

Abstract
Gene therapy for neuropathic pain requires efficient gene delivery to both central and peripheral nervous systems. We previously showed that an adenoassociated virus serotype 9 (AAV9) vector expressing short-hairpin RNA (shRNA) could suppress target molecule expression in the dorsal root ganglia (DRG) and spinal cord upon intrathecal injection. To evaluate the therapeutic potential of this approach, we constructed an AAV9 vector encoding shRNA against vanilloid receptor 1 (TRPV1), which is an important target gene for acute pain, but its role in chronic neuropathic pain remains unclear. We intrathecally injected it into the subarachnoid space at the upper lumbar spine of mice 3 weeks after spared nerve injury (SNI). Delivered shTRPV1 effectively suppressed mRNA and protein expression of TRPV1 in the DRG and spinal cord, and it attenuated nerve injury-induced thermal allodynia 10–28 days after treatment. Our study provides important evidence for the contribution of TRPV1 to thermal hypersensitivity in neuropathic pain and thus establishes intrathecal AAV9-mediated gene delivery as an investigative and potentially therapeutic platform for the nervous system.
Introduction
Neuropathic pain following peripheral nerve injury is characterized by a loss of afferent sensory function as well as hyperalgesia and allodynia,1 and available treatments are insufficient because the underlying mechanisms are poorly understood. Furthermore, a problem associated with centrally acting drugs is that they target receptors/channels that are widely expressed and are thus associated with a higher risk of adverse effects such as sedation, dizziness, somnolence, or loss of cognitive function.2,3,4
An alternative strategy for developing novel analgesics is to target receptors and ion channels that respond to noxious stimuli at peripheral terminals of primary sensory neurons, whose cell bodies are located in the dorsal root ganglia (DRG), to produce hyperexcitability and spontaneous firing in neuropathic pain. These receptors and ion channels are selectively or primarily expressed in primary sensory neurons in the periphery,5 which reduces the potential for side effects when they are specifically targeted by drugs. As a treatment target, vanilloid receptor 1(TRPV1) has been well characterized as a sensory transducer in nociceptors and shown to contribute to the development of mechanical and/or heat hypersensitivity under various acute pain conditions.6 However, how TRPV1 contributes to chronic hypersensitivity induced by nerve injury (i.e., neuropathic pain) remains unclear, although various TRPV1 antagonists are under clinical development based on the hypothesis that TRPV1 is involved in persistent pain, especially in thermal hyperalgesia in inflammatory pain states.7 Further development of clinical therapeutics will be therefore facilitated by the elucidation of the role of TRPV1 in neuropathic pain; however, such an investigation first requires the development of a technology to reliably and specifically modulate TRPV1 activity in vivo in the peripheral nervous system/central nervous system (CNS).
RNA interference has emerged as a potent tool to investigate the specific effects of proteins by suppressing their expression, and recent work in the field of drug delivery has addressed the delivery of synthetic short interfering RNA (siRNA) across the blood–brain barrier. Nonviral systems including hydrodynamic injection, cationic liposomes,8 nanoparticles,9 and lipid conjugation10 provide effective and relatively safe delivery of nucleic acids to the CNS. In comparison with viral vectors, however, they still show low transduction efficiency, insufficient inhibition of gene expression and a short duration of therapeutic effects; therefore, viral vectors may be preferable for treating chronic pain states. In particular, viral delivery of short-hairpin RNA (shRNA) expression cassettes that support efficient and long-lasting RNA interference is expected open new frontiers for molecular therapy of the CNS.11 We previously reported that intrathecal administration of shRNA expressed by an adenoassociated virus serotype 9 (AAV9) vector produced long-term suppression of target gene expression in both the DRG and spinal cord without neurologic side effects or toxicity,12 which suggests that this approach can be used to modulate the expression of genes involved in neuropathic pain states. Thus, in the present study, we used intrathecal administration of AAV9 encoding shRNA targeting TRPV1 in a mouse model of spared nerve injury (SNI) to test the hypothesis that TRPV1 plays a critical role in neuropathic pain.
Results
Design, selection, and validation of siRNA sequences against TRPV1 for construction of AAV9 shTRPV1
Seven candidate siRNA sequences for TRPV1 were designed from the TRPV1 coding sequence (NM_001001445.1, Figure 1a). The silencing efficacy of these sequences was determined using a luciferase assay in vitro as described below. Subsequently, we selected the one siRNA sequence out of seven candidates that had the highest efficiency, and then two sequences were designed for inclusion in the shRNA expression vector (Figure 1b).
Figure 1.

Selection of siRNA-E sequence for insertion into the AAV9 vector. (a) Small interfering RNA sequences targeting TRPV1. (b) Short-hairpin RNA (shRNA) sequences targeting TRPV1. (c) Schematic representation of the plasmid vector used in vitro. This plasmid encodes firefly luciferase and a fusion renilla luciferase and TRPV1. (d) Relative TRPV1 expression level in vitro as determined by renilla/firefly ratio. Data are presented as means ± SEM. (e) The AAV9-shTRPV1 vector, including the shRNA sequence driven by the human polymerase III human U6 promoter and hGH inserted downstream of the shRNA sequence for the AAV9 genome detection assay, is shown. AAV, adenoassociated virus; hGH, human growth hormone; HSV-TK, herpes simplex virus-thymidine kinase; ITR, inverted terminal repeat; SC, scramble; SV, Simian virus; TRPV1, transient receptor potential vanilloid 1.
The selected seven siRNA and two shRNA molecules were tested individually for knockdown efficiency in cultured human embryonic kidney (HEK)293T cells that were transfected with a plasmid vector encoding of renilla luciferase with TRPV1 in its 3′ untranslated region as well as firefly luciferase as a transfection control (Figure 1c). Among the seven siRNA sequences, siRNA-6 reduced renilla luciferase activity (used as a proxy for TRPV1 expression) by ~90% relative to firefly luciferase activity, whereas the other sequences reduced renilla luciferase activity by ~70–80%. Renilla luciferase activity was not significantly altered by transfection with siRNA containing a scrambled sequence. Two putative shRNA sequences were therefore designed based on the siRNA-6 sequence. Between the two shRNA sequences, shRNA-A was more effective, as it reduced renilla luciferase activity by ~95% relative to firefly luciferase activity (Figure 1d) and was thus selected for further experiments and inserted downstream of the human polymerase III U6 promoter in the AAV9 vector (Figure 1e).
TRPV1 mRNA expression is significantly increased in the lumbar DRG and spinal cord following SNI
We first confirmed the development of hypersensitivity in the hindpaw of SNI mice (Figure 2a).13 Behavioral testing was performed on the lateral side of the plantar hindpaw, which is completely innervated by the spared sural nerve.
Figure 2.

Time course of mechanical, cold, and heat sensitivity following spared nerve injury. (a) Schematic representation of the spared nerve injury model showing ligation of the tibial and common peroneal nerves distal to the trifurcation. Spared nerve injury resulted in immediate mechanical (b) and cold (c) hypersensitivity. Latencies to heat stimulation at 50 and 55 °C decreased gradually and were significantly lower at 3 weeks after nerve injury than at (d and e) baseline. (f) Quantitative reverse transcription polymerase chain reaction analysis showed that trpv1 expression at 3 weeks after nerve injury was 179 ± 23% (P < 0.05) in the DRG and 147 ± 10% (P < 0.05) in the spinal cord relative to tissues from uninjured mice. Data are presented as means ± SEM (n = 6–18 mice per group; P < 0.05). trpv1, transient receptor potential vanilloid 1.
