

Abstract
The advent of complete-genome genotyping across phenotype cohorts has provided a rich source of information for bioinformaticians. However the search for SNPs from this data is generally performed on a study-by-study case without any specific hypothesis of the location for SNPs that are predictive for the phenotype. We have designed a method whereby very large SNP lists (several gigabytes in size), combining several genotyping studies at once, can be sorted and traced back to their ultimate consequence in protein structure. Given a working hypothesis, researchers are able to easily search whole genome genotyping data for SNPs that link genetic locations to phenotypes. This allows a targeted search for correlations between phenotypes and potentially relevant systems, rather than utilizing statistical methods only. HyDn-SNP-S returns results that are less data dense, allowing more thorough analysis, including haplotype analysis. We have applied our method to correlate DNA polymerases to cancer phenotypes using four of the available cancer databases in dbGaP. Logistic regression and derived haplotype analysis indicates that ~80 SNPs, previously overlooked, are statistically significant. Derived haplotypes from this work link POLL to breast cancer and POLG to prostate cancer with an increase in incidence of 3.01- and 9.6-fold, respectively. Molecular dynamics simulations on wild-type and one of the SNP mutants from the haplotype of POLL provide insights at the atomic level on the functional impact of this cancer related SNP. Furthermore, HyDn-SNP-S has been designed to allow application to any system. The program is available upon request from the authors.
Keywords: SNP search, Biomarkers, Molecular dynamics, DNA polymerases, Cancer
1. Introduction
In recent years, the amount of genomic data on disease phenotypes has increased exponentially. The decreasing cost of genetic testing, along with the future promise of personalized medicine has resulted in a boom in individual genomic data [1–5]. Most bioinformatic techniques determine clusters of mutations that may be followed and used as a diagnostic tool in various diseases [6–9]. Traditional analysis of genome wide association studies (GWAS) focus on a single phenotype, and aim to find SNPs that show statistically significant association with the phenotype in any of the measured genes. In most cases these analysis do not have an a priori hypothesis of the locations of the SNPs. Therefore, very stringent statistical criteria are needed to obtain SNPs that are predictive, resulting in only a small number of SNPs being identified. Few studies have leveraged the vast information generated to identify new SNPs with clear functional impact on disease onset [10–13].
Moreover, tracking a mutation resulting from these SNPs through transcription and translation to their ultimate effects in a cell is largely left to the scientific community at large. In addition, correlating a mutation to a phenotype is a daunting task for researchers who typically work at a cellular level. Most biochemists or molecular biologists have a biosystem of interest, and broad sweeping GWAS studies are typically intractable for their purposes. It was our intent to create a tool that would allow a user to query genomic-level data for their system of interest. There have been examples previously where this has been done on a single GWAS study, but not on combined data sets [14]. To this end, we developed an algorithm whereby a researcher can directly query one or several GWAS studies for their gene region of interest. We term this method hypothesis driven SNP search (HyDn-SNP-S). The software returns all SNP mutations within their gene region, along with information on which phenotype the mutation occurs in. Further statistical methods can then be applied to the GWAS data. Additionally, by returning a focused set of mutations, tracking the consequences of the mutations through the RNA and protein levels becomes trivial.
The workflow shown in Fig. 1 outlines the process used in HyDn-SNP-S. Researchers can select one or many GWAS for the phenotypes of interest, apply the SNP-Phenotype program to search for their gene of interest, and further analysis can be carried out as desired. Our program returns both intronic and exonic SNPs, allowing the investigation of impact on RNA, gene processing or protein sequence. The simplicity of the software implementation makes this method ideal for researchers not comfortable with large-scale bioinformatic analyses, or those who lack the resources to perform such studies.
Fig. 1.
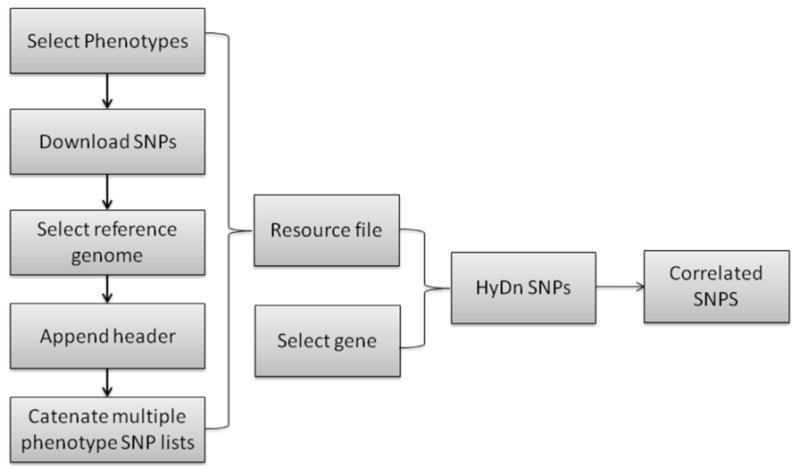
Flowchart of the HyDn-SNPs method. Upon development of a hypothesis, researchers select GWAS studies with relevant phenotypes, and obtain locations of the genes of interest. Following application of the algorithm, SNPs can be separated by intronic or exonic. Further analysis can be performed by in vitro validation or computational studies.
