

Abstract
The rhabdovirus Maraba has recently been characterized as a potent oncolytic virus. In the present study, we engineered an attenuated Maraba strain, defined as MG1, to express a melanoma-associated tumor antigen. Its ability to mount an antitumor immunity was evaluated in tumor-free and melanoma tumor-bearing mice. Alone, the MG1 vaccine appeared insufficient to prime detectable adaptive immunity against the tumor antigen. However, when used as a boosting vector in a heterologous prime-boost regimen, MG1 vaccine rapidly generated strong antigen-specific T-cell immune responses. Once applied for treating syngeneic murine melanoma tumors, our oncolytic prime-boost vaccination protocol involving Maraba MG1 dramatically extended median survival and allowed complete remission in more than 20% of the animals treated. This work describes Maraba virus MG1 as a potent vaccine vector for cancer immunotherapy displaying both oncolytic activity and a remarkable ability to boost adaptive antitumor immunity.
Introduction
Oncolytic viruses (OVs) specifically infect, replicate in and kill malignant cells, leaving normal tissues unaffected. Several OVs have reached advanced stages of clinical evaluation for the treatment of various neoplasms.1 Data from clinical trials and preclinical models have demonstrated that these viral agents alone or in combination with standard cancer therapies hold great promise for improved therapeutic efficacy.2,3,4
We have previously reported that a naturally attenuated vesicular stomatitis virus (VSVΔM51), a prototypical rhabdovirus, is a compelling oncolytic agent due to its safety profile and the lack of pre-existing neutralizing antibodies in human populations, a practical problem associated with several other OV platforms.5,6,7,8 To expand our current array of safe and potent OVs from the Rhabdoviridae, we have recently identified several new vesiculoviruses that display strong oncolytic activities.9,10 Specifically, the Maraba virus showed the broadest oncotropism in vitro and specific genetic modifications were shown to dramatically improved its tumor selectivity and reduced its virulence in normal cells. In vivo, this attenuated strain, called MG1, demonstrated potent antitumor activity in xenograft and syngeneic tumor models in mice, with superior therapeutic efficacy than VSVΔM51.9
Data accumulated over the past several years has revealed that antitumor efficacy of OVs not only depends on their direct oncolysis but may also depend on their ability to stimulate antitumor immunity.11 This immune-mediated tumor control seems to play a critical role in the overall efficacy of OV therapy. Indeed, tumor-specific adaptive immune cells can patrol the tissues and destroy tumor cells that have been missed by the OV. Moreover, their memory compartment can prevent tumor recurrence.
Various strategies have been developed to improve OV-induced antitumor immunity.12 Some groups have genetically engineered OV expressing immunostimulatory cytokines. An herpes simplex and a vaccinia virus expressing granulocyte-macrophage colony-stimulating factor have respectively reached phase III and IIB of the clinical evaluation for cancer therapy while a VSV expressing interferon (IFN)-β has just entered phase I.1 Another strategy, defined as oncolytic vaccine, consists of expressing a tumor antigen from the OV. Previously, we and others have demonstrated that VSV could also be used as a cancer vaccine vector.13,14 When applied in an heterologous prime-boost setting to treat a murine melanoma model, our VSV oncolytic vaccine not only induced an increased tumor-specific immunity but also a concomitant reduction in antiviral adaptive immunity. As a result, the therapeutic efficacy was dramatically improved with an increase of both median and long-term survivals.13
Given that both VSV and Maraba are classified as vesiculoviruses, we hypothesized that Maraba MG1 could share the vaccine properties of VSV. In the present study, we demonstrated that oncolytic Maraba MG1 can be used as an oncolytic vaccine platform. While unable to prime detectable responses against a melanoma-associated antigen, Maraba MG1 vaccine displayed a potent ability to boost pre-existing tumor-specific CD4+ and CD8+ T-cell immunity. When applied to the treatment of syngeneic murine melanoma tumor models, Maraba MG1-mediated recall immunization resulted in a dramatic extension of the median survival with complete remission in more than 20% of the animals treated. Together with our previous study involving VSV,13 this work confirmed that vesiculoviruses can be potent vaccine vectors for cancer immunotherapy displaying both oncolytic activity and a remarkable ability to boost adaptive cell immunity.
Results
Characterizing the oncolytic activity of Maraba MG1 in vitro and its oncotropism in vivo in the murine B16-F10 melanoma model
The oncolytic property of Maraba virus has recently been characterized in five melanoma-derived cell lines from the NCI-60 panel.9 However, the B16-F10 metastatic melanoma cell line, used as the tumor model in the present study, was not included. Before evaluating the therapeutic efficacy of Maraba MG1 in tumor-bearing mice, we evaluated its ability to lyse B16-F10 cells in vitro. For this purpose, a monolayer culture of B16-F10 melanoma cells was infected with MG1-GFP at a multiplicity of infection of 0.01 and cell viability and viral replication were measured. By visualizing green fluorescent protein (GFP) expression, Maraba infection was noticeable at 12 hours and spread through the whole culture within 24 hours (Figure 1a). A burst of virus production was achieved within 20 hours (Figure 1b), along with clearance of the B16-F10 cell population (Figure 1c and visible field on Figure 1a). These data demonstrated that Maraba MG1 was able to infect, replicate in and kill B16-F10 melanoma cells.
Figure 1.

Maraba MG1 infects, replicates in and kill B16-F10 melanoma cells in vitro. A monolayer culture of murine B16-F10 melanoma cells was incubating with Maraba MG1-GFP at a multiplicity of infection of 0.01. (a) Maraba virus infects B16-F10 cells and spread across the culture (GFP signal) leading to complete cell population clearance within 36 hours (visible field). (b) Replication of Maraba MG1-GFP illustrated by viral titers (pfu/ml) in B16-F10 culture supernatant. (c) Population of viable B16-F10 cells, monitored by MTT assay, in absence or presence of Maraba MG1-GFP. (b,c) Curves from a representative experiment realized in triplicate where each timepoint is illustrated as mean ± SD. GFP, green fluorescent protein; pfu, plaque-forming units.
