnetic cell sorting. To separate CD133-expressing cells, cancer cells were subjected to immunomagnetic separation using a MACS human CD133 microbead kit (Miltenyi Biotec) in a sterile manner according to the manufacturer's protocol. To determine the purity of the separation, cells were stained with antibodies for human CD133-PE (Miltenyi Biotec) and analyzed by BD FACS Aria flow cytometer.
Western blot analysis. To determine the expression level of the TfR, western blot analysis was performed. Twenty micrograms of total cellular protein lysates from noncancerous normal human fibroblast cell line IMR-90 and immunomagnetically sorted CD133+ and CD133− population of HT-29 cells were separated by 8% SDS-polyacrylamide gel electrophoresis, transferred to nylon membrane, and hybridized with anti-human TfR (Zymed Laboratories, San Francisco, CA) antibody. In the western blots, an antibody recognizing glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Trevigen, Gaithersburg, MD) was utilized as an internal control for equal protein loading.
Immunofluorescence imaging. HT-29, U251, and U87 cells were seeded at 5.5 × 104 cells per chamber of a 4-chamber Falcon culture slide (BD Biosciences Discovery Labware, Bedford, MA). Twenty-four hours later, cells were transfected with scL-GFP (0.5 µg DNA/chamber). Forty-eight hours after transfection, cells were fixed in PBS containing 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) and incubated with rabbit polyclonal anti-CD133 antibodies (Abcam) followed by Texas Red-conjugated secondary sheep anti-rabbit IgG antibodies (Abcam). After incubation, slides were mounted with ProLong anti-fade kit (Molecular Probes, Eugene, OR) and the cells were observed with an Olympus IX71 inverted epifluorescent microscope (Olympus, Center Valley, PA).
Live cell imaging of in vitro transfection. HT-29 cells were seeded at 4.0 × 105 cells per dish on 50 mm glass bottom microwell dishes (MatTek, Ashland, MA). Twenty-four hours later, cells were labeled with CD133-PE antibodies by replacing the medium with the antibody working stock solution. After incubation for 15 minutes in a CO2 incubator at 37 °C, the antibody-containing medium was gently replaced with an equal volume of warm fresh medium and cells were transfected with the scL-6FAM-ODN (1 µmol/l ODN). A scrambled ODN that was labeled with 6-carboxyfluorescein phosphoramidite (6FAM-ODN, 5′-6FAM-CTAGCCATGCTTGTC-3′) was synthesized and purified by reverse phase HPLC (TriLink Biotechnologies, San Diego, CA). Screening of this ODN sequence against the GenBank Database indicated that no significant match was found with any sequence in the database. Immediately after adding the complexes, cells were imaged for 17 hours using total internal reflection fluorescence microscopy on an inverted Nikon Eclipse TE2000E microscope controlled by Element software (Nikon, Melville, NY). Images were analyzed with the same software.
In vitro transfection and cell viability. For in vitro cell survival studies, immunomagnetically sorted CD133+ or CD133− cells (HT-29, HCT-116, U251, and U87) were plated in a 96-well plate (2 × 103 per well). Twenty-four hours later, the cells were transfected with scL-p53 complex prepared as described above. After transfection for 48 hours, cell viability was determined by the XTT assay (Polysciences, Warrington, PA) following the manufacturer's protocol. The IC50 value, the drug concentration resulting in 50% cell kill, was interpolated from the graph of the log of drug concentration versus the fraction of surviving cells.
Induction of exogenous p53 expression in CSCs and non-CSCs. To assess the expression of exogenous wtp53 in CSCs and non-CSCs, the scL complex was prepared as described above except that plasmid GFP-p53 (Addgene plasmid 12091; Addgene, Cambridge, MA) was used as the payload. This plasmid carries genes encoding for both GFP and human wtp53.30 HT-29 cells (6.0 × 105 cells/100 mm dish) were transfected with scL-GFP-p53 at doses of 10 µg DNA/dish. At 24 hours after transfection, the cells were harvested with trypsin, stained with monoclonal antibodies for human CSCs (CD133, CD166, and CD44), and sorted by BD FACS Aria cell sorter. To determine the expression level of the GFP-p53 fusion protein, the sorted cells were lysed. Western blot analysis was performed as described above with anti-GFP (Millipore, Bedford, MA) and anti-human p53 (Calbiochem, San Diego, CA) antibodies. An antibody recognizing GAPDH was utilized as an internal control for equal protein loading.