The database of genotypes and phenotypes (dbGaP, http://www.ncbi.nlm.nih.gov/gap) was developed to “archive and distribute the results of studies that have investigated the interaction of genotype and phenotype”. Data is distributed in the form of either raw genotyping data tagged with individual specific data such as gender, race and onset of phenotype; or as catenated lists of SNPs. This repository provides an ideal source of GWAS data useful to researchers with a targeted interest. Users are able to freely download the sets of SNPs, in a standard format for use with our software. Any phenotype of interest that is represented in this database would be a possible point of study for a HyDn-SNP-S study.
In this contribution, we present the development of HyDn-SNP-S and its application to search for cancer related SNPs on all human DNA polymerases. These enzymes are involved in all processes related to DNA replication, repair and recombination. The efficiency and fidelity of these processes are critical since errors can lead to carcinogenesis. Numerous studies indicate that mutations in DNA polymerases affect characteristics ranging from fidelity, to nucleotide incorporation rate, to cell proliferation [15–26]. However, a direct link has not been established between these mutations and cancer onset.
Our results uncovered a large number of cancer related SNPs on DNA polymerases. Statistical analysis on selected studies reveals for the first time the possibility that DNA POLL could be a major contributor to cancer risk. Molecular dynamics simulations were performed on wild-type and a SNP mutant on POLL to further investigate the functional impact of the mutation.
2. Methods
In this section we describe the algorithm to search for disease related SNPs based on a given hypothesis and its implementation in an easy to use software package. Subsequently we describe the statistical methods to determine the association of the SNPs with the phenotype. This is followed by a description of the graph analysis of the resulting data from HyDn-SNP-S for the present studies. Finally, the details of molecular dynamics (MD) simulations on DNA polymerase lambda structures are described.
2.1. HyDn-SNP-S
SNP collections for each phenotype are obtained from studies deposited on the database of genotypes and phenotypes (dbGaP). A header is appended to each data set declaring the phenotype associated with each individual study. Mutations listed relative to the HuRef and Celera genomes are removed, as we are working within the frame of reference of the GrCh37 human genome reference build. All files containing the SNP collections are subsequently catenated into a single searchable resource file. This single file allows HyDn-SNP-S to search all relevant SNPs for every phenotype that was included in the resource file in one single run. Following the generation of this resource file, the program HyDn SNPs was used to search for mutations within the gene region of interest. Users enter the chromosome, and gene range for searching, and point the program to the resource file. Sample resource files are available with the HyDn SNPS download. Further information and instructions are available in the documentation for this program. Any SNPs found that match the chromosome and gene location range are deposited into a results file. This file lists all the SNP associated information, such as ss and rs number, allele, chromosome, chromosomal location, contig number, and contig location, and type of chip used in the original genotyping experiment. These can then be categorized by location; intronic, exonic, or at a splice site. For our purposes, exonic SNPs were then compared to reference SNPs to ascertain the extent of prior investigation, as well as relative allele frequency in the natural population. The consequence of any given SNP was determined either by use of the reference SNP database, or in the case of previously unreported SNPs, translated by use of a DNA codon table in conjunction with the gene sequence and protein sequence. HyDnSNPs is available upon request from the authors.
2.2. Statistical analysis
We utilized four publically available case/control genome wide association studies (GWAS) from dbGAP (access request #1961) across multiple cancer types (including breast, melanoma, lung and prostate cancers) [27–32] to determine if SNPs or haplotypes constructed from SNPs in our genes of interest (genes coding for all human polymerases) are associated with any of these disease phenotypes. Additionally, we determined if any synergistic results across multiple databases exist that may imply a common cancer genesis. Multiple genetic modes of inheritance were examined: additive, dominant, recessive and genotypic in a covariate-adjusted logistic regression analysis associating each SNP with the disease phenotype. The maximum likelihood estimate of the posterior probabilities of haplotypes for each observation was produced using the EM algorithm. Score statistics for the association of the haplotypes with the cancer phenotype were constructed using these posterior probabilities. We use the R package “haplo.stats” to implement these haplotype functions [33]. Logistic regression is also used to estimate the association of a haplotype with the disease phenotype, given the genetic context. As we focus on the SNPs in only a few genes, we avoid issues with multiple testing, which are burdensome when trying to evaluate the association between genetic markers and a disease phenotype when measuring thousands or millions of genetic variants.
2.3. Graph analysis
For ease of visual analysis, the data resulting from the HyDn-SNP-S search has been transformed into edge-node format to allow visual interpretation of the networks of phenotypes and polymerases involved in tumorigenesis. Frequently more than one polymerase was found to have single point mutations within a cohort of cancer patients; network analysis allows for easy visual interpretation. Edge-node tables were csv formatted for use in Gephi [34], visualization was performed with a Fruchterman–Reingold [35] algorithm using an area of 15,000 and a gravity of 7.0. Nodes and edges were weighted by degree; for these analyses, weight was the number of mutations occurring between each phenotype and polymerase.
