

Abstract
Recent clinical trials have demonstrated that treatment with selective serotonin reuptake inhibitors (SSRIs) after stroke enhances motor functional recovery; however, the underlying mechanisms remain to be further elucidated. We hypothesized that daily administration of the clinical drug citalopram would produce these functional benefits via enhancing neurovascular repair in the ischemic peri-infarct region. To test this hypothesis, focal ischemic stroke was induced in male C57/B6 mice by permanent ligation of distal branches of the middle cerebral artery to the barrel cortex and 7-min occlusion of the bilateral common carotid arteries. Citalopram (10 mg/kg, i.p.) was injected 24 hrs after stroke and daily thereafter. To label proliferating cells, bromo-deoxyuridine was injected daily beginning 3 days after stroke. Immunohistochemical and functional assays were performed to elucidate citalopram-mediated cellular and sensorimotor changes after stroke. Citalopram treatment had no significant effect on infarct formation or edema 3 days after stroke; however, citalopram-treated mice had better functional recovery than saline-treated controls 3 and 14 days after stroke in the adhesive removal test. Increased expression of brain derived neurotrophic factor was detected in the peri-infarct region 7 days after stroke in citalopram-treated animals. The number of proliferating neural progenitor cells and the distance of neuroblast migration from the sub-ventricular zone towards the ischemic cortex were significantly greater in citalopram-treated mice at 7 days after stroke. Immunohistochemical staining and co-localization analysis showed that citalopram-treated animals generated more new neurons and microvessels in the peri-infarct region 21 and 28 days after stroke. Taken together, these results suggest that citalopram promotes post-stroke sensorimotor recovery likely via enhancing neurogenesis, neural cell migration and the microvessel support in the peri-infarct region of the ischemic brain.
Keywords: Ischemic stroke, SSRI, Citalopram, Neurogenesis, Angiogenesis
Introduction
Stroke is the fourth-leading cause of death and the primary cause of long-term disability in the United States; however, there are limited effective treatments for stroke patients (Towfighi and Saver, 2011). Stroke patients often suffer from post-stroke depression, which can be treated with anti-depressant drugs to alleviate depressive symptoms (Anderson et al., 1994, Wiart et al., 2000, Bilge et al., 2008). Enhanced functional recovery observed in post-stroke depression patients treated with selective serotonin reuptake inhibitors (SSRI) prompted a number of clinical trials looking for further therapeutic benefits of anti-depressant administration after stroke in non-depressed as well as depressed patients (Acler et al., 2009, Chollet et al., 2011, Mikami et al., 2011a).
Clinical Data suggest that anti-depressant treatments after stroke enhance motor functional recovery independent of their anti-depressant activity. Three months of treatment with the SSRI fluoxetine after stroke significantly improved patient scores in the Fugl-Meyer motor scale and the motor portion of the National Institutes of Health stroke scale (NIHSS) (Chollet et al., 2011). Similarly, in another clinical study, administration of the SSRI citalopram for four months after stroke improved NIHSS performance, unrelated to mood (Acler et al., 2009). Fluoxetine and the tricyclic anti-depressant nortriptyline increased scores in the modified Rankin scale, which measures independence in activities of daily life (Chollet et al., 2011, Mikami et al., 2011). These studies provide a foundation supporting the use of anti-depressants for improving stroke recovery. However, the mechanisms behind these therapeutic benefits need to be further elucidated in order to improve treatment and develop more targeted and specific therapeutics.
SSRIs have been noted for their effect on neural progenitor proliferation in the hippocampus, dentate gyrus, and sub-ventricular zone (SVZ). Neuronal progenitor proliferation and migration after stroke to the damaged area may contribute to long-term recovery (Lindmark and Hamrin, 1988, Kreuzberg et al., 2010). Brain derived neurotrophic factor (BDNF) is physiologically and pathologically important in the control of survival, proliferation and migration of neural progenitor cells in the SVZ. Chronic SSRI treatment increases the expression of neurotrophic factors in the brain, particularly BDNF mRNA and protein (Balu et al., 2008). BDNF activates the tyrosine kinase receptor TrkB that mediates down-stream signaling cascades involved in growth and survival including phosphotidylinositol-3 kinase/ Akt pathways, the c-AMP response element (CREB) and the Ras/Mitogen-activated protein kinase pathways (Russo-Neustadt and Chen, 2005). Enhanced BDNF expression is believed to modulate neuronal plasticity, survival, and cell-signaling pathways during development, and contribute to axonal plasticity, learning, memory, and sensorimotor recovery (Mizuno et al., 2000, Schabitz et al., 2004).
Repair of the neurovasculature after stroke is an important step in restoring brain function (Ohab et al., 2006). Vascular regeneration after stroke is also a potential target for SSRI mediated therapies. BDNF-TrkB receptor activation and downstream Akt has been shown to promote endothelial cell survival (Kermani and Hempstead, 2007). Therefore, SSRI treatments may contribute to new vessel formation and promote survival of existing vessels after ischemic insult (Greene et al., 2009). The present investigation tested whether administration of the SSRI drug citalopram after stroke could enhance sensorimotor functional recovery in parallel with neurovascular repair in the ischemic peri-infarct region. Citalopram was selected in the investigation based on its clinical applications and functional benefits after stroke evaluated by the NIH stroke score (Acler et al., 2009).
Experimental Procedures
Middle cerebral artery occlusion
The Institutional Animal Care and Use Committee at Emory University approved all in vivo experimental procedures. The focal ischemic stroke targeted to the right barrel cortex was induced as previously described (Li et al., 2007, Ogle et al., 2012) with some modifications. Briefly, adult male C57 mice (Charles River Labs; Wilmington, MA) weighing 20-25g were anesthetized with 4% chloral hydrate. A distal branch of right middle cerebral artery (MCA) supplying the barrel cortex was permanently ligated by 10-0 suture and the bilateral common carotid arteries (CCA) were occluded for 7-min and then reperfused. Animal body temperature was maintained at 37 ± 0.5°C using a heating pad controlled by the temperature control unit (Thermocare; Incline Village, NV) during the surgery and in an environmental controlled incubator after surgery until they recovered from the anesthesia. The mortality rate due to surgery and anesthesia was equal to or less than 10% in this investigation. Fully recovered animals were then returned to their home cages with free access to food and water.
Drug administration
All animals were subjected to the same MCA occlusion (MCAO) procedure and were randomized to saline or citalopram treatment groups after stroke. Researchers were blinded to experimental groups. Citalopram (10 mg/kg) was diluted in sterile saline and injected intra-peritoneally (i.p.) 24 hrs after stroke and then daily for 7,14, 21, or 28 days. This chronic drug administration paradigm was chosen due to previous research suggesting that SSRI's effect on depression was due to delayed neurochemical mechanisms and potentially by increasing BDNF levels (Stahl, 1998, Balu et. al., 2008). In addition, the 24-hr treatment window after stroke provides a clinically relevant paradigm for stroke therapy. In neuroprotection experiments, Citalopram was administered 30 min after stroke and then daily for 3 days until sacrifice at day 3 (n=20, 10 per group). Bromo-deoxyuridine (BrdU)was diluted in sterile saline (5 mg/ml) and was injected i.p. (10 mg/kg) beginning 72 hrs after stroke and then daily until sacrifice unless otherwise indicated.