We subsequently evaluated the ability of Maraba virus to selectively infect B16-F10 melanoma cells in vivo. Immunocompetent mice bearing B16-F10 lung metastases, as well as tumor-free controls, were inoculated intravenously (i.v.) with MG1-GFP. Infectious particles were quantified in the lungs and in the secondary lymphoid organs (spleen and inguinal lymph nodes) at 24 hours and 48 hours after virus injection (Figure 2a). Maraba MG1 was detected at high titers in the spleen 24 hours after inoculation (5 log plaque-forming units (pfu)/g tissue) but cleared from the organ by 48 hours (<2 log pfu/g, Figure 2b). The kinetic of splenic virus clearance was similar in both tumor-free and tumor-bearing animals (Figure 2b). MG1 was rarely detectable in the lymph nodes (<2 log pfu/g, Figure 2c), independently of the presence of tumor, suggesting a poor distribution or rapid clearance of Maraba in the lymph nodes. While MG1 was cleared from the lungs of tumor-free mice at 48 hours, a substantial level of virus was still persisting at that time in the mice with B16-F10 lung metastases (Figure 2d, mean ± SD = 3.01 log pfu/g ± 1.84 at 48 hours). Analysis of the MG1 titer at early timepoint after infection revealed a large virus uptake in the lungs which was significantly higher in presence of metastases: 4.26 log pfu/g ± 0.40 in tumor-free controls versus 5.84 log pfu/g ± 0.19 in tumor-bearing mice at 2 hours (Figure 2d). In the lungs with metastases, MG1 titer increased by 8 times between 2 and 12 hours, reaching 6.75 log pfu/g ± 0.18 at 12 hours, before progressively decreasing (Figure 2d). In contrast, in tumor-free lungs, the MG1 load did not increase and displayed a quick clearance (Figure 2d). These observations illustrate the ability of MG1 to selectively target and replicate at the tumor site.
Figure 2.

Maraba MG1 selectively replicates at the B16-F10 melanoma metastases site in vivo. (a) C57Bl/6 mice received 2.5 × 105 syngeneic B16-F10 cells by intravenous injection in order to establish syngeneic lung melanoma metastases. Fourteen days after tumor challenge, MG1-GFP was administered by intravenous injection at a dose of 109 pfu. To address Maraba tumor selectivity, virus replication in tumor-free mice was compared to replication in lung metastases-bearing animals. Viral titers were quantified at various timepoints after infection in the lungs (d), where metastases are located in tumor-bearing animals, as well as in secondary lymphoid organs: (b) spleen and (c) inguinal lymph nodes. Titers were quantified in three samples per tissue and illustrated as mean ± SD. **P < 0.01, ***P < 0.001. GFP, green fluorescent protein; NS, nonsignificant; pfu, plaque-forming units.
Maraba MG1-hDCT alone is insufficient to improve the therapeutic outcome and to induce antitumor immunity
Although Maraba MG1 could lyse B16-F10 cells in vitro and replicates at the tumor site in vivo, i.v. administration of MG1-GFP at its highest tolerable dose (109 pfu—Brun et al.)9 did not result in an improvement of survival in a 5-day-old B16-F10 lung metastatic model, compared to untreated control animals (Figure 3a). This result was not entirely surprising as previous studies have shown that B16 cells are responsive to type I IFNs. Considering the sensitivity of attenuated VSV to such response,15 it was likely that type I IFNs may also protect tumor cells from Maraba MG1 oncolysis. We validated the hypothesis in vitro by demonstrating that Maraba MG1 was unable to kill B16-F10 cells that have been pre-exposed to IFN-β (Supplementary Figure S1). As a consequence, the induction of the interferon response following Maraba MG1 replication at the tumor site may limit the extent of its oncolytic action. One way to broaden antitumor spectrum and activity of oncolytic therapy is to increase the antitumor immunity induced during oncolysis. To this end, we engineered Maraba MG1 to express the human form of the melanoma-associated antigen dopachrome tautomerase (termed MG1-hDCT, see Supplementary Figure S2a,b for the cloning strategy and characterization of transgene expression) that is highly homologous to its murine counterpart expressed by B16-F10 cells. We postulated that overexpression of dopachrome tautomerase (DCT) by the Maraba vector during oncolysis might enhance antigen-specific CD8+ T-cell responses leading to improved overall therapeutic outcomes. Disappointingly, survival was not extended when mice were treated with MG1-hDCT 5 days after tumor engraftment as compared to MG1-GFP or mock treatments (Figure 3a).
Figure 3.
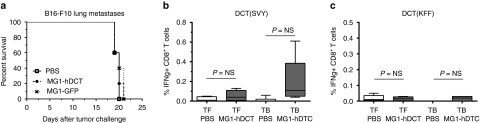
Maraba MG1-hDCT alone is insufficient to improve the therapeutic outcome and to induce antitumor immunity. (a) C57Bl/6 mice received 2.5 × 105 syngeneic B16-F10 cells by intravenous injection (i.v.) in order to establish syngeneic lung melanoma metastases. Five days after tumor challenge, Maraba MG1 expressing either GFP or the melanoma antigen hDCT was administered i.v. at a dose of 109 pfu; untreated mice received phosphate-buffered saline (PBS). Despite viral replication at the tumor site, MG1-hDCT failed to increase mouse survival as illustrated by Kaplan–Meier curves (n = 5 per group). (b,c) C57Bl/6 mice received or not 2.5 × 105 syngeneic B16-F10 cells by i.v. injection in order to establish syngeneic lung melanoma metastases. Five days later, tumor-free (TF) and tumor-bearing (TB) mice were administered i.v. with 109 pfu MG1-hDCT or PBS. Nine days after Maraba injection, immune responses against the tumor antigen DCT were measured in the blood. (b) Percentage of CD8+ T cells secreting IFN-γ after ex vivo exposure to the MHC-I restricted DCT immunodominant epitope SVYDFFVWL (SVY). (c) Percentage of CD4+ T cells secreting IFN-γ after ex vivo exposure to the MHC-II restricted DCT immunodominant epitope KFFHRTCKCTGNFA (KFF). Box plots representing 25–75 percentile including median and whiskers illustrating the range between minimal and maximal values (n = 5 mice per group). P value considered nonsignificant (NS) when >0.05. DCT, dopachrome tautomerase; GFP, green fluorescent protein; IFN-γ, interferon-gamma; pfu, plaque-forming units.
Our immunological analyses indicated that DCT-specific CD8+ or CD4+ T-cell responses induced by MG1-hDCT were barely detectable either in tumor-bearing or in tumor-free mice (Figure 3b,c) suggesting that Maraba MG1 is a weak vaccine vector for priming significant T-cell responses against the transgene antigen.
