

Abstract
Autophagy may contribute to ischemia-induced cell death in the brain, but the regulation of autophagic cell death is largely unknown. Nuclear factor kappa B (NF-κB) is a regulator of apoptosis in cerebral ischemia. We examined the hypothesis that autophagy-like cell death could contribute to ischemia-induced brain damage and the process was regulated by NF-κB. In adult wild-type (WT) and NF-κB p50 knockout (p50−/−) mice, focal ischemia in the barrel cortex was induced by ligation of distal branches of the middle cerebral artery. Twelve to 24 h later, autophagic activity increased as indicated by enhanced expression of Beclin-1 and LC3 in the ischemic core and/or penumbra regions. This increased autophagy contributed to cell injury, evidenced by terminal deoxynucleotidyltransferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL) co-staining and a protective effect achieved by the autophagy inhibitor 3-methyladenine. The number of Beclin-1/TUNEL-positive cells was significantly more in p50−/− mice than in WT mice. Neuronal and vascular cell death, as determined by TUNEL-positive cells co-staining with NeuN or Collagen IV, was more abundant in p50−/− mice. Immunostaining of the endothelial cell tight junction marker occludin revealed more damage to the blood–brain barrier in p50−/− mice. Western blotting of the peri-infarct tissue showed a reduction of Akt-the mammalian target of rapamycin (mTOR) signaling in p50−/− mice after ischemia. These findings provide the first evidence that cerebral ischemia induced autophagy-like injury is regulated by the NF-κB pathway, which may suggest potential treatments for ischemic stroke.
Keywords: autophagy, cerebral ischemia, NF-kappaB, neurovascular unit, blood-brain barrier, Akt, the mammalian target of rapamycin (mTOR)
INTRODUCTION
Autophagy is an evolutionary mechanism conserved in eukaryotic cells that starts with the formation of an autophagosome, enclosed within a double membrane that engulfs part of the cytoplasm. In mammalian cells, autophagosomes undergo a maturation process by fusing with the endocytic compartment and lysosomes (Eskelinen, 2005). In a broad sense, autophagy can be defined as a cellular process involved in the breakdown of intracellular proteins and organelles via lysosomal degradation. There are three pathways for autophagy in mammalian cells: microautophagy, chaperone-mediated autophagy and macroautophagy. The last one is the most common and active form of autophagy that many investigations, including the present study, have focused on and referred to. Autophagy is active at a basal level in most of the cells and this reflects its role in cell homeostasis by regulating the turnover of long-lived proteins and getting rid of damaged structures. Autophagy can be stimulated by various stress signals such as nutrient depletion. During nutrient shortage, autophagy provides the constituents required to maintain the metabolism essential for survival (Lum et al., 2005). In addition to its survival role, autophagy has also been implicated as a cell death pathway in recent years (Shacka et al., 2008). It is commonly agreed that, under pathological conditions, autophagy can act as a double-edged sword, contributing either to cell survival or cell damage (Shintani and Klionsky, 2004; Shaw and Kirshenbaum, 2008).
Cerebral ischemia induces cell death via necrosis and apoptosis or mixed “hybrid” death mechanisms that result in concurrent necrosis and apoptosis in the same cells (Wei et al., 2004, 2006). Some believe that in addition to necrosis and apoptosis, autophagy is a third type of cell death mechanism (Rami et al., 2008; Balduini et al., 2009). Until recently, however, whether autophagy is a pro-survival mechanism or contributes to cell death after cerebral ischemia has not been well defined. The mechanism regulating ischemia-stimulated autophagy is largely unknown. The present investigation aimed to understand whether ischemia-triggered autophagy contributed to brain tissue damage and whether it was regulated by an intracellular signaling pathway, specifically nuclear factor kappa B (NF-κB).
NF-κB regulates the expression of genes involved in a broad range of biological processes. In the central nervous system, NF-κB plays a dual role in neuronal survival following nerve injury. It participates in the regulation of apoptotic and inflammatory genes (Pizzi and Spano, 2006). A possible role of NF-κB genes in ischemia-induced activities was originally illustrated in clinical studies examining the expression of NF-κB family members in brain tissues of patients who died after stroke. Increased expression of RelA and p50 (NF-κB1) was detected in the ischemic and penumbra regions, implicating a regulatory role of these NF-κB subunit genes after ischemic stroke (Terai et al., 1996; Nurmi et al., 2004). In animal models of transient and permanent occlusion of the middle cerebral artery (MCA), the expression of NF-κB members p65 and p50 increased after cerebral ischemia (Stephenson et al., 2000). Several neuroprotective reagents showed an effect of decreasing NF-κB expression and/or activity, suggesting that NF-κB contributes to ischemic brain damage (Chen et al., 2009; Garg et al., 2010; Wang et al., 2010). In loss-of-function experiments, inhibition or knockdown of NF-κB members such as RelA and p50 leads to decreased brain damage in experimental stroke models (Harari and Liao, 2010).
However, the role of NF-κB in ischemic pathogenesis is much more complicated and conflicting data have been reported in similar stroke models. For example, in p50-deficient mouse increased neurodegeneration including apoptotic cell death and infarct formation and reduced regenerative activity were observed after transient and permanent MCA occlusion (Duckworth et al., 2006; Li et al., 2008a). Our own investigations demonstrated a neuroprotective role of the NF-κB p50 activity after ischemic stroke and during the development of the neonatal brain (Lu et al., 2006, Li et al., 2008a). Currently, available data suggest that the outcome of NF-κB expression and activation in the ischemic brain may depend on interplays among the multiple functions of the specific NF-κB family members and the balance between their proinflammatory effects and antiapoptotic effects, as well as the location and levels of NF-κB expressions (Harari and Liao, 2010). Furthermore, this balance may be under different regulations for neurons and other cell types in the ischemic core and penumbra regions, at different times. The fate of any given cell also depends (in addition to other factors) on both the subunit composition and temporospatial expression of NF-κB (Nijboer et al., 2008). To better understand the functional complexity of the NF-κB signaling in the ischemic brain, revealing of additional effects of NF-κB family members on cellular events is necessary and may help to explain the conflicting roles of this key regulatory mechanism in the post-ischemic brain.
A direct cross-talk between the NF-κB signaling and autophagic activity has been regarded as a new frontier for deciphering the role of autophagy in diverse biological processes (Xiao, 2007). In the NF-κB regulation of autophagy, the Akt-mTOR activity may play a mediator role, and both negative and positive roles of its signaling have been reported (Xiao et al., 2006; Degtyarev et al., 2008; Mehrpour et al., 2010). To better understand the cell death mechanism after ischemic stroke, the present investigation on wild-type (WT) and p50 knockout mice explored the role of the NF-κB subunit in autophagy of the ischemic brain. We also inspected the potential role of the mTOR activity in ischemia-induced autophagy-like cell death.
EXPERIMENTAL PROCEDURES
Experimental animals
Adult wild-type (WT, B6, 129PF2) and p50 knockout (p50−/−, B6, 129P-Nfkb1) mice (male, 2.5–3 month-old, body weight = 25–30 g; Jackson Laboratories, Bar Harbor, ME, USA) were used in the experiments. Animals were maintained at room temperature (~23 °C) with a 12-h light/dark cycle in the pathogen-free Laboratory Animal Center with free access to food and water. N ≥ 6 per experimental group in the in vivo experiments.
Barrel cortex ischemic stroke in mice
Animal experiments and surgery procedures were approved by the University Animal Research Committee and met National Institutes of Health (NIH) standards. Surgical procedures were modified from a previously described protocol (Li et al., 2008b). Briefly, mice were subjected to 3% isoflurane anesthesia in a mixture of 70% N2O and 30% O2. The MCA branches supplying the right barrel cortex were permanently ligated by a 10-0 suture (Surgical Specialties Co., Reading, PA, USA). This was accompanied by 10-min ligation of the bilateral common carotid artery (CCA). During surgery and recovery periods, body temperature was monitored with a rectal probe and maintained at 37.0±0.5 °C using a temperature control unit and heating pad. In sham-operated animals, the same procedure was performed without ligation of the MCA and CCAs. Animals were euthanized by decapitation between 12 h to 7 days after ischemic stroke, depending on specific experimental designs. The brain was immediately removed and preserved in OCT (optimum cutting temperature) compound (Sakura Finetek, Inc., Torrance, CA, USA) at −80 °C for further processing.
Immunofluorescence staining
Flash frozen brains were sliced into coronal sections of 10-lm thick using a cryostat vibratome (Ultapro 5000, St. Louis, MO, USA). After air drying, slices were fixed in 10%-buffered formalin phosphate for 10 min, followed by treatments in a −20 °C ethanol: acetic acid (2:1) solution for 12 min, in 0.2% Triton-100 for 5 min and washed with phosphate buffered saline (PBS) three times between each step. Slides were blocked in 1% gelatin from cold water fish (Sigma, St. Louis, MO, USA) diluted in PBS at room temperature for 1 h, and subsequently incubated with primary antibodies diluted in PBS overnight at 4 °C. Primary antibodies used for single or double staining were as follows: rabbit anti-Beclin-1 (1:200; Chemicon, Temecula, CA, USA), mouse anti-NeuN (1:400; Chemicon), rabbit polyclonal anti-glial fibrillary acidic protein (GFAP; 1:500; Sigma), goat anti-Collagen IV (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA, USA) and mouse anti-occludin (1:500, Invitrogen, Carlsbad, CA, USA). After rinsing with PBS, brain sections were then treated with secondary antibody Cy3-conjugated anti-mouse, anti-rabbit or anti-goat IgG (Jackson ImmunoResearch, West Grove, PA, USA) for 1.5 h at room temperature. Hoechst 33342 (Molecular Probes, Carlsbad, CA, USA) was applied to stain all the nuclei as a background staining. The brain sections were mounted and cover-slipped, imaged and photographed using either a fluorescence microscope (IX61, Olympus, Tokyo, Japan) or a confocal microscope (BX61; Olympus). At least five brain sections from five animals were included in each experimental group for staining examinations.
