

Abstract
Duchenne muscular dystrophy is a monogenic disease potentially treatable by gene replacement. Use of recombinant adeno-associated virus (AAV) will ultimately require a vascular approach to broadly transduce muscle cells. We tested the impact of preexisting AAV antibodies on microdystrophin expression following vascular delivery to nonhuman primates. Rhesus macaques were treated by isolated limb perfusion using a fluoroscopically guided catheter. In addition to serostatus stratification, the animals were placed into one of the three immune suppression groups: no immune suppression, prednisone, and triple immune suppression (prednisone, tacrolimus, and mycophenolate mofetil). The animals were analyzed for transgene expression at 3 or 6 months. Microdystrophin expression was visualized in AAV, rhesus serotype 74 sero-negative animals (mean: 48.0 ± 20.8%) that was attenuated in sero-positive animals (19.6 ± 18.7%). Immunosuppression did not affect transgene expression. Importantly, removal of AAV binding antibodies by plasmapheresis in AAV sero-positive animals resulted in high-level transduction (60.8 ± 18.0%), which is comparable with that of AAV sero-negative animals (53.7 ± 7.6%), whereas non-pheresed sero-positive animals demonstrated significantly lower transduction levels (10.1 ± 6.0%). These data support the hypothesis that removal of AAV binding antibodies by plasmapheresis permits successful and sustained gene transfer in the presence of preexisting immunity (natural infection) to AAV.
Introduction
Duchenne muscular dystrophy (DMD) is the most common, severely debilitating childhood form of muscular dystrophy. The disease is caused by mutations in the DMD gene,1,2 which follows an X-linked recessive inheritance pattern. The size of this gene1 creates an exceptionally large target for spontaneous germ-line mutations (1 in 10,000 sperm or eggs). Based on pooled data from worldwide newborn screening studies and the most recent study,3 the revised estimate of DMD incidence at birth is ~1:5,000 newborn males. Newborn screening can never eliminate the disease, emphasizing the importance of finding an effective treatment.
Dystrophin plays a central role in muscle function and integrity; specifically, it provides a scaffold for a number of important proteins that form the dystrophin-glycoprotein complex, linking the subsarcolemmal cytoskeleton to the extracellular matrix in skeletal muscle and cardiomyocytes.4 Mutant dystrophin hinders stability of the dystrophin-glycoprotein complex, weakening the sarcolemmal membrane, leading to muscle cell injury and muscle fiber loss with replacement by connective tissue and fat.4 Gene replacement by mini- or micro-dystrophins (micro-dys) delivered by recombinant adeno-associated virus (rAAV) represents an approach showing promise in proof-of-principle studies in mouse and dog.5,6,7,8,9 These smaller dystrophin transgenes were designed to accommodate for the genome packaging limit of AAV (<5 kb) while maintaining much of the functional features of dystrophin.
Safety–tolerability clinical gene transfer trials for muscular dystrophies have so far been limited to direct intramuscular delivery trials.10,11,12 Efficacy will require a different strategy reaching multiple muscle groups best achieved through the circulation. In mdx mice, we have demonstrated that rAAV can deliver micro-dys9,13 using an isolated limb perfusion model through the femoral artery. Outcome parameters indicative of efficacy include reduction in central nucleation, improved tetanic force, and increased resistance to eccentric contraction.9,13 These studies set the stage for a more ambitious model in the nonhuman primate (NHP) translatable to the clinic.14 This approach was modeled after the mdx mice studies with targeted vascular delivery to one or a small group of contiguous muscles.9 In the present study, we used the gastrocnemius as the target muscle for delivery because we could thread the catheter along to the sural artery through femoral access and achieve consistent results that were dose dependent in the macaque.15
In addition to proof of principle for our vascular delivery model, the impetus for the current study was our experience in clinical gene transfer trials of mini-dystrophin and α-sarcoglycan.10,11,12 In DMD, we found that transduction efficiency was limited by T-cell–mediated responses to transgene by two different mechanisms.12 The first was a T-cell response mounted against novel epitopes presented by the mini-dystrophin transgene in an area of the patient's deletion. A second mechanism emerged unexpectedly demonstrating that revertant fibers had presented novel epitopes downstream of patient mutations eliciting antigen-specific T-cell responses before and after gene transfer. In the LGMD2D study, five of the six patients demonstrated robust transgene expression, whereas subject 6 had no measurable expression. The only parameter that was unique to subject 6 was the presence of preexisting binding and neutralizing antibodies to AAV1 before gene transfer.10 Others have reported similar findings of poor transgene expression when vector has been administered in the presence of AAV neutralizing antibody titers,16,17,18 and recent gene transfer clinical trials have enrolled subjects negative for anti-AAV antibodies.19,20 In a rhesus macaque with endogenous dystrophin, it is difficult to fully address the problem of transgene T-cell immunity; however, the potential impedance of AAV antibodies on transgene expression and ways to circumvent this could be tested directly. Moreover, any confounding responses due to transgene immunity are minimized in this model.
We created an experimental paradigm closely simulating the clinical scenario where we attempted gene transfer in rhesus macaques with preexisting immunity to rAAV rhesus serotype 74 ((rAAVrh.74) anti-AAV binding antibody titers and presumed T-cell immunity to rAAVrh.74), in addition to sero-negative animals. The AAVrh.74 serotype is endogenous to the rhesus macaque and was isolated from mesenteric lymph nodes and subsequently from the spleen. We have previously reported that this serotype of AAV results in highly efficient muscle transduction in mice and rhesus macaque when delivered via the vasculature.9,15 In addition to anatomical and immunological correlates to humans, the NHP was chosen for these studies because of ease of direct removal of preexisting AAV antibodies using plasmapheresis, a well-established method to treat autoimmune disease,21 in contrast to pharmacologic immunosuppression. Moreover, AAV naturally infects NHPs like its human host. Findings from studies in this report have potential clinical applicability.