Infarct volume of the ischemic brain
Infarct volume was assessed with a sample size of ten animals per group. Aniamls were randomly assigned (10 and 10) to citalopram and saline groups and injected i.p. with the appropriate solution 30 min, 24 and 48 hrs after MCAO. The mortality rate of 10% due to anesthesia and/or surgery resulted in the animal number of 9 in each group for analysis. The animals were sacrificed 72 hrs post-stroke for ischemic infarct size assessment as previously described (Ogle et al., 2012). Briefly, animals were sacrificed under anesthesia; brains were removed and then sliced into 1-mm thick coronal sections. Brain sections were then stained with 2% 2,3,5-Triphenyltetrazolium chloride (TTC) solution at 37°C for 10 min and were then placed into 10% buffered formalin. After 24 hrs, brain sections were scanned and images imported into Image J software (NIH, Bethesda, MD, USA), where the stroke infarct, ipsilateral, contralateral, and total area were measured by a blinded researcher. The infarct volume (mm3) and indirect infarct volume ratio were calculated. Student's t test was used to detect differences between the saline control and citalopram groups.
Edema Measurement
Edema or water content of the brain was assessed 72 hrs after MCAO. Animals were randomly assigned (n=6 per group) to citalopram or saline treatment groups and injected i.p. with the appropriate solution 30 min, 24 and 48 hrs after MCAO. After 72 hrs, the brains were removed and secrtioned into three 2-mm thick sections. The contralateral and ipsilateral hemispheres were separated and each was weighed on a piece of pre-weighed tin foil to determine the wet weight. This procedure took less than 30 sec to complete. According to other studies, the water loss during this 30 sec accounts for less than 0.3% of the wet weight of the hemisphere (Ito et al., 1979). Therefore, the water loss during the procedure was considered negligible. Sections wer then dehydrated at 110°C for 48hrs and the dry weight was measured. The water content was evaluated by calculating the difference between the wet and dry weights and then dividing by the wet weight and multiplying by 100 (Ito et al., 1979).
Behavioral assessment
The adhesive removal behavioral test was employed to measure changes in sensorimotor function by placing a small, round adhesive onto the distal forepaw of the right or left leg alternately and measuring the amount of time in seconds it takes for the mouse to remove the adhesive as previously described (Bouet et al., 2009, Freret et al., 2009, Ogle et al., 2012). In animals with damage to the right cortex, the left paw function is expected to develop functional deficits and the right paw function is used as a control. Animals underwent 3 training sessions of three trials each prior to the baseline adhesive removal assessment one day before stroke. Animals were numbered but were not randomized to experimental groups prior to stroke. After MCAO animals were randomly placed into control and treatment groups and injected i.p. with either saline or citalopram beginning 24 hrs after stroke and daily until sacrifice at 14 day after stroke. Both latency and removal times were further assessed at 3 and 14 days post-stroke by a blinded experimenter. The data was analyzed by comparing fold change (post-stroke removal time (sec)/ baseline removal time (sec)) between treatment groups and over time. Data was expressed using fold-change to account for individual variability and learning. To analyze the data using two-way repeated measured ANOVA, only animals tested at baseline, 3 days and 14 days post-stroke were included in the analyses (n=11 per group/per time point). Researchers were blinded to experimental groups in order to reduce experimental error and biases.
Immunohistochemical staining
Brains were frozen immediately after sacrifice in optimal cutting temperature (OCT) media (Sakura Finetek; Torrance, CA) at −80°C. Sections were cut at 10-μm thickness from frozen brains on a cryostat microtome (Leica Biosystems; Buffalo Grove, IL). Slides were fixed for 5 min in 10% buffered formalin, washed in phosphate buffered saline (PBS) three times, then incubated for 15 min in −20°C methanol. After air-drying for 5 min, slides were rehydrated in PBS for 1 min, and then incubated in 2N HCl at 37°C for 1 hr. Sections were neutralized by washing in borate buffer three times for 5 min each wash. The slides were then incubated in 0.2% TritonX-100 for 45 min, and then washed in PBS three times. After washing, slides were incubated in 1% fish gelatin (Sigma, St. Louis, MO) for 30-60 min. Sections were incubated with the appropriate primary antibody overnight at 4°C: anti-neuronal nuclei (NeuN, Millipore; Billerica, MA), anti-collagen IV (Millipore), anti-BrdU (AbD Serotec; Raleigh, NC), anti-Glut-1 (Millipore), and anti-GFAP (Neomarkers; Fremont, CA). Slides were then washed and incubated with the appropriate conjugate secondary antibody for 45 min at 37°C: donkey anti-mouse Cy5, donkey anti-rat, Cy3 donkey anti-rabbit (Jackson Immunoresearch; West Grove, PA), and donkey anti-goat 488- (Invitrogen; Grand Island, NY). Slides were washed three times in PBS and cover-slipped prior to imaging under a fluorescent microscope (Olympic BX61) employing slide book Software (Olympus America, inc).
In staining for doublecortin (DCX), Iba1, and BrdU positive cells, animals were perfused with 0.9% saline (pH 7.4) followed by 10% buffered formalin, brains were removed and placed in formalin for 24 hours, then placed in 30% sucrose solution. Brains were frozen at −20°C in OCT and were cut into 14-μm thick sections on a cryostat (Leica Biosystems; Buffalo Grove, IL). The slides were placed in formalin to fix for 10 min, washed in PBS 3×, fixed in methanol 2× for 7 min, incubated in 0.2% TritonX-100/PBS for 5 min, and 1% fish gel for 30-60 min. Slides were incubated in the primary antibody against the following proteins overnight at 4°C: DCX (Santa Cruz Biotechnology, Santa Cruz, CA), Iba1 (Biocare Medical; Concord, CA), and BrdU (Santa Cruz Biotechnology). Secondary antibody was applied for 45 min at 37°C: anti-goat 488 (Invitrogen). Slides were co-stained with BrdU as described above. Double immunoflorescence staining was utilized to determine the phenotype of BrdU positive nuclei. Cell counting was performed by an individual who was blinded to experimental groups.
Western blot analysis
After MCAO animals were randomized to control and treatment groups (n=5 per group) and injected i.p. with either saline or citalopram beginning 24 hrs after stroke and daily for 7 days. Animals were then sacrificed upon finishing the 7 day treatment. Tissues were collected from the peri-infarct region of the cortex to detect changed in BDNF levels. To identify peri-infarct tissue, brains were sectioned into 1-mm coronal sections using a brain matrix. The ischemic core is easily distinguished from the surrounding peri-infarct tissue due to the pale or white appearance. The peri-infarct tissue located 1.2 mm from the edge of the ischemic core was dissected and immediately frozen on dry ice for further processing. Proteins were isolated, electrophoresed, and immunoblotted as previously described (Ogle et al., 2012). Western blots were probed using primary antibodies: anti-BDNF (Santa Cruz Biotechnology) and anti-β-Actin (Sigma) and HRP-labeled secondary antibodies.