Within 3 weeks after nerve injury, the mice exhibited tactile hyperalgesia in response to probing by Von Frey filaments, cold hyperalgesia in response to acetone, and thermal hyperalgesia in response to contact with a hotplate (50 or 55 °C; Figure 2b–e). The behavioral hypersensitivity lasted until 8 weeks after SNI. However, there was no decrease in the latency of withdrawal to radiant heat stimulation, as reported previously (data not shown).13
The expression of trpv1 was investigated in the ipsilateral L5 and L6 DRGs and the ipsilateral L1-L2 spinal cord (which receives input from the L5 and L6 spinal nerves), before and after SNI by quantitative reverse transcription polymerase chain reaction (RT-PCR) analysis. Three weeks after SNI, the relative trpv1 expression as a percentage of gapdh expression was 179 ± 23% in the DRG and 147 ± 10% in the spinal cord relative to tissues from uninjured mice (Figure 2f), which indicates that trpv1 was upregulated by SNI.
Thermal analgesic effect of AAV9 encoding shRNA against TRPV1 in mice with SNI
AAV9 vectors targeting TRPV1 (AAV9-shTRPV1) or superoxide dismutase 1 (AAV9-shSOD1), or phosphate-buffered saline (PBS) in the vehicle-only group, were injected intrathecally at the second lumbar vertebra of mice 3 weeks after SNI (Figure 3a). Because we previously demonstrated that intrathecal injection of AAV9-shSOD1 did not produce behavioral changes in mice,14 AAV9-shSOD1 was used as a negative control vector in this study. To analyze the distribution of AAV9 in the DRG or spinal cord after intrathecal injection, we performed genomic PCR to detect human growth hormone cassette (Figure 1e), whose sequence had been inserted into the AAV9 genome. Based on the detection of the human growth hormone sequence, higher levels of AAV9 genomic DNA were present in the upper thoracic and lumbar DRGs and lower thoracic and lumbar spinal cord than in other regions (Figure 3b).
Figure 3.
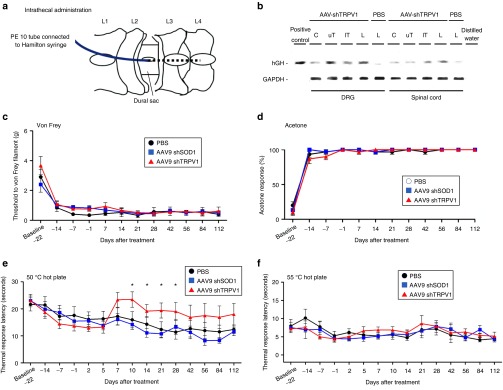
TRPV1 knockdown via intrathecal delivery of AAV9-shTRPV1 partially attenuates spared nerve injury (SNI)-induced thermal hyperalgesia. (a) Three weeks after injury, a PE-10 catheter was inserted into the subarachnoid space caudally between the L2 and L3 vertebrae following fenestration, and AAV9 or phosphate-buffered saline (PBS) was injected using a Hamilton syringe connected to the catheter. (b) One week after intrathecal injection of AAV9 at the lumbar spine level, high levels of AAV9 genomic DNA were detected in the upper thoracic and lumbar DRGs and lower thoracic and lumbar spinal cord, whereas low expression was observed at other levels. (c) Withdrawal threshold for von Frey filaments, (d) response to acetone application and latency of response to (e) 50 °C and (f) 55°C heat stimulation. No significant differences were observed among the three groups for withdrawal thresholds in response to (c) mechanical, (d) cold, and (f) 55 °C heat stimulation. (e) However, a partial but significant attenuation of thermal hyperalgesia in response to 50 °C heat stimulation was observed for animals injected with AAV9-shTRPV1 from day 10 to day 28 relative to the other two groups. Data are represented as means ± SEM (n = 9 mice per group; P < 0.05). AAV, adenoassociated virus; Cs, cervical; lTs, lower thoracic; Ls, lumbar; PBS, phosphate-buffered saline; SOD1, superoxide dismutase 1; TRPV1, transient receptor potential vanilloid 1; uTs, upper thoracic.
To determine whether the knockdown of TRPV1 alleviated neuropathy-induced hindpaw hypersensitivity, the Von Frey, acetone, and hot plate assays were performed at various time points after intrathecal injection of the AAV9 vector. The paw withdrawal thresholds in response to mechanical (Figure 3c), cold (Figure 3d) and 55 °C thermal stimuli (Figure 3f) showed no obvious change in each group after treatment, and there were no significant differences among the three groups except for in the response to thermal stimulation at 50 °C. In the 50 °C thermal hot plate test, the latency to hindpaw withdrawal in the AAV9-shTRPV1 group was significantly increased from day 10 to day 28 after treatment, relative to the other two groups (Figure 3e).
TRPV1 expression in the spinal cord and DRG, and treatment efficacy after lumbar intrathecal administration of AAV9 shRNA against TRPV1
Quantitative RT-PCR analysis showed that the expression of trpv1 in the AAV9-shTRPV1-treated group was ~55% lower in the DRGs and 95% lower in the lumbar spinal cord relative to PBS-treated animals at 4 weeks after injection (Figure 4a). The knockdown effect appeared to be specific for the target gene trpv1, given that the mRNA levels of gapdh and transthyretin (ttr) did not change (Figure 4b). Furthermore, a western blot analysis confirmed significantly decreased TRPV1 protein levels in the lumbar DRGs and spinal cord of AAV9-shTRPV1-treated mice at the same time point (Figure 4c).
Figure 4.

Intrathecal administration of AAV9-shTRPV1 specifically suppresses trpv1 mRNA and TRPV1 protein expression in vivo. (a) Quantitative reverse transcription polymerase chain reaction (RT-PCR) analysis of trpv1 mRNA expression in lumbar dorsal root ganglia (DRG) and spinal cord in mice 4 weeks after intrathecal injection of phosphate-buffered saline (PBS) (control), AAV9-shSOD1 or AAV9-shTRPV1. trpv1 mRNA expression was significantly (*P < 0.05) inhibited in the lumbar DRG and spinal cord of the AAV9-shTRPV1-treated group relative to the two other groups. (b) Quantitative RT-PCR analysis of the expression of non-targeted ttr and gapdh mRNA in the lumbar DRG and spinal cord at 4 weeks after the treatment. No significant differences were found in the expression of ttr and gapdh mRNA among the three groups. Data are presented as means ± SEM (n = 4 or 5 mice per group; *P < 0.05). (c) TRPV1 protein levels in the lumbar DRG and spinal cord of two mice from each group, as assessed by Western blot analysis 4 weeks after treatment. A robust reduction of TRPV1 protein levels in both the DRG and spinal cord was observed in the AAV9-shTRPV1-treated group. (d) and (e) Relationship between thermal analgesia and trpv1 mRNA expression in the lumbar DRG and spinal cord. Analgesia in response to 50 °C thermal stimulation was significantly correlated with trpv1 mRNA expression in the lumbar DRG (Y = −36.64X+32.70; R2: 0.94, P < 0.01) and spinal cord (e, Y = −17.2X+11.91; R2: 0.79, P < 0.01).