Animal models. All animal experiments were performed in accordance with and under approved Georgetown University GUACUC protocols. Xenograft models of human colorectal (HT-29) and brain (U251 and U87) cancers were used in this study. HT-29 xenograft tumors were induced in female athymic nude (nu/nu) mice (5–6 weeks old, Harlan Sprague-Dawley, Indianapolis, IN) by subcutaneously inoculating 1.0 × 105 magnetically sorted CD133+ HT-29 cells suspended in Matrigel collagen basement membrane (BD Biosciences) on the lower back above the tail (two sites per mouse). U251 xenograft tumors were induced similarly by subcutaneously injecting 1.0 × 106 U251 cells in female athymic nude mice. For the intracranial brain tumor model, 5.0 × 105 U87 cells were stereotactically injected into the right hemisphere of female athymic nude mice.49 Syngeneic mouse models of 1) subcutaneous breast tumor (EO771) and 2) lung metastases (B16F10) were used in this study. EO771 tumors were induced by subcutaneously injecting 2.5 × 105 cells near the fat pad of the fourth mammary gland in the lower abdomen of female C57BL/6 mice (8–12 weeks old, Jackson Laboratory, Bar Harbor, ME). For the B16F10 mouse model, 1.0 × 105 cells were injected i.v. via the tail vein of 8–10 weeks old female C57BL/6 mice. Chemically produced lung tumors were induced by oral gavage with 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone plus benzo[a]pyrene in female A/J mice (5 weeks old, Jackson Laboratory).50
In vivo tumor targeting study. When subcutaneous xenografts of human colon and brain tumors were at least 250 mm3, the mice were injected i.v. three times within 24 hours (at 0, 12, and 24 hours) for HT-29, and once for U251 with 100 µg/mouse/injection of 6FAM-ODN complexed with scL (scL-6FAM-ODN), untargeted Lip (Lip-6FAM-ODN), or as free 6FAM-ODN. In another study, mice bearing subcutaneous HT-29 xenografts were injected once with 100 µg of GFP plasmid DNA/mouse complexed with scL (scL-GFP), Lip (Lip-GFP), or as free GFP. In a U87 intracranial xenograft model, tumor-bearing mice received a single i.v. injection with 100 µg/mouse/injection of 6FAM-ODN complexed with scL (scL-6FAM-ODN). In the mouse syngeneic and chemically induced tumor models, mice were i.v. injected once with 100 µg/mouse/injection of 6FAM-ODN complexed with scL (scL-6FAM-ODN), Lip (Lip-6FAM-ODN), or as free 6FAM-ODN. At 24 hours (for 6FAM-ODN) or 64 hours (for GFP) after the last injection, tumor, liver, kidney, lung, heart, and spleen were imaged with the Maestro in vivo fluorescence imaging system (CRi, Woburn, MA). The resulting acquisition images were analyzed and the fluorescence signal intensity was quantified with the vendor's software (Maestro 2.10.0). After imaging, single-cell suspensions were obtained from the tumors by collagenase digestion in Hank's balanced solution containing 1 mg/ml collagenase (Sigma) and 2 mmol/l DNase I (Sigma) for 1 hour at 37 °C. The fractionated cells were passed through a 70-μm cell strainer and washed with PBS. To identify the CSC population, single cells were stained with antibodies against CSC markers. Antibodies used in this study are monoclonal antibodies recognizing human CD133-PE, human SSEA-1, human Nestin, human Nanog-Alexa Fluor 647, human Msi1, mouse CD133-PerCP-eFluor 710 (eBioscience, San Diego, CA), mouse CD24-eFluor 450 (eBioscience), mouse CD34-PE (BioLegend), human/mouse CD44-APC/Cy7 (BioLegend), mouse Sca-1-APC (BioLegend), mouse c-Kit-APC/Cy7 (BioLegend), mouse CD45-PE/Cy7 (BioLegend), and mouse hematopoietic lineage eFluor 450 cocktail (eBioscience). The labeled cells were analyzed by BD FACS Aria flow cytometer.
In vivo efficacy study. Subcutaneous HT-29 tumor xenografts were induced in female athymic nude mice (5–6 weeks old) as described above. At 20 days after injection, the tumors averaged 250–300 mm3. The mice were randomized into groups (10–20 mice per group). The targeted liposome complex (scL) carrying wt p53 plasmid DNA was i.v. injected as a bolus two times per week via the tail vein. Thirty microgram of plasmid DNA (1.5 mg DNA/kg body weight) was administered per injection per mouse. A total of 12 injections were administered to the animals over the course of the experiment. The in vivo response was evaluated based upon the changes in tumor volume over time. The size of each tumor was measured weekly and tumor volume (L × W × H) in mm3 calculated. The in vivo efficacy was evaluated by assessing apoptosis of the tumor cells using cell cycle analysis. Percentage of CD133+ CSCs in tumors was monitored by FACS at multiple time points. The relative number of CSCs was calculated from tumor size and percentage of CD133+ CSCs.