Results
Optimization of muscle-directed isolated focal limb perfusion with rAAVrh.74.CMV.eGFP
We used rAAVrh.74.CMV.eGFP vector with the eGFP reporter transgene to determine the optimal delivery volume to achieve efficient transduction with the least variability. This included volumes of 0.5, 1.0, and 2.5 ml/kg delivered to 18 rhesus macaques (n = 6 per group) negative for AAVrh.74 antibodies (end point titer <1:50). Dosing was consistent between groups at 2 × 1012 vg/kg. A fluoroscopically guided catheter was introduced into the femoral artery at the apex of the femoral triangle and advanced to the femoral–sural bifurcation and into the sural artery, which feeds the lateral and medial heads of the gastrocnemius (Figure 1a; upper and lower panels). Two tourniquets proximal and distal to the gastrocnemius compartmentalized the region for vector delivery. End point analysis was completed 3 weeks after vector administration at which time both the treated and contralateral gastrocnemius muscles were removed, cut into blocks (~1.0 × 0.75 cm), and snap frozen. In addition to muscle tissue, vital organ tissues (heart, lung, liver, kidney, spleen, gonad, and lymph nodes) were collected for vector biodistribution analysis. The percentage of muscle fibers expressing eGFP was quantified across the entire muscle (see Figure 2 for quantification scheme) and revealed that all treatment groups had efficient transduction of the gastrocnemius muscle (Figure 1b); however, the group treated using a delivery volume of 2.5 ml/kg demonstrated the greatest expression with the least variability (51.5 ± 9.5% fibers; Figure 1c). Expression levels and biodistribution were confirmed with quantitative polymerase chain reaction (PCR) (Figure 1d). Vector genome (vg) copies were detected in the target muscle 3.2 × 104 ± 6.3 × 104 vg copies/µg genomic DNA (range of values was equivalent to 0.2–1 vg/nucleus). Off-target detection of vector genomes was highest in the spleen, a highly vascularized organ for which AAVrh.74 possesses some tropism.
Figure 1.

Optimizing vector/transgene delivery to the gastrocnemius. (a) Upper and lower panels show fluoroscopic images defining the lower extremity vasculature at the level of the thigh and gastrocnemius with a catheter tip at the femoral art. sural art. junction (arrow head). The lower panel shows the placement of the proximal tourniquet (between the two clamps) and contrast-blush of the gastrocnemius vasculature (oval). (b) Gastrocnemius specimen from (animal no. 03D347) treated with 2 × 10 12 vg/kg of AAVrh.74.CMV.eGFP with 2.5 ml/kg administration volume at necropsy with insets showing direct fluorescence images of eGFP expression and the percentage of muscle fiber counts found in the identified regions. (c) Summary graph of muscle fiber counts following vector/transgene (eGFP) administration comparing vector administration volumes—2.5, 1.0, and 0.5 ml/kg. Graph bars represent transgene expression mean values ± SEM; n = 6 per group. (d) Summary graph depicting vector biodistribution levels in several tissues at the time of necropsy. The dotted line references 100 transgene copies per microgram of host DNA. Graph bars represent tissue mean values ± SEM; data represent tissues from the nine animals treated with eGFP. The highest values were found in the treated gastrocnemius. The next highest values were found in the spleen, which is a tissue the vector exhibits tropism for. The remaining tissue expression levels, including contralateral control muscle, are below 100 transgene copies per microgram of host DNA. AAVrh.74, adeno-associated virus, rhesus serotype 74.
Figure 2.

Transgene expression quantification scheme for target tissue analysis. Each gastrocnemius was separated into medial and lateral heads, which were cut into 16–20 blocks. Each block was sectioned and the transgene expression was quantified by counting the percentage of positive muscle fibers in four randomly selected 10× images taken under fluorescent microscopy.
Impact of AAV immunity on micro-dys expression in NHP muscle
The overall study design consisted of a three-tiered stratification scheme to compare the efficiency of vascular delivery of micro-dys in the presence or absence of preexisting antibody immunity with AAVrh.74 (Supplementary Figure S1a). Thirty-six rhesus macaques were divided equally into AAVrh.74 antibody–negative (sero-negative) and AAVrh.74 antibody–positive (sero-positive) groups based on baseline testing at least 4 weeks before treatment. Animals were categorized as sero-negative with a binding antibody titer <1:50 and animals were considered sero-positive with a binding antibody titer ≥1:50 (range of end point titers: 1:400 to 1:3,200 for AAVrh.74 sero-positive (Supplementary Figure S2)) based on both animal studies16,22 and clinical trials.10,18 This stratification allowed the comparison of sero-negative and sero-positive (naturally infected) animals to gene transfer with AAVrh.74.
To evaluate the potential effects of immunosuppression (IS) in our model, sero-negative and sero-positive animals were further stratified into one of the three IS arms: (i) no IS, (ii) prednisone alone, and (iii) triple therapy (prednisone, tacrolimus, and mycophenolate mofetil (MMF)) (Supplementary Figure S1a). Shown to prolong ambulation in DMD patients, prednisone therapy is considered to be the standard of care.23 Treatment with AAV may elicit additional T-cell responses to the AAV capsid and as some have suggested may require adjunctive T-cell treatments to modulate this response.24 Accordingly, we chose to test the addition of T-cell inhibitors currently used clinically, tacrolimus and MMF along with prednisone as an additional cohort termed triple therapy. Therefore, the final study design included AAVrh.74-binding antibody–positive and AAVrh.74-binding antibody–negative animals further stratified into one of the three groups: 1) no immune suppression (IS), (ii) prednisone alone, and (iii) triple therapy (prednisone, tacrolimus, and MMF) (n = 6 per group, n = 3 at 3-month end point and n = 3 at 6-month end point) (Supplementary Figure S1a). The third stratification was to assess transgene expression over time, in particular at 12 or 24 weeks.
To validate the effectiveness of our IS regimens, a lymphocyte proliferation assay was conducted at baseline and after 6 weeks of immunosuppression treatment in a cohort of NHPs and demonstrated effective downregulation of lymphocyte proliferation (Supplementary Figure S3a,b). Peripheral blood mononuclear cells (PBMCs) were isolated for interferon-γ (IFN-γ) enzyme-linked immunospot (ELISpot) analysis to detect AAV capsid– or micro-dys.FLAG–specific T cells and serum for total AAVrh.74 antibody enzyme-linked immunosorbent assays were collected at two baseline bleeds and every 2 weeks thereafter (Supplementary Figure S1b). Chemistries were collected on a monthly basis (Supplementary Figure S1b and Table S1), and physical exams were conducted weekly. No abnormal chemistry values were noted.
On the day of treatment, the left lower limb of the macaque (4–8 kg animals) was perfused with 2 × 1012 vg/kg rAAVrh.74.MCK.micro-dys.FLAG in 2.5 ml/kg saline through a catheter advanced into the sural artery and compartmentalized gastrocnemius using our published protocol.14 Three or 6 months after transfer, the macaques were euthanized, and the entire gastrocnemius muscle was removed, cut into blocks (~1.0 × 0.75 cm), and snap frozen. Micro-dys.FLAG expression was visualized by immunofluorescence using an anti-FLAG antibody.15 As previously described, a FLAG tag consisting of eight amino acids (DYKDDDDK) was fused to the C-terminus of micro-dys to track expression in the presence of endogenous dystrophin in the NHP.14,15 The contralateral gastrocnemius served as a control. A comprehensive quantification scheme was used and resulted in the analysis of 30,000–42,000 muscle fibers per animal (Figure 2). This allowed us to generate a more precise representation of expression throughout the entire muscle rather than a snapshot of a given area. An example of the variability of expression that likely resulted from vascular distribution variability in the muscle is demonstrated by a representative schematic heat map (Supplementary Figure S4). This is an important point for translation given the inherent variability of vascular supply in various muscles or even in between subjects. Comparing overall AAV antibody–positive and AAV antibody–negative groups irrespective of immunosuppression regimen, efficient expression of micro-dys.FLAG (Figure 3a,b) was visualized in AAV sero-negative animals (mean: 48.02 ± 20.78%) with significantly less expression noted in AAV sero-positive animals (19.61 ± 18.66%) (P ≤ 0.001, three-way analysis of variance).