Terminal deoxynucleotidyltransferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL) staining
A TUNEL staining kit (DeadEnd Fluorometric TUNEL system, Promega, Madison, WI, USA) was used to visualize cell death in 10-u.m coronal frozen sections, as per kit instructions. To identify neuronal cell death and vasculature damage, TUNEL-staining slides were then incubated with mouse anti-NeuN or goat anti-Collagen IV antibody overnight. Results were visualized by the Olympus fluorescence microscope.
Quantification of immunocytostained cells
For systematic random sampling in design-based stereological cell counting, six coronal brain sections per mouse were selected, spaced 120 μm apart across the region of interest in each animal (from bregma + 0.5 mm to bregma −0.5 mm). For multistage random sampling, six fields per brain section were randomly chosen in the ischemic border region under 20× magnification of a light microscope or in confocal images. Cell counts were performed using ImageJ (National Institutes of Health, Bethesda, MD, USA), compiled, and analyzed. Courting was performed under blind condition to the genotype of mice.
Western blot analysis
Tissue samples were taken from the peri-infarct region of the cortex and proteins were extracted by homogenization in protein lysis buffer (25 mM Tris–HCl, containing 150 mM NaCl, 5 mM EDTA, 0.1% SDS, 2 mM sodium orthovanadate, 100 mM NaF, 1% Triton, leupeptin, aprotinin, and pepstatin, pH = 7.4). Homogenate was centrifuged at 13,000 rpm for 20 min to pellet the insoluble fraction and the supernatant was collected. The protein concentration of each sample was determined using the Bicinchoninic Acid Assay (Sigma). Proteins from each sample (50 μg) were separated by SDS–polyacrylamide gel electrophoresis and electrophoretically transferred to a polyvinylidene difluoride (PVDF) membrane (BioRad, Hercules, CA, USA). Membranes were blocked in 7% evaporated milk, diluted in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) at room temperature for at least 2 h, and then incubated overnight at 4 °C with one of the following primary antibodies: rabbit anti-Beclin-1, light chain 3 (LC3), P-AKt, P-S6K1, P-4E-BP1 and phospho-S6R (P-S6R) (1:500–1:4000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA). Mouse β-actin antibody (Sigma) was used for protein loading control. After primary antibody incubation, membranes were washed with TBS-T and incubated with alkaline phosphatase-conjugated anti-mouse or anti-rabbit IgG antibodies (Promega, Madison, WI, USA) for 2 h at room temperature. Membranes were washed with TBS-T followed by three washes with TBS; signals were detected by the addition of 5-bromo-4-chloro-3-indolylphosphate/nitroblue tetrazolium (BCIP/NBT) solution (Sigma), then quantified and digitally analyzed using the ImageJ program (NIH). The intensity of each band was measured and subtracted from the background. The expression ratio of target proteins was normalized against β-actin. At least five animals per group were used for brain sample collections in Western blot analysis.
Statistical analysis
Multiple comparisons were performed using one-way analysis of variance (ANOVA) followed by the Tukey test for post hoc analysis. GraphPad data analysis software (GraphPad Software, Inc., La Jolla, CA, USA) was used for statistical analysis and making original line and bar graphs. Mean values are reported with the standard deviation (SD) which provides the information of the dispersion of the measured data set around the mean (Altman and Bland, 2005; Schlattmann and Dirnagl, 2010). Changes were identified as significant if the P value was less than 0.05.
RESULTS
Increased autophagy after cerebral ischemia and its regulation by NF-κB
To determine whether cerebral ischemia stimulated autophagy in the brain, we first examined the expression levels of Beclin-1, also known as autophagy-related gene (Atg) 6, and of microtubule-associated protein LC3 in the mouse brain. Both genes are autophagy-related proteins and concurrent increases of both genes indicate autophagic activity (Martinet et al., 2006). In the Beclin-1 immunoreactivity assay of the WT brain, the level was low in the normal cortex of sham-operated mice (Fig. 1). Acutely (3 h) after the focal ischemia in the barrel cortex, neurons in the ischemic core started to show nuclear condensation in immunohistochemical examination (Fig. 1). Using specific antibodies, a few Beclin-1 and TUNEL-positive cells could be seen inside and around the ischemic core. Starting from 6 h after ischemia, Beclin-1 levels were significantly augmented and reached a plateau level from 12 to 24 h after ischemia (Fig. 1). The granular, homogeneous staining pattern of Beclin-1-positive cells was confined almost exclusively to the region of the ischemic core and border regions. The increase in Beclin-1 immunoreactivity gradually subsided 3 days after ischemia and returned to the basal level 7 days after ischemia (Fig. 1).
Fig. 1.
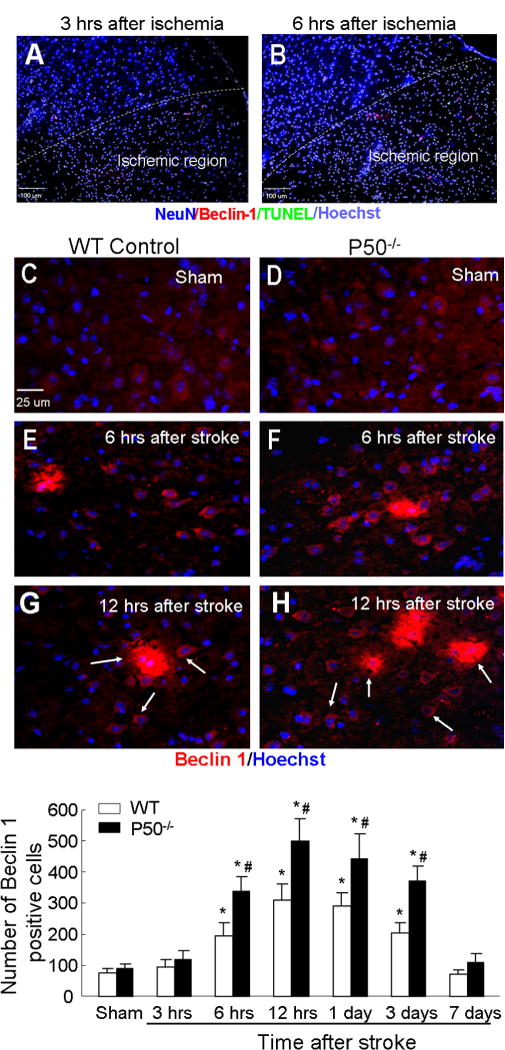
Ischemia-enhanced Beclin-1 immunoreactivity in the brain of WT and NF-κB p50 knockout mice. Immunohistochemical staining was performed at different time points after barrel cortex ischemia in cortex brain sections. (A, B) Demonstration of the ischemic and non-ischemic regions distinguishable by staining with specific antibodies at early time points of 3 and 6 h after barrel cortex ischemia in WT mice. The NeuN/Hoechst 33342 nuclear staining shows condensed nuclei in the ischemic core. A few TUNEL-positive (green) and Beclin-1-positive (red) cells can be seen in the ischemic and surrounding regions. (C–H) Beclin-1 staining (red) was examined in brain sections of sham control, 12 and 24 h post-ischemia from WT and p50−/− mice. Hoechst 33342 (blue) staining revealed nuclei of all cells. There was very weak Beclin-1 immunoreactivity in sham-operated mice (C, D). Increased beclin-1-poistive cells were seen 6 (E, F) and 12 h (G, H) after ischemia. (I) The bar graph shows quantitative assessment of Beclin-1-positive cells in the penumbra. Cell counts show increased numbers of Beclin-1-positive cells in WT and p50−/− mice from 6 h after ischemia. Even higher Beclin-1 staining was seen in p50−/− mice from 6 h to 3 days after ischemia. N = 5 animals in each group. Data are expressed as mean ± SD. *P < 0.05 compared with sham controls, #P < 0.05 compared with WT group. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
To understand the role of NF-κB in the ischemia-induced autophagy, we examined the Beclin-1 expression in p50 knockout (p50−/−) mice. As in the WT brain, the Beclin-1 immunoreactivity was low in sham-operated p50−/− mice (Fig. 1). Following a similar time course in WT mice, Beclin-1 expression in the ischemic and peri-infarct region of p50−/− mice was significantly enhanced from 6 h after ischemia, and reached a peak level at 12 h after the focal ischemia (Fig. 1). The increased level of Beclin-1 expression, however, was much higher in the absence of p50 compared to WT mice (Fig. 1). The increased Beclin-1 expression was confirmed using Western blot analysis. The protein levels of Beclin-1 in WT mice significantly increased 3 days after ischemia, while in p50−/− mice, it was augmented from 12 to 24 h after ischemia and significantly higher than that in WT mice (Fig. 2).