Systematic random sampling, cell counting, and microvessel density
After MCAO animals were randomly placed into control and treatment groups (7 per group/per time point) and injected i.p. with either saline or citalopram beginning 24 hrs after stroke and daily for 21 or 28 days. Every 20th brain section across the region of interest was chosen for imaging and cell counting (6 sections per animal, with sections more than 200 μm apart) (Treins et al., 2002). For multistage random sampling, six 40× images from the cortical region supplied by the right MCA were captured in each sampled section. The region of interest was defined as a 1.2 mm region within the perfusion territory of the MCA. Data is represented as the total number of cells per animal in the region of interest.
For neurogenesis, cell counts were performed in the peri-infarct region defined as a 1.2 mm area surrounding the ischemic core. From each animal, 6 sections were selected and imaged. The total number of NeuN-positive cells and those cells determined to be double-labeled by Z-stack imaging with BrdU and NeuN were counted for all sections. Only BrdU positive cells shown by Z stack imaging to overlay with NeuN were counted as BrdU/NeuN positive (Rakic, 2002). The number of double-labeled BrdU-positive and NeuN-positive cells were added and divided by the total number of NeuN-positive cells and expressed as a percentage of new neurons. Angiogenesis was similarly measured as BrdU and collagen IV double positive vessels under Z stack imaging. The total number of co-labeled vessels was divided by the number of collagen IV positive vessels and expressed as a percentage of proliferative vessels. The vessel density was measured using pixel threshold in Image J (NIH). Vessel density was reported as the percentage of vessel area per region of interest as previously described (Nih et al., 2012).
Neural progenito the SVZ and cell migration
After MCAO animals were randomly placed into control and treatment groups (n=5 per group) and injected i.p. with either saline or citalopram beginning 24 hrs after stroke and daily for 7 days. Proliferating cells in the SVZ and their migration towards the ischemic cortex were captured in a series of 6-10 images, depending on the distance of migration, at 10× or 20× magnification with 8 sections analyzed per animal. Analyses were performed on coronal brain section images taken within the region of interest: the SVZ migration tract and peri-infarct region. Cells co-labeled with BrdU and DCX were counted as newly formed neural progenitors. Migration distance (μm) was evaluated from the SVZ to the furthest co-labeled DCX and BrdU positive cell, determined by Z-stack imaging (Nih et al., 2012). Cell counting was performed by an individual who was blinded to experimental groups.
Statistical analyses
All analyses were performed using Graphpad Prism 4.0, Graphpad StatMate statistical software (GraphPad Software, Inc., La Jolla, CA), or IBM SPSS statistics software (IBM, Armonk, NY). Multiple comparisons were performed by one-way or two-way repeated measures ANOVA. Single comparisons were performed using Student's t-test. Changes were identified as significant if the p value was less than 0.05. Mean values are reported with the standard error of the mean (SEM) or standard deviation (SD) as specified.
Results
Citalopram administration after stroke has no significant effect on infarct formation in mice
To determine whether citalopram administration post-stroke could impact ischemic infarct volume, adult male mice were treated 30 min after the focal barrel cortex ischemic stroke with citalopram (10 mg/kg, i.p.) and at 24hr intervals for the next 2 days. The citalopram dosage was chosen based on previous evidence that 10 mg/kg citalopram delivered by i.p. injection to mice increased BDNF levels in the cerebral cortex (Balu et al., 2008). At day 3 after stroke, animals were sacrificed and brain sections were subjected to staining for the measurement of infarct formation. For stroke animals receiving saline-injection, the infarct volume was 5.69±0.79 mm3 (n = 9; Mean±SD). Citalopram-treated stroke animals had an infarct volume of 6.21±0.64 mm3 of total brain volume (n = 9). There was no statistical difference between the two groups (t(16)=0.51, p=0.62).).
Citalopram administration has no effect on cerebral edema 72 hrs after stroke
To determine whether citalopram administration decreases cerebral edema, mice were treated at 30 min, 24 and 48 hrs after stroke with citalopram (10mg/kg; i.p.; n=6 per group). At 72 hrs after stroke, the water content was calculated using methods previously described (Ito et al., 1979). Treatment with citalopram did not significantly affect edema (n=6 per group; p=0.99; two-way ANOVA) (Fig. 1D).
Figure 1. Citalopram administration does not attenuate infarct volume.
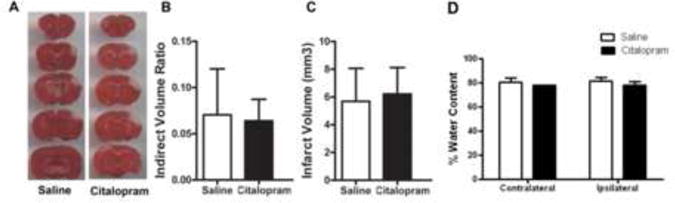
Citalopram was administered at 10 mg/kg (i.p.) 30 min after stroke and again at 24 and 48 hrs after stroke. A. Animals were sacrificed at 3 days after stroke. Infarct analysis was performed by staining brain sections with 2,3,5-triphenyltetrazolium chloride (TTC, red) for live mitochondria in brain cells. The white color area represents dead cells, those negative for TTC staining. B. Indirect volume analyses revealed no significant difference in infarct volume between the saline stroke control and citalopram treatment groups. (n=9 per group; Mean ± SD, p=0.61). C. Students T test revealed no significant differences in infarct volume (mm3) between the saline and citalopram treatment groups. (n=9 per group; Mean ± SD, p=0.73). Water content was measured using the dry-weight method. D. A two-way ANOVA test revealed no significant effect of treatment on cerebral edema after 72 hours. (n=6 per group; Mean ± SD, p=0.10).
Although we could no identify morphological neuroprotection by administration of citalopram, according to previous clinical data, we hypothesized that citalopram might provide long-term functional benefits. This idea is supported by observations that brain infarct volume may not necessary correlate with functional recovery (Schabitz et al., 2004).