Maraba MG1 is a potent vector for boosting DCT-specific responses
We have previously reported that another oncolytic rhabdovirus, vesicular stomatitis virus (VSV), is highly effective as a booster of pre-existing immunity.13 We speculated that Maraba virus might share the same biological property. To test this possibility, we chose a recombinant adenoviral vector expressing hDCT (Ad-hDCT) as a priming vector due to its ability to induce strong primary CD8+ T-cell responses. In this case, Ad-hDCT was administered by intramuscular injection (i.m.) at a dose of 2 × 108 pfu, as optimized previously.13,16 DCT-specific T-cell responses were measured at day 10 after Ad injection in the blood, which corresponds to the peak time of the primary response elicited by Ad vectors. As expected, no DCT-specific T-cell response was detected in control mice injected with the empty Ad vector (Figure 4a) while Ad-hDCT administration generated a strong DCT-specific CD8+ T-cell response (% IFN-γ-secreting CD8+ T cells: mean ± SEM = 6.05% ± 0.6, Figure 4a,b).
Figure 4.

Maraba MG1 is a potent vector for boosting DCT-specific response. (a) C57Bl/6 mice received 2 × 108 pfu of Ad-hDCT by intramuscular injection (DCT prime) before receiving, or not, 109 pfu of VSV-hDCT or Maraba MG1-hDCT by intravenous injection (DCT boost). Negative controls received 2 × 108 pfu of Ad-empty. DCT-specific T-cell responses were measured 10 days after Ad (peak of the prime response) and 5 days after VSV/MG1 (peak of the boost response). Immune responses represented as the percentage of CD8+ T cells secreting IFN-γ after ex vivo exposure to SVY peptide corresponding to the MHC-I restricted immunodominant epitope of DCT (n = 5 mice for Ad-empty and Ad-hDCT + VSV-hDCT groups, n = 10 mice for Ad-hDCT and Ad-hDCT + MG1-hDCT groups). (b) Representative dot plots of DCT-specific CD8+ T-cell responses measured in Ad-hDCT and Ad-hDCT + MG1-hDCT immunized mice. Box plots representing 25–75 percentile including median and whiskers illustrating the range between minimal and maximal values. *P < 0.05 and ***P < 0.001. Ad, adenoviral vector; DCT, dopachrome tautomerase; IFN-γ, interferon-gamma; pfu, plaque-forming units.
As demonstrated previously,13 i.v. administration of 109 pfu of VSV-hDCT 12 days after Ad-hDCT priming generated a strong systemic secondary CD8+ T-cell response (Ad-hDCT + VSV-hDCT: 19.32% ± 2.72; Figure 4a). Interestingly, the same dose of MG1-hDCT elicited an even stronger recall response confirming that rhabdoviruses share a common characteristic as boosting vaccine vectors (Ad-hDCT + MG1-hDCT: 30.59% ± 3.49; Figure 4a,b). We subsequently tested other routes of Maraba administration including i.v., i.m., and intranasal (i.n.). Results indicated that the i.v. route was far superior to other routes in boosting CD8+ T-cell response demonstrating systemic increases in DCT-specific immune responses in both blood and spleen (Figure 5a,c): 30.59% ± 3.49 by i.v. versus 4.73% ± 1.52 i.n. versus 13.84% ± 1.88 i.m. in the blood. In addition to CD8+ T cells, the MG1-hDCT was also able to boost systemic CD4+ T-cell responses following i.v. administration (Figure 5b,d): % IFN-γ–secreting CD4+ T-cells in the blood = 0.31% ± 0.12; Figure 5b.
Figure 5.

Systemic administration of Maraba MG1 vaccine is required for generating optimal tumor-specific boost responses. C57Bl/6 mice received 2 × 108 pfu Ad-hDCT by intramuscular injection (DCT prime). Twelve days later, 109 pfu of Maraba MG1-hDCT were administered through various routes (DCT boost): intravenous (i.v.), intranasal (i.n.), or intramuscular (i.m.). DCT-specific T-cell responses were measured 5 days after Maraba injection in the blood (a,b) and in the spleen (c,d). Immune response measured at the same timepoint in Ad-hDCT prime only animals is indicated (“No boost”). (a,c) Percentage of CD8+ T cells secreting IFN-γ after ex vivo exposure to the MHC-I restricted DCT immunodominant epitope SVY. (b,d) Percentage of CD4+ T cells secreting IFN-γ after ex vivo exposure to the MHC-II restricted DCT immunodominant epitope KFF. Box plots representing 25–75 percentile including median and whiskers illustrating the range between minimal and maximal values (n = 10 mice for the “i.v.” group, n = 5 for the “i.n.” and “i.m.” groups). P value considered nonsignificant (NS) when >0.05, *P < 0.05, **P < 0.01, and ***P < 0.001. Ad, adenoviral vector; DCT, dopachrome tautomerase; IFN-γ, interferon-gamma; pfu, plaque-forming units.
Maraba MG1 vaccine accelerates secondary CD8+ T-cell responses
The ability of Maraba MG1 vector to rapidly amplify CD8+ T-cell responses prompted us to evaluate the outcome of shortening the prime-boost interval on the DCT boost response. MG1-hDCT was injected i.v. at a dose of 109 pfu, 12, 9 or 4 days after Ad-hDCT prime. Interestingly, an interval as short as 9 days was not only sufficient to boost the secondary responses but actually elicited a much higher magnitude of DCT-specific CD8+ T cells than the 12-day interval (9-day interval: 37.53% ± 3.66 versus 12-day interval: 30.59% ± 3.49; Figure 6a). More surprisingly, a significant boost could be achieved within 4 days after Ad priming (Figure 6a, 11.99% ± 2.26) suggesting that Maraba MG1 may represent a unique boost vector for certain diseases where rapid amplification of CD8+ T cells is required.
Figure 6.