Figure 3.

Micro-dys.FLAG expression results correlate with antivector antibody levels on the day of vector administration. (a) Summary graph of percent muscle fiber expression of micro-dys.FLAG in vector sero-negative and vector sero-positive (− and +, repectivily) animals in the presence and absence of immune suppression (IS) (NIS: no immune suppression, Pred: prednisone only, or Trip: triple IS with prednisone, tacrolimus, and mycophenolate mofetil). Trip IS was started 2 weeks before vector administration and continued until the 12th week after vector administration. The Pred and Trip animals stratified to the 24-week cohorts continued to receive prednisone only until the end of the study. Animal tissues were harvested at 12 or 24 weeks after vector administration and assayed for the presence of micro-dys.FLAG expression by immunofluorescence. *Different from sero-negative group, P ≤ 0.001 by three-way analysis of variance (ANOVA); n = 3 for each group. (b) Representative images (top) of micro-dys.FLAG expression demonstrated in gastrocnemius muscles in AAV sero-negative animals (7–90 shown) and AAV sero-positive animals (06D054 shown) Scale bar = 200 µm. The inset is nontargeted contralateral gastrocnemius as negative control. Representative hematoxylin and eosin staining from isolated muscle tissue from treated animals revealed no evidence of tissue damage or cellular infiltration (bottom). Scale bar = 100 µm. (c) Summary box chart of vector binding antibody levels for each cohort represented in (a) measured on the day of vector administration. The red line represents the cohort mean, the black line represents the cohort median, and the box ends represent the 5 and 95% confidence intervals for the cohort; n = 3 for each group. (d) Regression analysis comparing individual animal vector antibody levels on the day of vector administration and transgene, micro-dys.FLAG expression sero-negative (blue) and sero-positive (red) animals.
In the sero-negative animal group, expression of micro-dys.FLAG did not improve with immunosuppression (prednisone alone or triple) compared with no IS (Figure 3a). Furthermore, transgene expression did not improve with extended treatment time in groups with end points at 12 versus 24 weeks (Figure 3a, three-way analysis of variance). In the AAV sero-positive animals, expression was decreased overall compared with AAV sero-negative animals, and the effects of immunosuppression in the antibody-positive group was indeterminate due to the strong effect of antibody levels on transgene expression (Figure 3a,c, three-way analysis of variance). There was no evidence of necrosis or mononuclear cell infiltration to account for the reduced expression in the AAV sero-positive group (Figure 3b). The only parameter that significantly associated with transgene expression was the level of anti-AAVrh.74 binding antibodies, which showed an inverse correlation with micro-dys.FLAG expression (Figure 3a,c). This was especially striking in the AAVrh.74 sero-positive group, where animals with the highest titer had the lowest level of expression (compare Figure 3a and Figure 3c). To further delineate these results, regression analysis comparing the percentage of fibers expressing micro-dys.FLAG in each animal with AAVrh.74 binding antibody titers (ratio at 1:800 dilution) revealed an inverse correlation (r2 = 0.39) (Figure 3d). Vector biodistribution was performed on AAVrh.74 sero-negative animals comparing 12- and 24-week time points. The target muscle and spleen contained the highest vg copy numbers, although the target muscle was statistically higher than the spleen (P ≤ 0.05) (Supplementary Figure S5).
To assess antigen-specific T-cell–mediated response against the micro-dys.FLAG and AAVrh.74 capsid proteins, PBMCs were isolated from each animal on a biweekly basis and assayed for IFN-γ ELISpot responses. Peptides were divided into three pools for both proteins as previously described.15 There were no significant responses to micro-dys.FLAG in any of the animals (Figure 4a). There were transient mild responses identified by the IFN-γ ELISpot assay to AAVrh.74 capsid in seven animals that were considered positive by exceeding a threshold of 50 spot-forming colonies/1 × 106 PBMCs (Figure 4b and Supplementary Table S2). There was no trend regarding serostatus or immunosuppression, and the responses were unsustained (Supplementary Table S2).
Figure 4.

Enzyme-linked immunosorbant spot assay for detection of transgene and/or capsid-specific T cells in Peripheral blood mononuclear cells (PBMCs). (a) Peptides for micro-dys.FLAG were separated into three pools. There were no significant responses for micro-dys or FLAG in any of the animals. Data represent PBMC studies from 18 treated sero-negative animals and 18 treated sero-positive animals. (b) Peptides for the AAVrh.74 capsid were separated into three pools. There were mild transient responses in a few animals that were unsustained at 4 and 6 weeks . IFN-γ, interferon-γ SFCs, spot-forming colonies. Data represent PBMC studies from 18 treated sero-negative animals and 18 treated sero-positive animals. AAVrh.74, adeno-associated virus, rhesus serotype 74.
Plasmapheresis before rAAV delivery circumvents the impact of AAVrh.74 binding antibodies
The strong reciprocal correlation of anti-AAVrh.74 antibody levels and micro-dys transgene expression (Figure 3d) coupled with the very limited T-cell response to vector or transgene provided a compelling argument to explore mechanisms for transient removal of antibodies before treatment. Plasmapheresis is a commonly used, effective procedure to remove offending antibodies in a variety of neurologic disorders21 and therefore would pose few obstacles for clinical application. We treated seven AAVrh.74 sero-positive rhesus macaques (end point titers 1:400 to 1:1,600) (Table 1) by performing two rounds of plasmapheresis (two rounds of three plasma volume exchanges over 2 consecutive days) and dosing the animal immediately following the second plasma exchange by isolated limb perfusion delivery of rAAVrh.74.micro-dys.FLAG to the gastrocnemius. For these studies to reflect worst-case scenario, no IS was used. The gastrocnemius was the targeted muscle and the protocol was the same as in the previous cohorts except that both limbs were treated (animal 00C170 is the exception-only one limb was treated). Animals were sacrificed after 3 months, analyzed for micro-dys.FLAG expression, and compared with the previously treated animals (AAVrh.74 sero-negative (n = 6) and AAVrh.74 positive (n = 6)) that were not treated with IS. The AAVrh.74 sero-positive animals that underwent plasmapheresis before vector administration demonstrated robust micro-dys.FLAG expression (60.8 ± 18.0%), which was equivalent to that achieved by treating AAVrh.74 sero-negative animals with micro-dys.FLAG and no immunosuppression (53.7 ± 7.6%) versus AAVrh.74 sero-positive animals that did not undergo plasmapheresis where expression was significantly lower (10.1 ± 6.0%; P ≤ 0.001) (Figure 5a). Histologically, there was no evidence of necrosis or mononuclear cell infiltration in the animals that underwent plasmapheresis and subsequent vector delivery (Figure 5b). The animals undergoing plasmapheresis (Figure 5c) had a significant >10-fold reduction of AAVrh.74 binding antibody levels (post- versus preplasmapheresis). The postplasmapheresis titer was not different from that of sero-negative animals (Figure 5c). There was a significant correlation between reduced antibody levels following plasmapheresis and micro-dys.FLAG expression (Figure 5d). Demonstrated in all groups, anti-AAVrh.74 binding antibodies were increased by 2 weeks after vector administration and remained elevated for the 12-week period. (Supplementary Figure S6). Of note, there was a rapid and robust increase in antibody levels in the two immune-positive groups, whereas, the sero-negative group had a modest antibody response following vector delivery. The strength and kinetics of the antibody responses stratify as predicted into a primary response for the sero-negative group and an amnestic memory response for both sero-positive groups. Importantly, the levels of AAVrh.74 binding antibodies in the postvector period had no effect on transgene expression in the sero-positive pheresed group.