Chronic citalopram treatment after stroke enhaces sensorimotor functional recovery
The distal MCA branch ligation targets the somatosensory cortex, primarily affecting whisker and forelimb function. The well defined cortical damage allowed the specific assessment of the sensorimoter function using the adhesive removal (sticky dot) test (Bouet et al., 2009, Freret et al., 2009). After pre-stroke training and baseline recording, mice were tested at days 3 and 14 after stroke and fold change from baseline was calculated. Although the test measures both sensory and motor deficits, the time for removal of the sticky dot from the mouse's paw is more associated with functional deficits in post-stroke animals than time to contact (latency)(Freret et al., 2009, Liu et al., 2012, Ogle et al., 2012). No differences were observed in left or right latency or right (non-affected paw) removal times between saline and citalopram treated animals. Three days after stroke, saline treated animals were significantly delayed in left paw adhesive removal compared to pre-stroke baseline (n=11 per group; t(20)=3.5, p=0.002). On the other hand, no significant delay was detected in citalopram treated animals (10 mg/kg, i.p. 30 min after stroke followed by daily injection thereafter)compared to pre-stroke baseline (n=11 per group; t(20)=1.8, p=0.09). When the left removal time was compared between saline and citalopram mice, however, no statistical difference was detected at this time point (n=11 per group; t(20)=1, p=0.33). After14 days, a 2-way repeated measures ANOVA showed that citalopram treatment had a significant effect on left paw adhesive removal fold change (Eta2 =0.709;Pobserved =0.95;n=11 per group; p=0.004), although not on left or right paw latency or right paw removal fold change (Fig. 2).
Figure 2. Citalopram treatment after stroke significantly enhances sensorimotor functional recovery.
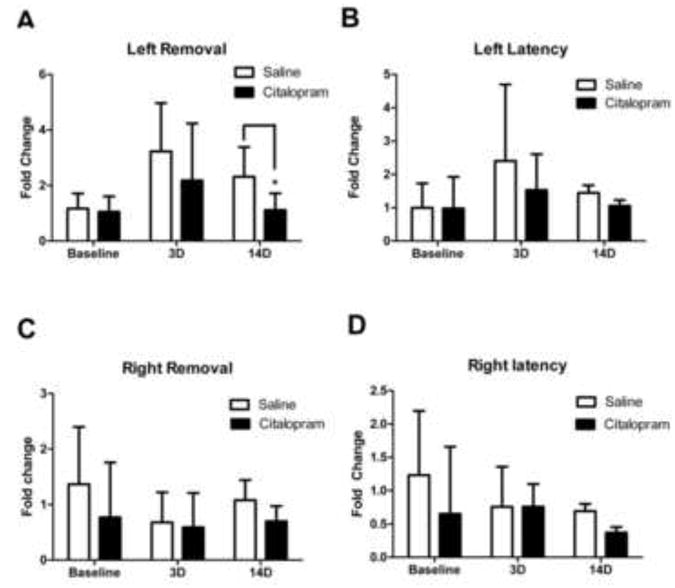
Citalopram or saline treatment was administered starting 24h after MCAO and then daily for 14 days. Sensorimotor function was assessed by the forelimb adhesive removal behavioral test. A. Three days after stroke significant deficits in adhesive removal time were detected by one-way ANOVA in saline-treated mice but not in citalopram-treated mice (n= 11 per group, Mean ± SD, # p<0.05 compared to baseline). After 14 days, citalopram-treated animals displayed improved performance of the sensorimotor task, significantly lower than saline-treated animals and not significantly different from baseline vs controls. 3D: 3 days after stroke; 14D: 14 days after stroke. (n=11 per group, Mean ± SD, * p=0.042, Pobserved =95%). B, C, and D. No significant differences in right or left latency or right removal times were detected by two-way Anova between citalopram and saline treated animals. (n=11 per group, Mean ± SD; p=0.075, p=0.22, and p=0.32, respectively.)
Citalopram treatment enhances neuroblast proliferation and migration after stroke
To understand the cellular mechanism of improved functional performance of citalopram-treated mice, we looked at the proliferation activity in the SVZ and measured migration of neuroblasts along the migration path to the ischemic cortex. The classical proliferation marker BrdU (10 mg/kg, i.p.) was injected daily beginning 3 days after stroke. To evaluate neuroblast migration to the ischemic cortex migrating neuroblasts were visualized along the SVZ-cortex migration path by DCX and BrdU double immunohistochemical staining (Fig. 3). Both stroke control and stroke mice treated with citalopram had neural progenitor migration from the SVZ germinal center; however, the number of DCX and BrdU double labeled cells in the white matter migration tract was significantly increased in the citalopram treated mice (n=5 per group; t(8)=4.4, p=0.009). In the citalopram-treated ischemic brain, DCX and BrdU double labeled cells were migrating significantly further from the SVZ and reaching areas closer to the infarct region compared to saline-treated stroke controls (n=5 per group; t(8)=4.8, p=0.002)(Fig. 3).
Figure 3. Citalopram enhances number and distance of neural precursor migration from the SVZ.
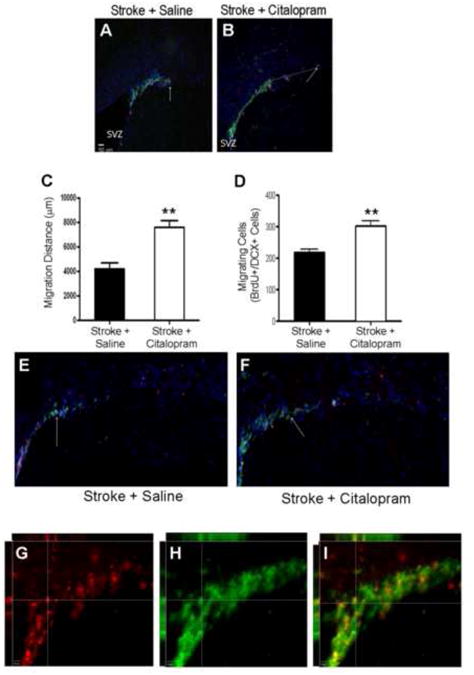
Immunohistochemical staining using specific cell markers was performed in brain sections 7 days after stroke. Migration of neural progenitors was visualized using doublecortin (DCX, green) and bromodeoxyuridine (BrdU, red) shows proliferating cells. A and B. Images of SVZ area show BrdU and DCX positive cells. These cells show different distribution (migration) in the migration track towards the ischemic cortex. Arrows point to some double labeled cells. C and D. The migration distance (μm) was measured using Image J. In the summarized bar graph in C, citalopram treatment showed increased neural progenitor migration distance as compared to control (n=5 per group, Mean ± SEM, ** p=0.009). The graph in D shows the number of BrdU and DCX double positive cells quantified using 3-D imaging. E and F. At 7 days post-stroke, citalopram treated animals showed an increase in the number of BrdU-positive neural progenitors compared to saline treated animals. (n=5 per group, Mean ± SEM, ** P=0.002 vs. control). G,H,I. 3-D Z stack image taken in 20× shows BrdU (G,red) and DCX (H, green) co-localization (I).
Citalopram treatment increases neurogenesis in the post-ischemic brain
In the peri-infarct region 21 and 28 days after stroke, some BrdU-positive cells overlapped with the mature neuronal cell marker NeuN (Fig. 4). These BrdU/NeuN double positive cells have been reported as newly formed neurons (Arvidsson et al., 2002). GFAP and Iba1 were stained with BrdU and NeuN individually to detect astrocytes and microglia. Sections taken from citalopram treated animals showed no triple labeled cells suggesting that those cells counted as BrdU/NeuN double labeled cells are likely not astrocytes or microglia. In the peri-infarct region of citalopram-treated mice, the number of BrdU/NeuN double positive cells and their ratio against the total NeuN-positive cells were significantly higher at 21 and 28 days (n=7 per group, p<0.05 and p<0.01, respectively; two-way ANOVA) compared to that in the saline-treated ischemic brain (Fig. 4).