To evaluate the efficacy of the AAV9-shTRPV1 treatment, we analyzed the relationship between the analgesic effect and the expression of trpv1 in the AAV9-shTRPV1 group. There was a significant correlation between the analgesic effect and trpv1 expression in the lumbar DRGs (Figure 4d; Y = −36.64X+32.70; R2: 0.94, P < 0.01) and spinal cord (Figure 4e; Y = −17.2X+11.91; R2: 0.79, P < 0.01).
Duration of silencing by shRNA against TRPV1 in the lumbar DRGs and spinal cord
We evaluated the duration of TRPV1 suppression by the AAV9-shTRPV1 vector by measuring the mRNA and protein levels of TRPV1 in the lumbar DRG and spinal cord. Quantitative PCR showed that trpv1 was reduced by 55 and 95% at 28 days, 36 and 5% at 56 days, and 37 and 0% at 84 days, respectively (Figure 5a). TRPV1 protein expression in both the lumbar DRGs and spinal cord was obviously inhibited at 28 days but returned to the baseline by 56 days after AAV9-shRNA injection (Figure 5b). These results indicate that the AAV9-shTRPV1 vector provides significant suppression of trpv1 mRNA and protein expression in the DRG and spinal cord for at least 1 month, but trpv1 mRNA and protein expression in the spinal cord return to baseline levels within by 2 months.
Figure 5.

Duration of shRNA-mediated silencing of TRPV1 in the lumbar dorsal root ganglia (DRGs) and spinal cord. (a) Significant suppression of trpv1 mRNA lasted for 3 months with a reduction of ~35% in the lumbar DRG. In lumbar spinal cord, a 95% reduction was observed at 1 month after injection, but trpv1 mRNA expression returned to the level observed in phosphate-buffered saline (PBS)-treated mice by 2 months after injection (* denotes significant difference in the DRG trpv1 mRNA expression, P < 0.05; ** denotes significant difference in spinal cord trpv1 mRNA expression, P < 0.05). (b) TRPV1 protein expression analysis based on western blot. TRPV1 protein expression in both the lumbar DRGs and spinal cord was obviously decreased at 28 days after AAV9-shTRPV1 injection but returned to the baseline by 56 days after injection. AAV, adenoassociated virus; P, PBS-treated mice; A, AAV9-shTRPV1-treated mice; TRPV1, transient receptor potential vanilloid 1.
Body weight and laboratory data
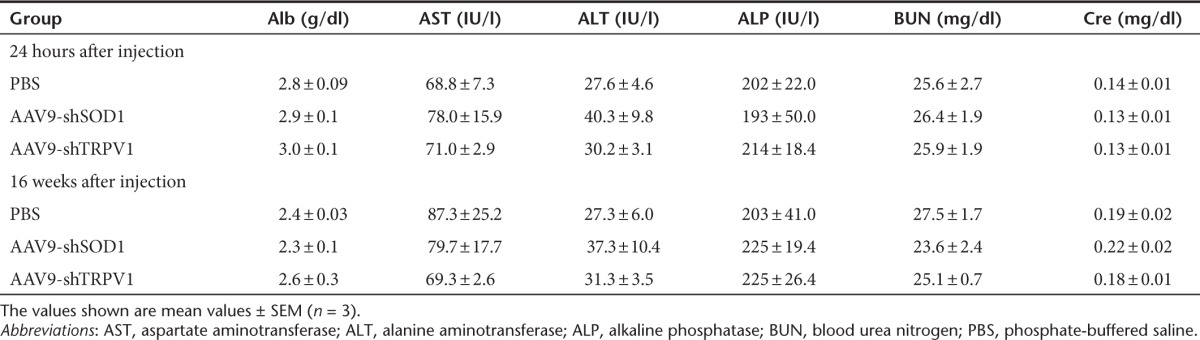
To assess the general health of our mice after the therapeutic injection, we observed them for 16 weeks after treatment. The body weight was similar among the three groups (Figure 6a). In addition, biochemical analysis of serum components including albumin, aminotransaminases, alkaline phosphatase, blood urea nitrogen, and creatinine did not show any marked abnormalities at both 24 hours and 16 weeks after AAV9 injection (Table 1).
Figure 6.

Assessment of body weight, inflammation, and immune response in phosphate-buffered saline (PBS), AAV9-shSOD1, and AAV9-shTRPV1-treated mice. (a) Throughout the 16-week experimental period, the body weight remained similar in the three treatment groups. Data are presented as means ± SEM (n = 9 mice per group). (b) Quantitative reverse transcription polymerase chain reaction analysis of ifnβ mRNA expression in lumbar dorsal root ganglia (DRG) and spinal cord in mice at 24 hours after intrathecal injection of PBS, AAV9-shSOD1, or AAV9-shTRPV1. There were no significant increases in ifnβ mRNA expression in the lumbar DRS and spinal cord of AAV9-shTRPV1-treated mice relative to PBS-treated mice. (c) Similarly, ifnβ mRNA expression after repeated injection of AAV9-shTRPV1 did not increase relative to that in PBS- or AAV9-shSOD1- injected mice. (d–o) Immunohistochemistry of CD8 expression in the lumbar DRG and spinal cord after either single (d–i) or repeated injection (j–o) of PBS, AAV9-shSOD1, or AAV9-shTRPV1. Few or no CD8+ cells were observed in the lumbar DRG or spinal cord after either single or repeated administration of AAV9-shTRPV1. Bar = 20 µm for d–f and j–l; bar = 100 µm for g–i and m–o.
Table 1. Laboratory data in each group at 24 hours and 16 weeks after injection.
Immune response to AAV9-shTRPV1
To evaluate potential inflammatory reactions in the lumbar DRGs and spinal cord after intrathecal injection of the AAV9-shTRPV1 vector, we measured the expression of interferon using quantitative RT-PCR 24 hours after injection. Injection of AAV9-shTRPV1 did not result in increased expression of interferon β (ifnβ) mRNA relative to the PBS-treated group in either DRG or spinal cord (Figure 6b). To further evaluate potential immune responses to repeat injection of AAV9-shTRPV1, ifnβ mRNA was also measured 24 hours after the second intrathecal injection, which was performed 28 days after the first injection. The repeated injection did not lead to a significant increase in ifnβ mRNA levels in either the lumbar DRGs or spinal cord (Figure 6c). In addition, we evaluated CD8 expression in the DRG and spinal cord to determine whether a cellular immune response to AAV9-shTRPV1 occurs after single or repeated injection. Immunohistochemistry indicated a lack of CD8+ cells in the lumbar DRG and spinal cord after either single administration (Figure 6d–i) or repeat administration of either AAV9-shTRPV1 or AAV9-shSOD1 (Figure 6j–o).