Table 1. Efficiency of plasmapheresis in removing AAVrh.74 binding antibody and level of transgene expression.
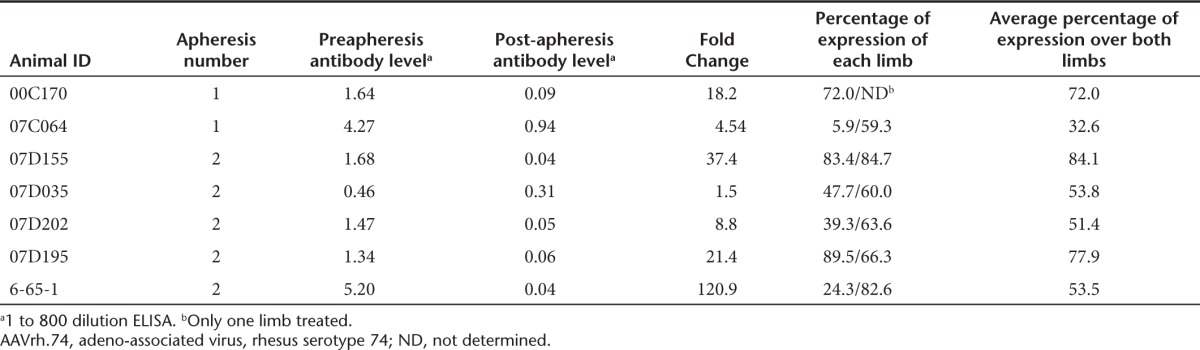
Figure 5.

Plasmapheresis reverses attenuation of transgene expression by vector binding antibodies present in AAVrh.74 sero-positive animals. (a) Summary graph of percent muscle fiber expression of micro-dys.FLAG in vector sero-negative, and vector sero-positive and vector sero-positive animals treated with plasmapheresis before vector administration (sero-negative n = 6, sero-positive n = 6, and sero-positive + plasmapheresis n = 7). Animal tissues were harvested at 12 weeks after vector administration and assayed for the presence of transgene, micro-dys.FLAG, expression by immunofluorescence. *Different from sero-negative group, P ≤ 0.001. **Different from immune cohort, P ≤ 0.001. (b) Representative image (animal no. 07D195) of micro-dys.FLAG expression demonstrated in gastrocnemius muscle (left). Scale bar = 200 µm. The inset is nontargeted contralateral gastrocnemius as negative control. Hematoxylin and eosin staining from isolated muscle tissue from treated animals revealed no evidence of tissue damage or cellular infiltration (right). Scale bar = 100 µm. (c) Summary box graph of vector binding antibody levels for sero-negative, immune, immune preplasmapheresis, and immune postplasmapheresis. The red line represents the cohort mean, the black line represents the cohort median, and the box ends represent the 5 and 95% confidence intervals for the cohort. (d) Regression analysis comparing the individual animal vector antibody levels on the day of vector administration and transgene, micro-dys.FLAG, expression. AAVrh.74, adeno-associated virus, rhesus serotype 74.
Discussion
The major findings in this study include the following: (i) Vascular delivery of AAVrh.74 to the gastrocnemius with either a reporter construct (eGFP) or a therapeutic gene (micro-dys) demonstrated broad and robust expression. (ii) A strong inverse correlation exists between micro-dys.FLAG gene expression and circulating antibodies to AAVrh.74, the serotype naturally infecting the rhesus macaque (Figure 3a). (iii) Benefits of immunosuppression were not realized by an observable improvement in transgene expression in the sero-negative animal group. Furthermore, any effects of IS on transgene expression in the sero-positive group were over shadowed by the effects of circulating AAVrh.74 binding antibodies. (iv) Removal of offending binding antibodies with plasmapheresis before vector administration reverses or attenuates the negative effects of circulating binding antibodies to AAVrh.74 capsid on transgene expression.
Of particular interest in considering therapeutic options for future gene transfer studies was whether treatment groups that received immunosuppression would have higher levels of micro-dys expression. Two approaches were used: prednisone alone or an aggressive regimen consisting of triple therapy (prednisone, tacrolimus, and MMF). Neither approach affected micro-dys.FLAG gene expression in AAVrh.74 antibody–positive or AAVrh.74 antibody–negative study groups. In contrast, clear evidence emerged that removal of circulating antibody to AAVrh.74 by plasmapheresis improved gene expression by fourfold, into a range with predictive therapeutic value for gene therapy. Following removal of antibody, gene expression levels were not significantly different from animals without evidence of preexisting AAVrh.74 antibodies, a group we labeled as sero-negative.
We and others have previously reported on the inhibitory effects of preexisting antibodies impeding muscle targeted transgene expression in clinical gene therapy trials10,11,18,19,25 and in NHPs following vascular delivery.15,16 This is highly relevant to clinical gene therapy studies with regard to patients exhibiting preexisting AAV antibodies to AAV and important for patients who may require later readministration of the same or similar vector and transgene following waning efficacy. In our LGMD2D trial, five of the six patients demonstrated robust gene expression.10,11 Patient 6 had no evidence of gene expression, and the only parameter that was unique in this subject was the presence of binding and neutralizing antibodies to the AAV1 vector before treatment. Although the possibility cannot be ruled out that an AAV capsid–specific T-cell response contributed to the lack of expression, several lines of evidence refute this. The capsid T-cell response that was noted in subject 6 was found very early on day 2 after transfer and subsided by day 14. This was significantly earlier than that was seen in any other patient who was considered to have a positive capsid T-cell response and consistent with a memory response. In addition, there was no evidence of MHCI upregulation in the treated muscle of subject 6, whereas the other five subjects who expressed the transgene had upregulation of MHCI. Finally, there was significantly less vector genomes present in the muscle in subject 6 consistent with failure of vector genome entry.