Maraba MG1 vaccine accelerates secondary CD8+ T-cell responses. C57Bl/6 mice received 2 × 108 pfu Ad-hDCT by intramuscular injection (DCT prime). At 12, 9 or 4 days after Ad-hDCT prime, MG1-hDCT was administered by intravenous injection at a dose of 109 pfu. DCT-specific T-cell responses were measured 5 days after Maraba injection in the blood. Immune response measured at the same timepoint in Ad-hDCT prime only animals is indicated (“No boost”). (a) Percentage of CD8+ T cells secreting IFN-γ after ex vivo exposure to the MHC-I restricted DCT immunodominant epitope SVY. (b) Percentage of CD4+ T cells secreting IFN-γ after ex vivo exposure to the MHC-II restricted DCT immunodominant epitope KFF. Box plots representing 25–75 percentile including median and whiskers illustrating the range between minimal and maximal values (n = 10 mice per group). P value considered nonsignificant (NS) when >0.05, *P < 0.05 and ***P < 0.001. Ad, adenoviral vector; DCT, dopachrome tautomerase; IFN-γ, interferon-gamma; pfu, plaque-forming units.
Additionally, while barely detectable by shortening the prime-boost interval to 4 days, the DCT-specific CD4+ T-cell boost response was maintained by reducing the interval from 12 to 9 days (12-day interval: 0.31% ± 0.12 versus 9-day interval: 0.32% ± 0.08 versus 4-day interval: 0.09% ± 0.03; Figure 6b).
Boosting with Maraba MG1-hDCT in a lung metastatic model generates potent antitumor immunity and extended survival
We subsequently evaluated the therapeutic efficacy of MG1-hDCT administered as a boosting vector in a lung metastatic model. Five days following i.v. injection of B16-F10 cells, animals received a sequential administration of Ad-hDCT and MG1-hDCT at a 9-day interval (Figure 7a). We confirmed that Ad-hDCT prime-MG1-hDCT boost vaccination generated a very strong DCT-specific CD8+ T-cell response (mean % IFN-γ+ CD8+ T cells = 27.54 ± 2.17, Figure 7b) that was 14 times higher than in nonboosted mice (1.95% ± 0.29 in Ad-hDCT group and 1.91% ± 0.59 in Ad-hDCT + MG1-GFP group, Figure 7b). Similarly, DCT-specific CD4+ T-cell responses were measured in MG1-hDCT boosted animals while rarely detected in primed only mice (mean % IFN-γ+ CD4+ T cells = 0.25% ± 0.06 in Ad-hDCT + MG1-hDCT group versus <0.05% in Ad-hDCT and Ad-hDCT + MG1-GFP groups, Figure 7c).
Figure 7.

Maraba MG1-hDCT administered in a heterologous prime-boost setting allowed to generate potent antitumor immunity and to extend survival of melanoma lung tumor-bearing animals. (a) C57Bl/6 mice were challenged intravenously with 2.5 × 105 B16-F10 cells in order to establish syngeneic lung melanoma metastases. Five days later, mice received 2 × 108 pfu Ad-hDCT by intramuscular injection. Control mice received empty Ad vector. Nine days after Ad injection, animals were administered intravenously with 109 pfu MG1-hDCT or its GFP control. Immune responses against the tumor antigen DCT were measured 5 days after Maraba injection in the blood and mouse survival was monitored daily. Percentage of CD8+ or CD4+ T cells reacting to SVY (b) or KFF (c) peptide exposure, respectively. Box plots representing 25–75 percentile including median and whiskers illustrating the range between minimal and maximal values. Pooled data from several experiments: n = 9 for Ad-empty group, n = 23 for Ad-hDCT group, n = 29 for Ad-hDCT + MG1-hDCT group, n = 11 for Ad-hDCT + MG1-GFP group. (d) Kaplan–Meier curves illustrating survival of treated melanoma lung-tumor bearing mice. Pooled data from several experiments: n = 16 for Ad-empty group, n = 23 for Ad-hDCT group, n = 30 for Ad-hDCT + MG1-hDCT group, n = 11 for Ad-hDCT + MG1-GFP group. (e) T-cell populations were selectively depleted to evaluate their respective therapeutic contribution. Kaplan–Meier curves illustrating survival of treated melanoma lung-tumor bearing mice (n = 5 per group). (-CD8) indicates CD8+ T-cells depletion while (-CD4) indicates CD4+ T-cells depletion. (f) Lungs extracted at day 19 from untreated and Ad-hDCT + MG1-hDCT treated mice. (g) Autoimmunity (vitiligo) induced in long-term survivors following Ad-hDCT + MG1-hDCT treatment. Pictures taken 280 days (mice on the left) or 220 days (mice on the right) after tumor challenge. P value considered nonsignificant (NS) when >0.05, *P < 0.05, **P < 0.01, and ***P < 0.001. Ad, adenoviral vector; GFP, green fluorescent protein; DCT, dopachrome tautomerase; IFN-γ, interferon-gamma; pfu, plaque-forming units.
Compared to Ad-empty control, Ad-hDCT or Ad-hDCT + MG1-GFP treatment slightly prolonged survival time (Figure 7d). However, boosting with the MG1-DCT vaccine dramatically improved the median survival, and more than 20% of mice were cured of their tumors (Figure 7d). Figure 7f is an example where decrease of the size and number of melanoma lung metastases was evident at 5 days after MG1-hDCT injection, coincident with the peak of secondary CD8+ T-cell responses.
To characterize the respective contribution of CD4+ and CD8+ T-cell responses to the therapeutic efficacy, each T-cell compartment was selectively depleted using corresponding antibodies. As shown on Figure 7e, depletion of the CD8+ T-cell population at the time of the boost abrogated the therapeutic benefit of MG1-hDCT administration. On the contrary, CD4+ T-cells depletion appeared not to affect significantly the therapeutic efficacy indicating that CD8+ T cells play a critical role in controlling tumor growth following the combination therapy with Ad-hDCT and MG1-hDCT.
We noted that all long-term survivors developed vitiligo of variable severity (Figure 7g), a manifestation of melanocyte destruction, reinforcing the potency of anti-DCT immune responses boosted by MG1-hDCT. Some long-term survivors were rechallenged with B16-F10 melanoma cells. As shown in the Supplementary Figure S3, none of them developed tumors illustrating the existence of anti-DCT immune memory that can protect from tumor recurrence.