Figure 4. Citalopram treatment increases new neurons in the peri-infarct region.
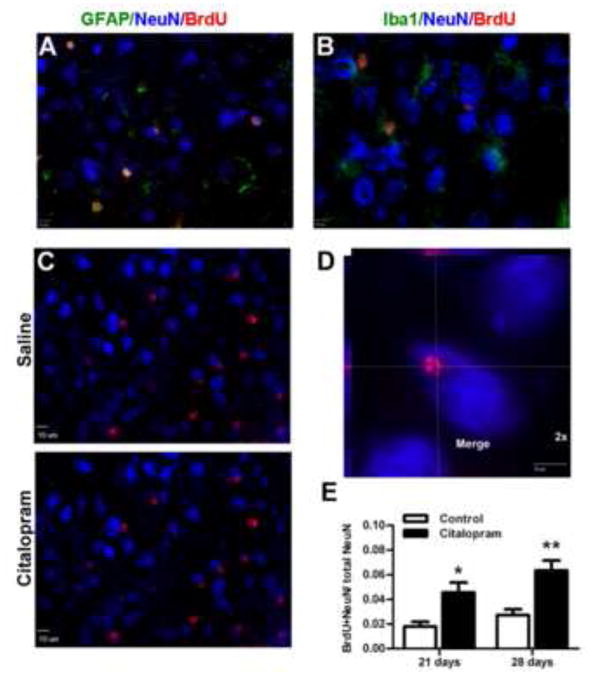
Neurogenesis was visualized using neuronal marker NeuN (blue) and BrdU (red). A. Astrocytes were labeled with GFAP (green) and B microglia were labeled with Iba1(green) along with NeuN and BrdU to illustrate that the quantified NeuN and BrdU co-labeled cells were not astrocytes or microglia. C. Saline treatment and citalopram treated animals have new neurons 28d after stroke. D. High power image in the peri-infarct cortex shows co-localization of NeuN and BrdU. E. Quantification of the number of co-labeled NeuN and BrdU labeled cells (new neurons) in the peri-infarct region. Both 21d and 28d citalopram treatment groups had significantly more new neurons in the peri-infarct region compared to the saline treated animals. (n=7 per group, Mean ± SEM, ** p<0.01, * p<0.05).
Citalopram treatment after stroke increases peri-infarct vessel density
To determine the effect of citalopram on the vasculature of the penumbra after stroke, brain sections were stained with collagen IV to mark the basal lamina of vessels in mice receiving BrdU injections. Citalopram-treated animals showed a strong trend of higher percentage of BrdU/Collagen IV double positive cells, although it did not reach statistical significance as determined by two-way ANOVA (n=7 per group; p=0.054) (Fig. 5D). Vessel density (vessel area relative to area of the region of interest), however, was significantly higher in the peri-infarct region of citalopram-treated animals compared to saline-treated animals at both 21 and 28 days using two-way ANOVA (n=7 per group; p<0.01 and p<0.05, respectively) (Fig. 5E).
Figure 5. Citalopram treatment enhances vessel representation in the peri-infarct region.
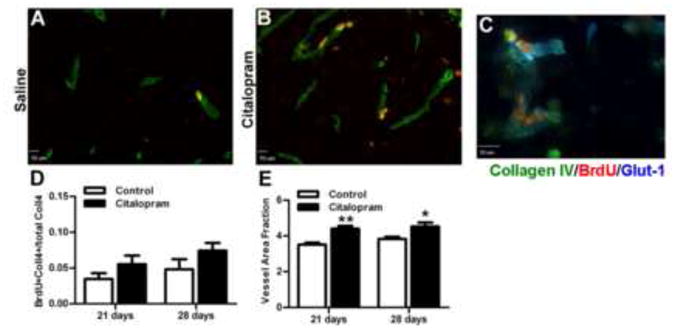
Vessels were visualized and counted using vessel basal lamina marker Collagen IV and dividing cells were marked using BrdU. A Immuno-staining for vessels (collagen IV, green) and newly divided cells (BrdU, red) after 28d saline treatment and B citalopram treatment. C. High power image in the peri-infarct shows endothelial cell marker (Glut-1,blue) co-labeled with Collagen IV, and BrdU showing vessel stain specificity. D. Quantification of the number of co-labeled Collagen IV and BrdU vessels signifying proliferating vessels in the peri-infarct region (n=7 per group, Mean ± SEM, p>0.05). D. Vessel density (vessel density/ area of interest) in the peri-infarct. Citalopram treated animals had significantly greater vessel area fraction compared to saline treated groups. (n=7 per group, Mean ± SEM,** p<0.01, * p<0.05).
Citalopram treatment after stroke increases BDNF expression in the peri-infarct cortex
Peri-infarct protein expression was assessed 7 days after stroke with or without citalopram treatment using Western blot analysis and students t test. Citalopram treatment induced significantly higher expression of BDNF compared to saline-treated animals (n=5 per group; t(7)=4.4, p=0.003) (Fig. 6).
Figure 6. Citalopram induces BDNF expression in the peri-infarct region.
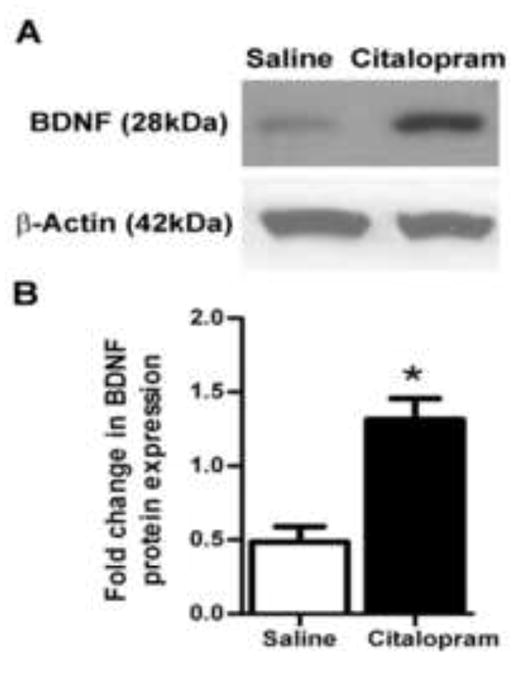
Western blot analysis of BDNF expression in the peri-infarct region (surrounding area 1.2 mm from the edge of the ischemic core). A Western blot gels performed 7 days after stroke. B. Summarized data from assays in A showing significant increase in BDNF protein expression in the peri-infarct region of the citalopram-treated brain (n=5, Mean ± SEM, *p=0.003 vs. control).