Fig. 2.
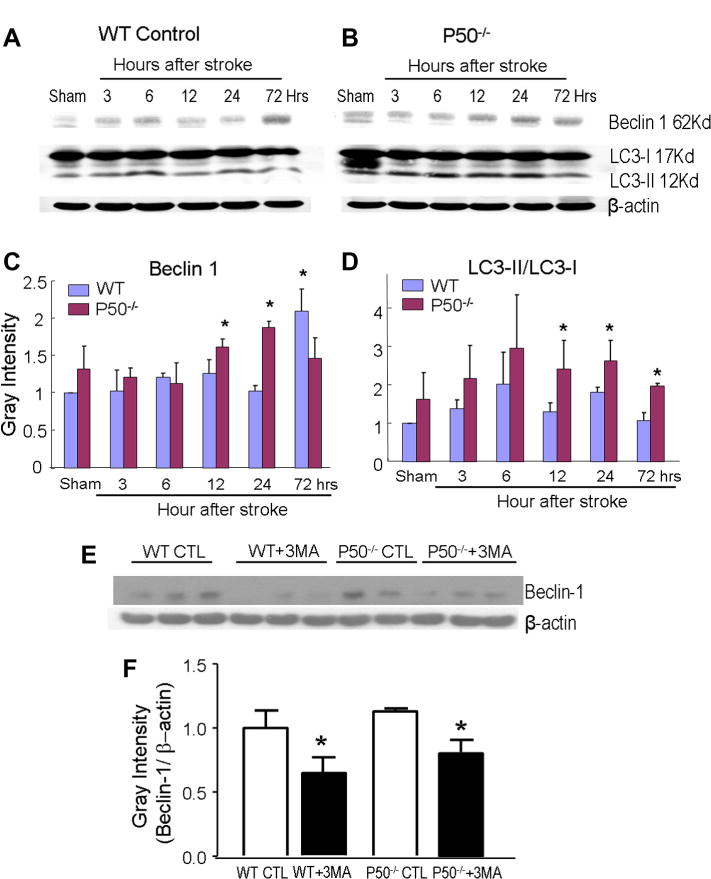
Increased expression of autophagic factors in the ischemic brain of WTand p50−/− mice. (A, B) Western blot analysis of Beclin-1 and LC3-II/LC3-I was performed in the ischemic/penumbra tissues from WT and p50−/− mice at 3–72 h following cerebral ischemia. (C and D) The immunoblotting diagrams show quantified results of protein expression of Beclin-1 and LC3-II/LC3-I, respectively. In densitometry analysis for each factor, gray intensity was normalized against β-actin and quantified using ImageJ software. From 12 to 24 h after ischemia, Beclin-1 and LC3 levels were significantly higher in p50−/− mice than that in WT mice. Beclin-1 expression, however was higher in WT mice 72 h after ischemia. (E, F) Effect of 3-MA on Beclin-1 protein expression in the ischemic brain measured by Western blot assay. Quantified data in the bar graph show significant reduction in Beclin-1 expression by 3-MA (1.2 mg/kg, i.c.v.) 24 h after ischemic and 3-MA treatment. CTL: ischemic control, 3MA: ischemia plus 3-MA treatment. N = 5 animals in each group, mean ± SD. *P < 0.05 compared with the WT group.
The autophagic activity was verified using another autophagy marker LC3 expression and the ratio of LC3-II/LC3-I. The LC3 expression increased in WT mice 6 h after ischemia, while a more pronounced increase in LC3 proteins was seen 12 and 24 h after ischemia (Fig. 2). The similar time course of increased Beclin-1 as well as LC3 levels supported an enhanced autophagic activity in the ischemic brain regions.
Our recent study shows that the autophagy inhibitor 3-methyladenine (3-MA) significantly decreases LC3/TUNEL double-positive cells, supporting the idea that cerebral ischemia-induced cell death is partly due to autophagic activities (Choi et al., 2012). To verify the contribution of enhanced autophagy to brain damage, 3-MA (1.2 mg/kg, i.c.v.) was administered soon after MCA occlusion and the effect of this treatment was assessed 24 h after stroke. In Western blot analysis, Beclin-1-positive cells in the peri-infarct region were significantly reduced by 3-MA treatment in both WT and p50−/− mice (Fig. 2E, F). In WT mice, the 3-MA treatment showed a trend of reducing infarct formation (indirect infarct volume = 19.1 ± 6.5 and 14.9 ± 5.5 mm3 for stroke control and stroke plus 3MA, n = 6 and 7, respectively, mean ± SD). The effect was significant when the comparison was analyzed by Student’s t test (P < 0.05); it was not statistically significant, however, when analyzed by the more rigorous one-way ANOVA multiple comparison analysis. On the other hand, the same 3-MA treatment significantly attenuated the infarct volume in P50−/− stroke mice (28.5 ± 6.0 and 19.8 ± 3.2 mm3 in stroke control and stroke plus 3-MA, respectively; P < 0.05, one-way ANOVA and Tukey’s post hoc correction, n = 7 per group). This observation is consistent with previous reports that 3-MA-sensitive autophagy contributes to ischemic brain injury and blocking this autophagic activity is neuroprotective in focal ischemic models (Tian et al., 2010; Choi et al., 2012).
Autophagy-like cell death and the role of NF-κB in the ischemic brain
Conflicting data have been reported regarding a causal relationship between increased autophagy and cell death after an ischemic insult (Levine and Yuan, 2005; Adhami et al., 2007). To understand whether the increased autophagy in the focal ischemia model was associated with ischemic injury or cell survival, we then tested cell death using the DNA fragmentation marker TUNEL staining. The TUNEL staining cannot distinguish between apoptosis and necrosis; however, it is a reliable marker for cell death (Wei et al., 2004, 2006). Cell death revealed by TUNEL-positive cells in the ischemia/peri-infarct regions was time-dependently increased, starting from early hours after ischemia and reached a peak level at 3 days after ischemia (Fig. 3). More cell death was seen in p50−/− mice, which was consistent with our previous investigation showing that the NF-κB signaling is neuroprotective after ischemic stroke (Li et al., 2008a). More interestingly, many TUNEL-positive cells were co-labeled with the autophagy marker Beclin-1 (Fig. 3). The autophagy associated cell death was significantly higher in p50−/− mice (Fig. 3). This type of TUNEL/Beclin-1 double-positive cells largely decreased by 7 days after ischemia when other forms of cell death (most likely apoptosis and/or hybrid cell death) were still easily detectable by TUNEL staining (Fig. 3).
Fig. 3.
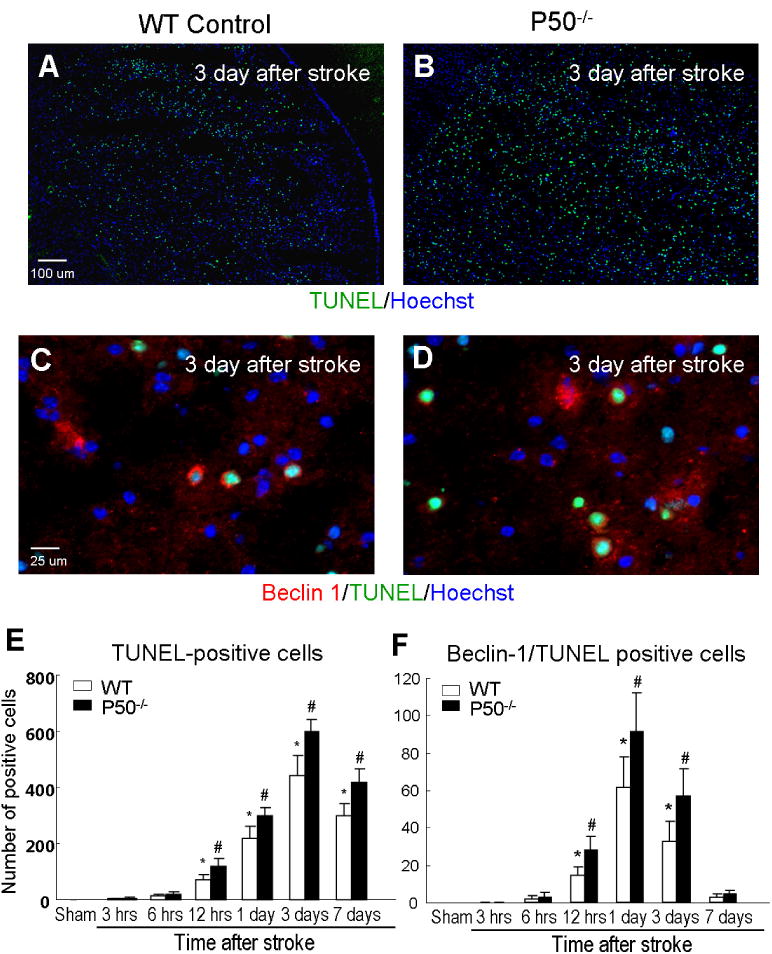
Ischemia-induced autophagy-like cell death in WT and p50−/− mice. (A–D) Immunostaining of Beclin-1 alone and co-stained with TUNEL in the penumbra region of the ischemic brain from WT and p50−/− mice. The images show the immunoreactivity of TUNEL-positive (green; A and B) and TUNEL/Beclin-1 (green/red; C and D)-positive cells. Hoechst 33342 (blue) was used to show the nuclei of all cells. (E, F) The bar graphs show cell counts of TUNEL-positive cells and Beclin-1/TUNEL double-positive cells at different times after ischemia. TUNEL-positive cells started to appear from 12 h after ischemia in both analyses and reached a peak around 1–3 days after ischemia. By 7 days after ischemia, general cell death was still relatively high while autophagic death (Beclin-1/TUNEL-positive cells) had already subsided. NF-κB p50 deletion increased cell death in both analyses. N = 5, mean ± SD. *P < 0.05 compared with sham controls, #P < 0.05 compared with WT group. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
To determine whether the increased autophagy-like cell death occurred on central neurons, brain sections were co-stained for the neuronal marker NeuN. In the peri-infarct region of WT mice, Beclin-1 and NeuN double-positive cells were markedly increased starting from 12 h after ischemia (Fig. 4). This autophagic activity in neurons declined toward basal level 3 days after ischemia. Knocking out the p50 gene significantly augmented the autophagic activity in neurons in two ways: it accelerated the neuronal autophagy to 6 h after ischemia and enhanced the activity thereafter until 3 days after ischemia (Fig. 4). Confocal microscopic imaging of ischemic brain sections stained with TUNEL, Beclin-1 and NeuN revealed triple-positive cells, supporting that autophagic activity was linked to neuronal cell death (Fig. 4). GFAP staining was performed to identify astrocytes, but we did not detect Beclin-1/GFAP double-positive cells under our experimental condition (data not shown).