Additionally, we evaluated the efficacy of (i) the two homologous prime-boost involving either Ad-hDCT or MG1-hDCT vaccines and (ii) the Ad-hDCT + VSV-hDCT heterologous prime-boost in the melanoma lung-metastasis model. The latter has previously shown efficacy against melanoma brain tumors13 but has never been applied yet to treat melanoma lung tumors. As shown in the Supplementary Figure S4, both Ad-hDCT and MG1-hDCT failed to boost their homologous prime and did not improve the therapeutic efficacy: median survival = 20 days upon MG1-hDCT treatment versus 23 days with MG1-hDCT + MG1-hDCT and 27 days with Ad-hDCT versus 28 days with Ad-hDCT + Ad-hDCT. When comparing the two oncolytic rhabdoviral vaccines, MG1-hDCT appeared more potent than VSV-hDCT to boost pre-existing DCT-specific CD8+ T-cell responses. This stronger antitumor immunity translated into an improved tumor outcome: 73.5 days upon Ad-DCT + MG1-hDCT treatment versus 44.5 days with Ad-hDCT + VSV-hDCT. As a consequence, these results favor Maraba MG1 over VSV as a candidate for further clinical evaluation of our oncolytic prime-boost approach.
Boosting with Maraba MG1-hDCT significantly improves therapeutic outcomes in an intracranial tumor model
Finally, we also evaluated the efficacy of our prime-boost strategy involving Maraba vaccine in a very challenging 5-day-old melanoma brain metastatic model. Immune responses resulting from the Ad-hDCT prime-MG1-hDCT booster treatment were again remarkably strong (29.11% ± 2.17 IFN-γ+ CD8+ T cells, Figure 8a) and of similar amplitude to that seen in lung-metastasis bearing mice (P = 0.61 versus Ad-hDCT + MG1-hDCT group in Figure 7b).
Figure 8.

Maraba MG1-hDCT efficiently boosted DCT-specific response in melanoma brain tumor-bearing mice and significantly extended their survival. C57Bl/6 mice were challenged with 103 B16-F10 cells by intracranial injection in order to establish syngeneic brain melanoma tumor. Five days later, mice received 2 × 108 pfu Ad-hDCT by intramuscular injection while control mice received an empty Ad vector. Nine days after Ad injection, animals were administered intravenously with 109 pfu MG1-hDCT. Mouse survival was monitored daily. (a) Percentage of CD8+ T cells secreting IFN-γ in response to DCT-specific SVY peptide exposure measured 5 days after Maraba injection in the blood. Box plots representing 25–75 percentile including median and whiskers illustrating the range between minimal and maximal values. Pooled data from surviving mice at day 19 after tumor challenge from several experiments: n = 2 for Ad-empty group (only 2 control mice out of 17 still alive at the time of immune analysis), n = 8 for Ad-hDCT group, n = 15 for Ad-hDCT + MG1-hDCT group. (b) Kaplan–Meier curves illustrating survival of treated melanoma brain-tumor bearing mice. Pooled data from several experiments: n = 17 for Ad-empty group, n = 10 for Ad-hDCT group, n = 15 for Ad-hDCT + MG1-hDCT group. **P < 0.01, and ***P < 0.001. Ad, adenoviral vector; DCT, dopachrome tautomerase; IFN-γ, interferon-gamma; pfu, plaque-forming units.
Ad-hDCT–mediated immunotherapy significantly improved survival of mice bearing melanoma brain metastasis, with a median extended from 15 days for Ad-empty controls to 25.5 days for the Ad-hDCT group (Figure 8b). The additional administration of a Maraba MG1-hDCT oncolytic booster further improved tumor outcome with a median survival reaching 42 days together with cures observed in 21.4% of treated animals (Ad-hDCT + MG1-hDCT group, Figure 8b).
Discussion
Cancer vaccines offer great promise for systemic cancer therapy and long-term tumor protection. While passive immunotherapy relies on repeated administration of short-lived molecules (antibody, cytokine) or immune cells, cancer vaccines aim at developing active antitumor immunity by stimulating patient's immune system to react against self antigens associated with the malignant phenotype. Due to immune tolerance, generating an adaptive antitumor immunity that results in a therapeutic benefit is challenging. To reach this goal, efforts are being made to identify immunogenic tumor antigens and characterize potent vaccine platforms. Several viral vaccine vectors are currently under clinical evaluation for cancer therapy. Candidates express immunogenic tumor-associated antigens (e.g., CEA, NY-ESO-1, 5T4, or PSA). These recombinant viruses are mostly replication-defective (e.g., TRICOM and TroVax poxviral vectors).17 In this context, therapeutic activity strictly relies on their ability to mount an efficient adaptive antitumor immunity.
While the efficacy of OV therapy relies on oncolytic viral replication in tumors, it very likely benefits from the induction of an antitumor immunity. Various strategies have been proposed to improve the therapeutic impact of these viruses by influencing these two aspects.12 We have recently identified Maraba virus as a new oncolytic vesiculovirus with more potent oncolytic activity than its prototypical cousin VSV.9,10 In order to stimulate tumor-specific adaptive immunity, we have developed the approach of inserting a transgene encoding a tumor antigen into the genome of an OV. However, similar to VSV-based oncolytic vaccines,13 Maraba MG1 expressing DCT (MG1-DCT) failed to induce meaningful DCT-specific CD8+ T-cell responses in tumor-free or tumor-bearing animals. We speculate that replicating OVs will prove to be weak priming vaccine vectors as the immune response against the viral vector is dominated by responses against highly immunogenic viral antigens that must be expressed for replication.8,13 To overcome this issue, we introduced our Maraba vaccine as a boosting vector in a heterologous prime-boost regimen. In this setting, the response against the tumor antigen transgene is a secondary response and is therefore more robust and can compete against the primary response against the vector itself.8,13
Our team and others have demonstrated that adenoviral vaccines are potent priming vectors capable of breaking immune tolerance.13,18,19,20 Coupled to Ad-hDCT, Maraba MG1-hDCT remarkably boosted Ad primed DCT-specific responses. When the combination vaccine was applied for treatment of melanoma tumor-bearing mice, tumor-specific T-cell responses generated continued to be potent despite tumor-induced immune suppression. In fact, we have previously demonstrated that using oncolytic VSV as a tumor vaccine booster leads to larger antitumor immune responses in tumor-bearing animals,13 and now show that this is even further enhanced when using Maraba MG1 virus as an oncolytic vaccine (Supplementary Figure S4). Systemic delivery of the OV appeared critical for its vaccine potential implying that this route allowed for engagement of antigen-presenting cells with a strong ability to boost immune responses. Regarding the strong uptake of MG1-hDCT in the spleen, this lymphoid tissue could be involved in the phenomenon. Investigations are ongoing in the laboratory. As well, Maraba virus replication in the tumor bed may change the local tumor microenvironment and affect its related immunosuppression. Tumor vasculature shutdown and/or local induction of an immunostimulatory cytokine storm have repeatedly been associated with OV tumor infection and may contribute to the phenomena.21,22,23,24,25,26 In the B16-F10 murine melanoma model, Maraba oncolysis had no impact on overall survival. This observation may not be surprising given the relatively weak and ephemeral replication of Maraba at the tumor site. While B16-F10 cells were permissive for Maraba infection in vitro, it would appear that B16-F10 cells were poorly permissive to Maraba in vivo. However, Maraba MG1 oncolysis previously showed potent efficacy for treating other mouse models of cancer9 indicating that this relatively poor oncolytic effect is model specific. Importantly, the oncolytic vaccine approach introduced in a heterologous prime-boost setting compensated for the transient viral oncolysis. The strong tumor-specific cytotoxic T-cell immunity induced a dramatically extended survival. Despite the aggressiveness of the melanoma tumor models used, Ad prime-Maraba MG1 boost vaccine combination allowed long-term complete remission in more than 20% of the animals treated.