Discussion
The present investigation shows that the clinical anti-depression drug citalopram provides functional benefits when administrated after ischemic stroke. Citalopram did not significantly reduce infarct formation measured by TTC staining or cerebral edema measured by dry-weight measure, suggesting that this functional benefit is not mainly due to neuroprotection. Interestingly, citalopram treatment stimulated neuroblast proliferation, enhanced cell migration from the SVZ to the peri-infarct region, and increased new neurons and microvessel density in the peri-infarct region 7-28 days after the ischemic insult. The increased expression of BDNF protein in the peri-infarct cortex occurs in parallel with the cellular changes, implying a potential signaling pathway underlying the improved tissue repair process.
Recent clinical trials have found that SSRI treatment enhanced motor recovery after stroke (Acler et al., 2009, Chollet et al., 2011, Mikami et al., 2011); however, the mechanisms behind this clinical functional recovery are not characterized. In treating depression, SSRIs increase neurotrophic factors and subsequently stimulate neurogenesis and synaptogenesis (Rantamaki et al., 2007, Castren, 2009). In a SSRI post-stroke clinical trial, patients were treated with citalopram for three months and then monitored for up to a year. The subjects treated with SSRIs had better scores for motor recovery, supporting that SSRIs may foster a continuous and long-lasting recovery (Mikami et al., 2011b). We hypothesized that SSRIs might function in a similar manner after stroke by enhancing neurotrophic factors during the recovery process, promoting neurovascular repair. To the best of our knowledge, this is the first investigation to directly examine the potential functional benefits and cellular/molecular mechanisms underlying the citalopram therapy for ischemic stroke.
The stroke model utilized in the current study produces a small focal ischemic stroke in the mouse whisker barrel cortex and forelimb motor cortex. The stroke model mimics the most prevalent human stroke, a small infarct involving 4.5-14% of the ipsilateral hemisphere (Carmichael, 2005). The adhesive removal behavioral task is a sensitive indicator of sensorimotor deficits caused by sensorimotor cortex damage after MCA occlusion-induced focal ischemia (Schabitz et al., 2007, Mohajerani et al., 2011). Three days after stroke, citalopram-treatment seems to abrogate the significant sensorimotor deficits caused by the distal focal ischemia model while no gross differences are observed in infarct volume or cerebral edema in these animals. Since the region of infarct that contributes to functional deficits in forepaw function is relatively small, it was not surprising that citalopram was able to show functional benefits without significant reduction in the gross infarct volume. The functional behavior test may be more sensitive than the gross infarct analysis. At 14 days after stroke, citalopram-treated animals were significantly faster at removing the adhesive, showing enhanced motor recovery compared to saline treated animals. Right paw removal times were also measured in our experiments as a control. Our study is limited by the use of a single behavior test and future studies would benefit from the use of other functional recovery measures. Additionally, differences in cognitive recovery or learning after citalopram treatment were not evaluated in this study. Future investigations should evaluate long-term behavioral benefits after drug treatment has concluded.
SSRIs such as citalopram are blood-brain barrier permeable and are designed to enhance monoaminergic neurotransmitters at synaptic clefts (Gardier et al., 1996, Russo-Neustadt and Chen, 2005). Serotonin increase leads to a time-dependent upregulation in neurotrophic factors, namely BDNF (Balu et al., 2008, Castren, 2009). Additionally, citalopram (10 mg/kg i.p) given to rodents with occipital bulbectomies, a well established depression model, restored the neural precursor proliferation and neural differentiation (Jaako-Movits et al., 2006). According to the neurotrophin depression theory, downstream activation of BDNF-regulated signaling pathways is the primary mediator of the SSRI anti-depressant effect (Russo-Neustadt and Chen, 2005). Enhanced serotonin and BDNF levels stimulate the formation of new neurons in the dentate gyrus and the SVZ, which are areas known to undergo adult neurogenesis (Jacobs et al., 2000, Schabitz et al., 2007). BDNF may also play a positive role in the survival and migration of SVZ-derived neural progenitors. The observed migration of progenitors to the peri-infarct region is a natural response to focal ischemia. This endogenous response was enhanced at 7 days by citalopram treatment (Lindmark, 1988, Li et al., 2008). The increased neural precursor cells in the migration track of citalopram treated animals along with the motor recovery seen at 14 days after stroke suggest that neurogenesis may play an important role in motor recovery. The enhanced endogenous migration in citalopram-treated mice may explain why functional recovery was observed at 14 days after stroke. Additionally, citalopram administration after ischemia up-regulated BDNF protein expression after 7 days in the peri-infarct cortex and enhanced the number of new neurons present in the peri-infarct region after 21 and 28 days treatment. Whether these new neuronal cells can form functional neural networks remains to be tested, yet the sensorimotor improvement indicates some degree of tissue repair and/or plasticity must take place for the observed functional recovery.
Neurogenesis and neuroblast migration are highly associated with the vasculature after stroke. Remodeling of vessels in the peri-infarct region after stroke may play a role in the observed recruitment of newly born neurons from the SVZ to the peri-infarct region (Ohab et al., 2006). Endothelial cell survival and vascular proliferation are enhanced by BDNF (Chen et al., 2003), which was increased with citalopram treatment. Vessel regeneration and protection are very important for the overall stroke outcome. In a recent clinical trial, pro-angiogenic plasma factors after stroke correlated with a better NIHSS outcome, where the presence of anti-angiogenic factors in patient plasma predicted a worse long-term recovery (Navarro-Sobrino et al., 2011). We show that vessel density in the peri-infarct region was significantly increased by citalopram, which may imply that there was either less endothelial damage and/or enhanced endothelial proliferation in the citalopram treatment group. Counting of BrdU-labeled vessels did not produce a statistical difference between groups, although there was a strong trend for more angiogenesis in the citalopram group. Augmented localization of new neurons and higher vessel density in the peri-infarct region should contribute to regeneration and repair of damaged cortex that leads to improved sensorimotor function with citalopram treatment.
SSRIs are FDA-approved, inexpensive, clinically accessible and safe drugs for use by both depressed and non-depressed stroke patients (Wiart et al., 2000, Gainotti et al., 2001). SSRI antidepressants were administered to patients following stroke for 12 weeks and mortality was assessed 9 years later. This study showed that treatment during the first 6 months post-stroke significantly increased the survival of both depressed and non-depressed patients. This study suggests that antidepressant treatment can modify post-stroke mortality risk from cardiovascular disease and recurrent strokes (Jorge et al., 2003). Potential side effects of Celexa (sitalopram hydrobromide) as reported by the FDA include serotonin syndrome, which can cause racing heartbeat and high or low blood pressure among several other symptoms. However, as measured by several clinical trials for stroke and depression, citalopram has not been reported to significantly increase blood pressure or pulse rate (Pederson et al., 1982). Taken togetherour findings and these previous studies suggest that citalopram treatment promotes the restoration of motor function and may provide sustained recovery after stroke. In addition, we provide novel evidence suggesting that citalopram promotes functional recovery via increased neurovascular regeneration and these results are consistent with SSRI studies indicating that anti-depressants function through delayed mechanisms (Rantamaki et al., 2007). In congruence with several clinical trials, our data support the idea that post-stroke treatment using SSRIs such as citalopram could potentially offer a new, safe avenue for stroke therapy.