Discussion
It is well known that several members of the transient receptor potential (TRP) family function as sensory transducers in nociceptive neurons and play important roles in the development of pathological hypersensitivity under various pain conditions. In particular, TRPV1, which is activated by various exogenous stimuli such as capsaicin, heat, protons, and spider toxins, is expressed in most small-diameter sensory fibers, i.e., unmyelinated C fibers and finely myelinated Aδ fibers, whose physiological role is to detect noxious stimuli.14 Several inflammatory conditions increase the expression of TRPV1 and even increase the density of TRPV1-positive axons.15 Conversely, in neuropathic pain states, the role of TRPV1 is controversial, even though analgesic effects of TRPV1 antagonists have been reported. Although the total expression level of TRPV1 in the DRG tends to decrease in most neuropathic pain models, e.g., chronic constriction injury,16 partial nerve ligation,17 and spinal nerve ligation,18 TRPV1 is upregulated in surviving DRG somata after partial nerve ligation17 and L4 DRG neurons after L5 spinal nerve ligation surgery.19 In human neuropathic pain states, TRPV1 expression increases in spared peripheral nerves after injury.20
In the present study, we found that trpv1 mRNA expression increased by 79 and 47% in the lumbar DRGs and spinal cord, respectively, at 3 weeks after SNI relative to the uninjured condition (Figure 2f). Our observations are consistent with previous findings that activation of TRPV1 in spared DRG neurons after nerve injury results in hyperalgesia.21 In addition to the peripheral axons of primary neurons, TRPV1 is also found in the central terminals of nociceptive neurons that synapse on neurons in the dorsal horn of the spinal cord.22 Activation of presynaptic TRPV1 in the dorsal horn by peripheral nerve injury results in increased synaptic release of glutamate and neuropeptides.23,24 Furthermore, postsynaptic TRPV1 in GABAergic spinal interneurons mediates neuropathic mechanical allodynia and disinhibition of nociceptive circuitry including spinothalamic projections to the ventral posterior lateral nucleus of the thalamus.25 Because neuropathic TRPV1 upregulation occurs in both the DRG and spinal cord, downregulation of TRPV1 in the peripheral nervous system and CNS may provide effective treatment of persistent pain. In the present study, intrathecal AAV9-mediated delivery of shRNA provided significant silencing of TRPV1 not only in the peripheral nervous system but also in the CNS, thereby alleviating nerve injury-induced hyperalgesia in a mouse model of peripheral neuropathic pain.
The excitation, desensitization, and neurotoxicity produced by capsaicin, which is a ligand for the TRPV1 receptor, are known to have analgesic effects. Additionally, systemic administration of resiniferatoxin, which is an ultrapotent TRPV1 agonist, to adult rats desensitizes TRPV1-positive fibers to produce long-lasting relief of thermal hyperalgesia in a neuropathic pain model.26 However, it has also been shown that genetic knockout of TRPV1 does not affect hypersensitivity following nerve injury,27 even though TRPV1 antagonists have been shown to reduce hypersensitivity in neuropathic pain models.28 Accordingly, given the conflicting findings in the literature and lack of consensus in the field, we pursued the AAV-mediated knockdown of TRPV1 via RNA interference to confirm the contribution of TRPV1 to neuropathic pain behavior, particularly pain produced by noxious heat and also to develop a platform for potential therapeutic intervention.
Several previous studies reported that RNAi-mediated suppression of TRPV1 in sensory afferent components including the DRG and spinal cord alleviated both mechanical and thermal hyperalgesia in animal models of neuropathic pain;25,29 however, the present study showed that specific knockdown of TRPV1 reversed thermal hyperalgesia but did not affect mechanical or cold hyperalgesia. The discrepancy between our findings and those of previous studies may be the result of differences in the nerve injury models, as different animal models of neuropathic pain have been found to yield different degrees of hypersensitivity.30 Our findings suggest that tactile and cold hypersensitivity are mediated by molecules other than TRPV1, which is logical given the specific physiological role of TRPV1 as a heat sensor.27
In contrast to the improvement observed at 50 °C, we found that AAV9-shTRPV1 injected intrathecally did not ameliorate thermal hyperalgesia at 55 °C, possibly because the response at 55 °C may rely on a different population of ion channels. Consistent with these findings, TRPV1 has been shown to be essential for the development of sensitization to thermal stimuli during inflammation but not for the normal sensation of noxious heat.31 The detection of noxious heat has been proposed to be mediated by several ion channels in addition to TRPV1, including TRPV2, TRPV3, TRPV4, TRP melastatin 2 (TRPM2), and TRPM3.32,33,34,35,36 Heat-evoked TRPV1 currents in vitro are activated at a threshold of >40 °C, increase linearly up to 50 °C, and decrease at temperatures higher than 50 °C. Although only a few receptors other than TRPV1, such as TRPM3 and ANO1,36,37 contribute to heat sensation in the temperature range of 43–50 °C, even a partial reduction of TRPV1 expression would be expected to decrease the nocifensive response to heat in this temperature range. However, it remains unclear what molecules are involved in heat sensation, thermal nociception and hyperalgesia at temperatures higher than 50 °C, although it is possible that TRPV1 plays a minimal role because TRPV1-independent inflammatory hyperalgesia at 56 °C was previously observed in rats treated with resiniferatoxin and shown to be mediated by TRPV2.38 Although that study and others27showed that TRPV2 was activated by noxious heat at temperatures higher than 50 °C in vivo, TRPV2 was recently shown not to be essential for heat hypersensitivity in a study using TRPV2 knockout mice.39 Other possible candidate channels include TRPV3 and TRPM3, which were individually shown in knockout mice to contribute to thermal hypersensitivity at >52 °C but not 50 °C.36,40 Thus, our findings shed light on the currently controversial role of TRPV1 in neuropathic pain by demonstrating that nerve injury-induced overexpression of TRPV1 indeed plays an important role in persistent pain but only in thermal hyperalgesia in response to moderate heat. Our findings also indicate that a single administration of AAV9-shTRPV1 leads to robust and relatively long-lasting suppression of TRPV1 in SNI mice (in which TRPV1 expression is significantly increased), thus resulting in a significant antinociceptive effect.
Gene therapy using AAV vectors has been increasingly applied to treat refractory neurologic disorders, and human clinical trials have suggested that recombinant AAV vectors are safe and well tolerated.41,42,43 We previously reported that intrathecal delivery of AAV9 in mice was associated with little or no immunogenicity, inflammation or toxicity in terms of neurologic dysfunction or micro-RNA maturation.12 In the current study, the interferon-β mRNA expression profile and lack of CD8+ inflammatory cells suggests a relative lack of inflammation in the DRGs and spinal cord after either a single or repeated intrathecal injection of the AAV9-shTRPV1 vector. This finding is consistent with previous reports in which AAV9 was reported to lower immunoreactivity than other AAV serotypes.44 However, the lack of an immune response in mice does not necessarily exclude the potential for immunogenicity in humans. Therefore, future investigations of the immune response to AAV9 are necessary in primates to validate the safety of the AAV9 vector for human clinical trials, and it may be necessary to engineer capsid sequences to prevent potential immune responses.45
In the present study, we did not observe adverse side effects on liver function or deterioration of the general condition after viral vector injection. However, in situations where side effects of shRNA expression or gene suppression are suspected or expected, strategies for tuning shRNA expression, such as the use of a tetracycline-inducible promoter for regulated expression, may be applied to increase safety.46 Such approaches allow expression of the shRNA to be switched on temporarily through the systemic addition of doxycycline and switched off when necessary (e.g., when the silencing effect is excessive or shRNA toxicity is observed) by withdrawal of doxycycline. Thus, the silencing effect on the target gene after intrathecal injection of shRNA-AAV9 may be adjusted on demand through the use of an inducible promoter. In the present study, we did not assess the therapeutic window of the shRNA encoded by the viral vector, although we confirmed that the gene-silencing effect in the DRG and spinal cord persisted for as long as 4 weeks in our mice; we therefore expect that this technique could be used for stable, long-term analgesia. Given our demonstration of AAV9's high affinity for central and peripheral nervous systems, effective transduction to drive long-term stable expression of transgenes in the DRG and spinal cord and apparent lack of systemic immunogenicity, we propose that intrathecal administration of AAV9 can be developed for clinical application to various neurologic disorders such as neurodegenerative diseases and cancer pain as well as neuropathic pain after nerve injury.