In the current study, we developed an experimental paradigm to explore these potential therapeutic scenarios. We selected the rhesus macaque animal model because of the anatomical similarities between the monkey and human extremities that enabled experimental vascular delivery via isolated focal limb perfusion establishing preclinical safety and possibility of dosing. In addition, rhesus macaques demonstrate a high incidence of anti-AAVrh.74 antibodies representing a potential natural barrier to gene delivery and expression.16 This provided an important rationale for correlating muscle fiber transduction percentages in the current study with total AAVrh.74 antibody titers pre- and postplasmapheresis treatment.
The experimental paradigm chosen for these studies has practical implications for clinical gene therapy studies for the muscular dystrophies. Clinically meaningful results will most likely be the product of widespread vector delivery reaching multiple muscle groups with a high level of gene expression per muscle. Until the numbers of expressing muscle fibers and level of transgene expression are validated, we can only speculate on how much dystrophin or micro-dys will be required to produce a clinically meaningful result. Based on our recent exon 51 skipping clinical trial, we were able to show improved ambulation with a sustained effect beyond 1 year with increase in dystrophin-positive fibers ranging from 43 to 52%; this is a feasible target for initiating clinical gene therapy trials.26 In the study designed here, however, we were less concerned about widespread delivery throughout the extremities and preferred to be highly focused on targeting a single muscle group, the gastrocnemius, enabling consistency in gene expression following vascular delivery. In this way, we could more definitively correlate gene expression to circulating antibody levels as we have shown in this report.
Taken together, these results suggest that meaningful gene transfer may be accomplished in the face of previous capsid exposure. Although discussed in the literature as a potential means to circumvent the deleterious effects of preexisting vector binding antibodies,27 the application of plasmapheresis in a gene transfer study has not been utilized in a systematic study as illustrated here. Plasmapheresis is a process whereby whole blood is removed from the subject, plasma is separated, and the remainders of whole blood constituents are returned with the addition of a replacement fluid—in this study 5% human albumin. Plasmapheresis is indicated in a number of clinical conditions some caused by the acute overproduction of offending antibodies.21 Given our findings of attenuated transgene expression in AAV sero-positive NHPs, we removed the offending antibodies with plasmapheresis and found that subsequent transgene expression was preserved comparable with sero-negative levels over a period of 12 weeks. We used two rounds of plasmapheresis (three plasma exchange volumes per day over 2 days) to achieve AAVrh.74 binding antibody levels approaching those measured in sero-negative animals and performed vector administration immediately following the second plasmapheresis to minimize the inhibitory effects on transgene expression21 due to “antibody rebound”. This phenomenon is caused by two mechanisms. The first is related to sequestering of most IgG in the interstitial space (up to 65%) and not necessarily in the circulation. As the circulation levels of IgG are reduced by plasmapheresis, the sequestered pool acts to replenish these levels. Second, de novo synthesis by B cells is known to replace antibody levels at a rapid rate. Therefore, under some clinical conditions, the use of pharmacologic regulation of B-cell function may influence immunity (or autoimmunity) in the case of diseases like myasthenia gravis or systemic lupus erythematosus.21 We did not find this necessary in the present study as the B-cell response may be too slow in onset to affect transgene expression as delivered in the current protocol.
Based on the evidence presented in this work, it is possible that transient removal of preexisting vector antibodies at the time of vector delivery would be sufficient to lead to long-term transgene expression in the clinical arena, but this will require testing in clinical trials before we can be sure. The practical application of plasmapheresis for patients requiring readministration of vector is an attractive consideration.
Materials and Methods
Animals and treatments. All procedures were approved by The Research Institute at Nationwide Children's Hospital Institutional Animal Care and Use Committee. All NHPs were housed in twos or threes to promote socialization.
Rhesus macaques and serostatus. Vendors provided serum from rhesus macaques before animal purchase for determination of AAVrh.74 binding antibody titers—the vector used throughout this study—that was originally isolated from mesenteric lymph nodes of a rhesus macaque at Nationwide Children's Hospital. Based on AAVrh.74 total antibody serostatus, we separated and housed purchased animals in cohorts of immune-negative (sero-negative) and immune-positive (sero-positive) groups. Animals were categorized as AAV negative with a total antibody titer <1:50 dilution and were considered positive at ≥1:50. On arrival to the Research Institute at Children's Hospital, the animals were bled and serum retested for the presence of AAVrh.74 binding antibodies. Animals that had sero-converted to positive since the original screen were deemed positive and housed accordingly.
Micro-dys.FLAG vector construction. The human micro-dys.FLAG cassette contained the (R4-R23/Δ71–78) domains as previously described, and the FLAG epitope (DYKDDDDK) was added to the C-terminus as a translational fusion by PCR.7 The cDNA was codon optimized for human usage and synthesized by GenScript (Piscataway, NJ). It includes a consensus Kozak sequence, an SV40 intron, and synthetic polyadenylation site (53 bp). An MCK promoter/enhancer (GenBank Accession No. M21390) derived sequence was used to drive muscle-specific gene expression. The promoter was synthesized by GenScript, following derivation from previous work28,29 with some modifications. It is composed of the mouse MCK enhancer (206 bp) fused to the 351 bp MCK promoter (-351-0 MCK). After the promoter, the 53 bp endogenous mouse MCK exon1 (untranslated) was added for efficient transcription initiation. This inclusion has been shown to improve expression with other promoters, including CMV and troponin.30,31 Salva et al.32 have also shown that the addition of 50 bp from MCK exon1 improves expression. MCK exon 1 was followed by the SV40 late 16S/19S splice signals (97 bp) and a small 5′UTR (61 bp). The intron and 5′ UTR are derived from plasmid pCMVβ (Clontech (Takara Bio), Otsu, Japan).9
The MCK micro-dys.FLAG expression cassette was cloned between AAV2 ITRs using flanking Xba I restriction enzyme sites in a plasmid derived from pCMVβ (Clontech). Msc I / Sma I restriction enzyme digestions were used to confirm ITR integrity.
rAAV vector production. rAAV vectors were produced by a modified cross-packaging approach, whereby the AAV type 2 ITRs can be packaged into multiple AAV capsid serotypes.33 Production was accomplished using a standard 3-plasmid DNA CaPO4 precipitation method using HEK293 cells. HEK293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% cosmic calf serum, penicillin, and streptomycin. The production plasmids were as follows: (i) pAAV.MCK.micro-dys.FLAG or pAAV.CMV.eGFP, (ii) rep2-cap8 modified AAV helper plasmids encoding cap serotype isolate rh.74, and (iii) an adenovirus type 5 helper plasmid (pAdhelper) expressing adenovirus E2A, E4 ORF6, and VA I/II RNA genes. A quantitative PCR-based titration method was used to determine an encapsidated vector genome titer utilizing a Prism 7500 Taqman detector system (PE Applied Biosystems (Life Technologies), Grand Island, NY).34 The primer and fluorescent probe targeted the MCK or CMV promoters and were as follows: MCK forward primer, 5-CCC GAG ATG CCT GGT TAT AAT T-3; MCK reverse primer, 5-GCT CAG GCA CAG GTG TTG-3; and MCK probe, 5- FAM-CCA GAC ATG TGG CTG CTC CCC C-TAMRA-3; CMV forward primer, 5- TGG AAA TCC CCG TGA GTC AA-3; CMV reverse primer, 5- CAT GGT GAT GCG GTT TTG G-3; and CMV probe, 5- FAM-CCG CTA TCC ACG CCC ATT GAT G-TAMRA-3.