Highlights.
Citalopram treatment enhances neural progenitor migration
Citalopram treatment enhances neurogenesis in the peri-infarct cortex
Citalopram treatment increases vessel peri-infarct representation
Citalopram treatment increases BDNF expression in the peri-infarct region
Citalopram treatment after stroke promotes sensorimotor recovery
Acknowledgments
This work was supported by National Institutes of Health Grants NS045810, NS062097, NS058710; and an American Heart Association EIA award 0840110N.
Abbreviations
- SVZ
sub-ventricular zone
- SSRI
selective serotonin reuptake inhibitor
- FMMS
Fugl-Meyer motor scale
- NIHSS
National Institutes of Health stroke scale
- MCA
middle cerebral artery
- MCAO
middle cerebral artery occlusion
- BDNF
brain derived neurotrophic factor
- CCA
common carotid ateries
- TTC
2,3,5-triphenyltetrazolium chloride
- DCX
doublecortin
- BrdU
bromodeoxyuridine
- NeuN
neuronal nuclei
- TrkB
TrkB tyrosine kinase
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acler M, Robol E, Fiaschi A, Manganotti P. A double blind placebo RCT to investigate the effects of serotonergic modulation on brain excitability and motor recovery in stroke patients. J Neurol. 2009;256:1152–1158. doi: 10.1007/s00415-009-5093-7. [DOI] [PubMed] [Google Scholar]
- Anderson CS, Jamrozik KD, Broadhurst RJ, Stewart-Wynne EG. Predicting survival for 1 year among different subtypes of stroke. Results from the Perth Community Stroke Study. Stroke. 1994;25:1935–1944. doi: 10.1161/01.str.25.10.1935. [DOI] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- Balu DT, Hoshaw BA, Malberg JE, Rosenzweig-Lipson S, Schechter LE, Lucki I. Differential regulation of central BDNF protein levels by antidepressant and non-antidepressant drug treatments. Brain Res. 2008;1211:37–43. doi: 10.1016/j.brainres.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilge C, Kocer E, Kocer A, Turk Boru U. Depression and functional outcome after stroke: the effect of antidepressant therapy on functional recovery. Eur J Phys Rehabil Med. 2008;44:13–18. [PubMed] [Google Scholar]
- Bouet V, Boulouard M, Toutain J, Divoux D, Bernaudin M, Schumann-Bard P, Freret T. The adhesive removal test: a sensitive method to assess sensorimotor deficits in mice. Nat Protoc. 2009;4:1560–1564. doi: 10.1038/nprot.2009.125. [DOI] [PubMed] [Google Scholar]
- Carmichael ST. Rodent models of focal stroke: size, mechanism, and purpose. NeuroRx. 2005;2:396–409. doi: 10.1602/neurorx.2.3.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castren E. [Neural plasticity and recovery from depression] Duodecim. 2009;125:1781–1786. [PubMed] [Google Scholar]
- Chen J, Zhang ZG, Li Y, Wang Y, Wang L, Jiang H, Zhang C, Lu M, Katakowski M, Feldkamp CS, Chopp M. Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann Neurol. 2003;53:743–751. doi: 10.1002/ana.10555. [DOI] [PubMed] [Google Scholar]
- Chollet F, Tardy J, Albucher JF, Thalamas C, Berard E, Lamy C, Bejot Y, Deltour S, Jaillard A, Niclot P, Guillon B, Moulin T, Marque P, Pariente J, Arnaud C, Loubinoux I. Fluoxetine for motor recovery after acute ischaemic stroke (FLAME): a randomized placebo-controlled trial. Lancet Neurol. 2011;10:123–130. doi: 10.1016/S1474-4422(10)70314-8. [DOI] [PubMed] [Google Scholar]
- Freret T, Bouet V, Leconte C, Roussel S, Chazalviel L, Divoux D, Schumann-Bard P, Boulouard M. Behavioral deficits after distal focal cerebral ischemia in mice: Usefulness of adhesive removal test. Behav Neurosci. 2009;123:224–230. doi: 10.1037/a0014157. [DOI] [PubMed] [Google Scholar]
- Gainotti G, Antonucci G, Marra C, Paolucci S. Relation between depression after stroke, antidepressant therapy, and functional recovery. J Neurol Neurosurg Psychiatry. 2001;71:258–261. doi: 10.1136/jnnp.71.2.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardier AM, Malagie I, Trillat AC, Jacquot C, Artigas F. Role of 5-HT1A autoreceptors in the mechanism of action of serotoninergic antidepressant drugs: recent findings from in vivo microdialysis studies. Fundam Clin Pharmacol. 1996;10:16–27. doi: 10.1111/j.1472-8206.1996.tb00145.x. [DOI] [PubMed] [Google Scholar]
- Greene J, Banasr M, Lee B, Warner-Schmidt J, Duman RS. Vascular endothelial growth factor signaling is required for the behavioral actions of antidepressant treatment: pharmacological and cellular characterization. Neuropsychopharmacology. 2009;34:2459–2468. doi: 10.1038/npp.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito U, Ohno K, Nakamura R, Suganuma F, Inaba Y. Brain edema during ischemia and after restoration of blood flow. Measurement of water, sodium, potassium content and plasma protein permeability. Stroke. 1979;10:542–547. doi: 10.1161/01.str.10.5.542. [DOI] [PubMed] [Google Scholar]
- Jaako-Movits K, Zharkovsky T, Pedersen M, Zharkovsky A. Decreased hippocampal neurogenesis following olfactory bulbectomy is reversed by repeated citalopram administration. Cell Mol Neurobiol. 2006;26:1559–1570. doi: 10.1007/s10571-006-9090-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs BL, van Praag H, Gage FH. Adult brain neurogenesis and psychiatry: a novel theory of depression. Mol Psychiatry. 2000;5:262–269. doi: 10.1038/sj.mp.4000712. [DOI] [PubMed] [Google Scholar]
- Jorge RE, Robinson RG, Arndt S, Starkstein S. Mortality and poststroke depression: a placebo-controlled trial of antidepressants. Am J Psychiatry. 2003;160:1823–1829. doi: 10.1176/appi.ajp.160.10.1823. [DOI] [PubMed] [Google Scholar]
- Kermani P, Hempstead B. Brain-derived neurotrophic factor: a newly described mediator of angiogenesis. Trends Cardiovasc Med. 2007;17:140–143. doi: 10.1016/j.tcm.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuzberg M, Kanov E, Timofeev O, Schwaninger M, Monyer H, Khodosevich K. Increased subventricular zone-derived cortical neurogenesis after ischemic lesion. Exp Neurol. 2010;226:90–99. doi: 10.1016/j.expneurol.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Li WL, Yu SP, Ogle ME, Ding XS, Wei L. Enhanced neurogenesis and cell migration following focal ischemia and peripheral stimulation in mice. Dev Neurobiol. 2008;68:1474–1486. doi: 10.1002/dneu.20674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lu Z, Keogh CL, Yu SP, Wei L. Erythropoietin-induced neurovascular protection, angiogenesis, and cerebral blood flow restoration after focal ischemia in mice. J Cereb Blood Flow Metab. 2007;27:1043–1054. doi: 10.1038/sj.jcbfm.9600417. [DOI] [PubMed] [Google Scholar]