Taking into account our previous work involving VSV,13 we confirmed here with Maraba MG1 that vesiculoviruses can be potent vaccine vectors for cancer immunotherapy, displaying both oncolytic activity and an impressive ability to boost adaptive cell immunity. Their association with Ad vaccine in a heterologous prime-boost regimen appeared impressively efficient regarding the therapeutic benefit. However, this setting can still be optimized. Numerous variations need to be evaluated. Among them, it might be possible to extend the prime-boost interval. Due to constraints linked to the aggressiveness of the tumor, such an option remained inaccessible with the B16-F10 melanoma model. As cells infected by the oncolytic vaccine overexpressed the targeted antigen, they represent a particularly immunogenic target for the antigen-specific cytotoxic T cells. Extending the interval should allow one to have reduced populations of these effector T cells at the time of the boost. This should allow for an extended window of viral spread with potentially increased boost efficacy. Another strategy for improvement could consist of replacing the replication-deficient Ad vector with another heterologous oncolytic vaccine vector. Also, we could modulate the immune system with drugs prior to, or along with, the oncolytic prime-boost vaccination. As a proof-of-concept, we recently succeeded in dramatically enhancing the therapeutic efficacy of the Ad prime-VSV boost protocol by administering an HDAC inhibitor along with the booster vaccination.27
Thus, our oncolytic Maraba vaccine appeared to have unprecedented boosting ability. Its combination with replication-deficient adenoviral vaccine vectors showed impressive therapeutic efficacy illustrating its therapeutic potential. We have now shown that MG1 Maraba virus displays greater oncolytic potency9 as well as an enhanced ability to boost antitumoral immunity leading to enhanced therapeutic impact relative to oncolytic VSV (Supplementary Figure S4). Our group is currently conducting a preclinical safety study of the Ad + Maraba MG1 oncolytic prime-boost approach in nonhuman primates. Clinical evaluations in patients with advanced tumor expressing a characterized tumor-associated antigen will follow.
Materials and Methods
Mice. Female C57BL/6 mice (8–10 weeks old at study initiation) were purchased from Charles River Laboratory (Wilmington, MA) and housed in a specific pathogen-free facility. All animal studies complied with Canadian Council on Animal Care guidelines and were approved by McMaster University's Animal Research Ethics Board.
Recombinant viruses. Recombinant viruses involved in the study have been described previously. Ad-empty and Ad-hDCT are replication-deficient adenoviruses (E1/E3-deletion) based on the human serotype 5.28,29 hDCT transgene encodes the full-length human melanoma antigen DCT. Ad-empty has no transgene. Recombinant Maraba and VSV were generated by transgene insertion between the G and L viral genes. VSV-hDCT derives from the wild-type Indiana strain of the VSV.16,30 MG1-GFP and MG1-hDCT derive from the attenuated strain MG1 of Maraba virus.9 Maraba MG1-hDCT has been developed for the purpose of the study.
Cell culture. Murine melanoma B16-F10 cells (expressing the murine DCT antigen) were grown in F11-MEM containing 10% fetal bovine serum, 2 mmol/l l-glutamine, 1 mmol/l sodium pyruvate and vitamin solution, 0.01 mmol/l nonessential amino-acids, 50 mmol/l β-Mercaptoethanol, 100 U/ml penicillin, and 100 mg/ml streptomycin (all from Invitrogen, Grand Island, NY). Vero cells were cultured in alpha-MEM containing 10% fetal bovine serum, 2 mmol/l l-glutamine, 100 U/ml penicillin, and 100 mg/ml streptomycin (all from Invitrogen, Grand Island, NY).
Cell transduction. 8 × 105 B16-F10 cells per well were seeded in six-well plates. The monolayer cell culture was transduced with Maraba MG1-GFP at a multiplicity of infection (MOI) of 0.01. Incubation was performed in 100 µl for 1 hour before adding 2.9 ml of culture medium. A 200 µl of culture supernatant were collected for virus titration at 8, 12, 24, and 36 hours and followed by cell imaging under fluorescent microscope. Virus titer in culture supernatant were determined by plaque assay on Vero cells using standard techniques. Viability of infected cells was evaluated at each timepoint by MTT assay.
MTT assay. A cell suspension of 2 × 106 B16-F10 cells per ml was incubated with MG1-GFP at MOI 0.01 for an hour under shaking. 24 × 103 cells were then seeded per well of a 96-well plate in 200 µl culture medium. At 1.5 hour prior to timepoint, 10 µl soluble MTT at 5 mg/ml (Sigma-Aldrich, Oakville, ON) was added and incubated for 3 hours. Formazan salts, resulting from the conversion of MTT by alive cells, were resuspended in dimethylsulfoxide and their concentration determined by optical density at 570 nm.
Mice infection/immunization. Rhabdoviruses Maraba MG1-GFP, MG1-hDCT and VSV-hDCT were injected intravenously (i.v.) in 200 µl phosphate-buffered saline at a dose of 109 pfu. For adenovirus injection, mice were anesthetized in a sealed chamber containing 5% inhalation isoflurane. Adenoviral vectors Ad-empty and Ad-hDCT were administered intramuscularly (i.m.) at a total dose of 2 × 108 pfu (1 × 108 pfu in 50 µl phosphate-buffered saline per quadriceps).