2.4. MD simulations
MD was performed on wild-type and the R438W mutant of DNA POLL in the binary and ternary conformations (PDBID: 1RZT, 2PFQ) using NAMD. The simulations were performed using a parallel build of NAMD [36] employing the CHARMM [37] force field on the XSEDE Teragrid. The structures were solvated, and appropriate counterions were added to reach 0.5 mM NaCl. A timestep of one femtosecond was used, a Langevin thermostat was used to maintain temperature at 300 K, and a Nose–Hoover Langevin combination method was used to control pressure. The systems were solvated with TIP3P water, neutralized with counter ions and subjected to 1000 steps of conjugate gradient minimization and temperature ramped to 300 K. After equilibration, the systems were run for at least 14 ns of production time. Frames from the trajectories were written every 1 ps. The solvation boxes included a 15 Å pad on each face of the box. Long range electrostatics were calculated using particle mesh Ewald [38], and van der Waals were calculated with a nonbonded cutoff of 8 Å and a switching function between 7 and 8 Å
Correlation analysis by residue was carried out for each system using the ptraj module of Amber11, across the entire simulation. An all residue correlation was performed and difference plots were calculated using Mathematica [39]. Correlation between the mutated residue and the residues in Loop 1 were also calculated and plotted. Generalized masked Delaunay analysis was carried out using the TimeScapes software from the D.E. Shaw group [40]. Trajectories were prepared using VMD [41], and all solvent and nucleic acids were excluded from analysis. A sliding window of 5% of the total number of frames was used, and total events per frame were calculated and plotted against frame number.
3. Results and discussion
The HyDn-SNP-S method returns results from whole genome genotyping studies rapidly, far faster than traditional bioinformatic methods. Pre-screening with HyDn-SNP-S dramatically decreases the time required to perform statistical analysis on GWAS data, by excluding all mutations not relevant to a researcher’s hypothesis. As proof of concept four genotyping studies have been statistically analyzed following application of the HyDn-SNP-S method. Additionally, one mutation, both determined to be statistically significant and of structural interest was subjected to molecular dynamics studies and consequent analysis.
Upon searching four cancer phenotype studies (melanoma, breast, lung and prostate cancer) [11, 29–32, 45] for mutations in all polymerase genes, a total of 708 mutations were found. Of these mutations 491 were intronic, and 217 were exonic. Additionally, four of the exonic mutations were found to be at splice sites. As per the workflow described above, all four searches were carried out simultaneously, and results were available within a few minutes. Following application of the HyDn-SNP-S analysis, the four studies were subjected to traditional biostatistical analysis. The focused nature of the search allows for relaxation of the more stringent mathematical methods, and facilitates more thorough analysis of the resulting mutations. Haplotype analysis on whole genome genotyping data is frequently not performed as the combinatorial nature of these studies across all mutations would be prohibitively computationally expensive. As the dataset used for analysis following the HyDn SNP-S method has significantly reduced complexity, these targeted studies can detect mutations of moderate significance that would be overlooked in traditional bioinformatic analyses and perform these searches more rapidly than is typically possible.
Logistical regression and haplotype analysis was performed on these studies to determine statistical significance. The prostate cancer case/control database examined yielded 69 SNPs in the genes of interest. Eleven of them were statistically significantly associated with prostate cancer status for at least one genetic model. The melanoma cancer case/control database examined yielded 215 SNPs in the genes of interest. Twenty-six of them were significantly associated with melanoma case/control status for at least one genetic model after controlling for age and gender. The breast cancer case/control database examined yielded 100 SNPs in the genes of interest. Twenty-two of them were statistically significantly associated with prostate cancer status for at least one genetic model. The lung cancer case/control database examined yielded 51 SNPs in the genes of interest. Twenty of them were statistically significantly associated with prostate cancer status for at least one genetic model. Table S1 reports all of the significant SNPs, their p-value and corresponding POL gene.
Analysis was performed to determine the association between the derived haplotypes from each gene and disease status. No haplotypes were predictive of disease status for the lung cancer study nor the melanoma study using any of the three genetic models. However, the haplotypes constructed from SNPs on POLL were borderline significant for the breast cancer study using a recessive (p-value = 0.048) or additive (p-value = 0.091) model formulation. This haplotype is constructed from two SNPs: rs3730477 (C > T; R438W) and rs3730463 (A > C: T221P). The odds ratios from individual significant and borderline significant contrasts within each model type are reported below. In the case of the additive model, for each additional C–A haplotype observed, the odds of breast cancer are multiplied by 1.15 (p-value = 0.029). Similarly, for each additional C–C haplotype observed, the odds of breast cancer are multiplied by 0.812 (p-value = 0.062), i.e., a protective genotype. For the recessive model, having 0 or 1 copy of the C–A haplotype results in the odds of breast cancer being multiplied by 0.829 relative to having 2 copies of the C–A haplotype (p = 0.026). Having 0 or 1 copy of the C–C haplotype results in the odds of breast cancer being multiplied by 3.01 relative to having two copies of the C–C haplotype (p = 0.099). The haplotypes constructed from SNPs on the PolG genes were significant for prostate cancer. This haplotype is constructed from three SNPs: rs3087374, rs2351000 and rs2247233. The odds ratios from individual significant contrasts within each model type are reported below. For the recessive model, having 0 or 1 copy of the G–T–G haplotype results in the odds of prostate cancer being multiplied by 1.33 relative to having 2 copies of the G–T–G haplotype (p = 0.005). Having 2 copies of the G–C–A haplotype results in 9.64 of the odds of prostate cancer as compared to having 0 or 1 copy of the G–C–A haplotype (p = 0.008).