- Lindmark B. Evaluation of functional capacity after stroke with special emphasis on motor function and activities of daily living. Scand J Rehabil Med Suppl. 1988;21:1–40. [PubMed] [Google Scholar]
- Lindmark B, Hamrin E. Evaluation of functional capacity after stroke as a basis for active intervention. Presentation of a modified chart for motor capacity assessment and its reliability. Scand J Rehabil Med. 1988;20:103–109. [PubMed] [Google Scholar]
- Liu Z, Li Y, Zhang L, Xin H, Cui Y, Hanson LR, Frey WH, Chopp M. Subacute intranasal administration of tissue plasminogen activator increases functional recovery and axonal remodeling after stroke in rats. Neurobiol Dis. 2012;45:804–809. doi: 10.1016/j.nbd.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikami K, Jorge RE, Adams HP, Jr, Davis PH, Leira EC, Jang M, Robinson RG. Effect of antidepressants on the course of disability following stroke. Am J Geriatr Psychiatry. 2011;19:1007–1015. doi: 10.1097/JGP.0b013e31821181b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno M, Yamada K, Olariu A, Nawa H, Nabeshima T. Involvement of brain-derived neurotrophic factor in spatial memory formation and maintenance in a radial arm maze test in rats. J Neurosci. 2000;20:7116–7121. doi: 10.1523/JNEUROSCI.20-18-07116.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohajerani MH, Aminoltejari K, Murphy TH. Targeted mini-strokes produce changes in interhemispheric sensory signal processing that are indicative of disinhibition within minutes. Proc Natl Acad Sci U S A. 2011;108:E183–191. doi: 10.1073/pnas.1101914108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Sobrino M, Rosell A, Hernandez-Guillamon M, Penalba A, Boada C, Domingues-Montanari S, Ribo M, Alvarez-Sabin J, Montaner J. A large screening of angiogenesis biomarkers and their association with neurological outcome after ischemic stroke. Atherosclerosis. 2011;216:205–211. doi: 10.1016/j.atherosclerosis.2011.01.030. [DOI] [PubMed] [Google Scholar]
- Nih LR, Deroide N, Lere-Dean C, Lerouet D, Soustrat M, Levy BI, Silvestre JS, Merkulova-Rainon T, Pocard M, Margaill I, Kubis N. Neuroblast survival depends on mature vascular network formation after mouse stroke: role of endothelial and smooth muscle progenitor cell co-administration. Eur J Neurosci. 2012;35:1208–1217. doi: 10.1111/j.1460-9568.2012.08041.x. [DOI] [PubMed] [Google Scholar]
- Ogle ME, Gu X, Espinera AR, Wei L. Inhibition of prolyl hydroxylases by dimethyloxaloylglycine after stroke reduces ischemic brain injury and requires hypoxia inducible factor-1alpha. Neurobiol Dis. 2012;45:733–742. doi: 10.1016/j.nbd.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohab JJ, Fleming S, Blesch A, Carmichael ST. A neurovascular niche for neurogenesis after stroke. J Neurosci. 2006;26:13007–13016. doi: 10.1523/JNEUROSCI.4323-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen OL, Kragh-Sorensen P, Bjerre M, Overo KF, Gram LF. Citalopram, a selective serotonin reuptake inhibitor: clinical antidepressive and long-term effect-a phase II study. Psychopharmacology (Berl) 1982;77:199–204. doi: 10.1007/BF00464566. [DOI] [PubMed] [Google Scholar]
- Rakic P. Adult neurogenesis in mammals: an identity crisis. J Neurosci. 2002;22:614–618. doi: 10.1523/JNEUROSCI.22-03-00614.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantamaki T, Hendolin P, Kankaanpaa A, Mijatovic J, Piepponen P, Domenici E, Chao MV, Mannisto PT, Castren E. Pharmacologically diverse antidepressants rapidly activate brain-derived neurotrophic factor receptor TrkB and induce phospholipase-Cgamma signaling pathways in mouse brain. Neuropsychopharmacology. 2007;32:2152–2162. doi: 10.1038/sj.npp.1301345. [DOI] [PubMed] [Google Scholar]
- Russo-Neustadt AA, Chen MJ. Brain-derived neurotrophic factor and antidepressant activity. Curr Pharm Des. 2005;11:1495–1510. doi: 10.2174/1381612053764788. [DOI] [PubMed] [Google Scholar]
- Schabitz WR, Berger C, Kollmar R, Seitz M, Tanay E, Kiessling M, Schwab S, Sommer C. Effect of brain-derived neurotrophic factor treatment and forced arm use on functional motor recovery after small cortical ischemia. Stroke. 2004;35:992–997. doi: 10.1161/01.STR.0000119754.85848.0D. [DOI] [PubMed] [Google Scholar]
- Schabitz WR, Steigleder T, Cooper-Kuhn CM, Schwab S, Sommer C, Schneider A, Kuhn HG. Intravenous brain-derived neurotrophic factor enhances poststroke sensorimotor recovery and stimulates neurogenesis. Stroke. 2007;38:2165–2172. doi: 10.1161/STROKEAHA.106.477331. [DOI] [PubMed] [Google Scholar]
- Stahl SM. Mechanism of action of serotonin selective reuptake inhibitors. Serotonin receptors and pathways mediate therapeutic effects and side effects. J Affect Disord. 1998;51:215–235. doi: 10.1016/s0165-0327(98)00221-3. [DOI] [PubMed] [Google Scholar]
- Towfighi A, Saver JL. Stroke declines from third to fourth leading cause of death in the United States: historical perspective and challenges ahead. Stroke. 2011;42:2351–2355. doi: 10.1161/STROKEAHA.111.621904. [DOI] [PubMed] [Google Scholar]
- Treins C, Giorgetti-Peraldi S, Murdaca J, Semenza GL, Van Obberghen E. Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-i kinase/target of rapamycin-dependent signaling pathway. J Biol Chem. 2002;277:27975–27981. doi: 10.1074/jbc.M204152200. [DOI] [PubMed] [Google Scholar]
- Wiart L, Petit H, Joseph PA, Mazaux JM, Barat M. Fluoxetine in early poststroke depression: a double-blind placebo-controlled study. Stroke. 2000;31:1829–1832. doi: 10.1161/01.str.31.8.1829. [DOI] [PubMed] [Google Scholar]