In a separate experiment, anticancer efficacy in CSCs and non-CSCs was evaluated by assessing apoptosis of the tumor cells using Annexin V-pacific blue (Biolegend) staining double-labeled with CSC markers CD133-PE/CD44-APC; EpCAM-PerCP-Cy5.5/CD166, or human Msi1. For this study, mice bearing subcutaneous HT-29 xenograft tumors were i.v. injected with scL-p53 (60 µg DNA/mouse/injection) three times within 24 hours via the tail vein when the tumors averaged 800 mm3. Tumors were harvested 12 hours after the last injection. Single-cell isolation and antibody staining were performed as described above and cells were analyzed by FACS.
Statistical analysis. Results are presented as the mean ± SD. All experiments were repeated at least three times. The statistical significance was determined by using the analysis of variance (SigmaPlot, Systat Software, San Jose, CA). P values of <0.05 were considered significant.
Acknowledgments
The authors thank to Karen Creswell at the Flow Cytometry Shared Resource at Georgetown University Medical Center for her assistance. They also thank Lauren Kelehan, Gary S Laevsky, and Jesse B DeWitt from Nikon Instruments Inc. for their assistance with TIRF imaging. This study was supported in part by NCI grant 5R01CA132012-02 (E.H.C.), a research grant from SynerGene Therapeutics Inc. (K.F.P.), and National Foundation for Cancer Research grant HU0001 (E.H.C.). This study was conducted in part using the Flow Cytometry & Cell Sorting Shared Resource, the Preclinical Imaging Research Laboratory, Microscopy & Imaging Shared Resource, Tissue Culture & Biorepository for Cell Lines and Biofluids Shared Resource, and Animal Core Facilities supported by NCI Cancer Center Support grant and US Public Health Service grant 2P30-CA-51008 and 1 S10 RR 15768-01. This investigation was performed in part in a facility constructed with support from Research Facilities Improvement grant C06RR14567 from the National Center for Research Resources, NIH. E.H.C. and K.F.P. are two of the inventors of the described technology, for which several patents owned by Georgetown University have been issued. The patents have been licensed to SynerGene Therapeutics Inc. for commercial development. E.H.C. owns equity interests in SynerGene Therapeutics Inc. and serves as a nonpaid scientific consultant to SynerGene Therapeutics Inc. The other authors declare no conflict of interest.
References
- Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- Park CY, Tseng D, Weissman IL. Cancer stem cell-directed therapies: recent data from the laboratory and clinic. Mol Ther. 2009;17:219–230. doi: 10.1038/mt.2008.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- Kim CF, Jackson EL, Woolfenden AE, Lawrence S, Babar I, Vogel S, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell. 2005;121:823–835. doi: 10.1016/j.cell.2005.03.032. [DOI] [PubMed] [Google Scholar]
- Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–10951. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- Arap W, Pasqualini R, Ruoslahti E. Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science. 1998;279:377–380. doi: 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16:510–520. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wu JJ, Huang L. Nanoparticles targeted with NGR motif deliver c-myc siRNA and doxorubicin for anticancer therapy. Mol Ther. 2010;18:828–834. doi: 10.1038/mt.2009.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low PS, Kularatne SA. Folate-targeted therapeutic and imaging agents for cancer. Curr Opin Chem Biol. 2009;13:256–262. doi: 10.1016/j.cbpa.2009.03.022. [DOI] [PubMed] [Google Scholar]
- Cheng PW. Receptor ligand-facilitated gene transfer: enhancement of liposome-mediated gene transfer and expression by transferrin. Hum Gene Ther. 1996;7:275–282. doi: 10.1089/hum.1996.7.3-275. [DOI] [PubMed] [Google Scholar]
- Xu L, Pirollo KF, Tang WH, Rait A, Chang EH. Transferrin-liposome-mediated systemic p53 gene therapy in combination with radiation results in regression of human head and neck cancer xenografts. Hum Gene Ther. 1999;10:2941–2952. doi: 10.1089/10430349950016357. [DOI] [PubMed] [Google Scholar]