Immunosuppression regimen and clinical monitoring. Immunosuppressive agents in this study include the following: (i) prednisone (0.75 mg/kg/day), (ii) MMF (50 mg/kg/day), and (iii) tacrolimus (2 mg/kg/day). Dosing started at 2 weeks before vector administration and included two cohorts (prednisone only and triple IS—prednisone, tacrolimus, and MMF). Dosing ended for tacrolimus and MMF at 12 weeks, and prednisone was continued for the full 24 weeks. Tacrolimus blood levels were monitored and found to vary from 2 to 11 ng/ml between animals where the suggested therapeutic range is 5–20 ng/ml.35 MMF levels were not followed due to a lack of commercial laboratory willingness to take on NHP serum in their facility.
T-cell proliferation assay. In a small cohort of NHPs, PBMCs were isolated using a Ficoll gradient, counted, and resuspended in media containing RPMI 1640, 10% fetal calf serum, 50 mU/ml penicillin, and 50 mg/ml streptomycin (P/S). Cells were seeded in triplicate and treated with 10 mg/ml Concanavalin A as a positive control or with superoxide dismutase as a negative control for 2 days in a 37 °C 5% CO2-humidified sterile tissue culture incubator. At the end of the second day, cells were given 1 mCi/well 3H thymidine for 18 hours, after which the cells were washed, harvested, and counted in a scintillation counter.
Isolated focal limb perfusion in rhesus macaques. Rhesus macaques were sedated with intramuscular telazol (3–6 mg/kg), intubated, and secured to a heated procedure table at 37 °C. General anesthesia was administered with isoflurane (in oxygen) 1–4% during the procedure. The left groin was shaved with extension to the mid-thigh and prepped with povidine iodine solution followed by 95% ethanol. A groin incision was made over the femoral bundle, and the femoral artery was isolated. The femoral artery was catheterized using fluoroscopy-guided 3-0 F catheter sheath (Cook Medical, Bloomington, IN). The animal was heparinized with 50 U/kg heparin before the insertion and advancement of a 3-0 F catheter to the sural branch of the popliteal artery. Before vector administration, a prevector flush of saline (2.5 ml/kg) was given over 1 minute. This was immediately followed by occluding blood flow to the extremity using a standard phlebotomy tourniquet placed proximal to the tip of the catheter, which was typically right above the knee. A second tourniquet was placed at the base of the gastrocnemius. Vector was infused over 60 seconds at a dose of 2 × 1012 vg/kg in 2.5 ml/kg of Tris-buffered saline. The extremity remained isolated from the circulation for 10 minutes. A postvector flush (2.5 ml/kg) was infused over 1 minute, and then the tourniquets were released. The catheter was removed and direct pressure was applied to the wound for 10 minutes to control bleeding and the wound was closed with 4-0 vicryl suture.
Gene expression analyses. All animals treated by isolated focal limb perfusion were sacrificed at 3 or 6 months, the time at which the entire gastrocnemius muscle was removed and blocked (0.75–1.0 × 0.5 cm blocks). Muscles were embedded in 7% gum tragacanth and flash frozen in isopentane cooled in liquid nitrogen. Cryostat sections (12 µm) were incubated with anti-FLAG polyclonal primary antibody (Sigma-Aldrich, St Loius, MO; F7425) at a dilution of 1:175 in blocking buffer (phosphate-buffered saline (PBS), 10% goat serum, and 0.1% Triton x-100) for 1 hour at 25 °C in a humid chamber. Sections were then washed three times with PBS, each for 20 minutes and reblocked. Visualization was achieved by incubation for 45 minutes at 25 °C with an Alexa 568 goat anti-rabbit antibody at a 1:300 dilution (Molecular Probes (Life Technologies), Grand Island, NY). Sections were washed three times with PBS for 20 minutes and mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA). Fluorescence staining was visualized using a Zeiss Axioskop2 Plus Microscope, and images were captured with a Zeiss AxioCam MRC5 camera (Carl Zeiss Microscopy, Thornwood, NY). The number of fibers with sarcolemmal staining was expressed as percentage of all fibers. Means were obtained by counting four 10× fields from each muscle block (16–20 blocks per head of gastrocnemius) (Figure 2).
Quantitative PCR to detect genome copy number. Taqman quantitative PCR was used to quantify the number of vector genome copies compared with contralateral control tissue and nontargeted organs as previously described.15,34 A vector-specific primer probe set amplified a portion of the unique sequence of the CMV promoter within the CMV.eGFP cassette. The rhesus erythropoietin gene was used as an internal control to normalize for genomic input and confirm the absence of PCR inhibitors in the sample DNA. To detect the MCK.micro-dys cassette, the following primers were used: uDys 5′ F–GCT CTA AAA GCT GCG GAA TTG TA-3′ and uDys 5′ R-CGT CCT CCC TTT CAT AAC AAT CC-3′, and probe uDys P-FAM/CGG CCG CCA-ZEN-CCA TGC TG-3IABkFQ/ (IDT). Copy number is reported as vector genomes per microgram of genomic DNA.
IFN-γ ELISpot analysis. ELISpot assays were performed on fresh PBMCs, which were added at a concentration of 2 × 105/well in duplicate wells of a 96-well flat-bottom membrane-plate (Millipore, Billerica, MA). Three peptide pools were used for the AAVrh.74 capsid protein (Genemed Synthesis, San Antonio, TX), containing 34–36 peptides, each 18 amino acids long and overlapping by 11 residues. Three peptide pools encompassing the micro-dys.FLAG protein (Genemed Synthesis) were used as previously described15 Concanavalin A (Sigma) served as a positive control and 0.25% dimethylsulfoxide as a negative control. Peptides were added directly to the wells at a final concentration of 1 µg/ml in 200 µl of AIM-HS (Aim-V lymphocyte media (Invitrogen (Life Technologies), Grand Island, NY) supplemented with 2% human AB serum (Gemini-BioScience BLCL medium (RPMI 1640 (Gibco (Life Technologies), Grand Island, NY)) supplemented with 10% fetal bovine serum (Gibco) and Pen Strep(Gibco)). Monkey IFN-γ ELISpot kits were purchased from U-CyTech (Utrecht, The Netherlands). After the addition of PBMCs and peptides, the plates were incubated at 37 °C for 48 hours and then developed according to the manufacturer's protocol. IFN-γ-spot formation was counted using a Cellular Technologies Systems analyzer (Cleveland, OH).