A literature search indicates that only one of these statistically evaluated mutations has been explored in vitro [42]. Experimental analysis of the mutations we report here is outside the scope of this work. However, the mutations arising from these SNPs present interesting targets for further experimental studies.
The data resulting from a HyDn-SNP-S search can not only be discussed at the molecular level, and in the context of predictive power, but due to the nature of these studies, on a much broader basis. Relating many phenotypes to many polymerases generates a network of data best represented by an edge-node interactive diagram. Using the number of SNPs as a weighting property, it is possible to broadly examine the complete network of phenotype–polymerase interactions. Fig. 2 shows a flattened version of this data, limited to statistically explored phenotype data, which is available in interactive form online at http://www.chem.wayne.edu/cisnerosgroup/gexf-js2/index2.html. Both polymerases and phenotypic studies are represented as nodes, sized according to SNP density. By clicking on a node, all connections to that node will be listed. Clicking on any of those connections will return all connections to that selected node.
Fig. 2.

Edge-node network of the HyDn-SNPs results. Phenotypes and polymerases are shown as nodes, edges are weighted by total number of SNPs connecting each phenotype to each polymerase. This is also available as an interactive map at http://www.chem.wayne.edu/cisnerosgroup/gexf-js2/index2.html.
The interactive map allows investigation of the network associations of various phenotypes and polymerases and the complete list of connections is available from the dropdown menu at the top of the page. A complete list of all connections is available upon request from andres@chem.wayne.edu. This file also includes the translated mutations. Many diseases are not caused by a single point mutation, but rather by a collection of factors. As the formatting for the results of HyDn-SNP-S is well suited to network analysis, and additional data can be garnered as desired from the genotyping studies, this approach may have critical importance in searching for combinations of factors that may be predictive for disease. Due to the targeted nature of the search, there is a significant reduction in the analytic space and thus, more thorough analysis can be performed. The haplotype described above is one example; individually the two mutations would have been overlooked by traditional analysis, but in combination they are strongly predictive.
To further validate that hypothesis driven analysis of whole genome genotyping data is valuable to researchers, we sought to study a mutation with statistical significance that would have been overlooked by traditional methods. Of the two mutations that comprise the haplotype linking POLL to breast cancer, only the mutation R438W is in the polymerase domain. This position is not close to the active site, but it is within 14 Å of Loop 1, which has been shown to be critical for fidelity [43]. The R438W SNP mutation has been previously shown to contribute to decreased fidelity in vitro, increased mutation frequency, and generation of chromosomal abnormalities [44]. An eightfold increase in inaccurate substitutions was observed in base substitution assays and karyotypic analysis of several cell lines carrying this mutation also reported a high level of spontaneous or IR-induced chromosomal aberrations. With ample evidence to suggest a molecular basis for these results, we selected DNA polymerase lambda R438W for further study.
Four MD simulations were performed, using crystal structures 1RZT and 2PFQ. These structures were selected as they represent the binary and ternary complexes of Pol lambda, respectively. The binary complex includes Pol lambda and the template DNA, the ternary complex includes Pol lambda with both the template and incoming nucleotide. The change in Loop 1 conformation between the two structures is shown in Fig. 3. Panel A overlays the binary and ternary complexes, Loop 1 is shown in purple to illustrate the alteration in conformation. Panel B shows a closer view of the loop conformations, indicating both binary and ternary conformations. Panels B and C illustrate the relative proximity to the R438W mutation. As the structure transits between the two loop conformations, the mutation ranges from roughly 12.6 Å to 14.3 Å away. A video illustrating the position of the mutation and the visual interpolation of the binary and ternary conformations is available as Movie S1. Mutations in this loop have been shown to have no effect on catalytic rate while simultaneously increasing the number of misincorporations, thus Loop 1 is critical for polymerase fidelity [43]. Following a 14 ns simulation, correlation analysis was carried out to determine whether the residues in Loop 1 were affected by the mutation.
Fig. 3.

(A) Overlay of Polλ in the binary and ternary conformations. DNA is shown in light blue, and the Loop 1 is shown in purple. (B) Differences in Loop 1 orientation between the two conformations. Distance between position 438 and Loop 1 following an interpolation between the two structures at its furthest (Panel D) and closest (Panel C) approaches. (For interpretation of the references to color in this artwork, the reader is referred to the web version of the article.)