- Bartlett DW, Su H, Hildebrandt IJ, Weber WA, Davis ME. Impact of tumor-specific targeting on the biodistribution and efficacy of siRNA nanoparticles measured by multimodality in vivo imaging. Proc Natl Acad Sci USA. 2007;104:15549–15554. doi: 10.1073/pnas.0707461104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Hong K, Kirpotin DB, Colbern G, Shalaby R, Baselga J, et al. Anti-HER2 immunoliposomes: enhanced efficacy attributable to targeted delivery. Clin Cancer Res. 2002;8:1172–1181. [PubMed] [Google Scholar]
- Xu L, Tang WH, Huang CC, Alexander W, Xiang LM, Pirollo KF, et al. Systemic p53 gene therapy of cancer with immunolipoplexes targeted by anti-transferrin receptor scFv. Mol Med. 2001;7:723–734. [PMC free article] [PubMed] [Google Scholar]
- Xu L, Huang CC, Huang W, Tang WH, Rait A, Yin YZ, et al. Systemic tumor-targeted gene delivery by anti-transferrin receptor scFv-immunoliposomes. Mol Cancer Ther. 2002;1:337–346. [PubMed] [Google Scholar]
- Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, et al. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci USA. 2006;103:6315–6320. doi: 10.1073/pnas.0601755103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SH, Rait A, Pirollo KF, Zhou Q, Yenugonda VM, Chinigo GM, et al. Tumortargeting nanodelivery enhances the anticancer activity of a novel quinazolinone analogue. Mol Cancer Ther. 2008;7:559–568. doi: 10.1158/1535-7163.MCT-07-0548. [DOI] [PubMed] [Google Scholar]
- Pirollo KF, Rait A, Zhou Q, Hwang SH, Dagata JA, Zon G, et al. Materializing the potential of small interfering RNA via a tumor-targeting nanodelivery system. Cancer Res. 2007;67:2938–2943. doi: 10.1158/0008-5472.CAN-06-4535. [DOI] [PubMed] [Google Scholar]
- Pirollo KF, Zon G, Rait A, Zhou Q, Yu W, Hogrefe R, et al. Tumor-targeting nanoimmunoliposome complex for short interfering RNA delivery. Hum Gene Ther. 2006;17:117–124. doi: 10.1089/hum.2006.17.117. [DOI] [PubMed] [Google Scholar]
- Pirollo KF, Rait A, Zhou Q, Zhang XQ, Zhou J, Kim CS, et al. Tumor-targeting nanocomplex delivery of novel tumor suppressor RB94 chemosensitizes bladder carcinoma cells in vitro and in vivo. Clin Cancer Res. 2008;14:2190–2198. doi: 10.1158/1078-0432.CCR-07-1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rait AS, Pirollo KF, Xiang L, Ulick D, Chang EH. Tumor-targeting, systemically delivered antisense HER-2 chemosensitizes human breast cancer xenografts irrespective of HER-2 levels. Mol Med. 2002;8:475–486. [PMC free article] [PubMed] [Google Scholar]
- Xu L, Frederik P, Pirollo KF, Tang WH, Rait A, Xiang LM, et al. Self-assembly of a virus-mimicking nanostructure system for efficient tumor-targeted gene delivery. Hum Gene Ther. 2002;13:469–481. doi: 10.1089/10430340252792594. [DOI] [PubMed] [Google Scholar]
- Daniels TR, Bernabeu E, Rodríguez JA, Patel S, Kozman M, Chiappetta DA, et al. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim Biophys Acta. 2012;1820:291–317. doi: 10.1016/j.bbagen.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senzer N, Nemunaitis J, Nemunaitis D, Bedell C, Edelman G, Barve M, et al. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol Ther. 2013;21:1096–1103. doi: 10.1038/mt.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd SD, Tsai KY, Jacks T. An intact HDM2 RING-finger domain is required for nuclear exclusion of p53. Nat Cell Biol. 2000;2:563–568. doi: 10.1038/35023500. [DOI] [PubMed] [Google Scholar]
- Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63:136–151. doi: 10.1016/j.addr.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Curr Opin Biotechnol. 2007;18:460–466. doi: 10.1016/j.copbio.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Duru N, Fan M, Candas D, Menaa C, Liu HC, Nantajit D, et al. HER2-associated radioresistance of breast cancer stem cells isolated from HER2-negative breast cancer cells. Clin Cancer Res. 2012;18:6634–6647. doi: 10.1158/1078-0432.CCR-12-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan CT. Cancer stem cells: controversial or just misunderstood. Cell Stem Cell. 2009;4:203–205. doi: 10.1016/j.stem.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. 2006;12:1167–1174. doi: 10.1038/nm1483. [DOI] [PubMed] [Google Scholar]