Although further studies are needed for optimization, our findings therefore establish that TRPV1 overexpression is responsible for a subset of thermal hyperalgesia in neuropathic pain that can be reversed by minimally invasive intrathecal AAV9-mediated RNA interference. Moreover, we believe that this technique can be developed as a platform technology to treat various other neuropathic pain conditions by modifying the shRNA sequence to target other molecules, including neurotrophic factors (e.g., nerve growth factor, neurotrophin-3, or brain-derived neurotrophic factor), inflammatory cytokines (e.g., tumor necrosis factor, interferon, or interleukin), ion channels (e.g., voltage-gated sodium channels and calcium channels), and neurotransmitters (e.g., calcitonin gene-related peptide or substance P), which may prove to be more attractive therapeutic targets than TRPV1 in the treatment of neuropathic pain. The present study thus demonstrates the power of intrathecal AAV-mediated gene transfer as an investigative and potentially therapeutic tool in the nervous system.
Materials and Methods
Plasmid construct. The TRPV1 coding sequence was subcloned in-frame from a TRPV1 plasmid, which was a kind gift from Dr. Michael Xi Zhu, into the HinDIII-XbaI sites of the pBluescriptII KS (+) vector (Stratagene, La Jolla, CA). The pBluescriptII KS (+)-TRPV1 product was then subcloned in-frame into the XhoI sites of psiCHECK-2 (Promega, Madison, WC). We confirmed based on quantitative PCR and a luciferase assay that HEK293T cells stably expressed both dual luciferase and TRPV1 after transfection of the psiCHECK-2 TRPV1 plasmid (Figure 1c).
Cell culture and dual luciferase assay for selecting the shRNA sequence. HEK293T cells were transfected with both the psiCHECK-2 vector encoding TRPV1 and each siRNA sequence using Lipofectamine Plus reagent (Life Technologies, Rockville, MD) in Opti-MEM (Invitrogen, Carlsbad, CA). After 48 hours, cells were maintained in D-MEM (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), penicillin (100 units/ml) and streptomycin (100 µg/ml; Invitrogen). Seven siRNA sequences targeting TRPV1 were designed: siRNA-1 (nucleotide 604–624), siRNA-2 (nucleotide 1,229–1,249), siRNA-3 (nucleotide 1,295–1,315), siRNA-5 (nucleotide 1,727–1,747), siRNA-6 (nucleotide 1,734–1,754), siRNA-7 (nucleotide 1,868–1,888) were newly designed. siRNA-4 was previously published.30 Cells were washed two times in PBS and harvested using Passive Lysis Buffer (Promega), followed by freezing at −80 °C for 15 minutes. The lysate was processed to measure firefly and renilla luciferase activity, and relative luminescence units were determined using a luminometer (GL-200: Standard type, Microtec, Chiba, Japan) and Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's instructions. Among the seven candidate siRNAs, the sequence showing the highest suppression of renilla luciferase activity (siRNA-6) was selected. Subsequently, referring to the construct of siRNA-6, two shRNA sequences were designed (Figure 1b) and investigated based on luciferase assay in order to select for the shRNA to be expressed from AAV9 vector. We also investigated which shRNA is most silencing efficiency.
Construction, production, and titration of AAV9-shTRPV1. The shRNA-A cassette and anti-SOD1 shRNA cassette as a negative control sequence were prepared as previously described.12,47 Each shRNA cassette was cloned downstream of the human polymerase III U6 promoter in the AAV9 vector plasmid (Stratagene). The recombinant AAV9 vector was produced and purified as previously reported (Figure 1e).12 HEK293 cells at ~70% confluence were transfected with the AAV9 packaging plasmid pRep2/Cap9 (gift from Dr. James M Wilson, University of Pennsylvania, Philadelphia, PA) and adenovirus helper plasmid (Stratagene) at a ratio of 1:1:1. At 6 hours after transfection, the culture medium was replaced with fresh medium, and the cells were incubated for 48 hours. The cells were then harvested from the culture dishes and pelleted by centrifugation, resuspended in PBS, and subjected to three rounds of freeze thawing. Cell debris was then pelleted by centrifugation at 1,200g for 15 minutes. AAV vectors were purified using ammonium sulfate precipitation and iodixanol (Axis-Shield) continuous gradient centrifugation. Size-exclusion chromatography was performed using an AKTA Explorer 100 HPLC system (GE Healthcare, Japan Corporation, Tokyo, Japan) equipped with a 2-mL sample loop. A Superdex 200 10/300 GL column (GE Healthcare) was equilibrated with minor histocompatibility antigen buffer (3.3 mmol/l Mes, 3.3 mmol/l Hepes, 3.3 mM NaOAc, 50 mmol/l NaCl, pH 6.5). The vector-containing fractions were loaded onto the column at a flow rate of 0.5 ml/minute, and the eluate was collected as 0.5 ml fractions over the duration of one column volume (23 ml). AAV peak fractions were identified by 280/260 nm absorbance and real-time quantitative polymerase chain reaction using vector-specific primers. The purified AAVs were then concentrated further by using Amico Ultra-4 tubes (Ultracel-30k; Merck Millipore Japan, Tokyo, Japan) to a final concentration of 2 × 1012 genome copies/ml, as determined by quantitative polymerase chain reaction. The genome copy number was calculated by TaqMan PCR (Applied Biosystems, Foster City, CA). The vectors were treated with Benzonase and digested with proteinase K (Wako Pure Chemical Industries, Osaka, Japan) for 1 hour and purified by phenol-chloroform extraction. The TaqMan primers and probe were designed as follows: forward primer: 5′-CAGGCTGGTCCAACTCCTA-3′, reverse primer: 5′-GCAGTGGTTCACGCCTGTAA-3′, and probe: 5′-TACCCACCTTGGCCTC-3′. The designed TaqMan PCR fragment was located in the human growth hormone poly A site in the vector.
Animals. Eight-week-old female Institute of Cancer Research mice weighing 25–35 g were used. Animals were kept under standard laboratory conditions with free access to standard laboratory food and tap water. The body weight of the mice was measured every week. All animal experiments were performed in accordance with the Ethical and Safety Guidelines for Animal Experiments of Tokyo Medical and Dental University and approved by the Animal Experiment Committee of Tokyo Medical and Dental University (#81213).
Animal model of neuropathic pain. Mice underwent surgery involving ligation of the tibial and common peroneal nerve according to the SNI model.13 Animals were anesthetized by intraperitoneal injection of chloral hydrate (0.5 mg per g body weight). The surgical site (left leg) was shaved and disinfected. An incision was made on the lateral mid-thigh, and the underlying muscles were separated to expose the sciatic nerve. The common peroneal and tibial nerves were then tight-ligated with 8.0 silk and sectioned distal to the ligation to remove 2–4 mm of the distal nerve stump (Figure 2a). Great care was taken to avoid any contact with or stretching of the intact sural nerve. The muscles and skin were closed in two layers using 4.0 monofilament nylon sutures. After the operation, animals were allowed to recover in their own cages.