Fig. 4.
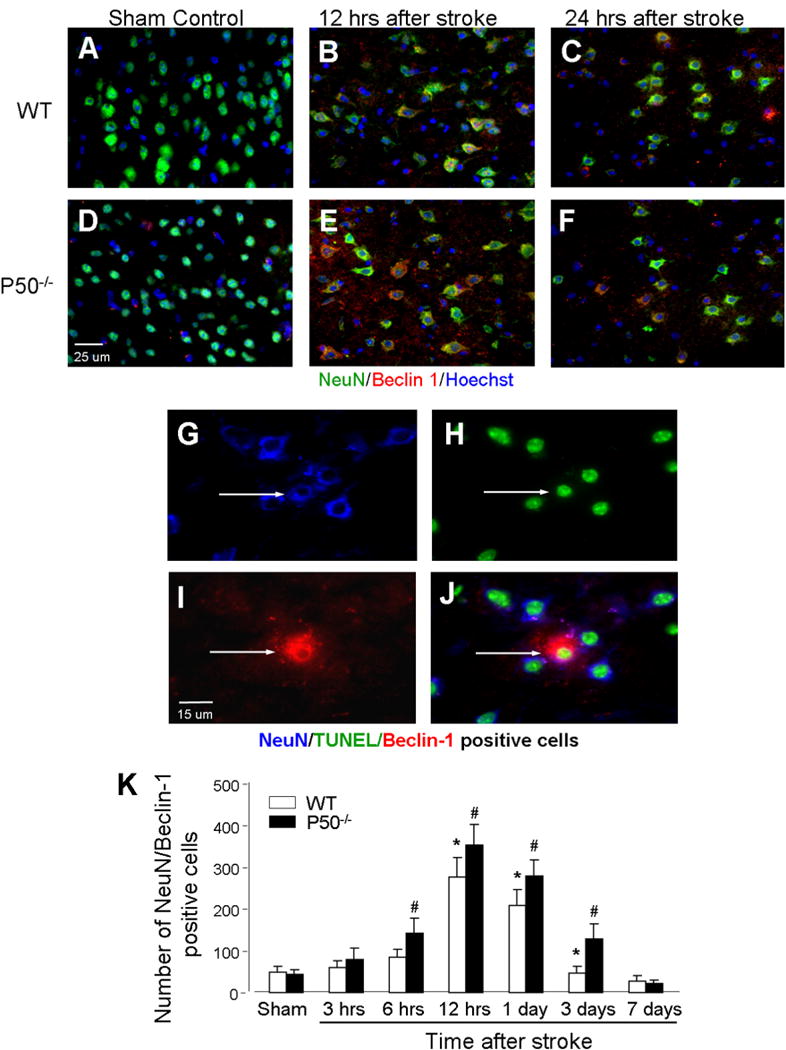
Autophagy-like neuronal cell death after ischemia and worsening effect of p50 deletion. (A–F) Immunohistochemical staining of NeuN (green) as well as Beclin-1 (red) staining was performed in the brain sections from WT and p50−/− mice at different time points before and after ischemia. Blue color was Hoechst 33342 staining to reveal the nuclei of all cells. (G–J) Images show triple staining of NeuN/Beclin 1/TUNEL (arrows), indicative of autophagy-like neuronal death. (K) The bar graph shows cell counts of NeuN/Beclin-1 double-positive cells at different times and the comparison between WT and p50−/− mice. At each time point from 6 h to 3 days after ischemia, significantly more double-positive cells were detected in p50−/− mice. The autophagic neuronal death returned to control levels 7 days after ischemia. N = 5 animals in each group, mean ± SD. *P < 0.05 compared with sham controls, #P < 0.05 compared with WT group. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Autophagy associated vasculature injury and the role of the NF-κB p50 subunit
Collagen IV is a component of the basal lamina that ensheathes endothelial cells; it has been used as a specific marker for cerebral microvessels (Li et al., 2008c; Lathia et al., 2010). In WT and p50−/− mice, collagen IV staining decreased gradually for at least 3 days after ischemia, suggesting vasculature damage in ischemic/peri-infarct regions (Fig. 5). In p50−/− mice, this decrease was significantly more evident at 1 and 3 days after ischemia (Fig. 5).
Fig. 5.
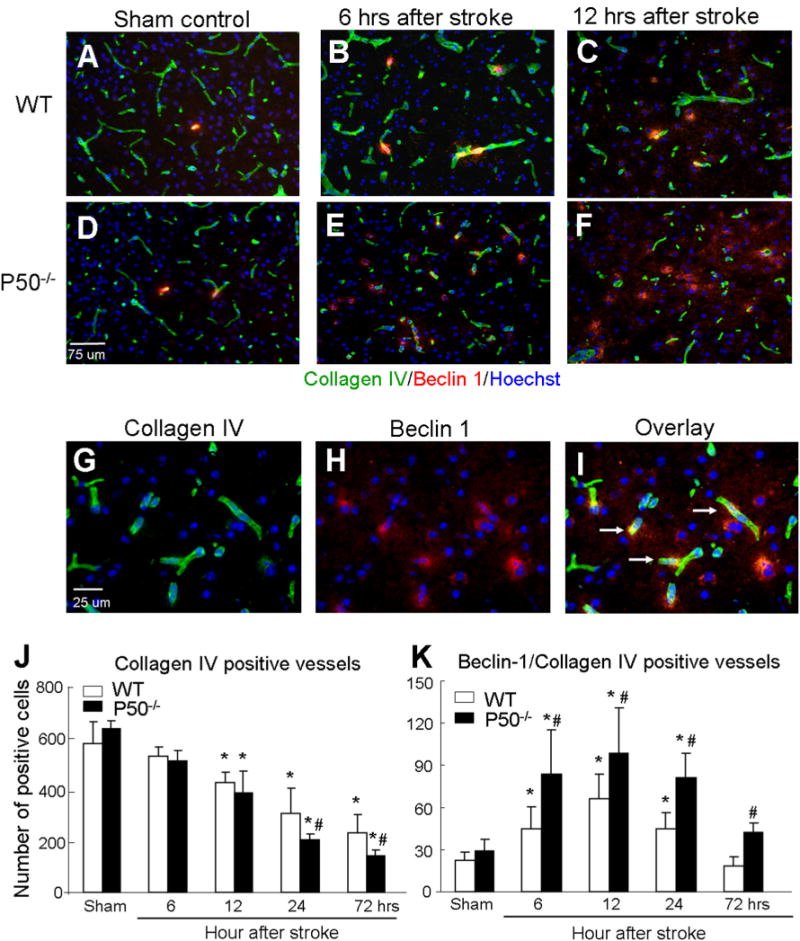
Autophagic microvessel injury after ischemia and worsening effect of p50 deletion. (A–F) Immunostaining of Beclin-1 (red) and Collagen IV (green) was performed in brain sections of sham-operated and post-ischemia from WT and p50−/− mice. Nuclei were stained with Hoechst 33342. (G–I) Enlarged images show co-localization of Beclin1 and Collagen IV (arrows). (J, K) Cell counts in bar graphs show gradual reduction of Collagen IV-positive cells after cerebral ischemia. More reduction of Collagen IV staining was seen in p50−/− mice 24 and 72 h after ischemia. In contrast, Beclin-1/Collagen IV double-positive cells were increased after ischemia and reached to a peak level at 12 h after ischemia in both WT and p50−/− mice. More Beclin-1/Collagen IV double-positive cells were detected in p50−/− mice. At 72 h after ischemia, the double-positive cells were already back to control level while significantly higher numbers of these cells were detected in p50−/− mice. N = 5 animals in each group, mean ± SD. *P < 0.05 compared with sham controls, #P < 0.05 compared with the WT group. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
To assess whether autophagy-like cell death is a mechanism of the ischemia-induced microvessel damage, we co-stained brain sections for Beclin-1 and collagen IV. An increased number of vascular cells double-positive to Becline-1 and collagen IV were detected in both WT and p50−/− mice, and much more Beclin-1/collagen IV-positive staining was seen in brain sections from p50−/− mice from 6 h to 3 days after ischemia (Fig. 5). The triple staining of TUNEL/Beclin-1/collagen IV demonstrated that the autophagic activity was associated with vascular injury (Fig. 6).