- Beier D, Schulz JB, Beier CP. Chemoresistance of glioblastoma cancer stem cells–much more complex than expected. Mol Cancer. 2011;10:128. doi: 10.1186/1476-4598-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris CC. Structure and function of the p53 tumor suppressor gene: clues for rational cancer therapeutic strategies. J Natl Cancer Inst. 1996;88:1442–1455. doi: 10.1093/jnci/88.20.1442. [DOI] [PubMed] [Google Scholar]
- Li H, Lochmüller H, Yong VW, Karpati G, Nalbantoglu J. Adenovirus-mediated wild-type p53 gene transfer and overexpression induces apoptosis of human glioma cells independent of endogenous p53 status. J Neuropathol Exp Neurol. 1997;56:872–878. doi: 10.1097/00005072-199708000-00005. [DOI] [PubMed] [Google Scholar]
- Yu W, Pirollo KF, Yu B, Rait A, Xiang L, Huang W, et al. Enhanced transfection efficiency of a systemically delivered tumor-targeting immunolipoplex by inclusion of a pH-sensitive histidylated oligolysine peptide. Nucleic Acids Res. 2004;32:e48. doi: 10.1093/nar/gnh049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman M, Chang EH, Zhou Q, Pirollo KF. Nanodelivery of MRI contrast agent enhances sensitivity of detection of lung cancer metastases. Acad Radiol. 2009;16:627–637. doi: 10.1016/j.acra.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirollo KF, Dagata J, Wang P, Freedman M, Vladar A, Fricke S, et al. A tumor-targeted nanodelivery system to improve early MRI detection of cancer. Mol Imaging. 2006;5:41–52. [PubMed] [Google Scholar]
- Yang C, Rait A, Pirollo KF, Dagata JA, Farkas N, Chang EH. Nanoimmunoliposome delivery of superparamagnetic iron oxide markedly enhances targeting and uptake in human cancer cells in vitro and in vivo. Nanomedicine. 2008;4:318–329. doi: 10.1016/j.nano.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannuti A, Foreman K, Rizzo P, Osipo C, Golde T, Osborne B, et al. Targeting Notch to target cancer stem cells. Clin Cancer Res. 2010;16:3141–3152. doi: 10.1158/1078-0432.CCR-09-2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge ME, Lum L. Drugging the cancer stem cell compartment: lessons learned from the hedgehog and Wnt signal transduction pathways. Annu Rev Pharmacol Toxicol. 2011;51:289–310. doi: 10.1146/annurev-pharmtox-010510-100558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J, Shen J, Liu L, Xia F, Xu C, Duan G, et al. Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factor pathway in human hepatocellular carcinoma. Hepatology. 2012;56:1004–1014. doi: 10.1002/hep.25745. [DOI] [PubMed] [Google Scholar]
- Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol. 2007;17:165–172. doi: 10.1016/j.cub.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonardo E, Frias-Aldeguer J, Hermann PC, Heeschen C. Pancreatic stellate cells form a niche for cancer stem cells and promote their self-renewal and invasiveness. Cell Cycle. 2012;11:1282–1290. doi: 10.4161/cc.19679. [DOI] [PubMed] [Google Scholar]
- LaBarge MA. The difficulty of targeting cancer stem cell niches. Clin Cancer Res. 2010;16:3121–3129. doi: 10.1158/1078-0432.CCR-09-2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo Y, Scopelliti A, Cammareri P, Todaro M, Iovino F, Ricci-Vitiani L, et al. Bone morphogenetic protein 4 induces differentiation of colorectal cancer stem cells and increases their response to chemotherapy in mice. Gastroenterology. 2011;140:297–309. doi: 10.1053/j.gastro.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402. doi: 10.1016/j.stem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Hadjipanayis CG, Fellows-Mayle W, Deluca NA. Therapeutic efficacy of a herpes simplex virus with radiation or temozolomide for intracranial glioblastoma after convection-enhanced delivery. Mol Ther. 2008;16:1783–1788. doi: 10.1038/mt.2008.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conaway CC, Wang CX, Pittman B, Yang YM, Schwartz JE, Tian D, et al. Phenethyl isothiocyanate and sulforaphane and their N-acetylcysteine conjugates inhibit malignant progression of lung adenomas induced by tobacco carcinogens in A/J mice. Cancer Res. 2005;65:8548–8557. doi: 10.1158/0008-5472.CAN-05-0237. [DOI] [PubMed] [Google Scholar]