Intrathecal treatment after SNI. Three weeks after nerve injury, when obvious behavioral hypersensitivity was observed, all mice were given intrathecal injections of AAV9-shTRPV1, AAV9-shSOD1, or PBS. After induction of anesthesia using intraperitoneal injections of chloral hydrate (0.5 mg per g body weight) and ketamine hydrochloride (0.05 mg per g body weight), each animal was shaved and disinfected. The mice were placed in the prone position, and a partial laminectomy of the caudal portion of the second lumbar vertebra and the rostral portion of the third lumbar vertebra were performed. The dura mater was exposed and punctured using a 27-gauge needle. Subsequently, a 10-mm PE-10 catheter was connected to a 10-µl Hamilton syringe, Hamilton Company (Watertown, MA) and inserted caudally into the subarachnoid space between the second and third lumbar vertebrae (Figure 3a). The tip of the catheter was located approximately at the level of L4. A volume of 10 µl (2 × 1012 vector genomes per microliter) of AAV9-shTRPV1, AAV9-shSOD1, or PBS was injected slowly over a 2-minute period. After the catheter was removed, the incision was sutured, and the mice were maintained in the head-up position and allowed to recover on a heating pad. Four weeks after the injection, all of the mice were euthanized, and the lumbar spinal cord and ipsilateral L5 and L6 DRGs were harvested for analysis.
AAV vector genomic DNA PCR. The spinal cord was obtained in separate segments from C6 to C8, T2 to T4, T10 to T12, and L1 to L3. The DRGs were also collected from bilateral C6 to C8, bilateral T2 to T4, bilateral T10 to T12, and bilateral L1 to L3. Whole genomic DNA was extracted from the harvested tissues. Vector genomic DNA was amplified using primers and probes targeting the human growth hormone cassette and yielding a 84-bp fragment. Forward 5′-CAGGCTGGTCCAACTCCTA-3′, reverse 5′-GCAGTGGTTCACGCCTGTAA-3′ served as the primer set, and 5′-TACCCACCTTGGCCTC-3′ served as the probe.
Measurement of mRNA expression by quantitative RT-PCR. Total RNA was extracted from the harvested tissues, including the lumbar DRG and the lumbar segment of the spinal cord, using Isogen (Nippon Gene, Tokyo, Japan). The RNA samples obtained from the lumbar spinal cord were collected from the lumbar region located at the first to second vertebral level. The RNA samples obtained from the DRG were obtained from the L5 and L6 DRG. DNase I-treated total RNA (0.5 µg) was reverse transcribed using SuperScript III Reverse Transcriptase (Invitrogen). The cDNA was amplified using the quantitative TaqMan system on a Light Cycle 480 Real-Time PCR Instrument (Roche, Basel, Switzerland), according to the manufacturer's protocol. As an internal control, gapdh cDNA was also quantitatively amplified using a TaqMan primer and probe set (Applied Biosystems). Primer and probe sets for mouse trpv1 and mouse ifnβ were also designed by Applied Biosystems. The ratio of trpv1 mRNA expression to gapdh mRNA expression was calculated to estimate the shRNA silencing efficiency.
Western blotting. The lumbar spinal cord located between the first and second vertebrae and ipsilateral L5 and L6 DRGs were isolated carefully from treated mice. The tissues were homogenized in cold homogenization buffer containing 0.1% sodium dodecylsulfate, 1% sodium deoxycholate, 1% Triton X-100, and 1 mmol/l phenylmethylsulfonyl fluoride together with a protein inhibitor cocktail (Roche, Penzberg, Germany). Five micrograms of extracted protein from each sample were mixed with Laemmli sample buffer (BioRad, Hercules, CA), denatured at 37 °C for 60 minutes, and separated on a 15% sodium dodecylsulfate–polyacrylamide gel electrophoresis. The separated proteins were transferred to a polyvinylidene difluoride membrane (BioRad) and then incubated with specific primary antibodies, including a rabbit anti-TRPV1 antibody (1:1,000; Transgenic, Kumamoto, Japan) and a mouse anti-glyceraldehyde-3-phosphate dehydrogenase monoclonal antibody (Biodesign, Saco, ME). After incubation, the membranes were rinsed and incubated with a 0.1% solution of horseradish peroxidase-conjugated secondary antibodies, including goat anti-rabbit HRP IgG and goat anti-mouse HRP IgG (Thermo Science, Rockford, IL). The protein–antibody interactions were visualized using the SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Science).
Immunohistochemistry. Mice were sacrificed at 24 hours after single or repeated injection of AAV9-shTRPV1, AAV9-shSOD1, or PBS by transcardiac perfusion of PBS for 5 minutes at room temperature, followed by 4% paraformaldehyde in PBS for 15 minutes at 4 °C. After perfusion, the lumbar spinal cord and DRG were immediately removed and postfixed in 4% paraformaldehyde in PBS at 4 °C overnight. After postfixation, the samples were transferred to PBS containing 30% sucrose and dehydrated for 3 days. The tissues were then embedded in low melting temperature agarose (BM Equipment, Tokyo, Japan) in PBS. Frozen sections ~20 µm thick were sectioned from the L5 DRG and the spinal cord at the first lumbar vertebral level from mice in each group and then incubated for 30 minutes at room temperature in a blocking solution (5% normal goat serum). The sections were then incubated with a rabbit polyclonal anti-CD8 antibody (1:500, Abcam, Tokyo, Japan) for 24 hours at 4 °C. Following incubation, the sections were washed and incubated for 30 minutes at room temperature with a biotinylated secondary antibody (1:200, Vector Laboratories, Ontario, Canada) in 0.5% horse serum. The sections were then incubated with ABC reagent (Vector Laboratories) for 30 minutes, followed by incubation with a peroxidase substrate solution for 2 minutes. After washing, the tissue sections were counter stained with hematoxylin for 1 minute.
Sensory behavioral tests. Animals were placed in Plexiglas boxes with dimensions of 9.5 × 21 × 25 cm and allowed to acclimate to the testing environment. The boxes were then placed on an elevated perforated plastic surface for a minimum of 30 minutes prior to all behavioral tests.48 Two observers who were blind to the experimental group assignments performed the behavioral testing once a week and at specific time points for 16 weeks in total.
Tactile threshold. Mechanical sensitivity was measured by applying a series of calibrated von Frey filaments (0.02–8 g) to the plantar aspect of the hindpaw. Each filament was applied once to each mouse. Beginning with the 1-g filament, each filament was applied perpendicular to the hind paw for 4–6 seconds. A brisk withdrawal of the hind paw indicated a positive response, and a lack of withdrawal indicated a negative response. The filament testing was repeated two times, and at least two responses to the filament out of the three trials indicated an overall positive response. If the mouse demonstrated an overall positive response, the filament with the next lower force was applied as described above. If no overall positive response was observed (0/3 or 1/3 responses), the filament with the next higher force was applied as described above. Once the threshold was determined (i.e., from response to no response, or vice versa) the responses to the next five filaments were recorded to determine the median withdrawal threshold.
Response to acetone. Using a plastic tube connected to a 1-ml syringe, and without touching the skin, 100 µl of acetone were applied to the plantar surface of the hindpaw. Acetone was applied five times to each paw at an interval of at least 30 seconds, and the number of brisk foot withdrawals in response to the acetone application was recorded.