As shown in Fig. 4, the binary complex shows little change in correlation between the wild-type and mutant structures. Conversely, the ternary complex shows high correlation and anti-correlation in two regions. The highest points of correlation are between residues at positions 438 and 569, as well as between 438 and 420. Residues showing the greatest change in correlation in the ternary complex were mapped to the structure and colored orange as shown in Fig. 4C. It is notable that a majority of these residues are on Loop 1. To further understand the impact of the SNP mutation on Loop 1, the correlation data between position 438 and all residues in Loop 1 was extracted and plotted. Fig. 4D shows that although there is higher correlation in the ternary complex between the wild-type and mutant, both complexes show altered correlation between the wild-type and mutant. The sum of these analyses suggests that the introduction of the R438W mutation alters the overall correlation pattern in the ternary complex, but more importantly, directly affects the motions of Loop 1 in the both complexes. As Loop 1 regulates fidelity, and transit between the two conformations shown in Fig. 3 is required for catalysis, the SNP leading to the R438W mutation likely has direct effects on polymerase activity in vitro and in vivo.
Fig. 4.

Correlation difference plots for the binary (A) and ternary (B) conformations relative to the wild type. Increases in correlation are shown in orange, while increases in anti-correlated motions are shown in blue. In both cases, alterations in the correlation plots are visible, more notably in the ternary complex. The highest values from the ternary complex correlation plots were mapped back to the residues affected, and are colored orange in Panel C. Notably many of these residues are on Loop 1. Panel D shows the individual correlation values for each of the residues in Loop 1. While the binary complex shows moderate alteration on several, the ternary complex shows considerable differences for several residues, particularly between residues 469 and 472. (For interpretation of the references to color in this artwork, the reader is referred to the web version of the article.)
In addition to the correlation analysis, generalized masked Delaunay (GMD) analysis was performed to determine the impact, if any, on the overall activity of the simulations. The results are shown in Fig. 5, events are plotted on the Y-axis, and frame number is plotted on the X-axis. GMD defines events as persistent motions across the masked Delaunay reduced representation of the protein structure. Panels A and B show the wild type activity for both the binary and ternary complexes, with average activity levels of roughly 0.2 events per frame. These patterns are typical of stable simulations, where no major rearrangement is occurring. The alteration in the correlation plots combined with the stability of the GMD indicates that the mutation induces only local alterations in activity.
Fig. 5.

GMD plots for the binary and ternary complex simulations in both wild type and mutant form. No drastic differences are apparent between the four simulations indicating that all four are showing the same general level of physical activity. This indicates that the overall motions of the polymerase are not perturbed. In light of the data presented in Fig. 4, this indicates that the significant alterations in conformational space are restricted to the Loop 1 region.
The sum of the correlation and GMD analysis indicates that the R438W mutation appears to modify only the movement of Loop 1, while the overall dynamics are not significantly altered. This provides context for the experimental work by Terrados et al. [44]. Their experiments indicated that the R438W mutation increases the error rate of Pol lambda, but does not alter the overall rate of polymerization. Our results indicate that the R438W mutation alters only the behavior of Loop 1, while leaving the overall conformational motions of Pol lambda unperturbed. This would agree with the behavior observed experimentally. The R438W mutation alters the behavior of Loop 1, thus decreasing fidelity, while the overall behavior of the polymerase is unaffected, allowing it to maintain a normal rate of polymerization.
4. Conclusions
We have developed a powerful method that allows researchers to interact with whole genome genotyping data in a focused, hypothesis driven way. By allowing researchers to find data on their own systems of interest, we will expedite the study of any mutations that may logically be connected to a phenotype. Also, the focused nature of these searches will allow more thorough statistical analysis, and appropriate recognition to combinations of factors that would be difficult to fully assess in an extremely broad GWAS analysis. By applying this methodology to our system of interest we were able make the first direct statistical link between DNA polymerases and cancer, define two haplotypes with strong predictive power, and trace a cancer-associated mutation to a structural effect in the translated protein and investigate its functional impact by computational simulations.
Supplementary Material
Acknowledgments
Computational time from TeraGrid and Wayne State C&IT, as well as access to genome data from dbGaP under access request #1961 is gratefully acknowledged. This work was funded by Wayne State University. Research support to collect data and develop an application to support this project was provided by 3P50CA093459, 5P50CA097007, 5R01ES011740, and 5R01CA133996.
Appendix A. Supplementary data
Supplementary material related to this article can be found, in the online version, at http://dx.doi.org/10.1016/j.dnarep.2013.06.001.
Footnotes
Conflict of interest statement
The authors declare that there are no conflicts of interest.