Fig. 6.
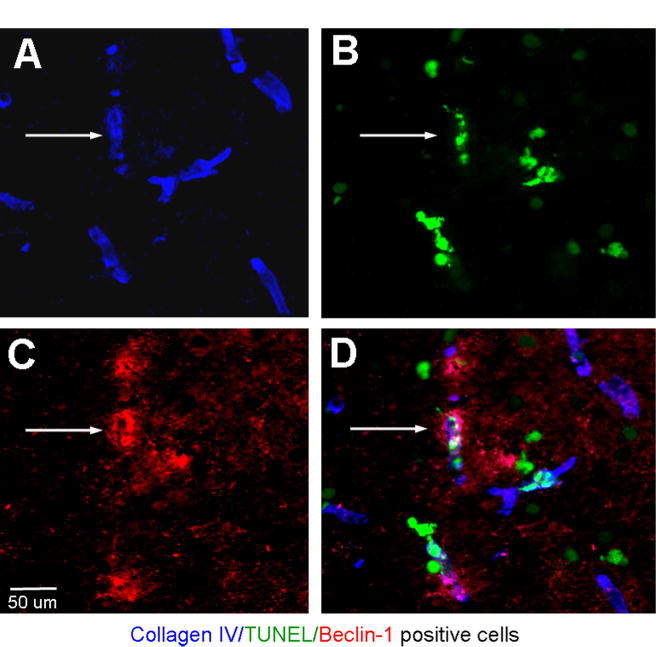
Ischemia-induced autophagic microvessel deterioration. (A–D) Immunohistochemical triple staining of collagen IV, Beclin-1 and TUNEL in the penumbra region 12 h after ischemia in a brain section from a p50−/− mouse. Collagen IV (blue) staining show consistent morphology of microvessels. Some of collagen IV-positive cells were co-labeled with TUNEL (green) and Beclin-1 (red) (arrows). The triple-positive staining indicated autophagic vasculature damage. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The autophagic injury to vasculature cells indicated a disruption of the integrity of the blood–brain barrier (BBB). To test this hypothesis, endothelial tight junction was visualized by occludin immunostaining. In WT and p50−/− sham-operated mice, occludin was strongly expressed in the cerebral vasculature and there was no significant difference in the quantity of endothelial tight junctions between the two groups (Fig. 7). At all time points from 6 h to 3 days after ischemia, occludin staining in the ischemic and peri-infarct regions was far more intense in WT mice than that in p50−/− mice (Fig. 7). Twenty-four hours after ischemia, WT mice had about twice the expression area of occludin than p50−/− mice, suggesting more severe damage to the BBB in the absence of NF-κB p50 subunit (Fig. 7).
Fig. 7.
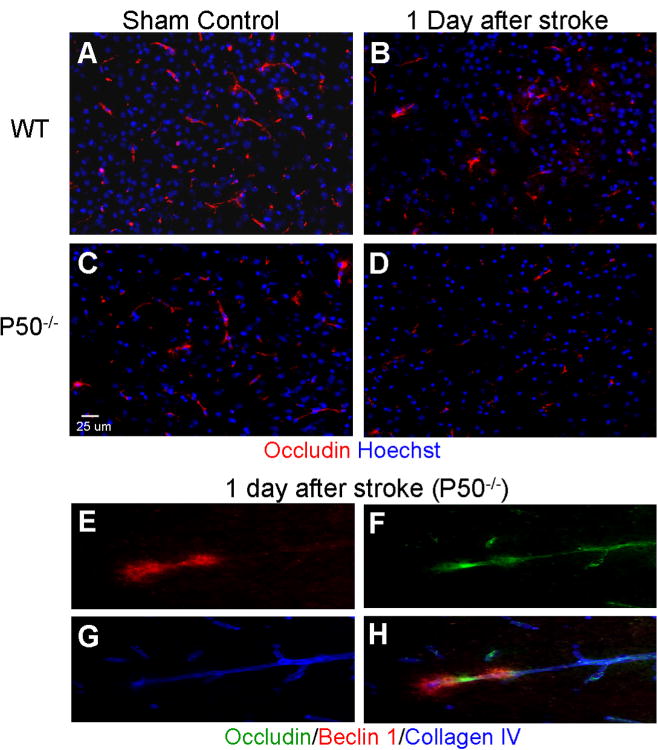
Autophagic injury to the blood-brain barrier (BBB) and worsening effect of deleting p50. (A–D) BBB integrity was evaluated using immunostaining of occludin (red) in WT and p50−/− mice 24 h after ischemia. Hoechst 33342 staining (blue) showed nuclei of all cells. Occludin staining exhibited strong occludin expression in the cerebral vasculature of control animals, while the focal ischemia noticeably reduced occluding expression in the penumbra region of WT and p50−/− mice. Even less occluding immunoreactivity was seen in the p50−/− brain (B, D). (E–H) In the confocal image panels of triple staining of occluding (green), Becline-1 (red) and Collagen IV (blue), the overlapping of the three markers illustrated autophagic damage to BBB. Representative of five animals in each group. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Regulation of Akt-mTOR signaling factors in the WT and NF-κB p50 knockout ischemic brain
To understand the mechanistic basis for our observations of increased levels of autophagy in p50−/− ischemic mice, we examined the Akt-mTOR signaling pathway, a known negative regulator of autophagy (Sarbassov et al., 2005). The focal ischemia in WT mice significantly increased phosphorylated Akt 12 h later, suggesting activation of this protective pathway. On the other hand, phosphorylation of Akt in the brain of p50−/− mice decreased after ischemia (Fig. 8). mTOR signaling plays an inhibitory role in regulation of autophagy (Mehrpour et al., 2010). The phosphorylation status of the well-characterized mTOR substrate, 70 kDa S6 kinase 1 (S6K1), was used as a readout of its activity. The total S6K1 (S6K1) in penumbra tissues was gradually and significantly decreased from 6 to 72 h after ischemia in WT and p50−/− mice (Fig. 9A–C). Although phosphorylation of S6K1 (p-S6K1/S6K1) decreased in the brain of both animal groups, the reduction was significantly greater in p50−/− mice from 12 to 72 h after ischemia (Fig. 9D). This diminished mTOR signaling activity was in line with the elevated autophagy in the ischemic brain and consistent with augmented autophagy in p50−/− mice. We also looked at the downstream target of S6K1, the 40S ribosomal protein S6 (S6R). The level of S6R in the penumbra of WT and p50−/− mice followed the same trend as S6K1, showing significant decreases from 24 to 72 h after ischemia (Fig. 9E, G). The phosphorylation of this protein in WT mice remained stable until an increase at 72 h after ischemia (Fig. 9H). However, p-S6R decreased in p50−/− mice compared with WT controls (Fig. 9H), consistent with augmented autophagy in the absence of p50.
Fig. 8.
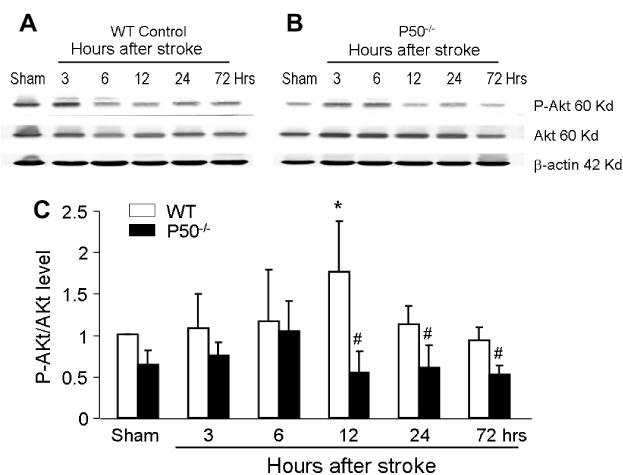
Up-regulation of Akt signaling after ischemia and impaired regulation in p50−/− mice Western blot analysis of the Akt-mTOR signaling pathway at different time points after cerebral ischemia. (A, B) Representative electropherograms show the expression levels of phospho-Akt (p-Akt) and total Akt in the penumbra region of the WTand p50−/− brain. Densitometric analysis provided a quantified comparison of the p-Akt/Akt ratio in the two groups of animals at different time points. Gray intensity was normalized against β-actin. The Akt phosphorylation, representative of activation of the pathway, was almost doubled at 12 h after ischemia in WT mice followed by less prominent bust significant enhancement of p-Akt compared to p50−/− mice up to 72 h after ischemia. The increased Akt activation was completely absent in p50−/− mice. N = 5 for each group. *P < 0.05 vs. sham; #P < 0.05 vs. WT.
Fig. 9.