Response to noxious heat stimulus. Responses to noxious heat were determined using a hot plate (NISSIN, Saitama, Japan). The mice were placed in a transparent plastic chamber on a 50 or 55 °C metal hot plate to measure the latency of paw flinching, licking or withdrawal. A maximum cutoff of 30 seconds was used to prevent tissue damage. A 5-minute interval between consecutive stimulations of the same hind paw was employed. Testing was performed three times on left lateral plantar hindpaw, and the withdrawal latencies were averaged. In addition, we defined analgesia as a change in the latency of withdrawal from the 50 °C hot plate after injection of AAV9-shTRPV1, relative to the preinjection latency.
Statistical analyses. All data are presented as the mean ± standard error of the mean (n = 3–9). We performed the statistical analysis using two-way analysis of variance with repeated measures for comparisons among the three groups for all experiments. In case of a significant treatment effect, pairwise comparisons were performed in a post hoc analysis with Bonferroni adjustment. The group size was n = 6–12. Significance was defined as P < 0.05.
Acknowledgments
We thank Michael Xi Zhu (Department of Integrative Biology and Pharmacology, The University of Texas Health Science Center at Houston Medical School) for kindly providing the TRPV1 plasmid and Miho Sekiguchi (Department of Orthopedic Surgery, Fukushima Medical University School of Medicine) and Igor Spigelman (Division of Oral Biology & Medicine, School of Dentistry, The University of California, Los Angeles) for technical support. This work was supported in part by a Grant-in-aid for Scientific Research (C) from the Japan Society for the Promotion of Science and a Grant-in-aid from the General Insurance Association of Japan.
References
- Campbell JN, Meyer RA. Mechanisms of neuropathic pain. Neuron. 2006;52:77–92. doi: 10.1016/j.neuron.2006.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan TF, Chen SR, Pan HL. Effect of the {mu} opioid on excitatory and inhibitory synaptic inputs to periaqueductal gray-projecting neurons in the amygdala. J Pharmacol Exp Ther. 2005;312:441–448. doi: 10.1124/jpet.104.074633. [DOI] [PubMed] [Google Scholar]
- Wilson SP, Yeomans DC, Bender MA, Lu Y, Goins WF, Glorioso JC. Antihyperalgesic effects of infection with a preproenkephalin-encoding herpes virus. Proc Natl Acad Sci USA. 1999;96:3211–3216. doi: 10.1073/pnas.96.6.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier A, Latrémolière A, Mauborgne A, Bourgoin S, Kayser V, Cesselin F, et al. Attenuation of pain-related behavior in a rat model of trigeminal neuropathic pain by viral-driven enkephalin overproduction in trigeminal ganglion neurons. Mol Ther. 2005;11:608–616. doi: 10.1016/j.ymthe.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Clapham DE. TRP channels as cellular sensors. Nature. 2003;426:517–524. doi: 10.1038/nature02196. [DOI] [PubMed] [Google Scholar]
- Levine JD, Alessandri-Haber N. TRP channels: targets for the relief of pain. Biochim Biophys Acta. 2007;1772:989–1003. doi: 10.1016/j.bbadis.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Pogatzki-Zahn EM, Shimizu I, Caterina M, Raja SN. Heat hyperalgesia after incision requires TRPV1 and is distinct from pure inflammatory pain. Pain. 2005;115:296–307. doi: 10.1016/j.pain.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Yokota T, Iijima S, Kubodera T, Ishii K, Katakai Y, Ageyama N, et al. Efficient regulation of viral replication by siRNA in a non-human primate surrogate model for hepatitis C. Biochem Biophys Res Commun. 2007;361:294–300. doi: 10.1016/j.bbrc.2007.06.182. [DOI] [PubMed] [Google Scholar]
- Baigude H, McCarroll J, Yang CS, Swain PM, Rana TM. Design and creation of new nanomaterials for therapeutic RNAi. ACS Chem Biol. 2007;2:237–241. doi: 10.1021/cb7000582. [DOI] [PubMed] [Google Scholar]
- Nishina K, Unno T, Uno Y, Kubodera T, Kanouchi T, Mizusawa H, et al. Efficient in vivo delivery of siRNA to the liver by conjugation of alpha-tocopherol. Mol Ther. 2008;16:734–740. doi: 10.1038/mt.2008.14. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Lebron E, Denovan-Wright EM, Nash K, Lewin AS, Mandel RJ. Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington's disease transgenic mice. Mol Ther. 2005;12:618–633. doi: 10.1016/j.ymthe.2005.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai T, Enomoto M, Machida A, Yamamoto M, Kuwahara H, Tajiri M, et al. Intrathecal shRNA-AAV9 inhibits target protein expression in the spinal cord and dorsal root ganglia of adult mice. Hum Gene Ther Methods. 2012;23:119–127. doi: 10.1089/hgtb.2012.035. [DOI] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Calixto JB, Kassuya CA, André E, Ferreira J. Contribution of natural products to the discovery of the transient receptor potential (TRP) channels family and their functions. Pharmacol Ther. 2005;106:179–208. doi: 10.1016/j.pharmthera.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Chan CL, Facer P, Davis JB, Smith GD, Egerton J, Bountra C, et al. Sensory fibres expressing capsaicin receptor TRPV1 in patients with rectal hypersensitivity and faecal urgency. Lancet. 2003;361:385–391. doi: 10.1016/s0140-6736(03)12392-6. [DOI] [PubMed] [Google Scholar]
- Schäfers M, Geis C, Svensson CI, Luo ZD, Sommer C. Selective increase of tumour necrosis factor-alpha in injured and spared myelinated primary afferents after chronic constrictive injury of rat sciatic nerve. Eur J Neurosci. 2003;17:791–804. doi: 10.1046/j.1460-9568.2003.02504.x. [DOI] [PubMed] [Google Scholar]
- Hudson LJ, Bevan S, Wotherspoon G, Gentry C, Fox A, Winter J. VR1 protein expression increases in undamaged DRG neurons after partial nerve injury. Eur J Neurosci. 2001;13:2105–2114. doi: 10.1046/j.0953-816x.2001.01591.x. [DOI] [PubMed] [Google Scholar]
- Rasband MN, Park EW, Vanderah TW, Lai J, Porreca F, Trimmer JS. Distinct potassium channels on pain-sensing neurons. Proc Natl Acad Sci USA. 2001;98:13373–13378. doi: 10.1073/pnas.231376298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuoka T, Tokunaga A, Tachibana T, Dai Y, Yamanaka H, Noguchi K. VR1, but not P2X(3), increases in the spared L4 DRG in rats with L5 spinal nerve ligation. Pain. 2002;99:111–120. doi: 10.1016/s0304-3959(02)00067-2. [DOI] [PubMed] [Google Scholar]