Tumor challenge. For lung metastases engraftment, 2.5 × 105 B16-F10 cells were injected i.v. in 200 µl saline water. For brain tumors, mice were anesthetized by inhalation of 5% isoflurane through a nose cone and engrafted by intracranial (i.c.) injection of 103 B16-F10 cells in 2 µl phosphate-buffered saline using a stereotactic frame as previously described.13 Mice were monitored daily and euthanized for tissue harvest or upon signs of morbidity.
Tissue homogenates. To measure virus replication in vivo, tissues (lungs, spleen, and inguinal lymph nodes) were harvested 1–3 days after i.v. inoculation of Maraba MG1-GFP, weighted and immediately frozen at −80 °C. After thawing, each tissue was homogenized in 1 ml phosphate-buffered saline using an Ultra Turrax T25 homogenizer (14,000 rpm for 10 seconds). Viral titers in homogenates were quantified by plaque assay on Vero cells and expressed as log pfu/g of tissue.
Peptides. Peptides corresponding to the immunodominant epitopes of DCT: (i) DCT180–188 SVYDFFVWL/“SVY” that binds to H-2Kb; 100% conserved between human and murine DCT and (ii) DCT89–102 KFFHRTCKCTGNFA/“KFF” from human DCT that binds to I-Ab; differing from the murine epitope in position 92: His (human) → Asn (murine). Peptides were synthesized by Biomer Technologies (San Francisco, CA).
Antibodies. Monoclonal antibodies used in flow cytometry assays: anti-CD16/CD32 (clone 2.4G2) to block Fc receptors, anti-CD3 (clone 145-2C11), anti-CD8a (clone 53–6.7), and anti-CD4 (clone RM4-5) for detecting cell surface markers, anti-IFN-γ (clone XMG1.2) for intracellular cytokine staining. All reagents were purchased from BD Pharmingen (Mississauga, ON). For T-cell depletion, 200 µg of anti-CD8a (clone 2.43) and anti-CD4 (clone GK1.5) antibodies were injected in the peritoneal cavity three times at 2 days interval starting 2 days before MG1-hDCT administration. Selective T-cell depletion was confirmed by flow cytometry on blood samples (data not shown).
Detection of antigen-specific T-cell responses. DCT-specific T-cell responses were measured 10 days post-prime and 5 days post-boost as followed. Peripheral blood mononucleated cells or splenocytes were incubated in complete RPMI (RPMI medium supplemented with fetal calf serum 10%, Penicillin-Streptomycin 1% and l-glutamine 1%) with SVY peptide (2 µg/ml) or KFF peptide (15 µg/ml) for DCT-specific CD8+ or CD4+ T-cell (re-)stimulation, respectively. Incubation was performed in incubator (37 °C, 5% CO2, 95% humidity) for 5 hours, with brefeldin A (1 μg/ml, GolgiPlug BD Pharmingen) during the last 4 hours. Cells were treated with antibodies targeting CD16/CD32 before staining with fluorescent-labeled antibodies targeting T-cell surface markers. Then, cells were permeabilized and fixed with Cytofix/Cytoperm (BD Pharmingen) and stained for intracellular cytokines. Data were acquired using a FACSCanto flow cytometer with FACSDiva software (BD Pharmingen) and analyzed with FlowJo Mac software (Treestar, Ashland, OR).
Statistic analysis. GraphPad Prism version 6 for Windows (GraphPad Software, San Diego, CA) was used for graphing and statistical analyses. Immune responses data were graphed as box plots (25–75 percentile with median) with whiskers illustrating the range between minimal and maximal values. Student's two-tailed t-test were used to analyze immune data and compare virus titers. Survival curves were represented according to the Kaplan–Meier method, and compared using the log-rank test. Differences between groups were considered significant when P ≤ 0.05 (*P < 0.05, **P < 0.01 and ***P < 0.001).
SUPPLEMENTARY MATERIAL Figure S1. Induction of the interferon response in B16-F10 cells protects from Maraba MG1 infection Figure S2. Maraba vector construction and expression of DCT in MG1-hDCT transduced Vero cells. Figure S3. Tumor-specific immune memory resulting from the Ad-hDCT+MG1-hDCT treatment protects cured mice from tumor recurrence. Figure S4. Ad-hDCT+MG1-hDCT heterologous prime-boost induces stronger antitumor immunity and shows better therapeutic efficacy than homologous prime-boost strategies or than the heterologous prime-boost involving VSV-hDCT.
Acknowledgments
Supported by grants to B.D.L. from the Canadian Cancer Society and the Ontario Institute for Cancer Research and to Y.W. from the Canadian Institute for Health Research. The authors declared no conflict of interest.