References
- 1.The Human Genome Project: 10 years later. Lancet. 2010;375:2194. doi: 10.1016/S0140-6736(10)61012-4. [DOI] [PubMed] [Google Scholar]
- 2.Caskey CT. Presymptomatic diagnosis: a first step toward genetic health care. Science. 1993;262:48–49. doi: 10.1126/science.8211129. [DOI] [PubMed] [Google Scholar]
- 3.Caskey CT. Using genetic diagnosis to determine individual therapeutic utility. Annu Rev Med. 2010;61:1–15. doi: 10.1146/annurev-med-011209-132719. [DOI] [PubMed] [Google Scholar]
- 4.Peakall D, Shugart L. The Human Genome Project (HGP) Ecotoxicology. 2002;11:7. [PubMed] [Google Scholar]
- 5.Rossiter BJ, Caskey CT. Presymptomatic testing for genetic diseases of later life: pharmacoepidemiological considerations. Drugs Aging. 1995;7:117–130. doi: 10.2165/00002512-199507020-00006. [DOI] [PubMed] [Google Scholar]
- 6.Margaritte P, Bonaiti-Pellie C, King MC, Clerget-Darpoux F. Linkage of familial breast cancer to chromosome 17q21 may not be restricted to early-onset disease. Am J Hum Genet. 1992;50:1231–1234. [PMC free article] [PubMed] [Google Scholar]
- 7.King MC, Marks JH, Mandell JB. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302:643–646. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 8.King MC. Localization of the early-onset breast cancer gene. Hosp Pract (Off Ed) 1991;26:121–126. doi: 10.1080/21548331.1991.11705309. [DOI] [PubMed] [Google Scholar]
- 9.Austin MA, King MC, Vranizan KM, Krauss RM. Atherogenic lipoprotein phenotype. A proposed genetic marker for coronary heart disease risk. Circulation. 1990;82:495–506. doi: 10.1161/01.cir.82.2.495. [DOI] [PubMed] [Google Scholar]
- 10.Gudmundsson J, Sulem P, Gudbjartsson DF, Blondal T, Gylfason A, Agnarsson BA, Benediktsdottir KR, Magnusdottir DN, Orlygsdottir G, Jakobsdottir M, Stacey SN, Sigurdsson A, Wahlfors T, Tammela T, Breyer JP, McReynolds KM, Bradley KM, Saez B, Godino J, Navarrete S, Fuertes F, Murillo L, Polo E, Aben KK, van Oort IM, Suarez BK, Helfand BT, Kan D, Zanon C, Frigge ML, Kristjansson K, Gulcher JR, Einarsson GV, Jonsson E, Catalona WJ, Mayordomo JI, Kiemeney LA, Smith JR, Schleutker J, Barkardottir RB, Kong A, Thorsteinsdottir U, Rafnar T, Stefansson K. Genome-wide association and replication studies identify four variants associated with prostate cancer susceptibility. Nat Genet. 2009;41:1122–1126. doi: 10.1038/ng.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Hoover RN, Thomas G, Chanock SJ. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–874. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hutter CM, Young AM, Ochs-Balcom HM, Carty CL, Wang T, Chen CTL, Rohan TE, Kooperberg C, Peters U. Replication of breast cancer GWAS susceptibility loci in the Women’s Health Initiative African American SHARe study. Cancer Epidemiol Biomark Prev. 2011;20:1950–1959. doi: 10.1158/1055-9965.EPI-11-0524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manolio TA. Genomewide association studies and assessment of the risk of disease. N Engl J Med. 2010;363:166–176. doi: 10.1056/NEJMra0905980. [DOI] [PubMed] [Google Scholar]
- 14.Manjarrez-Orduño N, Marasco E, Chung SA, Katz MS, Kiridly JF, Simpfendorfer KR, Freudenberg J, Ballard DH, Nashi E, Hopkins TJ. CSK regulatory polymorphism is associated with systemic lupus erythematosus and influences B-cell signaling and activation. Nat Genet. 2012;44:1227–1230. doi: 10.1038/ng.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Copeland WC, Lam NK, Wang TS. Fidelity studies of the human DNA polymerase alpha. The most conserved region among alpha-like DNA polymerases is responsible for metal-induced infidelity in DNA synthesis. J Biol Chem. 1993;268:11041–11049. [PubMed] [Google Scholar]
- 16.Copeland WC, Wang TS. Mutational analysis of the human DNA polymerase alpha. The most conserved region in alpha-like DNA polymerases is involved in metal-specific catalysis. J Biol Chem. 1993;268:11028–11040. [PubMed] [Google Scholar]