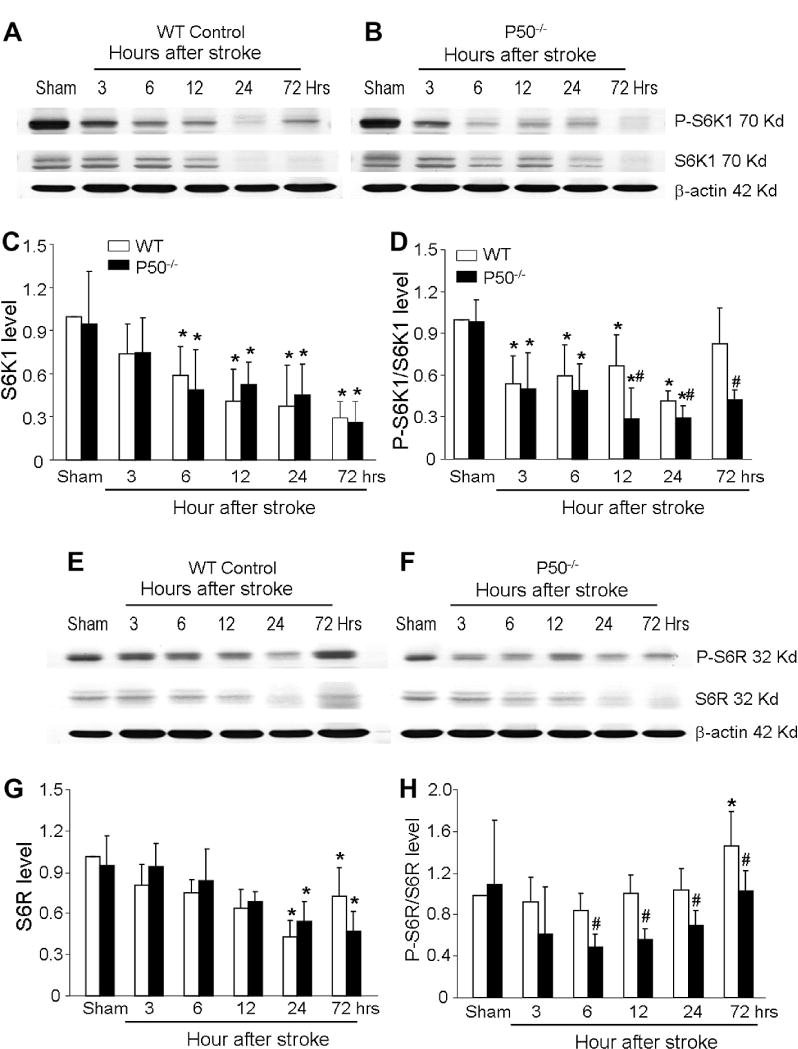
Changes in autophagic signaling in WT and p50−/− mice after focal ischemia. Western blot analysis of several autophagic signals in the post-ischemic brain of WT and p50−/− mice. (A, B) Electropherograms show the expression levels of total mTOR substrate S6K1 and phosphorylated S6K1. (C, D) Quantified comparisons are shown in the bar graph of the total S6K1 level (C) and the p-S6K1/S6K1 ratio (D) in the two groups of animals at different time points. Consistent with enhanced autophagic cell death in the area, the total S6K1 level and phosphorylated S6K1 (p-S6K1) were diminishing in the ischemic brain. Even more reduction in p-S6K1 was observed in p50−/− mice 12–72 h after ischemia. (E, F) mTOR substrate S6R in the post-ischemic WT and p50−/− brain. Electropherograms show the expression levels of phospho-S6R (p-S6R) and total S6R in the penumbra region of the ischemic brain. G and H. Quantified data of E and F. The total level of S6R gradually declined after ischemia, significant reduction was seen from 12 to 72 h after ischemia in both WTand p50−/− mice (G). The ratio of p-S6R/S6R in WT animals was about the same until it increased at 72 h after ischemia (H). This ratio was much lower in the p50−/− brain from 6 to 72 h after ischemia compared to WT controls. Gray intensity was normalized against the loading control of β-actin. N = 5 animals in each group. mean ± SD. *P < 0.05 vs. sham control, #P < 0.05 vs. WT group.
DISCUSSION
In the present study, we show that autophagy increases after focal ischemia in the ischemic and penumbra regions. The increased autophagic activity occurs in a delayed fashion, starting from 6 to 12 h after ischemia and can last for up to 3 days after ischemia. Therefore, the ischemia-induced autophagy was a sub-acute and delayed post-ischemic event. Cell death and autophagy assays agree that the increased autophagy is associated with dead or dying cells, although this autophagic mechanism is not prominent enough to have a significant impact on the infarct volume examination in the focal ischemia model. We have also delineated an important role of NF-κB signaling in the ischemia-stimulated autophagy in the brain. Deletion of the NF-κB p50 subunit increased the expression of Beclin-1 and LC3, as well as the number of Beclin-1/TUNEL-positive cells in the ischemic cortex. The increased autophagic-like cell death not only occurs in neurons, but also in microvessel or vascular endothelial cells in p50−/− animals. In addition, our work is the first report linking p50 activity to the integrity of the BBB in autophagy, showing that p50-deficient mice suffer even greater disruption of the BBB than WT mice. The damage to neuronal and microvessel cells thus disrupts the neurovascular unit that is critically vital for normal neurovasculature infrastructure and function (Han and Suk, 2005). Lastly, we found that, in response to ischemia, Akt-mTOR signaling is down-regulated in p50-deleted mice. This decrease is consistent with increased autophagy and cell death; however, a cause-effect 0 relationship between these molecules and the autophagy-like cell death remains to be verified using more specific approaches such as gain and loss of function studies of gene manipulations.
It might be possible that the coincident presence of autophagy and cell death markers in the cells is misinterpreted as a cause-effect relationship. A close examination on the time courses of these events, however, does not support this assumption. Our Beclin-1 staining and TUNEL staining data showed that the development of autophagy proceeds and peaked several hours before massive cell death after ischemia in the same brain areas. This time difference of several hours in the evolution of these two events is highly consistent with the idea that the increased cell death is a consequence, at least in part, of the enhanced autophagic activity after cerebral ischemia. Whether autophagy kills cells via interaction with an apoptosis pathway or via an entirely independent pathway is not well understood (Hamacher-Brady et al., 2006). Moreover, since autophagy has the function of cleaning up injured/deteriorated proteins and cellular structures, we cannot completely rule out the possibility that the TUNEL-positive cells represent cases where autophagy failed to protect cells from death, although again the earlier occurrence of autophagy does not agree with this speculation. Since the PI3K/Akt signaling is a survival mechanism after ischemic stroke (Zhao et al., 2006), the reduced phosphorylated Akt observed in p50−/− mice may also contribute together with the increased autophagy to ischemia-induced cell death.
Recent studies have suggested that hypoxic/ischemic insults are a powerful stimulus for autophagic cell death in the brain (Adhami et al., 2006; Rami et al., 2008). Although a role for autophagic-mediated cell death following cerebral ischemia has been controversial (Levine and Yuan, 2005; Adhami et al., 2007), several reports have provided convincing evidence that this form of cell death is indeed induced by ischemia and that it does contribute to neuronal cell death. For example, using Atg7-deficient mice, Koike and colleagues presented perhaps the first in vivo evidence of autophagy-mediated neuronal cell death in the neonatal mammalian brain after hypoxic-ischemic injury (Koike et al., 2008). Blocking autophagy with pathway inhibitors also demonstrated the contribution of autophagy to tissue damage resulting from focal ischemia (Wen et al., 2008; Puyal et al., 2009). The present investigation provides new evidence for autophagic death in several cell types in the ischemic brain of adult animals.
Following injury in the brain, NF-κB has been shown to regulate both inflammatory response and apoptotic cell death. Of the five known NF-κB subunits in mammals, p50, p52, p65 (RelA), RelB and cRel, p50 is believed to be the most predominant (Xiao, 2007). Previous research in our laboratory has shown that ischemic infarct volume and apoptotic cell death are potentiated in p50−/− mice after focal ischemia (Li et al., 2008a). The present investigation now suggests a novel function for NF-κB in the cortical parenchyma: inhibition or attenuation of ischemia-induced autophagy. This is evidenced by marked elevations of Beclin-1 and LC3 expression in p50−/− mice. Therefore, the NFκB pathway can not only regulate apoptosis, it also participates in the regulation of post-ischemic autophagic cell death. In support of this latter finding, in tumor cell lines NF-κB activation inhibits autophagy in response to challenge by Tumor Necrosis Factor-α (TNF-α; Djavaheri-Mergny et al., 2007). Consistently, inhibition of NF-κB results in an enhancement of starvation-induced autophagy (Fabre et al., 2007).
A major pathway for the regulation of autophagy occurs through the protein kinase mTOR (Wullschleger et al., 2006). One well-characterized pathway for mTOR activation involves the insulin/insulin-like growth factor (IGF) receptor-induced PI3-kinase, with subsequent activation of Akt. Activated mTOR enhances protein translation by phosphorylating a number of substrates including S6K1 and 4E-BP1 (Zeng and Zhou, 2008). In this study, we observed reduced Akt-mTOR signaling in p50−/− mice after cerebral ischemia, which may contribute to the activation of autophagic cell death. Therefore, the mechanism by which NF-κB deletion enhances autophagy may involve repression of mTOR. These results are consistent with previously published findings showing that NF-κB activation mediates repression of autophagy in TNF-α-treated Ewing sarcoma cells, which is associated with an NF-κB-dependent activation of mTOR (Djavaheri-Mergny et al., 2006).