Supplementary Material
References
- Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329–336. doi: 10.1038/nm.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karapanagiotou EM, Roulstone V, Twigger K, Ball M, Tanay M, Nutting C, et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin Cancer Res. 2012;18:2080–2089. doi: 10.1158/1078-0432.CCR-11-2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington KJ, Hingorani M, Tanay MA, Hickey J, Bhide SA, Clarke PM, et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin Cancer Res. 2010;16:4005–4015. doi: 10.1158/1078-0432.CCR-10-0196. [DOI] [PubMed] [Google Scholar]
- Miest TS, Yaiw KC, Frenzke M, Lampe J, Hudacek AW, Springfeld C, et al. Envelope-chimeric entry-targeted measles virus escapes neutralization and achieves oncolysis. Mol Ther. 2011;19:1813–1820. doi: 10.1038/mt.2011.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CL, Twigger KR, Vidal L, De Bono JS, Coffey M, Heinemann L, et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther. 2008;15:911–920. doi: 10.1038/gt.2008.21. [DOI] [PubMed] [Google Scholar]
- Law M, Hollinshead R, Smith GL. Antibody-sensitive and antibody-resistant cell-to-cell spread by vaccinia virus: role of the A33R protein in antibody-resistant spread. J Gen Virol. 2002;83 Pt 1:209–222. doi: 10.1099/0022-1317-83-1-209. [DOI] [PubMed] [Google Scholar]
- Chen Y, Yu DC, Charlton D, Henderson DR. Pre-existent adenovirus antibody inhibits systemic toxicity and antitumor activity of CN706 in the nude mouse LNCaP xenograft model: implications and proposals for human therapy. Hum Gene Ther. 2000;11:1553–1567. doi: 10.1089/10430340050083289. [DOI] [PubMed] [Google Scholar]
- Brun J, McManus D, Lefebvre C, Hu K, Falls T, Atkins H, et al. Identification of genetically modified maraba virus as an oncolytic rhabdovirus. Mol Ther. 2010;18:1440–1449. doi: 10.1038/mt.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney DJ, Lefebvre C, Allan K, Brun J, Sanaei CA, Baird S, et al. Virus-tumor interactome screen reveals ER stress response can reprogram resistant cancers for oncolytic virus-triggered caspase-2 cell death. Cancer Cell. 2011;20:443–456. doi: 10.1016/j.ccr.2011.09.005. [DOI] [PubMed] [Google Scholar]
- Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther. 2011;19:1008–1016. doi: 10.1038/mt.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pol J, Rességuier J, Lichty B. Oncolytic viruses: a step into cancer immunotherapy. Vir Adapt Treat. 2011;2012:1–21. [Google Scholar]
- Bridle BW, Li J, Jiang S, Chang R, Lichty BD, Bramson JL, et al. Immunotherapy can reject intracranial tumor cells without damaging the brain despite sharing the target antigen. J Immunol. 2010;184:4269–4275. doi: 10.4049/jimmunol.0901447. [DOI] [PubMed] [Google Scholar]
- Pulido J, Kottke T, Thompson J, Galivo F, Wongthida P, Diaz RM, et al. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat Biotechnol. 2012;30:337–343. doi: 10.1038/nbt.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–825. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- Bridle BW, Boudreau JE, Lichty BD, Brunellière J, Stephenson K, Koshy S, et al. Vesicular stomatitis virus as a novel cancer vaccine vector to prime antitumor immunity amenable to rapid boosting with adenovirus. Mol Ther. 2009;17:1814–1821. doi: 10.1038/mt.2009.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draper SJ, Heeney JL. Viruses as vaccine vectors for infectious diseases and cancer. Nat Rev Microbiol. 2010;8:62–73. doi: 10.1038/nrmicro2240. [DOI] [PubMed] [Google Scholar]
- Aurisicchio L, Mennuni C, Giannetti P, Calvaruso F, Nuzzo M, Cipriani B, et al. Immunogenicity and safety of a DNA prime/adenovirus boost vaccine against rhesus CEA in nonhuman primates. Int J Cancer. 2007;120:2290–2300. doi: 10.1002/ijc.22555. [DOI] [PubMed] [Google Scholar]
- Facciabene A, Aurisicchio L, Elia L, Palombo F, Mennuni C, Ciliberto G, et al. DNA and adenoviral vectors encoding carcinoembryonic antigen fused to immunoenhancing sequences augment antigen-specific immune response and confer tumor protection. Hum Gene Ther. 2006;17:81–92. doi: 10.1089/hum.2006.17.81. [DOI] [PubMed] [Google Scholar]
- Mennuni C, Calvaruso F, Facciabene A, Aurisicchio L, Storto M, Scarselli E, et al. Efficient induction of T-cell responses to carcinoembryonic antigen by a heterologous prime-boost regimen using DNA and adenovirus vectors carrying a codon usage optimized cDNA. Int J Cancer. 2005;117:444–455. doi: 10.1002/ijc.21188. [DOI] [PubMed] [Google Scholar]
- Breitbach CJ, De Silva NS, Falls TJ, Aladl U, Evgin L, Paterson J, et al. Targeting tumor vasculature with an oncolytic virus. Mol Ther. 2011;19:886–894. doi: 10.1038/mt.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LC, Lynn RC, Cheng G, Alexander E, Kapoor V, Moon EK, et al. Treating tumors with a vaccinia virus expressing IFNß illustrates the complex relationships between oncolytic ability and immunogenicity. Mol Ther. 2012;20:736–748. doi: 10.1038/mt.2011.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo Y, Sakai R, Ouchi M, Onimatsu H, Hioki M, Kagawa S, et al. Virus-mediated oncolysis induces danger signal and stimulates cytotoxic T-lymphocyte activity via proteasome activator upregulation. Oncogene. 2008;27:2375–2381. doi: 10.1038/sj.onc.1210884. [DOI] [PubMed] [Google Scholar]
- Errington F, Steele L, Prestwich R, Harrington KJ, Pandha HS, Vidal L, et al. Reovirus activates human dendritic cells to promote innate antitumor immunity. J Immunol. 2008;180:6018–6026. doi: 10.4049/jimmunol.180.9.6018. [DOI] [PubMed] [Google Scholar]
- Benencia F, Courrèges MC, Fraser NW, Coukos G. Herpes virus oncolytic therapy reverses tumor immune dysfunction and facilitates tumor antigen presentation. Cancer Biol Ther. 2008;7:1194–1205. doi: 10.4161/cbt.7.8.6216. [DOI] [PubMed] [Google Scholar]
- Benencia F, Courrèges MC, Conejo-García JR, Mohamed-Hadley A, Zhang L, Buckanovich RJ, et al. HSV oncolytic therapy upregulates interferon-inducible chemokines and recruits immune effector cells in ovarian cancer. Mol Ther. 2005;12:789–802. doi: 10.1016/j.ymthe.2005.03.026. [DOI] [PubMed] [Google Scholar]
- Bridle BW, Chen L, Lemay CG, Diallo JS, Pol J, Nguyen A, et al. HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol Ther. 2013;21:887–894. doi: 10.1038/mt.2012.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane C, Leitch J, Tan X, Hadjati J, Bramson JL, Wan Y. Vaccination-induced autoimmune vitiligo is a consequence of secondary trauma to the skin. Cancer Res. 2004;64:1509–1514. doi: 10.1158/0008-5472.can-03-3227. [DOI] [PubMed] [Google Scholar]
- Ng P, Beauchamp C, Evelegh C, Parks R, Graham FL. Development of a FLP/frt system for generating helper-dependent adenoviral vectors. Mol Ther. 2001;3 5 Pt 1:809–815. doi: 10.1006/mthe.2001.0323. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Stillman EA, Whitt MA, Rose JK. Recombinant vesicular stomatitis viruses from DNA. Proc Natl Acad Sci USA. 1995;92:4477–4481. doi: 10.1073/pnas.92.10.4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.