- Facer P, Casula MA, Smith GD, Benham CD, Chessell IP, Bountra C, et al. Differential expression of the capsaicin receptor TRPV1 and related novel receptors TRPV3, TRPV4 and TRPM8 in normal human tissues and changes in traumatic and diabetic neuropathy. BMC Neurol. 2007;7:11. doi: 10.1186/1471-2377-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Katsura H, Sakurai J, Kobayashi K, Yamanaka H, Dai Y, et al. Suppression of the p75 neurotrophin receptor in uninjured sensory neurons reduces neuropathic pain after nerve injury. J Neurosci. 2006;26:11974–11986. doi: 10.1523/JNEUROSCI.3188-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SJ, Burette A, Rustioni A, Valtschanoff JG. Vanilloid receptor VR1-positive primary afferents are glutamatergic and contact spinal neurons that co-express neurokinin receptor NK1 and glutamate receptors. J Neurocytol. 2004;33:321–329. doi: 10.1023/B:NEUR.0000044193.31523.a1. [DOI] [PubMed] [Google Scholar]
- Kosugi M, Nakatsuka T, Fujita T, Kuroda Y, Kumamoto E. Activation of TRPA1 channel facilitates excitatory synaptic transmission in substantia gelatinosa neurons of the adult rat spinal cord. J Neurosci. 2007;27:4443–4451. doi: 10.1523/JNEUROSCI.0557-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrini F, Salio C, Vergnano AM, Merighi A. Vanilloid receptor-1 (TRPV1)-dependent activation of inhibitory neurotransmission in spinal substantia gelatinosa neurons of mouse. Pain. 2007;129:195–209. doi: 10.1016/j.pain.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Kim YH, Back SK, Davies AJ, Jeong H, Jo HJ, Chung G, et al. TRPV1 in GABAergic interneurons mediates neuropathic mechanical allodynia and disinhibition of the nociceptive circuitry in the spinal cord. Neuron. 2012;74:640–647. doi: 10.1016/j.neuron.2012.02.039. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Bian D, Malan TP, Jr, Lai J, Porreca F. Lack of involvement of capsaicin-sensitive primary afferents in nerve-ligation injury induced tactile allodynia in rats. Pain. 1999;79:127–133. doi: 10.1016/s0304-3959(98)00187-0. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, et al. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Culshaw AJ, Bevan S, Christiansen M, Copp P, Davis A, Davis C, et al. Identification and biological characterization of 6-aryl-7-isopropylquinazolinones as novel TRPV1 antagonists that are effective in models of chronic pain. J Med Chem. 2006;49:471–474. doi: 10.1021/jm051058x. [DOI] [PubMed] [Google Scholar]
- Christoph T, Grünweller A, Mika J, Schäfer MK, Wade EJ, Weihe E, et al. Silencing of vanilloid receptor TRPV1 by RNAi reduces neuropathic and visceral pain in vivo. Biochem Biophys Res Commun. 2006;350:238–243. doi: 10.1016/j.bbrc.2006.09.037. [DOI] [PubMed] [Google Scholar]
- Klusáková I, Dubový P. Experimental models of peripheral neuropathic pain based on traumatic nerve injuries - an anatomical perspective. Ann Anat. 2009;191:248–259. doi: 10.1016/j.aanat.2009.02.007. [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- Jordt SE, McKemy DD, Julius D. Lessons from peppers and peppermint: the molecular logic of thermosensation. Curr Opin Neurobiol. 2003;13:487–492. doi: 10.1016/s0959-4388(03)00101-6. [DOI] [PubMed] [Google Scholar]
- Patapoutian A, Peier AM, Story GM, Viswanath V. ThermoTRP channels and beyond: mechanisms of temperature sensation. Nat Rev Neurosci. 2003;4:529–539. doi: 10.1038/nrn1141. [DOI] [PubMed] [Google Scholar]
- Moran MM, Xu H, Clapham DE. TRP ion channels in the nervous system. Curr Opin Neurobiol. 2004;14:362–369. doi: 10.1016/j.conb.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ. Thermosensation and pain. J Neurobiol. 2004;61:3–12. doi: 10.1002/neu.20079. [DOI] [PubMed] [Google Scholar]
- Vriens J, Owsianik G, Hofmann T, Philipp SE, Stab J, Chen X, et al. TRPM3 is a nociceptor channel involved in the detection of noxious heat. Neuron. 2011;70:482–494. doi: 10.1016/j.neuron.2011.02.051. [DOI] [PubMed] [Google Scholar]
- Cho H, Yang YD, Lee J, Lee B, Kim T, Jang Y, et al. The calcium-activated chloride channel anoctamin 1 acts as a heat sensor in nociceptive neurons. Nat Neurosci. 2012;15:1015–1021. doi: 10.1038/nn.3111. [DOI] [PubMed] [Google Scholar]
- Shimosato G, Amaya F, Ueda M, Tanaka Y, Decosterd I, Tanaka M. Peripheral inflammation induces up-regulation of TRPV2 expression in rat DRG. Pain. 2005;119:225–232. doi: 10.1016/j.pain.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Park U, Vastani N, Guan Y, Raja SN, Koltzenburg M, Caterina MJ. TRP vanilloid 2 knock-out mice are susceptible to perinatal lethality but display normal thermal and mechanical nociception. J Neurosci. 2011;31:11425–11436. doi: 10.1523/JNEUROSCI.1384-09.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moqrich A, Hwang SW, Earley TJ, Petrus MJ, Murray AN, Spencer KS, et al. Impaired thermosensation in mice lacking TRPV3, a heat and camphor sensor in the skin. Science. 2005;307:1468–1472. doi: 10.1126/science.1108609. [DOI] [PubMed] [Google Scholar]
- Suhy DA, Kao SC, Mao T, Whiteley L, Denise H, Souberbielle B, et al. Safe, long-term hepatic expression of anti-HCV shRNA in a nonhuman primate model. Mol Ther. 2012;20:1737–1749. doi: 10.1038/mt.2012.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janson C, McPhee S, Bilaniuk L, Haselgrove J, Testaiuti M, Freese A, et al. Clinical protocol. Gene therapy of Canavan disease: AAV-2 vector for neurosurgical delivery of aspartoacylase gene (ASPA) to the human brain. Hum Gene Ther. 2002;13:1391–1412. doi: 10.1089/104303402760128612. [DOI] [PubMed] [Google Scholar]
- Kaplitt MG, Feigin A, Tang C, Fitzsimons HL, Mattis P, Lawlor PA, et al. Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson's disease: an open label, phase I trial. Lancet. 2007;369:2097–2105. doi: 10.1016/S0140-6736(07)60982-9. [DOI] [PubMed] [Google Scholar]
- Boutin S, Monteilhet V, Veron P, Leborgne C, Benveniste O, Montus MF, et al. Prevalence of serum IgG and neutralizing factors against adeno-associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21:704–712. doi: 10.1089/hum.2009.182. [DOI] [PubMed] [Google Scholar]
- Bartel M, Schaffer D, Büning H. Enhancing the Clinical Potential of AAV Vectors by Capsid Engineering to Evade Pre-Existing Immunity. Front Microbiol. 2011;2:204. doi: 10.3389/fmicb.2011.00204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappel S, Matthess Y, Kaufmann M, Strebhardt K. Silencing of mammalian genes by tetracycline-inducible shRNA expression. Nat Protoc. 2007;2:3257–3269. doi: 10.1038/nprot.2007.458. [DOI] [PubMed] [Google Scholar]
- Yokota T, Miyagishi M, Hino T, Matsumura R, Tasinato A, Urushitani M, et al. siRNA-based inhibition specific for mutant SOD1 with single nucleotide alternation in familial ALS, compared with ribozyme and DNA enzyme. Biochem Biophys Res Commun. 2004;314:283–291. doi: 10.1016/j.bbrc.2003.12.098. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]