Anti-AAV binding titers. An enzyme-linked immunosorbent assay was performed to measure the level of circulating AAVrh.74 capsid binding antibody in plasma. Briefly, Immulon-4 96-well plates (ISC BioExpress, Kaysville, UT) were coated with 100 µl of 2 × 1010 vg/ml AAVrh.74 viral stock in carbonate buffer (pH 9.4; Pierce, Rockford, IL) per well. Plates were sealed overnight at 4 °C. Plates were blocked with 280 µl per well of a 5% nonfat dry milk and 1% normal goat serum (Invitrogen) in PBS for 3 hours at 25 °C. Rhesus plasma was diluted at a 1:50 ratio in solution identical to the blocking solution and 100 µl added in duplicate to both wells coated with AAVrh.74 particles in carbonate buffer and wells coated with carbonate buffer alone. Plates were incubated at 25 °C for 1 hour before being washed five times with 280 µl of PBS-T (0.05% Tween). Blocking solution was used again to dilute the secondary antibody, goat anti-Monkey IgG-HRP (Sigma) at a 1:10,000 dilution. Wells received 250 µl of the secondary antibody and were incubated at 25 °C for 30 minutes before being washed five times and blotted dry. Tetramethybenzidine (100 µl/well; Pierce) was added and incubated at 25 °C for 10 minutes in the dark, before the addition of 100 µl of 1 N H2S04 (Acros Organics, Geel, Belgium) to stop the reaction. The absorbance at 450 Å was measured using a Wallace 1420-050 Multilabel Counter (Perkin Elmer, Downers Grove, IL). Samples were considered positive if the absorbance at 450 Å average of the antigen-coated wells was three times greater than wells coated with carbonate buffer alone.
Alternatively, the above enzyme-linked immunosorbent assay analysis procedure was followed, but a dilution of 1–800 of rhesus plasma was used on blood drawn at the time of vector administration and subsequently on all remaining timed blood draws (Supplementary Figure S1b). This allowed a more sensitive separation of plasma AAVrh.74 antibody levels for comparison.
Plasmapheresis. On day 1 of plasmapheresis, rhesus macaques were sedated with intramuscular telazol (3–6 mg/kg), intubated and secured to a heated procedure table and anesthetized with isoflurane (in oxygen) 1–4%. An 18-gauge angiocatheter placed in each saphenous vein provided access (blood out) to the plasmapheresis unit and one angiocatheter placed in the cephalic vein provided return (blood in) to the animal. Once access was obtained, the animal was heparinized (50–100 U/kg), the catheter lines were secured and connected to the plasmapheresis unit (COBE Spectra Plasmapheresis System distributed by Terumo BCT, Lakewood, CO) following the manufacturer's recommendations, and then the process was started. The animal was monitored throughout the procedure to maintain an activated clotting time of ~2× the animals baseline value. The animal's whole blood, previously drawn 4 and 2 weeks before plasmapheresis and stored with the anticoagulant acid-citrate-dextrose at 4 °C, was used to prime the unit. Five percent of human albumin was used as the replacement fluid during the exchange on a ml per ml basis with processed plasma. Anticoagulation was managed with a continuous heparin infusion. We monitored and replaced plasma Ca+2 as needed during the procedure. The procedure was calculated to exchange three plasma volumes over ~2 hours. Importantly, the blood column in the Spectra unit was kept quite small to facilitate protein removal over cellular components. On completion of plasmapheresis, the angiocatheters were removed and bleeding controlled by direct pressure. The animals were then extubated, monitored until awake, and returned to their cages. On the following day, the procedure was repeated, but using the 2-week-old blood to prime the circuit. An early observation was made that during the procedure, the animal's core temperature and blood pressures drifted to low levels and were difficult to maintain. This was corrected by providing a low dose epinephrine drip (0.01–0.04 µg/kg/minute) to maintain blood pressures at low normal levels, which stabilized the core temperature as well. On completion of the second day's plasmapheresis of three plasma volumes (six plasma volumes over 2 days), the angiocatheters were removed, pressure was provided to control bleeding, and the animal was then prepared for vector administration to the gastrocnemius as described above.
SUPPLEMENTARY MATERIAL Figure S1. Stratification scheme and experimental timeline for nonhuman primate studies. Figure S2. Boxplot of AAVrh.74 total binding antibody levels 4 weeks before vector dosing. Figure S3. Lymphocyte proliferation assay was performed on peripheral blood mononuclear cells (PBMC's) isolated from 6 Rhesus macaques at baseline and after 6 weeks of daily immunosuppressive regimen. Figure S4. Representative schematic heat map of microdystrophin expression. Figure S5. Micro-dys vector biodistribution. Figure S6. AAVrh.74 binding antibody levels in sero-negative, sero-positive and sero-positive postplasmapheresis animals that were not treated with immune suppression therapy. Table S1. Serial biweekly animal chemistries. Table S2. Enzyme-linked immunosorbant spot assay detecting capsid-specific T cells in PBMCs—transient elevations of spot forming colonies.
Acknowledgments
We thank the Viral Vector Core at Nationwide Children's Hospital for vector production. We thank Maura Lienesch at CaridianBCT for plasmapheresis technical support. We thank Nancy Davis and Spencer Sprinkle for their technical support. This research was supported by the Children's Hospital Foundation (J.R.M.); National Institutes of Health U54 (1U54NS055958-01A1) to J.R.M., L.G.C., K.R.C., Z.S., and C.M.W.; Senator Paul D. Wellstone Muscular Dystrophy Cooperative Research Center Grant (1U54HD066409) from Nationwide Childrens Hospital, Muscular Dystrophy Association to J.R.M.; Jesse's Journey Foundation for Gene and Cell Therapy to J.R.M., L.R.K., L.G.C.; and Ruth L. Kirschstein NRSA postdoctoral fellowship (1F32AR055008) to L.R.K. The authors have nothing to disclose.