- 17.Daee DL, Mertz TM, Shcherbakova PV. A cancer-associated DNA polymerase β variant modeled in yeast causes a catastrophic increase in genomic instability. Proc Natl Acad Sci. 2010;107:157–162. doi: 10.1073/pnas.0907526106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lang T, Maitra M, Starcevic D, Li SX, Sweasy JB. A DNA polymerase β mutant from colon cancer cells induces mutations. Proc Natl Acad Sci USA. 2004;101:6074–6079. doi: 10.1073/pnas.0308571101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Longley MJ, Clark S, Yu Wai Man C, Hudson G, Durham SE, Taylor RW, Nightingale S, Turnbull DM, Copeland WC, Chinnery PF. Mutant POLG2 disrupts DNA polymerase gamma subunits and causes progressive external ophthalmoplegia. Am J Hum Genet. 2006;78:1026–1034. doi: 10.1086/504303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Longley MJ, Ropp PA, Lim SE, Copeland WC. Characterization of the native and recombinant catalytic subunit of human DNA polymerase gamma: identification of residues critical for exonuclease activity and dideoxynucleotide sensitivity. Biochemistry. 1998;37:10529–10539. doi: 10.1021/bi980772w. [DOI] [PubMed] [Google Scholar]
- 21.Matsuda T, Bebenek K, Masutani C, Rogozin IB, Hanaoka F, Kunkel TA. Error rate and specificity of human and murine DNA polymerase eta. J Mol Biol. 2001;312:335–346. doi: 10.1006/jmbi.2001.4937. [DOI] [PubMed] [Google Scholar]
- 22.Ohashi E, Bebenek K, Matsuda T, Feaver WJ, Gerlach VL, Friedberg EC, Ohmori H, Kunkel TA. Fidelity and processivity of DNA synthesis by DNA polymerase kappa, the product of the human DINB1 gene. J Biol Chem. 2000;275:39678–39684. doi: 10.1074/jbc.M005309200. [DOI] [PubMed] [Google Scholar]
- 23.Ohashi E, Ogi T, Kusumoto R, Iwai S, Masutani C, Hanaoka F, Ohmori H. Error-prone bypass of certain DNA lesions by the human DNA polymerase kappa. Genes Dev. 2000;14:1589–1594. [PMC free article] [PubMed] [Google Scholar]
- 24.Rogozin IB, Pavlov YI, Bebenek K, Matsuda T, Kunkel TA. Somatic mutation hotspots correlate with DNA polymerase eta error spectrum. Nat Immunol. 2001;2:530–536. doi: 10.1038/88732. [DOI] [PubMed] [Google Scholar]
- 25.Sweasy JB, Lang T, Starcevic D, Sun KW, Lai CC, DiMaio D, Dalal S. Expression of DNA polymerase β cancer-associated variants in mouse cells results in cellular transformation. Proc Natl Acad Sci USA. 2005;102:14350–14355. doi: 10.1073/pnas.0505166102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong SW, Wahl AF, Yuan PM, Arai N, Pearson BE, Arai K, Korn D, Hunkapiller MW, Wang TS. Human DNA polymerase alpha gene expression is cell proliferation dependent and its primary structure is similar to both prokaryotic and eukaryotic replicative DNA polymerases. EMBO J. 1988;7:37–47. doi: 10.1002/j.1460-2075.1988.tb02781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, Dong Q, Zhang Q, Gu X, Vijayakrishnan J, Sullivan K, Matakidou A, Wang Y, Mills G, Doheny K, Tsai YY, Chen WV, Shete S, Spitz MR, Houlston RS. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–622. doi: 10.1038/ng.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bishop DT, Demenais F, Iles MM, Harland M, Taylor JC, Corda E, Randerson-Moor J, Aitken JF, Avril M-F, Azizi E, Bakker B, Bianchi-Scarra G, Bressac-de Paillerets B, Calista D, Cannon-Albright LA, Chin-A-Woeng T, Debniak T, Galore-Haskel G, Ghiorzo P, Gut I, Hansson J, Hocevar M, Hoiom V, Hopper JL, Ingvar C, Kanetsky PA, Kefford RF, Landi MT, Lang J, Lubinski J, Mackie R, Malvehy J, Mann GJ, Martin NG, Montgomery GW, van Nieuwpoort FA, Novakovic S, Olsson H, Puig S, Weiss M, van Workum W, Zelenika D, Brown KM, Goldstein AM, Gillanders EM, Boland A, Galan P, Elder DE, Gruis NA, Hayward NK, Lathrop GM, Barrett JH, Newton Bishop JA. Genome-wide association study identifies three loci associated with melanoma risk. Nat Genet. 2009;41:920–925. doi: 10.1038/ng.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang X, Pankratz VS, Fredericksen Z, Tarrell R, Karaus M, McGuffog L, Pharaoh PDP, Ponder BAJ, Dunning AM, Peock S, Cook M, Oliver C, Frost D, Sinilnikova OM, Stoppa-Lyonnet D, Mazoyer S, Houdayer C, Hogervorst, Hooning FBL, Ligtenberg MJ, Spurdle A, Chenevix-Trench G, Schmutzler RK, Wappenschmidt B, Engel C, Meindl A, Domchek SM, Nathanson KL, Rebbeck TR, Singer CF, Gschwantler-Kaulich D, Dressler C, Fink A, Szabo CI, Zikan M, Foretova L, Claes K, Thomas G, Hoover RN, Hunter DJ, Chanock SJ, Easton DF, Antoniou AC, Couch FJ EMBRACE, GEMO, HEBON, kConFab. Common variants associated with breast cancer in genome-wide association studies are modifiers of breast cancer risk in BRCA1 and BRCA2 mutation carriers. Hum Mol Genet. 