The expression levels of Akt and mTOR signaling factors showed different or even opposite changes in the post-ischemic brain. In WT animals, Akt activation increased in the penumbra which was consistent with the role of a protective stress signal of Akt (Zhao et al., 2006). Since Akt may inhibit autophagy (Degtyarev et al., 2008), the increased Akt activity did not agree with increased autophagy observed in the ischemic brain. On the other hand, mTOR signaling was generally decreased in the ischemic brain. The interpretation of the opposite regulation of these associated pathways is complicated by the fact that mTOR is a part of two complexes: one is TORC1, which includes Raptor, signals to S6K and 4EBP1, controls protein synthesis and is rapamycin-sensitive; the second one is TORC2, which includes Rictor, signals to Akt-FoxO, and is rapamycin-insensitive (Guertin et al., 2006). The TORC2 or Akt-FoxO pathway actually shows inhibitory effect on mTOR signaling (Mammucari et al., 2008). This may at least partly explain reduced mTOR activity while Akt activity increases in the ischemic brain. The different time courses of Akt and mTOR activity changes may also suggest that mTOR is subjected to additional regulatory mechanisms under ischemic conditions.
It is now clear that neurons, astrocytes, vascular endothelial cells, and a number of stromal elements (i.e. microglia, oligodendrocytes, pericytes, and basal membranes) exist in close physical proximity and are considered a functional entity: neurovascular unit (Moroni and Chiarugi, 2009). Physiological interactions among neurovascular unit components participate in the regulation of cerebral microcirculation and are critical for normal brain function, as well as tissue repair after brain damage (Curin et al., 2006). A clear understanding of the molecular regulation of cell survival and death by the neurovascular unit will be essential for the design of efficacious treatments and therapy for ischemic brain injury. In this study, we demonstrate ischemia-mediated autophagic activity in the principal cells of the neurovascular unit, i.e. neurons and vasculature (endothelial) cells. Interestingly, we detected up-regulation of Beclin-1 in neurons, but not in astrocytes, which is consistent with a previous report (Rami et al., 2008). The ischemia-induced autophagy in neurons and microvessels was sufficient to disrupt the neurovascular unit. Consistently, the BBB was broken down as shown in this investigation.
The BBB is the specialized system of microvascular endothelial cells that shields the brain from blood-borne toxins, supplies brain tissues with nutrients, and filters harmful compounds from the brain back into the bloodstream. The close interaction between endothelial cells and other components of the neurovascular unit is critical to the proper functioning of the CNS (Persidsky et al., 2006). Prevention of the disruption of the BBB minimizes tissue damage following cerebral ischemia (Wang et al., 2009). The area of expression of the tight junction protein occludin, has been used as a reliable indicator of BBB integrity (Li et al., 2007; Fernandez-Lopez et al., 2012). We show here that, in response to ischemia, p50 is required to maintain the integrity of the BBB, as reflected in the reduced levels of occludin staining in p50−/− mice. The BBB disruption likely results in consecutively aggravated neuronal cell death. Based on our observations, it is suggested that autophagy and the NF-κB pathway may be manipulated for retaining the integrity of the BBB during and after ischemia.
CONCLUSION
Our study demonstrates that ischemic insult activates autophagy in a delayed and sub-acute fashion that likely exacerbates the ischemia-induced brain damage, specifically the neurovascular unit. Activation of NF-κB may play an anti-autophagic role in cerebral ischemia by regulating mTOR signaling. Thus, prevention of autophagy-like cell death by enhancing NF-κB and downstream signaling pathways may be a potential strategy for the treatment of ischemic stroke.
Acknowledgments
This work was supported by NIH Grants NS045810 (L.W.), NS062097 (L.W.), NS058710 (L.W.), NS057255 (S.P.Y.) and the American Heart Association Established Investigator Award (LW). It was also supported by the NIH Grant NS055077 to the ENNCF (Emory Neurology-NINDS Core Facility).
Abbreviations
- 3-MA
3-methyladenine
- ANOVA
analysis of variance
- BBB
blood–brain barrier
- CCA
common carotid artery
- EDTA
ethylenediaminetetraacetic acid
- GFAP
glial fibrillary acidic protein
- IGF
insulin-like growth factor
- LC3
light chain 3
- MCA
middle cerebral artery
- NF-κB
nuclear factor kappa B
- NIH
National Institutes of Health
- p-Akt
phospho-Akt
- mTOR
the mammalian target of rapamycin
- PBS
phosphate buffered saline
- p-S6R
phospho-S6R
- S6K1
S6 kinase 1
- SD
standard deviation
- SDS
sodium dodecyl sulfate
- TBS-T
Tris-Buffered Saline and Tween 20
- TNF-α
Tumor Necrosis Factor-α
- TUNEL
terminal deoxynucleotidyltransferase (TdT)-mediated dUTP-biotin nick end labeling
- WT
wild-type
References
- Adhami F, Liao G, Morozov YM, Schloemer A, Schmithorst VJ, Lorenz JN, Dunn RS, Vorhees CV, Wills-Karp M, Degen JL, Davis RJ, Mizushima N, Rakic P, Dardzinski BJ, Holland SK, Sharp FR, Kuan CY. Cerebral ischemia–hypoxia induces intravascular coagulation and autophagy. Am J Pathol. 2006;169:566–583. doi: 10.2353/ajpath.2006.051066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhami F, Schloemer A, Kuan CY. The roles of autophagy in cerebral ischemia. Autophagy. 2007;3:42–44. doi: 10.4161/auto.3412. [DOI] [PubMed] [Google Scholar]
- Altman DG, Bland JM. Standard deviations and standard errors. BMJ. 2005;331:903. doi: 10.1136/bmj.331.7521.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balduini W, Carloni S, Buonocore G. Autophagy in hypoxia–ischemia induced brain injury: evidence and speculations. Autophagy. 2009;5:221–223. doi: 10.4161/auto.5.2.7363. [DOI] [PubMed] [Google Scholar]
- Chen B, Liao WQ, Xu N, Xu H, Wen JY, Yu CA, Liu XY, Li CL, Zhao SM, Campbell W. Adiponectin protects against cerebral ischemia-reperfusion injury through anti-inflammatory action. Brain Res. 2009;1273:129–137. doi: 10.1016/j.brainres.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Choi KE, Hall CL, Sun JM, Wei L, Mohamad O, Dix TA, Yu SP. A novel stroke therapy of pharmacologically induced hypothermia after focal cerebral ischemia in mice. FASEB J. 2012;26:2799–2810. doi: 10.1096/fj.11-201822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curin Y, Ritz MF, Andriantsitohaina R. Cellular mechanisms of the protective effect of polyphenols on the neurovascular unit in strokes. Cardiovasc Hematol Agents Med Chem. 2006;4:277–288. doi: 10.2174/187152506778520691. [DOI] [PubMed] [Google Scholar]
- Degtyarev M, De Maziere A, Orr C, Lin J, Lee BB, Tien JY, Prior WW, van Dijk S, Wu H, Gray DC, Davis DP, Stern HM, Murray LJ, Hoeflich KP, Klumperman J, Friedman LS, Lin K. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008;183:101–116. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Souquere S, Pierron G, Codogno P. NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy. J Biol Chem. 2006;281:30373–30382. doi: 10.1074/jbc.M602097200. [DOI] [PubMed] [Google Scholar]
- Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Codogno P. Regulation of autophagy by NFkappaB transcription factor and reactives oxygen species. Autophagy. 2007;3:390–392. doi: 10.4161/auto.4248. [DOI] [PubMed] [Google Scholar]
- Duckworth EA, Butler T, Collier L, Collier S, Pennypacker KR. NF-kappaB protects neurons from ischemic injury after middle cerebral artery occlusion in mice. Brain Res. 2006;1088:167–175. doi: 10.1016/j.brainres.2006.02.103. [DOI] [PubMed] [Google Scholar]
- Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- Fabre C, Carvalho G, Tasdemir E, Braun T, Ades L, Grosjean J, Boehrer S, Metivier D, Souquere S, Pierron G, Fenaux P, Kroemer G. NF-kappaB inhibition sensitizes to starvation-induced cell death in high-risk myelodysplastic syndrome and acute myeloid leukemia. Oncogene. 2007;26:4071–4083. doi: 10.1038/sj.onc.1210187. [DOI] [PubMed] [Google Scholar]