Supplementary Material
References
- Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- Mendell JR, Shilling C, Leslie ND, Flanigan KM, al-Dahhak R, Gastier-Foster J, et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. 2012;71:304–313. doi: 10.1002/ana.23528. [DOI] [PubMed] [Google Scholar]
- Ervasti JM. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta. 2007;1772:108–117. doi: 10.1016/j.bbadis.2006.05.010. [DOI] [PubMed] [Google Scholar]
- Watchko J, O'Day T, Wang B, Zhou L, Tang Y, Li J, et al. Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum Gene Ther. 2002;13:1451–1460. doi: 10.1089/10430340260185085. [DOI] [PubMed] [Google Scholar]
- Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L, et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med. 2006;12:787–789. doi: 10.1038/nm1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- Yue Y, Ghosh A, Long C, Bostick B, Smith BF, Kornegay JN, et al. A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs. Mol Ther. 2008;16:1944–1952. doi: 10.1038/mt.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodino-Klapac LR, Janssen PM, Montgomery CL, Coley BD, Chicoine LG, Clark KR, et al. A translational approach for limb vascular delivery of the micro-dystrophin gene without high volume or high pressure for treatment of Duchenne muscular dystrophy. J Transl Med. 2007;5:45. doi: 10.1186/1479-5876-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Rodino-Klapac LR, Rosales XQ, Coley BD, Galloway G, Lewis S, et al. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Ann Neurol. 2010;68:629–638. doi: 10.1002/ana.22251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Rodino-Klapac LR, Rosales-Quintero X, Kota J, Coley BD, Galloway G, et al. Limb-girdle muscular dystrophy type 2D gene therapy restores alpha-sarcoglycan and associated proteins. Ann Neurol. 2009;66:290–297. doi: 10.1002/ana.21732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, et al. Dystrophin immunity in Duchenne's muscular dystrophy. N Engl J Med. 2010;363:1429–1437. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin PT, Xu R, Rodino-Klapac LR, Oglesbay E, Camboni M, Montgomery CL, et al. Overexpression of Galgt2 in skeletal muscle prevents injury resulting from eccentric contractions in both mdx and wild-type mice. Am J Physiol, Cell Physiol. 2009;296:C476–C488. doi: 10.1152/ajpcell.00456.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodino-Klapac LR, Montgomery CL, Mendell JR, Chicoine LG. AAV-mediated gene therapy to the isolated limb in rhesus macaques. Methods Mol Biol. 2011;709:287–298. doi: 10.1007/978-1-61737-982-6_19. [DOI] [PubMed] [Google Scholar]
- Rodino-Klapac LR, Montgomery CL, Bremer WG, Shontz KM, Malik V, Davis N, et al. Persistent expression of FLAG-tagged micro dystrophin in nonhuman primates following intramuscular and vascular delivery. Mol Ther. 2010;18:109–117. doi: 10.1038/mt.2009.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlbut GD, Ziegler RJ, Nietupski JB, Foley JW, Woodworth LA, Meyers E, et al. Preexisting immunity and low expression in primates highlight translational challenges for liver-directed AAV8-mediated gene therapy. Mol Ther. 2010;18:1983–1994. doi: 10.1038/mt.2010.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manno CS, Chew AJ, Hutchison S, Larson PJ, Herzog RW, Arruda VR, et al. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood. 2003;101:2963–2972. doi: 10.1182/blood-2002-10-3296. [DOI] [PubMed] [Google Scholar]
- Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- Flotte TR, Trapnell BC, Humphries M, Carey B, Calcedo R, Rouhani F, et al. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing a1-antitrypsin: interim results. Hum Gene Ther. 2011;22:1239–1247. doi: 10.1089/hum.2011.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese I, Chaudhry V, So YT, Cantor F, Cornblath DR, Rae-Grant A. Evidence-based guideline update: plasmapheresis in neurologic disorders: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2011;76:294–300. doi: 10.1212/WNL.0b013e318207b1f6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Calcedo R, Vandenberghe LH, Figueredo JM, Wilson JM. Impact of preexisting vector immunity on the efficacy of adeno-associated virus-based HIV-1 Gag vaccines. Hum Gene Ther. 2008;19:663–669. doi: 10.1089/hum.2008.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Moxley RT, Griggs RC, Brooke MH, Fenichel GM, Miller JP, et al. Randomized, double-blind six-month trial of prednisone in Duchenne's muscular dystrophy. N Engl J Med. 1989;320:1592–1597. doi: 10.1056/NEJM198906153202405. [DOI] [PubMed] [Google Scholar]
- Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet. 2011;12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- Mendell J, Rodino-Klapac LR, Sahenk Z, Roush K, Bird L, Lowes LP, et al. 2013Eteplirsen for the treatment of duchenne muscular dystrophy. Ann Neurolepub ahead of print). [DOI] [PubMed]
- Monteilhet V, Saheb S, Boutin S, Leborgne C, Veron P, Montus MF, et al. A 10 patient case report on the impact of plasmapheresis upon neutralizing factors against adeno-associated virus (AAV) types 1, 2, 6, and 8. Mol Ther. 2011;19:2084–2091. doi: 10.1038/mt.2011.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shield MA, Haugen HS, Clegg CH, Hauschka SD. E-box sites and a proximal regulatory region of the muscle creatine kinase gene differentially regulate expression in diverse skeletal muscles and cardiac muscle of transgenic mice. Mol Cell Biol. 1996;16:5058–5068. doi: 10.1128/mcb.16.9.5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa K, Sakamoto M, Miyagoe-Suzuki Y, Tanouchi A, Yamamoto H, Li J, et al. Adeno-associated virus vector-mediated gene transfer into dystrophin-deficient skeletal muscles evokes enhanced immune response against the transgene product. Gene Ther. 2002;9:1576–1588. doi: 10.1038/sj.gt.3301829. [DOI] [PubMed] [Google Scholar]
- Nikovits W, Jr, Mar JH, Ordahl CP. Muscle-specific activity of the skeletal troponin I promoter requires interaction between upstream regulatory sequences and elements contained within the first transcribed exon. Mol Cell Biol. 1990;10:3468–3482. doi: 10.1128/mcb.10.7.3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simari RD, Yang ZY, Ling X, Stephan D, Perkins ND, Nabel GJ, et al. Requirements for enhanced transgene expression by untranslated sequences from the human cytomegalovirus immediate-early gene. Mol Med. 1998;4:700–706. [PMC free article] [PubMed] [Google Scholar]
- Salva MZ, Himeda CL, Tai PW, Nishiuchi E, Gregorevic P, Allen JM, et al. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol Ther. 2007;15:320–329. doi: 10.1038/sj.mt.6300027. [DOI] [PubMed] [Google Scholar]
- Rabinowitz JE, Rolling F, Li C, Conrath H, Xiao W, Xiao X, et al. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol. 2002;76:791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark KR, Liu X, McGrath JP, Johnson PR. Highly purified recombinant adeno-associated virus vectors are biologically active and free of detectable helper and wild-type viruses. Hum Gene Ther. 1999;10:1031–1039. doi: 10.1089/10430349950018427. [DOI] [PubMed] [Google Scholar]
- McMaster P, Mirza DF, Ismail T, Vennarecci G, Patapis P, Mayer AD. Therapeutic drug monitoring of tacrolimus in clinical transplantation. Ther Drug Monit. 1995;17:602–605. doi: 10.1097/00007691-199512000-00010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.