2010;19:2886–2897. doi: 10.1093/hmg/ddq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waters KM, Le Marchand L, Kolonel LN, Monroe KR, Stram DO, Henderson BE, Haiman CA. Generalizability of associations from prostate cancer genome-wide association studies in multiple populations. Cancer Epidemiol Biomark Prev. 2009;18:1285–1289. doi: 10.1158/1055-9965.EPI-08-1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, Lin WM, Province MA, Kraja A, Johnson LA, Shah K, Sato M, Thomas RK, Barletta JA, Borecki IB, Broderick S, Chang AC, Chiang DY, Chirieac LR, Cho J, Fujii Y, Gazdar AF, Giordano T, Greulich H, Hanna M, Johnson BE, Kris MG, Lash A, Lin L, Lindeman N, Mardis ER, McPherson JD, Minna JD, Morgan MB, Nadel M, Orringer MB, Osborne JR, Ozenberger B, Ramos AH, Robinson J, Roth JA, Rusch V, Sasaki H, Shepherd F, Sougnez C, Spitz MR, Tsao MS, Twomey D, Verhaak RG, Weinstock GM, Wheeler DA, Winckler W, Yoshizawa A, Yu S, Zakowski MF, Zhang Q, Beer DG, Wistuba II, Watson MA, Garraway LA, Ladanyi M, Travis WD, Pao W, Rubin MA, Gabriel SB, Gibbs RA, Varmus HE, Wilson RK, Lander ES, Meyerson M. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–898. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yeager M, Orr N, Hayes RB, Jacobs KB, Kraft P, Wacholder S, Minichiello MJ, Fearnhead P, Yu K, Chatterjee N, Wang Z, Welch R, Staats BJ, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cancel-Tassin G, Cussenot O, Valeri A, Andriole GL, Gelmann EP, Tucker M, Gerhard DS, Fraumeni JF, Hoover R, Hunter DJ, Chanock SJ, Thomas G. Genome-wide association study of prostate cancer identifies a second risk locus at 8q24. Nat Genet. 2007;39:645–649. doi: 10.1038/ng2022. [DOI] [PubMed] [Google Scholar]
- 33.Sinnwell JP, Schaid DJ, Yu Z. haplo.stats: Statistical Analysis of Haplotypes with Traits and Covariates when Linkage Phase is Ambiguous. R Package Version 1. 2005 [Google Scholar]
- 34.Bastian M, Heymann S, Jacomy Gephi M. An Open Source Software for Exploring and Manipulating Networks. 2009 [Google Scholar]
- 35.Fruchterman TMJ, Reingold EM. Graph drawing by force-directed placement. Software: Pract Exp. 1991;21:1129–1164. [Google Scholar]
- 36.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 38.Wang H, Dommert F, Holm C. Optimizing working parameters of the smooth particle mesh Ewald algorithm in terms of accuracy and efficiency. J Chem Phys. 2010;133:034117. doi: 10.1063/1.3446812. [DOI] [PubMed] [Google Scholar]
- 39.Wolfram S, Mathematica A. System for Doing Mathematics by Computer. Addison Wesley; 1991. pp. 201–19334. [Google Scholar]
- 40.Willy Wriggers KAS, Shan Y, Piana S, Maragakis P, Kresten Lindorff-Larsen PJM, Gullingsrud J, Charles Rendleman A, Michael Eastwood RODP, David Shaw E. Automated event detection and activity monitoring in long time-scale molecular dynamics. J Chem Theory Comput. 2009;5:2595–2605. doi: 10.1021/ct900229u. [DOI] [PubMed] [Google Scholar]
- 41.Humphrey W, Dalke A, Schulten KVMD. Visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 42.Woodbridge P, Liang C, Davis RL, Vandebona H, Sue CM. POLG mutations in Australian patients with mitochondrial disease. Int Med J. 2013;43(2):150–156. doi: 10.1111/j.1445-5994.2012.02847.x. http://dx.doi.org/10.1111/j.1445-5994.2012.02847.x. [DOI] [PubMed] [Google Scholar]
- 43.Bebenek K, Garcia-Diaz M, Zhou RZ, Povirk LF, Kunkel TA. Loop 1 modulates the fidelity of DNA polymerase lambda. Nucleic Acids Res. 2010;38:5419–5431. doi: 10.1093/nar/gkq261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Terrados G, Capp JP, Canitrot Y, Garcia-Diaz M, Bebenek K, Kirchhoff T, Villanueva A, Boudsocq FO, Bergoglio VR, Cazaux C, Kunkel TA, Hoffmann JSB, Blanco L. Characterization of a natural mutator variant of human DNA polymerase λ which promotes chromosomal instability by compromising NHEJ. PLoS ONE. 2009;4:e7290. doi: 10.1371/journal.pone.0007290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amos CI, Wang LE, Lee JE, Gershenwald JE, Chen WV, Fang S, Kosoy R, Zhang M, Qureshi AA, Vattathil S, Schacherer CW, Gardner JM, Wang Y, Bishop DT, Barrett JH GenoMEL Investigators; MacGregor S, Hayward NK, Martin NG, Duffy DL Q-Mega Investigators; Mann GJ, Cust A, Hopper J AMFS Investigators. Brown KM, Grimm EA, Xu Y, Han Y, Jing K, McHugh C, Laurie CC, Doheny KF, Pugh EW, Seldin MF, Han J, Wei Q. Genome-wide association study identifies novel loci predisposing to cutaneous melanoma. Hum Mol Genet. 2011 Dec 15;20(24):5012–23. doi: 10.1093/hmg/ddr415. doi: 10.1093/hmg/ddr415. Epub 2011 Sep 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.