- Fernandez-Lopez D, Faustino J, Daneman R, Zhou L, Lee SY, Derugin N, Wendland MF, Vexler ZS. Blood–brain barrier permeability is increased after acute adult stroke but not neonatal stroke in the rat. J Neurosci. 2012;32:9588–9600. doi: 10.1523/JNEUROSCI.5977-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg P, Duncan RS, Kaja S, Koulen P. Intracellular mechanisms of N-acylethanolamine-mediated neuroprotection in a rat model of stroke. Neuroscience. 2010;166:252–262. doi: 10.1016/j.neuroscience.2009.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM. Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell. 2006;11:859–871. doi: 10.1016/j.devcel.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Hamacher-Brady A, Brady NR, Gottlieb RA. The interplay between pro-death and pro-survival signaling pathways in myocardial ischemia/reperfusion injury: apoptosis meets autophagy. Cardiovasc Drugs Ther. 2006;20:445–462. doi: 10.1007/s10557-006-0583-7. [DOI] [PubMed] [Google Scholar]
- Han HS, Suk K. The function and integrity of the neurovascular unit rests upon the integration of the vascular and inflammatory cell systems. Curr Neurovasc Res. 2005;2:409–423. doi: 10.2174/156720205774962647. [DOI] [PubMed] [Google Scholar]
- Harari OA, Liao JK. NF-kappaB and innate immunity in ischemic stroke. Ann N Y Acad Sci. 2010;1207:32–40. doi: 10.1111/j.1749-6632.2010.05735.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, Shibata M, Tadakoshi M, Gotoh K, Komatsu M, Waguri S, Kawahara N, Kuida K, Nagata S, Kominami E, Tanaka K, Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Chigurupati S, Thundyil J, Selvaraj PK, Mughal MR, Woodruff TM, Chan SL, Karamyan VT, Mattson MP, Arumugam TV. Pivotal role for beta-1 integrin in neurovascular remodelling after ischemic stroke. Exp Neurol. 2010;221:107–114. doi: 10.1016/j.expneurol.2009.10.007. [DOI] [PubMed] [Google Scholar]
- Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lu ZY, Ogle M, Wei L. Erythropoietin prevents blood–brain barrier damage induced by focal cerebral ischemia in mice. Neurochem Res. 2007;32:2132–2141. doi: 10.1007/s11064-007-9387-9. [DOI] [PubMed] [Google Scholar]
- Li J, Lu Z, Li WL, Yu SP, Wei L. Cell death and proliferation in NF-kappaB p50 knockout mouse after cerebral ischemia. Brain Res. 2008a;1230:281–289. doi: 10.1016/j.brainres.2008.06.130. [DOI] [PubMed] [Google Scholar]
- Li WL, Yu SP, Ogle ME, Ding XS, Wei L. Enhanced neurogenesis and cell migration following focal ischemia and peripheral stimulation in mice. Dev Neurobiol. 2008b;68:1474–1486. doi: 10.1002/dneu.20674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Ogle ME, Wallace GC, Lu ZY, Yu SP, Wei L. Erythropoietin attenuates intracerebral hemorrhage by diminishing matrix metalloproteinases and maintaining blood– brain barrier integrity in mice. Acta Neurochir Suppl. 2008c;105:105–112. doi: 10.1007/978-3-211-09469-3_22. [DOI] [PubMed] [Google Scholar]
- Lu ZY, Yu SP, Wei JF, Wei L. Age-related neural degeneration in nuclear-factor kappaB p50 knockout mice. Neuroscience. 2006;139:965–978. doi: 10.1016/j.neuroscience.2005.12.062. [DOI] [PubMed] [Google Scholar]
- Lum JJ, DeBerardinis RJ, Thompson CB. Autophagy in metazoans: cell survival in the land of plenty. Nat Rev Mol Cell Biol. 2005;6:439–448. doi: 10.1038/nrm1660. [DOI] [PubMed] [Google Scholar]
- Mammucari C, Schiaffino S, Sandri M. Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy. 2008;4:524–526. doi: 10.4161/auto.5905. [DOI] [PubMed] [Google Scholar]
- Martinet W, De Meyer GR, Andries L, Herman AG, Kockx MM. Detection of autophagy in tissue by standard immunohistochemistry: possibilities and limitations. Autophagy. 2006;2:55–57. doi: 10.4161/auto.2217. [DOI] [PubMed] [Google Scholar]
- Mehrpour M, Esclatine A, Beau I, Codogno P. Autophagy in health and disease. 1. Regulation and significance of autophagy: an overview. Am J Physiol Cell Physiol. 2010;298:C776–785. doi: 10.1152/ajpcell.00507.2009. [DOI] [PubMed] [Google Scholar]
- Moroni F, Chiarugi A. Post-ischemic brain damage: targeting PARP-1 within the ischemic neurovascular units as a realistic avenue to stroke treatment. FEBS J. 2009;276:36–45. doi: 10.1111/j.1742-4658.2008.06768.x. [DOI] [PubMed] [Google Scholar]
- Nijboer CH, Heijnen CJ, Groenendaal F, May MJ, van Bel F, Kavelaars A. A dual role of the NF-kappaB pathway in neonatal hypoxic-ischemic brain damage. Stroke. 2008;39:2578–2586. doi: 10.1161/STROKEAHA.108.516401. [DOI] [PubMed] [Google Scholar]
- Nurmi A, Lindsberg PJ, Koistinaho M, Zhang W, Juettler E, Karjalainen-Lindsberg ML, Weih F, Frank N, Schwaninger M, Koistinaho J. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke. 2004;35:987–991. doi: 10.1161/01.STR.0000120732.45951.26. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Ramirez SH, Haorah J, Kanmogne GD. Blood-brain barrier: structural components and function under physiologic and pathologic conditions. J Neuroimmune Pharmacol. 2006;1:223–236. doi: 10.1007/s11481-006-9025-3. [DOI] [PubMed] [Google Scholar]
- Pizzi M, Spano P. Distinct roles of diverse nuclear factor-kappaB complexes in neuropathological mechanisms. Eur J Pharmacol. 2006;545:22–28. doi: 10.1016/j.ejphar.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66:378–389. doi: 10.1002/ana.21714. [DOI] [PubMed] [Google Scholar]
- Rami A, Langhagen A, Steiger S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol Dis. 2008;29:132–141. doi: 10.1016/j.nbd.2007.08.005. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Schlattmann P, Dirnagl U. Statistics in experimental cerebrovascular research: comparison of more than two groups with a continuous outcome variable. J Cereb Blood Flow Metab. 2010;30:1558–1563. doi: 10.1038/jcbfm.2010.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shacka JJ, Roth KA, Zhang J. The autophagy–lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front Biosci. 2008;13:718–736. doi: 10.2741/2714. [DOI] [PubMed] [Google Scholar]
- Shaw J, Kirshenbaum LA. Molecular regulation of autophagy and apoptosis during ischemic and non-ischemic cardiomyopathy. Autophagy. 2008;4:427–434. doi: 10.4161/auto.5901. [DOI] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson D, Yin T, Smalstig EB, Hsu MA, Panetta J, Little S, Clemens J. Transcription factor nuclear factor-kappa B is activated in neurons after focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20:592–603. doi: 10.1097/00004647-200003000-00017. [DOI] [PubMed] [Google Scholar]
- Terai K, Matsuo A, McGeer EG, McGeer PL. Enhancement of immunoreactivity for NF-kappa B in human cerebral infarctions. Brain Res. 1996;739:343–349. doi: 10.1016/s0006-8993(96)01073-6. [DOI] [PubMed] [Google Scholar]
- Tian F, Deguchi K, Yamashita T, Ohta Y, Morimoto N, Shang J, Zhang X, Liu N, Ikeda Y, Matsuura T, Abe K. In vivo imaging of autophagy in a mouse stroke model. Autophagy. 2010;6:1107–1114. doi: 10.4161/auto.6.8.13427. [DOI] [PubMed] [Google Scholar]
- Wang J, Jiang C, Li X, Liu C, Cheng N, Hao Y. The protective mechanism of progesterone on blood–brain barrier in cerebral ischemia in rats. Brain Res Bull. 2009;79:426–430. doi: 10.1016/j.brainresbull.2009.05.018. [DOI] [PubMed] [Google Scholar]
- Wang L, Zhang X, Liu L, Yang R, Cui L, Li M. Atorvastatin protects rat brains against permanent focal ischemia and downregulates HMGB1, HMGB1 receptors (RAGE and TLR4), NF-kappaB expression. Neurosci Lett. 2010;471:152–156. doi: 10.1016/j.neulet.2010.01.030. [DOI] [PubMed] [Google Scholar]
- Wei L, Ying DJ, Cui L, Langsdorf J, Yu SP. Necrosis, apoptosis and hybrid death in the cortex and thalamus after barrel cortex ischemia in rats. Brain Res. 2004;1022:54–61. doi: 10.1016/j.brainres.2004.06.080. [DOI] [PubMed] [Google Scholar]
- Wei L, Han BH, Li Y, Keogh CL, Holtzman DM, Yu SP. Cell death mechanism and protective effect of erythropoietin after focal ischemia in the whisker-barrel cortex of neonatal rats. J Pharmacol Exp Ther. 2006;317:109–116. doi: 10.1124/jpet.105.094391. [DOI] [PubMed] [Google Scholar]
- Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy. 2008;4:762–769. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Xiao G, Rabson AB, Young W, Qing G, Qu Z. Alternative pathways of NF-kappaB activation: a double-edged sword in health and disease. Cytokine Growth Factor Rev. 2006;17:281–293. doi: 10.1016/j.cytogfr.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Xiao G. Autophagy and NF-kappaB: fight for fate. Cytokine Growth Factor Rev. 2007;18:233–243. doi: 10.1016/j.cytogfr.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M, Zhou JN. Roles of autophagy and mTOR signaling in neuronal differentiation of mouse neuroblastoma cells. Cell Signal. 2008;20:659–665. doi: 10.1016/j.cellsig.2007.11.015. [DOI] [PubMed] [Google Scholar]
- Zhao H, Sapolsky RM, Steinberg GK. Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol Neurobiol. 2006;34:249–270. doi: 10.1385/MN:34:3:249. [DOI] [PubMed] [Google Scholar]